

Abstract
Background:
Epidermal growth factor receptor signaling blockade increases CCL5 expression that regulate either the anti-tumor immune response or tumor progression. We investigated the potential role of CCL5/CCR5 axis in cetuximab-based treatment in metastatic colorectal cancer (mCRC) patients.
Patients and methods:
Genomic DNA was extracted from 491 samples of two different cohorts with KRAS wild-type mCRC from the FIRE-3 trial: an evaluation cohort of 244 patients receiving cetuximab plus FOLFIRI; and a control cohort of 247 patients receiving bevacizumab plus FOLFIRI. Single nucleotide polymorphisms (SNPs) of CCL5 and CCR5 genes were analyzed by PCR-based direct sequencing.
Results:
Patients in the evaluation cohort with any CCL5 rs2280789 G allele had shorter overall survival (OS) compared with those with the A/A variant (HR 1.56, P=0.024). Patients carrying any CCR5 rs1799988 T allele had a trend toward lower response rate than those with the C/C variant (68 vs. 81%, P=0.078). In the analysis based on primary tumor location (left-sided [L]: right-sided [R]), remarkable differences in outcomes were observed between patients with L-CCR5 SNPs C/C variant (L-C/C), L-any T, R-T/T and R-any C as follows: median OS, 38.5, 30.6, 27.1, 15.8 months, P<0.001; response rate, 91, 66, 92, 48%, P<0.001. Median OS for CCL5 SNPs including L-A/A, L-any G, R-A/A and R-any G groups were 38.3, 21.7, 21.9, 18.3 months, P<0.001. The findings were not significant in the control cohort.
Conclusion:
Genetic variants of CCL5 and CCR5 SNPs may predict outcomes in mCRC patients receiving cetuximab-based treatment depending on tumor location.
Keywords: CCL5, CCR5, cetuximab, metastatic colorectal cancer, primary tumor location
Introduction
Previously reported randomized trials have clearly proven the clinical impact of first-line chemotherapy plus either anti-epidermal growth factor receptor (EGFR) or anti-vascular endothelial growth factor (VEGF) antibodies for metastatic colorectal cancer (mCRC) [1,2]. According to the subgroup analyses regarding the tumor molecular subtype and primary tumor location, recent guidelines suggest that anti-EGFR antibodies cetuximab and panitumumab are preferred in RAS wild-type tumors located in the left side of the colon or rectum. Meanwhile, the anti-VEGF antibody bevacizumab is recommended for patients regardless of either RAS status or primary tumor location [3,4]. However, how to predict efficacy of the biologic agents has not been sufficiently clarified to date.
EGFR signaling blockade increases C-C motif chemokine ligand-5 (CCL5) expression, which attracts tumor-infiltrating leukocytes that regulate either the host-derived anti-tumor immunity or tumor progression [5–7]. A high degree of T cell infiltration in cancer tissue is known to be associated with a favorable prognosis in colorectal cancer. T helper type 1 (Th1) expresses C-C motif chemokine receptor-5 (CCR5) and C-X-C motif chemokine receptor-3 (CXCR3), whereas Th2 expresses CCR4; and CCR5- and CXCR3-expressing T cells are recruited in to the invasive margin as anti-tumor immune responses. CCL5 is expressed and localized within CD8+ T cells, while a CXCR3 ligand CXCL10/IP-10 is localized in tumor cells and macrophages within the invasive margin [8]. Stimulation of EGFR down-regulates CCL2, CCL5 and IP-10, while it increases CXCL8/IL-8 expression in normal cells. Conversely, EGFR signaling blockade produces opposite effects, with increased CCL2, CCL5, and IP-10, and reduced IL-8 expression. Inhibition of EGFR signaling might exert antitumor activity by favoring the recruitment of inflammatory cells and a more pronounced anti-tumor immune response, along with down-regulation of IL-8, which is an important growth factor for malignant epithelial cells [6,7]. Expression of IL-8/CXCR2 in the tumor microenvironment has been shown to play a critical role in progression and metastases with increased tumor angiogenesis in colon cancer [9]. In addition, the CCL5/CCR5 axis participates in VEGF-A production by inducing endothelial progenitor cell migration [10] (Fig. A. 1). Meanwhile, a novel immune escape in CRC via CCL5/CCR5 axis that enhances tumor’s ability to beat antitumor CD8+ T cells by forming infiltration of T-regular cells [11]. Nevertheless, it still remains unclear how EGFR signaling blockade regulate either the host-derived anti-tumor immunity or tumor progression via CCL5 activation in colorectal cancer patients.
Fig. A. 1.

Role of CCL5/CCR5 axis in EGFR signaling.
We therefore tested whether genetic polymorphisms in CCL5 and CCR5 genes could predict efficacy of cetuximab or bevacizumab plus FOLFIRI in first-line treatment in metastatic colorectal cancer patients from the FIRE-3 trial (Trial Registration: NCT00433927).
Materials and methods
Study design and patients
Two different cohorts with KRAS exon 2 wild-type mCRC from the randomized phase III FIRE-3 trial [1] were investigated in this study: an evaluation cohort of 244 patients receiving cetuximab plus FOLFIRI; and a control cohort of 247 patients receiving bevacizumab plus FOLFIRI. All patients fulfilled the eligible criteria; no history of prior treatment for mCRC, measurable or evaluable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.0, and signed informed consent. Cetuximab (400 mg/m2 followed by 250 mg/m2 thereafter) and bevacizumab (5 mg/kg) were administered weekly and every 2 weeks, respectively. FOLFIRI (irinotecan 180 mg/m2, 5-FU bolus 400 mg/m2, 5-FU infusion 2400 mg/m2, leucovorin 200 mg/m2) was administered every 2 weeks. Treatment was continued until any of the following occurred: disease progression, unmanageable toxicity, or patient refusal. The current study was approved by the Institutional Review Board of each participating site, and the molecular analyses were conducted at the University of Southern California/Norris Comprehensive Cancer Center in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. We were fully compliant with the Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK) guidelines.
Selection of candidate single-nucleotide polymorphisms (SNPs)
The two candidate SNPs of genes CCL5 and CCR5 were selected using any of the following criteria: i) SNP with biological significance according to a published literature review, ii) tagging SNPs selected by the HapMap genotype data with r2threshold = 0.8: http://snpinfo.niehs.nih.gov/snpinfo/snptag.php, or iii) minor allele frequency ≥10% in Caucasians (in the Ensembl Genome Browser: http://uswest.ensembl.org/index.html). Functional significance was predicted based on the functional single-nucleotide polymorphism (F-SNP) database: http://compbio.cs.queensu.ca/F-SNP/ (Table A. 1).
Table A. 1.
Candidate SNPs in CCL5/CCR5 pathway
| Genes rs number |
Allele location | Base exchange |
MAF† (CEU) |
Function of polymorphism | Forward / Reverse primer (5’−3’) |
|---|---|---|---|---|---|
|
CCL5 rs2280789 |
Intron Chromosome 17:35879999 |
A>G | 0.11 | Transcriptional regulation | F: ATCTCCCCAACATGAGTCCA R: CCATATGTCCTGTTGTCCTTGA |
|
CCR5 rs1799988 |
5’ prime UTR Chromosome 3:46370768 |
C>T | 0.47 | Transcriptional regulation | F: TGGGATGAGCAGAGAACAAA R: GGCGAAAAGAATCAGAGAACA |
MAF, Minor allele frequency; CEU, Caucasian; F. forward primer; R, reverse primer; Tag SNP, tagging SNP.
In Caucasians from the Ensembl Genome Browser: http://uswest.ensembl.org/index.html.
DNA extraction and genotyping
Genomic DNA was extracted from peripheral whole blood in patients of all cohorts using the QIAmp Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol (www.qiagen.com). The candidate SNPs were tested using PCR-based direct DNA sequence analysis by ABI 3100A Capillary Genetic Analyzer and Sequencing Scanner v1.0 (Applied Biosystems). Both forward and reverse primers used for amplification of extracted DNA are listed in Table A. 1. For quality control purposes, a randomly selected 10% of the samples was analyzed by direct DNA sequencing for each SNP, and the genotype concordance rate was of 99% or more. The investigators analyzing SNPs were blinded to the clinical data.
Statistical analysis
The primary endpoint of the current study was progression-free survival (PFS), and secondary endpoints were overall survival (OS) and objective response rate (ORR). PFS was defined as the interval between the date of randomization and the date of confirmed disease progression or death. OS was calculated from the date of randomization until the date of death from any cause. Data of patients without disease progression or death were censored at the date of last follow up. For patients who were lost to follow-up, data were censored at the date when the patient was last confirmed to be alive. The ORR was calculated from the number of patients who achieved complete response (CR) and partial response (PR) to treatment, according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0. Chi-square tests were used to examine the difference in baseline patient characteristics between two cohorts. Fisher’s exact test was applied to examine the associations between SNPs and response. Association between SNPs and PFS and OS were estimated by the Kaplan-Meier method and were compared using the log-rank test, with predictive or prognostic clinical factors and candidate SNPs that was identified by univariate analysis using codominant, dominant, or recessive genetic model if appropriate. Multivariable analysis using the Cox proportional hazards model was then conducted to re-evaluate factors influencing PFS and OS. The baseline demographic and clinical characteristics that remained statistically significantly associated with PFS and OS in multivariable analyses were included in the final models. We further performed subgroup analyses by primary tumor site, then built the models combining tumor site and SNPs according to the results from subgroup analyses. With 244 patients (206 PFS events) in the evaluation cohort, we would have 80% power to detect a minimum hazard ratio (HR) of 1.49 to 1.65 for a SNP with minor allele frequency of 0.1 to 0.5 on PFS using a two-sided 0.05 level log-rank test. The power would be greater than 80% in the control cohort with 247 patients (203 PFS events) when applying the same test and assuming the same allele frequencies. Analysis in the RAS wild-type patients was also performed. All analyses were carried out with SAS 9.4 (SAS Institute, Cary, NC, USA). All tests were 2-sided at a significance level of 0.050.
Results
Baseline patients and tumor characteristics
The median follow-up time, median PFS and OS were 34.1 months, 9.8 months and 29.7 months in the evaluation cohort; and 39.4 months, 10.2 months and 24.8 months in the control cohort, respectively. The baseline characteristics of the evaluation and control are summarized in Table A. 2. The associations between baseline characteristics and clinical outcome are summarized in Table A. 3 and 4 for the evaluation and control cohorts, respectively. In detail, right-sided location, RAS and BRAF mutant were significantly associated with shorter PFS and OS in the evaluation cohort. In the control cohort, higher ECOG, right-sided location and BRAF mutant were significantly correlated with shorter PFS and OS. Frequencies of genetic variants of the SNPs satisfied the Hardy–Weinberg equilibrium (P>0.01) using the exact test in the Haploview software version 4.2.
Table A. 2.
Baseline patients and tumor characteristics
| Cohort | Evaluation cohort (N=244) FIRE-3, FOLFIRI+CET arm |
Control cohort (N=247) FIRE-3, FOLFIRI+BEV arm |
P value * | ||
|---|---|---|---|---|---|
| N | % | N | % | ||
| Sex | |||||
| Male | 169 | 69 | 162 | 66 | 0.38 |
| Female | 75 | 31 | 85 | 34 | |
| Age (year) | |||||
| Median (range) | 64 (38–79) | 65 (31–76) | |||
| ≤ 65 | 130 | 53 | 130 | 53 | 0.89 |
| > 65 | 114 | 47 | 117 | 47 | |
| ECOG Performance status | |||||
| ECOG 0 | 124 | 51 | 133 | 54 | 0.50 |
| ECOG 1–2 | 120 | 49 | 114 | 46 | |
| Primary tumor site | |||||
| Right | 45 | 18 | 62 | 25 | 0.070 |
| Left | 194 | 80 | 179 | 72 | |
| Unknown | 5 | 2 | 6 | 2 | |
| Liver metastasis | |||||
| Yes | 84 | 34 | 81 | 33 | 0.70 |
| No | 160 | 66 | 166 | 67 | |
| Lung metastasis | |||||
| Yes | 94 | 39 | 94 | 38 | 0.92 |
| No | 150 | 61 | 153 | 62 | |
| Number of metastases | |||||
| 0–1 | 105 | 43 | 107 | 43 | 0.95 |
| ≥ 2 | 139 | 57 | 140 | 57 | |
| Primary tumor resected | |||||
| Yes | 203 | 83 | 213 | 86 | 0.47 |
| No | 39 | 16 | 34 | 14 | |
| Unknown | 2 | 1 | 0 | 0 | |
| Adjuvant history | |||||
| Yes | 50 | 20 | 45 | 18 | 0.49 |
| No | 192 | 79 | 202 | 82 | |
| Unknown | 2 | 1 | 0 | 0 | |
| RAS status | |||||
| Wild-type | 192 | 79 | 199 | 81 | 0.64 |
| Mutant | 38 | 16 | 35 | 14 | |
| Unknown | 14 | 6 | 13 | 5 | |
| BRAF status | 0.78 | ||||
| Wild-type | 214 | 88 | 214 | 87 | |
| Mutant | 22 | 9 | 24 | 10 | |
| Unknown | 8 | 3 | 9 | 4 | |
P value was based on Chi-square test, or the Wilcoxon test when appropriate.
Table A. 3.
Association of baseline characteristics with clinical outcome in the evaluation cohort
| N | Progression-free survival | Overall survival | |||||
|---|---|---|---|---|---|---|---|
| Median (95%CI), months | Univariate HR (95%CI) † | P value* | Median (95%CI), months | Univariate HR (95%CI) † | P value* | ||
| Sex | 0.004 | 0.13 | |||||
| Male | 169 | 10.5(9.3,12.2) | 1(Reference) | 30.6(23.9,38.5) | 1(Reference) | ||
| Female | 75 | 7.9(6.1,10.4) | 1.52(1.14,2.04) | 27.9(19.9,37.1) | 1.31(0.92,1.87) | ||
| Age (year) | 0.083 | 0.22 | |||||
| ≤ 65 | 130 | 10.3(9.3,12.2) | 1(Reference) | 29.8(23.9,38.5) | 1(Reference) | ||
| > 65 | 114 | 9.0(7.5,10.6) | 1.27(0.96,1.67) | 30.6(20.6,38.3) | 1.23(0.87,1.74) | ||
| ECOG Performance status | 0.36 | 0.051 | |||||
| ECOG 0 | 124 | 10.4(9.6,12.2) | 1(Reference) | 33.1(24.4,42.8) | 1(Reference) | ||
| ECOG 1–2 | 120 | 8.8(7.6,10.5) | 1.14(0.86,1.49) | 26.5(20.5,36.6) | 1.40(0.99,1.97) | ||
| Priary tumor site | <0.001 | <0.001 | |||||
| Right | 45 | 7.4(4.4,9.0) | 1(Reference) | 18.5(14.9,24.5) | 1(Reference) | ||
| Left | 194 | 10.4(9.6,11.8) | 0.51(0.36,0.72) | 36.6(29.8,42.8) | 0.41(0.27,0.61) | ||
| Liver metastasis | 0.21 | 0.021 | |||||
| Yes | 84 | 10.4(8.3,12.8) | 1(Reference) | 37.5(24.4,56.2) | 1(Reference) | ||
| No | 160 | 9.7(8.2,10.9) | 1.20(0.90,1.61) | 28.0(21.7,33.6) | 1.53(1.06,2.21) | ||
| Lung metastasis | 0.22 | 0.051 | |||||
| Yes | 94 | 9.0(7.4,10.6) | 1(Reference) | 27.9(18.3,36.4) | 1(Reference) | ||
| No | 150 | 10.4(9.2,11.8) | 0.84(0.64,1.12) | 33.1(24.4,40.9) | 0.71(0.51,1.01) | ||
| Number of metastases | 0.38 | 0.052 | |||||
| 0–1 | 105 | 10.2(9.0,12.2) | 1(Reference) | 36.6(25.2,40.9) | 1(Reference) | ||
| ≥ 2 | 139 | 9.6(8.1,11.1) | 1.13(0.86,1.49) | 27.9(20.6,33.8) | 1.40(0.99,1.98) | ||
| Primary tumor resected | 0.46 | 0.31 | |||||
| Yes | 203 | 9.7(8.5,10.6) | 1(Reference) | 30.0(24.4,38.3) | 1(Reference) | ||
| No | 39 | 10.4(5.7,14.0) | 0.86(0.58,1.28) | 28.0(15.3,59.0) | 1.27(0.80,2.04) | ||
| Adjuvant history | 0.32 | 0.93 | |||||
| Yes | 50 | 9.9(7.6,12.2) | 1(Reference) | 33.1(21.7,51.3) | 1(Reference) | ||
| No | 192 | 9.9(8.7,10.9) | 0.85(0.61,1.18) | 29.8(23.7,37.1) | 1.02(0.67,1.54) | ||
| RAS status | <0.001 | 0.004 | |||||
| Wild-type | 192 | 10.4(9.3,12.1) | 1(Reference) | 33.1(24.5,39.4) | 1(Reference) | ||
| Mutant | 38 | 6.0(5.1,8.5) | 1.90(1.31,2.76) | 19.1(15.9,27.6) | 1.85(1.19,2.90) | ||
| BRAF status | <0.001 | <0.001 | |||||
| Wild-type | 214 | 10.2(9.2,11.3) | 1(Reference) | 33.1(24.5,38.5) | 1(Reference) | ||
| Mutant | 22 | 3.4(1.9,8.6) | 2.61(1.65,4.15) | 12.9(5.2,23.8) | 2.69(1.60,4.52) | ||
P values<0.05 were shown in bold.
P value was based on the log-rank test for PFS and OS in the univariate analysis (†).
Table A. 4.
Association of baseline characteristics with clinical outcome in the control cohort
| Progression-free survival | Overall survival | ||||||
|---|---|---|---|---|---|---|---|
| N | Median (95%CI), months | Univariate HR (95%CI) † | P value* | Median (95%CI), months | Univariate HR (95%CI) † | P value* | |
| Sex | 0.16 | 0.64 | |||||
| Male | 162 | 10.3(9.7,11.7) | 1(Reference) | 24.2(20.6,28.0) | 1(Reference) | ||
| Female | 85 | 10.1(8.6,13.0) | 1.23(0.92,1.63) | 25.6(21.9,30.8) | 0.92(0.66,1.28) | ||
| Age (year) | 0.63 | 0.31 | |||||
| ≤ 65 | 130 | 10.1(9.1,12.0) | 1(Reference) | 23.1(19.0,28.4) | 1(Reference) | ||
| > 65 | 117 | 10.4(9.7,12.2) | 1.07(0.81,1.41) | 25.4(23.1,28.8) | 0.85(0.62,1.17) | ||
| ECOG Performance status | <0.001 | 0.001 | |||||
| ECOG 0 | 133 | 11.8(10.3,13.5) | 1(Reference) | 29.0(25.4,33.2) | 1(Reference) | ||
| ECOG 1–2 | 114 | 9.7(8.8,10.2) | 1.61(1.22,2.13) | 21.0(18.6,23.8) | 1.66(1.21,2.27) | ||
| Primary tumor site | 0.043 | 0.011 | |||||
| Right | 62 | 9.0(6.9,10.9) | 1(Reference) | 21.8(16.5,23.6) | 1(Reference) | ||
| Left | 179 | 10.7(9.8,12.2) | 0.72(0.52,1.00) | 27.4(23.7,30.8) | 0.64(0.45,0.91) | ||
| Liver metastasis | 0.58 | 0.033 | |||||
| Yes | 81 | 12.0(9.7,13.1) | 1(Reference) | 27.6(20.6,38.1) | 1(Reference) | ||
| No | 166 | 9.9(9.1,10.8) | 1.08(0.81,1.45) | 24.7(21.2,26.5) | 1.43(1.02,2.01) | ||
| Lung metastasis | 0.67 | 0.85 | |||||
| Yes | 94 | 9.9(8.9,11.5) | 1(Reference) | 26.1(23.2,28.4) | 1(Reference) | ||
| No | 153 | 10.8(9.7,12.3) | 1.06(0.80,1.42) | 23.1(19.5,28.6) | 1.03(0.74,1.43) | ||
| Number of metastases | 0.14 | 0.010 | |||||
| 0–1 | 107 | 11.9(9.9,13.0) | 1(Reference) | 27.6(21.3,36.0) | 1(Reference) | ||
| ≥ 2 | 140 | 9.8(9.0,10.8) | 1.23(0.93,1.62) | 23.7(21.0,26.4) | 1.50(1.09,2.07) | ||
| Primary tumor resected | 0.80 | 0.044 | |||||
| Yes | 213 | 10.1(9.6,11.5) | 1(Reference) | 25.4(22.3,28.4) | 1(Reference) | ||
| No | 34 | 10.8(8.8,14.6) | 0.95(0.62,1.45) | 21.2(16.7,26.5) | 1.60(0.99,2.58) | ||
| Adjuvant history | 0.93 | 0.61 | |||||
| Yes | 45 | 9.7(8.8,10.8) | 1(Reference) | 25.6(19.4,35.0) | 1(Reference) | ||
| No | 202 | 10.5(9.8,12.3) | 1.01(0.72,1.43) | 24.7(21.5,28.0) | 1.11(0.75,1.63) | ||
| RAS status | 0.89 | 0.23 | |||||
| Wild-type | 199 | 10.2(9.3,11.5) | 1(Reference) | 25.4(23.0,28.6) | 1(Reference) | ||
| Mutant | 35 | 10.9(8.9,13.9) | 0.97(0.64,1.47) | 19.0(16.7,26.5) | 1.33(0.83,2.15) | ||
| BRAF status | <0.001 | <0.001 | |||||
| Wild-type | 214 | 11.3(10.1,12.4) | 1(Reference) | 27.4(23.8,29.6) | 1(Reference) | ||
| Mutant | 24 | 6.3(3.6,7.8) | 3.43(2.19,5.39) | 13.7(7.8,19.5) | 2.74(1.74,4.31) | ||
P values<0.05 were shown in bold.
P value was based on the log-rank test for PFS and OS in the univariate analysis (†).
Association of clinical outcome and SNPs tested in the evaluation cohort
In the evaluation cohort, patients with any CCL5 rs2280789 G allele had shorter OS compared to those carrying the A/A variant in the univariate analysis (19.9 vs. 33.4 months, HR 1.56, 95% confidence interval (CI): 1.05–2.30, P=0.024); this effect was confirmed by the multivariable analysis (HR 1.57, P=0.030). Patients carrying any CCR5 rs1799988 T allele had a lower response rate (68 vs. 81%, P=0.078) and significantly shorter PFS in the multivariable analysis than those with the C/C variant (9.4 vs. 10.6 months, HR 1.40, 95%CI: 1.01–1.93, P=0.043) (Table 1).
Table 1.
Association between gene polymorphism and clinical outcome
| Tumor response | Progression-free survival | Overall Survival | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | CR+PR | SD+PD | P value* |
Median, months (95%CI) |
HR (95%CI)† | P value* |
HR (95%CI) † ‡ | P value* |
Median, months (95%CI) |
HR (95%CI)† | P value* |
HR (95%CI) † ‡ | P value* |
|
| Evaluation cohort | ||||||||||||||
| CCL5 | 0.21 | 0.22 | 0.23 | 0.024 | 0.030 | |||||||||
| rs2280789 | ||||||||||||||
| A/A | 192 | 123 (73%) | 45 (27%) | 10.3 (9.0, 11.1) | 1 (Reference) | 1 (Reference) | 33.4 (27.1, 38.5) | 1 (Reference) | 1 (Reference) | |||||
| A/G a | 42 | 23 (68%) | 11 (32%) | 9.2 (6.1, 12.2) | 1.23 (0.88, 1.72) | 1.24 (0.87, 1.76) | 19.9 (16.8, 25.2) | 1.56 (1.05, 2.30) | 1.57 (1.04, 2.35) | |||||
| G/G a | 8 | 2 (40%) | 3 (60%) | |||||||||||
| CCR5 | 0.049 | 0.25 | 0.13 | 0.59 | 0.32 | |||||||||
| rs1799988 | ||||||||||||||
| C/C | 65 | 42 (81%) | 10 (19%) | 10.6 (9.2, 13.3) | 1 (Reference) | 1 (Reference) | 36.4 (21.8, 44.1) | 1 (Reference) | 1 (Reference) | |||||
| C/T | 107 | 59 (63%) | 35 (37%) | 9.0 (7.5, 10.8) | 1.32 (0.94, 1.84) | 1.39 (0.98, 1.96) | 28.7 (20.4, 38.7) | 1.19 (0.78, 1.81) | 1.30 (0.85, 1.98) | |||||
| T/T | 62 | 41 (76%) | 13 (24%) | 10.5 (8.2, 13.3) | 1.22 (0.83, 1.80) | 1.41 (0.94, 2.11) | 26.5 (19.9, 37.1) | 1.26 (0.79, 2.00) | 1.42 (0.88, 2.30) | |||||
| 0.078 | 0.11 | 0.043 | 0.31 | 0.15 | ||||||||||
| C/C | 65 | 42 (81%) | 10 (19%) | 10.6 (9.2, 13.3) | 1 (Reference) | 1 (Reference) | 36.4 (21.8, 44.1) | 1 (Reference) | 1 (Reference) | |||||
| Any T | 169 | 100 (68%) | 48 (32%) | 9.4 (8.0, 10.9) | 1.28 (0.94, 1.75) | 1.40 (1.01, 1.93) | 27.6 (22.5, 33.6) | 1.21 (0.83, 1.79) | 1.34 (0.90, 1.98) | |||||
| Control cohort | ||||||||||||||
| CCL5 | 0.95 | 0.47 | 0.79 | 0.32 | 0.56 | |||||||||
| rs2280789 | ||||||||||||||
| A/A | 191 | 113 (64%) | 63 (36%) | 10.5 (9.7, 12.0) | 1 (Reference) | 1 (Reference) | 24.8 (22.3, 28.8) | 1 (Reference) | 1 (Reference) | |||||
| A/Ga | 48 | 27 (63%) | 16 (37%) | 9.8 (8.9, 11.5) | 1.13 (0.81, 1.57) | 0.95 (0.67, 1.36) | 23.1 (19.0, 28.4) | 1.20 (0.83, 1.74) | 0.89 (0.59, 1.33) | |||||
| G/Ga | 7 | 4 (67%) | 2 (33%) | |||||||||||
| CCR5 | 0.38 | 0.84 | 0.39 | 0.72 | 0.97 | |||||||||
| rs1799988 | ||||||||||||||
| C/C | 79 | 46 (68%) | 22 (32%) | 10.5 (9.6, 13.1) | 1 (Reference) | 1 (Reference) | 23.8 (20.1, 27.6) | 1 (Reference) | 1 (Reference) | |||||
| C/T | 104 | 57 (58%) | 41 (42%) | 10.3 (9.2, 11.9) | 1.10 (0.80, 1.52) | 1.25 (0.90, 1.74) | 24.8 (19.4, 28.1) | 0.96 (0.67, 1.37) | 0.98 (0.68, 1.42) | |||||
| T/T | 58 | 36 (67%) | 18 (33%) | 9.9 (8.8, 11.7) | 1.07 (0.74, 1.56) | 1.21 (0.83, 1.79) | 28.6 (22.3, 34.6) | 0.84 (0.55, 1.30) | 1.03 (0.66, 1.60) | |||||
| 0.45 | 0.57 | 0.17 | 0.61 | 0.97 | ||||||||||
| C/C | 79 | 46 (68%) | 22 (32%) | 10.5 (9.6, 13.1) | 1 (Reference) | 1 (Reference) | 23.8 (20.1, 27.6) | 1 (Reference) | 1 (Reference) | |||||
| Any T | 162 | 93 (61%) | 59 (39%) | 10.2 (9.2, 11.7) | 1.09 (0.81, 1.47) | 1.24 (0.91, 1.68) | 25.4 (21.5, 28.8) | 0.92 (0.66, 1.28) | 0.99 (0.71, 1.40) | |||||
CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease. P values<0.05 were shown in bold.
P value was based on the Fisher’s exact test for response, log-rank test in the univariate analysis
and Wald test in the multivariable analysis within Cox regression model
adjusted for sex, ECOG performance status, liver metastasis, resection of the primary tumors, RAS and BRAF status.
Grouped together for estimates of HR.
Subgroup analysis by primary tumor location showed that patients with any CCL5 rs2280789 G allele had shorter PFS and OS compared to those with A/A variant in both right and left-sided subgroups; however, neither of these effects reached statistical significance. For CCR5 rs1799988, the effects were in opposite direction between right and left-sided; the T/T variant was favorable in right-sided tumors, while T allele was unfavorable in left-sided tumors for tumor response, PFS and OS (Table 2). The phenomena were clearly identified in models combining SNPs and tumor location (Fig. 1). Statistically significant differences were shown among the groups consisting of primary tumor location and CCL5 or CCR5 SNPs (Model 1, Groups I–IV; Model 2, Groups I–IV). Similar effect was found in the RAS wild-type populations (Table 2). In model 1, any CCL5 rs2280789 G allele was associated with poor tumor response, shorter PFS and OS compared with the A/A variant regardless of tumor location, suggesting similar allelic characteristics between tumor locations. In model 2, patients with right-sided tumor and any CCR5 C allele (Group IV) had poor tumor response, shorter PFS and OS compared with those with a left-sided tumor and the CCR5 C/C variant (Group I), suggesting an opposite allelic effects between tumor locations (Table 2).
Table 2.
Gene polymorphism and clinical outcome by tumor location in the evaluation cohort
| Tumor response | Progression-free survival | Overall Survival | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | CR+PR | SD+PD | P value* |
Median, months (95%CI) |
HR (95%CI)† | P value* |
HR (95%CI) † ‡ | P value* |
Median, months (95%CI) |
HR (95%CI)† | P value* |
HR (95%CI) † ‡ | P value* |
|
| Right-sided subgroup | ||||||||||||||
| CCL5 rs2280789 |
0.25 | 0.93 | 0.54 | 0.35 | 0.14 | |||||||||
| A/A | 33 | 19(68%) | 9 (32%) | 7.8 (5.3,9.5) | 1 (Reference) | 1 (Reference) | 21.9(11.7, 27.1) | 1 (Reference) | 1 (Reference) | |||||
| Any G | 11 | 4 (44%) | 5 (56%) | 3.9 (1.4,14.0) | 1.03 (0.51, 2.09) | 1.30 (0.56, 2.99) | 18.3 (10.1,18.9) | 1.42 (0.64, 3.17) | 2.01 (0.80, 5.02) | |||||
| CCR5 rs 17 99988 |
0.012 | 0.072 | 0.058 | 0.022 | 0.26 | |||||||||
| C/C | 8 | 2 (25%) | 6 (75%) | 5.5 (1.4,14.2) | 1 (Reference) | 1 (Reference) | 13.7 (5.5, 21.9) | 1 (Reference) | 1 (Reference) | |||||
| C/T | 22 | 10(59%) | 7 (41%) | 7.2 (2.5,9.0) | 1.34 (0.57, 3.17) | 1.93 (0.75, 4.98) | 16.1 (7.1, 27.9) | 0.67 (0.27,1.67) | 0.60(0.23,1.54) | |||||
| T/T | 14 | 11(92%) | 1(8%) | 9.0 (5.9,14.1) | 0.64 (0.26,1.57) | 0.64 (0.21, 1.97) | 27.1 (16.4, 37.5) | 0.31 (0.11,0.88) | 0.38 (0.12,1.22) | |||||
| 0.013 | 0.037 | 0.052 | 0.013 | 0.22 | ||||||||||
| Any C | 30 | 12 (48%) | 13 (52%) | 6.5 (3.0,8.8) | 1 (Reference) | 1 (Reference) | 15.8 (10.3,18.9) | 1 (Reference) | 1 (Reference) | |||||
| T/T | 14 | 11(92%) | 1(8%) | 9.0 (5.9,14.1) | 0.52 (0.26,1.05) | 0.41(0.17, 1.01) | 27.1 (16.4, 37.5) | 0.41(0.18,0.91) | 0.54 (0.20,1.46) | |||||
| Left-sided subgroup | ||||||||||||||
| CCL5 rs2280789 |
0.82 | 0.44 | 0.42 | 0.14 | 0.19 | |||||||||
| A/A | 157 | 103 74%) | 36 (26%) | 10.5 (9.6,12.2) | 1 (Reference) | 1 (Reference) | 38.3 (30.6, 44.1) | 1 (Reference) | 1 (Reference) | |||||
| Any G | 36 | 20(71%) | 8 (29%) | 10.0 6.9,13.5) | 1.17 (0.78,1.74) | 1.19(0.78,1.82) | 21.7(18.1, 46.3) | 1.43 (0.89,2.31) | 1.39(0.85,2.28) | |||||
| CCR5 rs1799988 |
0.003 | 0.27 | 0.14 | 0.32 | 0.23 | |||||||||
| C/C | 56 | 40 (91%) | 4(9%) | 10.6 (9.9,14.1) | 1 (Reference) | 1 (Reference) | 38.5 (33.1, 49.8) | 1 (Reference) | 1 (Reference) | |||||
| C/T | 84 | 49 (64%) | 28 (36%) | 10.2 (7.7,12.2) | 1.29 (0.89,1.88) | 1.35(0.92,1.98) | 33.4 (23.7, 52.0) | 1.23 (0.75,1.99) | 1.36(0.83,2.24) | |||||
| T/T | 45 | 28 (72%) | 11 (28%) | 9.6 (6.5,14.0) | 1.36 (0.87, 2.13) | 1.56(0.98,2.49) | 27.6(17.3,41.2) | 1.52 (0.88,2.64) | 1.63(0.92,2.90) | |||||
| 0.001 | 0.11 | 0.063 | 0.22 | 0.12 | ||||||||||
| C/C | 56 | 40 (91%) | 4(9%) | 10.6 (9.9,14.1) | 1 (Reference) | 1 (Reference) | 38.5 (33.1, 49.8) | 1 (Reference) | 1 (Reference) | |||||
| Any T | 129 | 77(66%) | 39 (34%) | 10.0 (8.2,12.1) | 1.32 (0.93,1.87) | 1.41(0.98,2.01) | 30.6 (22.6, 40.9) | 1.32 (0.84,2.07) | 1.45(0.91,2.29) | |||||
| Combined tumor site and SNPs models | ||||||||||||||
| Model 1 | ||||||||||||||
| CCL5 by tumor location | 0.28 | 0.001 | 0.008 | <0.001 | <0.001 | |||||||||
| Group I | 157 | 103 (74%) | 36 (26%) | 10.5 (9.6,12.2) | 1 (Reference) | 1 (Reference) | 38.3 (30.6, 44.1) | 1 (Reference) | 1 (Reference) | |||||
| Group II | 36 | 20(71%) | 8 (29%) | 10.0 (6.9,13.5) | 1.17 (0.78,1.74) | 1.16 (0.76,1.76) | 21.7(18.1, 46.3) | 1.43 (0.89,2.31) | 1.45 (0.89, 2.37) | |||||
| Group III | 33 | 19(68%) | 9 (32%) | 7.8 (5.3,9.5) | 1.95 (1.30, 2.91) | 1.75 (1.15, 2.66) | 21.9(11.7, 27.1) | 2.45 (1.55, 3.88) | 2.33 (1.45, 3.74) | |||||
| Group IV | 11 | 4 (44%) | 5 (56%) | 3.9 (1.4,14.0) | 2.13(1.14,3.96) | 2.27(1.21,4.26) | 18.3 (10.1, 18.9) | 3.33(1.62,6.84) | 3.11 (1.49, 6.50) | |||||
| Model 2 | ||||||||||||||
| CCR5 by tumor location | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |||||||||
| Group I | 56 | 40 (91%) | 4(9%) | 10.6 (9.9,14.1) | 1 (Reference) | 1 (Reference) | 38.5 (33.1, 49.8) | 1 (Reference) | 1 (Reference) | |||||
| Group II | 129 | 77(66%) | 39 (34%) | 10.0 (8.2,12.1) | 1.30 (0.92,1.84) | 1.41 (0.99, 2.02) | 30.6(22.6, 40.9) | 1.32 (0.84,2.06) | 1.48 (0.94, 2.34) | |||||
| Group III | 14 | 11(92%) | 1(8%) | 9.0 (5.9,14.1) | 1.56 (0.84, 2.91) | 1.67 (0.88, 3.19) | 27.1 (16.4, 37.5) | 1.66 (0.78, 3.53) | 2.06 (0.93, 4.55) | |||||
| Group IV | 30 | 12 (48%) | 13 (52%) | 6.5 (3.0,8.8) | 3.00(1.85,4.86) | 2.87 (1.74, 4.72) | 15.8(10.3,18.9) | 3.99 (2.26, 7.06) | 3.86 (2.14, 6.97) | |||||
| Novel classification | 0.020 | <0.001 | 0.001 | <0.001 | <0.001 | |||||||||
| Group I | 161 | 104 (75%) | 35 (25%) | 10.6 (9.5,12.2) | 1 (Reference) | 1 (Reference) | 38.3 (30.6, 44.1) | 1 (Reference) | 1 (Reference) | |||||
| Group II | 37 | 23(72%) | 9 (28%) | 10.0 (6.9,13.4) | 1.20 (0.82,1.75) | 1.23 (0.81,1.85) | 24.4(18.1, 37.1) | 1.67(1.06,2.62) | 1.91 (1.16, 3.13) | |||||
| Group III | 29 | 11(46%) | 13 (54%) | 7.1 (2.9,9.0) | 2.51 (1.64, 3.84) | 2.26 (1.46, 3.51) | 15.8 (10.3, 18.9) | 3.65 (2.25, 5.92) | 3.26(1.95, 5.42) | |||||
| RAS wild-type subgroup | ||||||||||||||
| Model 1 | ||||||||||||||
| CCL5 by tumor location | 0.059 | <0.001 | 0.016 | <0.001 | <0.001 | |||||||||
| Group I | 122 | 89 (82%) | 20 (18%) | 10.9 (9.9,13.3) | 1 (Reference) | 1 (Reference) | 41.2 (33.8, 49.8) | 1 (Reference) | 1 (Reference) | |||||
| Group II | 29 | 16 (73%) | 6 (27%) | 10.0 (6.9,14.1) | 1.13(0.72,1.78) | 1.05 (0.65,1.69) | 20.3 (16.8, 46.3) | 1.79 (1.06, 3.03) | 1.98 (1.14, 3.42) | |||||
| Group III | 28 | 15 (65%) | 8 (35%) | 7.3 (4.4,9.5) | 2.23 (1.43, 3.49) | 1.91 (1.20, 3.04) | 16.5 (10.3, 27.1) | 3.04 (1.83, 5.05) | 2.87 (1.70, 4.85) | |||||
| Group IV | 7 | 2 (40%) | 3 (60%) | 3.9 (1.4, 15.0) | 2.10 (0.97, 4.55) | 2.16 (0.98, 4.77) | 18.3 (2.5, 18.9) | 5.40 (2.18, 13.36) | 4.63 (1.88, 11.42) | |||||
| Model 2 | ||||||||||||||
| CCR5 by tumor location | 0.002 | <0.001 | 0.009 | <0.001 | <0.001 | |||||||||
| Group I | 46 | 35 (92%) | 3 (8%) | 11.5 (9.9, 14.0) | 1 (Reference) | 1 (Reference) | 42.8 (33.8, 55.5) | 1 (Reference) | 1 (Reference) | |||||
| Group II | 99 | 66 (75%) | 22 (25%) | 10.5 (9.3, 13.6) | 1.13 (0.77, 1.67) | 1.17 (0.78, 1.75) | 38.7 (24.4, 52.0) | 1.13 (0.68, 1.87) | 1.18 (0.70, 1.97) | |||||
| Group III | 10 | 7 (88%) | 1 (13%) | 12.2 (1.9, 18.3) | 1.41 (0.68, 2.94) | 1.61 (0.77, 3.39) | 27.1 (5.9, 60.7) | 1.51 (0.61, 3.70) | 2.17 (0.85, 5.49) | |||||
| Group IV | 24 | 9 (47%) | 10 (53%) | 6.5 (2.9, 9.0) | 3.12 (1.81, 5.37) | 2.54 (1.43, 4.50) | 11.7 (7.1, 18.5) | 4.73 (2.53, 8.86) | 3.97 (2.08, 7.59) | |||||
| Novel classification | 0.005 | <0.001 | 0.005 | <0.001 | <0.001 | |||||||||
| Group I | 125 | 90 (83%) | 19 (17%) | 11.1 (10.0, 13.3) | 1 (Reference) | 1 (Reference) | 40.9 (33.4, 51.3 | 1 (Reference) | 1 (Reference) | |||||
| Group II | 29 | 17 (71%) | 7 (29%) | 10.3 (6.9, 14.0) | 1.19 (0.77, 1.84) | 1.16 (0.72, 1.86) | 19.9 (16.8, 37.1) | 1.94 (1.16, 3.24) | 2.41 (1.38, 4.23) | |||||
| Group III | 24 | 9 (47%) | 10 (53%) | 6.5 (2.9, 9.0) | 2.95 (1.83, 4.76) | 2.33 (1.40, 3.89) | 11.7 (7.1, 18.5) | 4.86 (2.83, 8.34) | 3.83 (2.16, 6.81) | |||||
CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease. P values<0.05 were shown in bold.
P value was based on the Fisher’s exact test for response, log-rank test in the univariate analysis
and Wald test in the multivariable analysis within Cox regression model
adjusted for sex, ECOG performance status, liver metastasis, resection of the primary tumors, RAS and BRAF status.
Fig. 1.

Models combining tumor location with CCL5 rs2280789 (Model 1) and CCR5 rs1799988 (Model 2).
We further developed a novel classification model composed of tumor location, and both CCL5 and CCR5 SNPs, which stratified patients into three categories: Group I, Group II and Group III (Fig. 2). Significant differences in tumor response, PFS and OS were observed among the three categories in both KRAS and RAS wild-type populations (Table 2, Fig. 3A-3C).
Fig. 2.

A novel classification composed of CCL5 rs2280789 and CCR5 rs1799988 based on tumor location regarding clinical outcome in the evaluation cohort, which was finally divided into three categories: Group I, Group II and Group III.
Fig. 3.
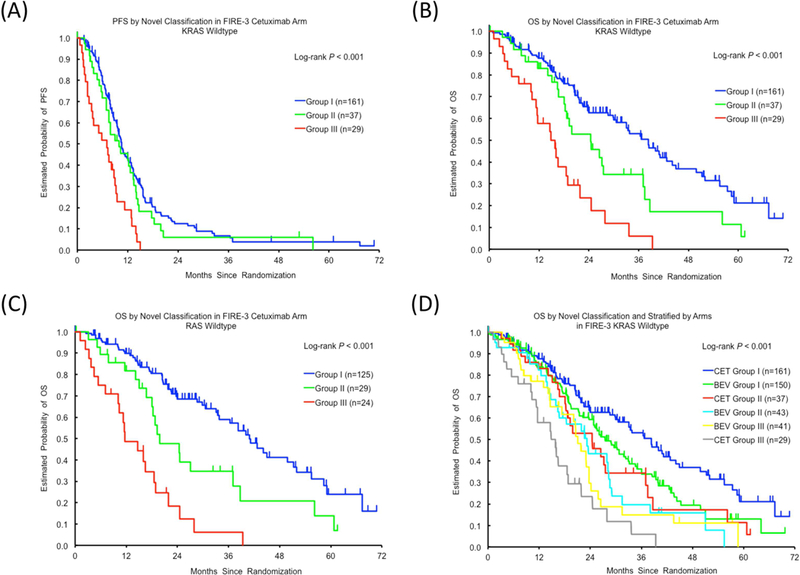
Progression-free survival (PFS) and overall survival (OS) according to the novel classification consisting of CCL5/CCR5 SNPs and tumor location in the evaluation cohort: (A) PFS in KRAS wild-type; (B) OS in KRAS wild-type; (C) OS in RAS wild-type. (D) OS by the novel classification and stratified by treatment arms in the FIRE-3 trial. CET, cetuximab; BEV, bevacizumab.
Association of clinical outcome and SNPs tested in the control cohort
In the control cohort, there was no significant association between SNPs and clinical outcomes in either univariate or multivariate analyses (Table 1). The novel classification model showed a significant association with OS for KRAS wild-type patients in only univariate analyses (P=0.025, adjusted P=0.20, Table A. 5). The interaction test for the classification groups and cohorts was then performed using the same multivariable Cox regression model for OS, which showed a significant association (P<0.001). Fig. 3D demonstrates K-M curves of the evaluation cohort along with the control cohort based on the classification. Accordingly, patient OS could be remarkably differentiated among the groups and cohorts, suggesting Group I in the evaluation cohort was likely to achieve the best OS, while Group III in the evaluation cohort tended to have poorer outcomes compared with the rest.
Table A. 5.
Models consisting SNPs and tumor location in the control cohort
| Tumor response | Progression-free survival | Overall Survival | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | CR+PR | SD+PD | P value* |
Median, months (95%CI) |
HR (95%CI)† | P value* |
HR (95%CI) † ‡ | P value* |
Median, months (95%CI) |
HR (95%CI)† | P value* |
HR (95%CI) † ‡ | P value* | |
| Model 1 | ||||||||||||||
| CCL5 by tumor location | 0.97 | 0.22 | 0.90 | 0.067 | 0.65 | |||||||||
| Group I | 142 | 85(64%) | 47(36%) | 11.1(9.9,12.8) | 1(Reference) | 1(Reference) | 27.4(22.3,33.2) | 1(Reference) | 1(Reference) | |||||
| Group II | 36 | 21(66%) | 11(34%) | 9.8(8.6,12.2) | 1.10(0.73,1.65) | 0.98(0.65,1.50) | 28.1(19.5,31.9) | 1.16(0.74,1.83) | 0.97(0.60,1.55) | |||||
| Group III | 45 | 25(63%) | 15(38%) | 8.8(6.6,12.5) | 1.38(0.95,2.01) | 1.13(0.77,1.67) | 23.0(15.9,23.8) | 1.58(1.04,2.39) | 1.22(0.77,1.92) | |||||
| Group IV | 17 | 9(60%) | 6(40%) | 9.3(5.2,12.3) | 1.49(0.85,2.60) | 0.93(0.51,1.70) | 20.6(14.1,23.1) | 1.77(0.96,3.27) | 0.78(0.37,1.66) | |||||
| Model 2 | ||||||||||||||
| CCR5 by tumor location | 0.76 | 0.20 | 0.46 | 0.075 | 0.30 | |||||||||
| Group I | 53 | 31(69%) | 14(31%) | 10.7(9.3,13.6) | 1(Reference) | 1(Reference) | 29.6(19.5,36.0) | 1(Reference) | 1(Reference) | |||||
| Group II | 122 | 71(61%) | 45(39%) | 10.8(9.7,11.9) | 1.08(0.75,1.54) | 1.22(0.84,1.76) | 27.4(21.9,30.3) | 1.08(0.72,1.63) | 1.25(0.82,1.91) | |||||
| Group III | 19 | 11(69%) | 5(31%) | 8.8(6.5,12.5) | 1.57(0.89,2.76) | 1.58(0.89,2.82) | 22.7(13.7,29.1) | 1.72(0.90,3.28) | 1.87(0.95,3.68) | |||||
| Group IV | 41 | 22(59%) | 15(41%) | 9.3(6.6,12.3) | 1.43(0.90,2.26) | 1.13(0.70,1.84) | 21.0(14.7,23.8) | 1.67(1.01,2.76) | 1.12(0.64,1.97) | |||||
| Novel classification | 0.73 | 0.16 | 0.47 | 0.025 | 0.20 | |||||||||
| Group I | 152 | 90(65%) | 48(35%) | 10.8(9.9,12.7) | 1(Reference) | 1(Reference) | 27.5(24.7,32.4) | 1(Reference) | 1(Reference) | |||||
| Group II | 43 | 23(61%) | 15(39%) | 9.7(8.3,11.5) | 1.27(0.88,1.84) | 1.26(0.87,1.84) | 23.2(15.9,28.4) | 1.48(0.97,2.25) | 1.48(0.95,2.30) | |||||
| Group III | 41 | 22(59%) | 15(41%) | 9.3(6.6,12.3) | 1.38(0.93,2.04) | 1.02(0.67,1.56) | 21.0(14.7,23.8) | 1.64(1.08,2.50) | 1.02(0.62,1.68) | |||||
| RAS wild-type subgroup | ||||||||||||||
| Model 1 | ||||||||||||||
| CCL5 by tumor location | 0.26 | 0.42 | 0.85 | 0.14 | 0.30 | |||||||||
| Group I | 116 | 71(66%) | 36(34%) | 10.8(9.8,12.7) | 1(Reference) | 1(Reference) | 27.5(23.7,33.2) | 1(Reference) | 1(Reference) | |||||
| Group II | 31 | 19(68%) | 9(32%) | 9.7(8.3,11.5) | 1.11(0.72,1.71) | 0.98(0.62,1.55) | 28.0(18.4,31.9) | 1.19(0.73,1.93) | 1.01(0.61,1.66) | |||||
| Group III | 35 | 19(61%) | 12(39%) | 8.8(6.2,12.5) | 1.40(0.92,2.15) | 1.12(0.72,1.73) | 23.0(14.2,23.8) | 1.70(1.06,2.72) | 1.28(0.76,2.14) | |||||
| Group IV | 10 | 3(33%) | 6(67%) | 9.0(1.6,15.9) | 1.29(0.63,2.67) | 0.78(0.35,1.72) | 20.6(7.1,58.7) | 1.35(0.62,2.97) | 0.53(0.20,1.37) | |||||
| Model 2 | ||||||||||||||
| CCR5 by tumor location | 0.52 | 0.37 | 0.66 | 0.15 | 0.77 | |||||||||
| Group I | 46 | 25(64%) | 14(36%) | 10.1(8.5,12.8) | 1(Reference) | 1(Reference) | 26.1(19.5,32.4) | 1(Reference) | 1(Reference) | |||||
| Group II | 99 | 62(66%) | 32(34%) | 11.1(9.7,12.0) | 1.02(0.69,1.49) | 1.19(0.80,1.78) | 28.1(24.7,33.2) | 0.88(0.57,1.35) | 1.12(0.71,1.77) | |||||
| Group III | 14 | 8(67%) | 4(33%) | 9.0(6.5,15.7) | 1.33(0.69,2.55) | 1.48(0.77,2.87) | 23.2(14.2,29.1) | 1.29(0.61,2.72) | 1.50(0.70,3.25) | |||||
| Group IV | 29 | 13(50%) | 13(50%) | 7.8(5.7,12.3) | 1.45(0.86,2.45) | 1.07(0.61,1.87) | 21.0(13.0,24.2) | 1.51(0.86,2.64) | 1.06(0.55,2.02) | |||||
| Novel classification | 0.21 | 0.21 | 0.58 | 0.069 | 0.49 | |||||||||
| Group I | 124 | 77(68%) | 37(32%) | 10.7(9.7,12.7) | 1(Reference) | 1(Reference) | 28.8(24.8,32.4) | 1(Reference) | 1(Reference) | |||||
| Group II | 34 | 18(60%) | 12(40%) | 9.7(8.3,11.8) | 1.21(0.80,1.83) | 1.24(0.82,1.89) | 23.6(14.8,28.6) | 1.31(0.82,2.11) | 1.35(0.82,2.22) | |||||
| Group III | 29 | 13(50%) | 13(50%) | 7.8(5.7,12.3) | 1.46(0.92,2.30) | 0.99(0.60,1.64) | 21.0(13.0,24.2) | 1.70(1.05,2.77) | 1.03(0.57,1.87) | |||||
CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease. P values<0.05 were shown in bold.
P value was based on the Fisher’s exact test for response, log-rank test in the univariate analysis
and Wald test in the multivariable analysis within Cox regression model
adjusted for sex, ECOG performance status, liver metastasis, resection of the primary tumors, RAS and BRAF status.
Discussion
Our data show the first evidence that SNPs of CCL5 and CCR5 genes are associated with clinical outcomes in first-line cetuximab plus FOLFIRI in patients with mCRC. In addition, the novel classification suggests that cetuximab-based treatment could be a possibility in right-sided mCRC, although the current guidelines do not recommend the use of anti-EGFR antibodies in a front-line setting in right-sided mCRC [3].
A potential role of CCL5/CCR5 in cancer progression has been demonstrated in various cancer types [8,12]. The CCL5/CCR5 axis is involved in the immune microenvironment and tumor progression through recruitment of specific immune cells by regulating either the host-derived anti-tumor immunity or tumor progression along with the concomitant chemokines [5,13,14]. A RNA expression analysis study of chemokines and chemokine receptors using RT-PCR in colorectal cancer demonstrated widely expressed CCL5 not only in cancer tissue but also in non-neoplastic mucosal tissues [8]. According to a clinicopathological study of colorectal liver metastases, T lymphocytes infiltrating into the invasive margin produced CCL5 delivered by tumor-infiltrating lymphocytes through CXCR3-mediated migration. By contrast, CCR5 was evident in all samples from CRC liver metastases though other CCL5 receptors, CCR1 and CCR3 were only localized on immune cells [15]. We thus expected that the CCL5/CCR5 signaling correlates with efficacy of the chemotherapeutic treatment for mCRC investigated in our study.
SNPs of CCL5 or CCR5 genes have been investigated in various cancers; however, the exact role of the alleles has not yet been clarified as pro-oncogenic action or response to chemotherapy in cancers, including colorectal cancer [16–23]. Considering genetic functionalities of SNPs, An et al. demonstrated that transcriptional regulation of CCL5 was primarily governed by CCL5 rs2280789 in the promoter region, of which the G allele corresponded with a strong decrease in transcriptional activity of CCL5 [24]. To our knowledge, there have been no published reports on the impact of the CCR5 rs1799988 on cancer susceptibility or its functional consequence on genes/proteins.
The most interesting findings in our study are that the clinical impact of CCR5 SNPs differed depending on primary tumor location, whereas CCL5 SNPs showed a similar trend regardless of tumor location. Furthermore, both the SNPs and primary tumor location had greater influence on OS than PFS. Considering not significant difference in PFS between cetuximab FOLFIRI arm and bevacizumab plus FOLFIRI arm in the FIRE-3 final RAS wild-type subgroup [1], the predictive value of those factors could not be neglected though its impact was smaller than prognostic value. The trend in OS and PFS was also confirmed in a pooled analysis consist of six randomized trials (CRYSTAL, FIRE-3, CALGB 80405, PRIME, PEAK and 20050181), comparing chemotherapy plus EGFR antibody with chemotherapy with or without bevacizumab [25]. As proposed in the study, it is interesting to note that a doublet plus EGFR antibody remains an option if treatment goal is tumor shrinkage; however, which still continues to confuse physician in treatment selection for RAS wild-type right-sided mCRC. In terms of tumor shrinkage, our novel classification could identify more clearly who benefit from chemotherapy plus EGFR antibody than tumor sidedness, albeit the trend in PFS and OS is similar between them. This suggests that the distribution of genetic variant of the SNPs and primary tumor location were not completely matching each other, and our novel classification seems to cover the ambiguity of primary tumor location in predicting tumor response. A recent study reported the in vivo distribution of the Human Immunodeficiency Virus/Simian Immunodeficiency Virus co-receptors, CXCR4, CCR3, and CCR5 in human and rhesus monkey [26]. In the study, the expression levels of co-receptors were higher in the proximal than the distal part of the GI tract, i.e., colon than rectum, which might be potentially regulated by the degree of cellular maturation and activation, and the concentration of the related chemokines. Although the findings regarding the co-receptor expression distributions are extremely informative, they should be confirmed by further studies of cancer patients. We therefore speculate that such mediators regulate CCR5 expression according to tumor location in CRC leading to the opposite outcomes. Meanwhile, CCR5 is known to participate in the pathogenesis of inflammatory bowel disease (IBD) [27]; and a recent study reported a correlation of CCR5 expression and β-arrestin2 expression in intestinal mucosa with leukocyte infiltration in IBD [28]. β-arrestin2 is a cellular soluble protein that negatively regulates G protein-coupled receptors such as CCR5 to be desensitized or internalized. In addition, the consensus molecular subtypes (CMSs) of colorectal cancer have been introduced as the most robust classifier for CRC based on biology but not outcome [29], of which CMS1 is categorized as microsatellite instability (MSI) immune harboring immune infiltration and activation. CMS1 has been shown to localize in right-sided more frequently than in left-sided CRC, and correlate with a worse prognosis in mCRC. Taken together, such variation in immune environment between right- and left-sided tumors may account for the diverse manner of CCR5 SNPs in the mechanism of action of cetuximab. Moreover, in a subgroup analysis of a randomized Phase III trial (CALGB/SWOG80405) comparing CET-based to BEV-based first-line chemotherapy, CMS1 was also frequently shown in right-sided tumors and revealed to be a potentially negative predictor of cetuximab [30]. We think the results could support the abovementioned our hypothesis. As next step in treatment for mCRC patients, immune checkpoint inhibitors targeting programmed death-1 (PD-1) and cytotoxic T-cell lymphocyte antigen-4 (CTLA-4) have demonstrated promising outcome, which focused on patients with DNA mismatch repair-deficient (dMMR)/MSI-high (MSI-H) mCRC [31,32]. Although crosstalk between EGFR signaling and PD-1 pathway still remains unclear, which urges us to develop possible combination of EGFR antibody and PD-1 and/or -CTLA-4 inhibitor regarding the poor prognosis in patients with dMMR/MSI-H mCRC. Recently, a phase Ib/II study reported the tolerability of combination of cetuximab and pembrolizumab in patients with RAS wild-type mCRC and no additional emergent toxicity was observed. The efficacy data in the ongoing phase II study is warranted [33].
The strengths of our study are as follows: the two cohorts derived from a randomized phase III trial (FIRE-3) comparing cetuximab-based with bevacizumab-based chemotherapy; and the consistent results between the KRAS wild-type and RAS wild-type populations in the evaluation cohort but not in the control cohort. As limitations of our study, we have to perform further preclinical and validation studies to explore the biology of CCR5 SNPs, which differed by primary tumor location for clinical outcome in cetuximab-based treatment. In addition, correlation between expression of CCL5/CCR5 within tumor and the related-gene polymorphisms may enhance our conclusions. Furthermore, the clinical role of the immune environment with specific modulators such as β-arrestin2 in different sided CRC should be explored in further studies.
In conclusion, genetic variants of CCL5 and CCR5 SNPs may predict outcomes in mCRC patients receiving CET-based first-line treatment depending on tumor location.
Highlights.
CCL5/CCR5 axis is involved in EGFR signaling blockade in tumor progression.
Impact of CCL5/CCR5 SNPs varies depending on tumor location in inhibiting EGFR.
Cetuximab-based treatment could be a possibility in right-sided colorectal cancer.
Acknowledgments
Mitsukuni Suenaga is the recipient of Takashi Tsuruo Memorial Fund. Martin D. Berger received a grant from the Swiss Cancer League (BIL KLS-3334-02-2014) and the Werner and Hedy Berger-Janser Foundation for cancer research. Yuji Miyamoto received a grant from Japan Society for the Promotion of Science (S2606).
Funding
This work was partly supported by the National Cancer Institute (grant number P30CA014089), The Gloria Borges WunderGlo Foundation-The Wunder Project, Dhont Family Foundation, San Pedro Peninsula Cancer Guild, Daniel Butler Research Fund and Call to Cure Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
The authors have no conflicts of interest to declare in this work.
References
- 1.Stintzing S, Modest DP, Rossius L, Lerch MM, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): a post-hoc analysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol 2016;17:1426–34. [DOI] [PubMed] [Google Scholar]
- 2.Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Fruth B, Meyerhardt JA, et al. Effect of First-Line Chemotherapy Combined With Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017;317:2392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.NCCN Clinical Practice Guideline: Colon Cancer ver. 1; 2018. Available from: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed March 14, 2018.
- 4.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386–422. [DOI] [PubMed] [Google Scholar]
- 5.Song A, Nikolcheva T, Krensky AM. Transcriptional regulation of RANTES expression in T lymphocytes. Immunol Rev 2000;177:236–45. [DOI] [PubMed] [Google Scholar]
- 6.Mascia F, Mariani V, Girolomoni G, Pastore S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol 2003;163:303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paul T, Schumann C, Rüdiger S, Boeck S, Heinemann V, Kächele V, et al. Cytokine regulation by epidermal growth factor receptor inhibitors and epidermal growth factor receptor inhibitor associated skin toxicity in cancer patients. Eur J Cancer 2014;50:1855–63. [DOI] [PubMed] [Google Scholar]
- 8.Musha H, Ohtani H, Mizoi T, Kinouchi M, Nakayama T, Shiiba K, et al. Selective infiltration of CCR5(+)CXCR3(+) T lymphocytes in human colorectal carcinoma. Int J Cancer 2005;116:949–56. [DOI] [PubMed] [Google Scholar]
- 9.Lee YS, Choi I, Ning Y, Kinouchi M, Nakayama T, Shiiba K, et al. Interleukin-8 and its receptor CXCR2 in the tumour microenvironment promote colon cancer growth, progression and metastasis. Br J Cancer 2012;106:1833–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang SW, Liu SC, Sun HL, Huang TY, Chan CH, Yang CY, et al. CCL5/CCR5 axis induces vascular endothelial growth factor mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 2015;36:104–14. [DOI] [PubMed] [Google Scholar]
- 11.Chang LY, Lin YC, Mahalingam J, Huang CT, Chen TW, Kang CW, et al. Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res 2012;72:1092–102. [DOI] [PubMed] [Google Scholar]
- 12.Barashi N, Weiss ID, Wald O, Wald H, Beider K, Abraham M, et al. Inflammation-induced hepatocellular carcinoma is dependent on CCR5 in mice. Hepatology 2013;58:1021–30. [DOI] [PubMed] [Google Scholar]
- 13.Wang SW, Liu SC, Sun HL, Huang TY, Chan CH, Yang CY, et al. CCL5/CCR5 axis induces vascular endothelial growth factor mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 2015;36:104–14. [DOI] [PubMed] [Google Scholar]
- 14.Balkwill F Cancer and the chemokine network. Nat Rev Cancer 2004;4:540–550. [DOI] [PubMed] [Google Scholar]
- 15.Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016;29:587–601. [DOI] [PubMed] [Google Scholar]
- 16.Sáenz-López P, Carretero R, Cózar JM, Romero JM, Canton J, Vilchez JR, et al. Genetic polymorphisms of RANTES, IL1-A, MCP-1 and TNF-A genes in patients with prostate cancer. BMC Cancer 2008;8:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorjgochoo T, Zheng Y, Gao YT, Ma X, Long J, Bao P, et al. No association between genetic variants in angiogenesis and inflammation pathway genes and breast cancer survival among Chinese women. Cancer Epidemiol 2013;37:619–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodelon C, Malone KE, Johnson LG, Malkki M, Petersdorf EW, McKnight B, et al. Common sequence variants in chemokine-related genes and risk of breast cancer in post-menopausal women. Int J Mol Epidemiol Genet 2013;4:218–27. [PMC free article] [PubMed] [Google Scholar]
- 19.Tahara T, Shibata T, Nakamura M, Yamashita H, Yoshioka D, Hirata I, et al. RANTES promoter genotype and gastric cancer risk in a Japanese population. Anticancer Res 2009;29:4265–69. [PubMed] [Google Scholar]
- 20.Gawron AJ, Fought AJ, Lissowska J, Ye W, Zhang X, Chow WH, et al. Polymorphisms in chemokine and receptor genes and gastric cancer risk and survival in a high risk Polish population. Scand J Gastroenterol 2011;46:333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai HT, Yang SF, Chen DR, Chan SE. CCL5–28, CCL5–403, and CCR5 genetic polymorphisms and their synergic effect with alcohol and tobacco consumptions increase susceptibility to hepatocellular carcinoma. Med Oncol 2012;29:2771–9. [DOI] [PubMed] [Google Scholar]
- 22.Duell EJ, Casella DP, Burk RD, Kelsey KT, Holly EA. Inflammation, genetic polymorphisms in proinflammatory genes TNF-A, RANTES, and CCR5, and risk of pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev 2006;15:726–31. [DOI] [PubMed] [Google Scholar]
- 23.Ying H, Wang J, Gao X. CCL5–403, CCR5–59029, and Delta32 polymorphisms and cancer risk: a meta-analysis based on 20,625 subjects. Tumour Biol 2014;35:5895–904. [DOI] [PubMed] [Google Scholar]
- 24.An P, Nelson GW, Wang L, Donfield S, Goedert JJ, Phair J, et al. Modulating influence on HIV/AIDS by interacting RANTES gene variants. Proc Natl Acad Sci U S A 2002;99:10002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold D, Lueza B, Douillard JY, Peeters M, Lenz HJ, Venook A, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol 2017;28:1713–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, He T, Talal A, Donfield S, Goedert JJ, Phair J. In vivo distribution of the human immunodeficiency virus/simian immunodeficiency virus coreceptors: CXCR4, CCR3, and CCR5. J Virol 1998;72:5035–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tokuyama H, Ueha S, Kurachi M, Matsushima K, Moriyasu F, Blumberg RS, et al. The simultaneous blockade of chemokine receptors CCR2, CCR5 and CXCR3 by a non-peptide chemokine receptor antagonist protects mice from dextran sodium sulfate-mediated colitis. Int Immunol 2005;17:1023–34. [DOI] [PubMed] [Google Scholar]
- 28.Ye X, Liu S, Hu M, Song Y, Huang H, Zhong Y. CCR5 expression in inflammatory bowel disease and its correlation with inflammatory cells and β-arrestin2 expression. Scand J Gastroenterol 2017;52:551–7. [DOI] [PubMed] [Google Scholar]
- 29.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lenz HJ, Ou FS, Venook AP, Hochster HS, Niedzwiecki D, Goldberg RM, et al. Impact of consensus molecular subtyping (CMS) on overall survival (OS) and progression free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 35, 2017. (suppl; abstr 3511). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018;36:773–9. [DOI] [PubMed] [Google Scholar]
- 32.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boland PM, Hutson A, Maguire O, Minderman H, Fountzilas C, Iyer RV. A phase Ib/II study of cetuximab and pembrolizumab in RAS-wt mCRC. J Clin Oncol 36, 2018. (suppl 4S; abstr 834). [Google Scholar]