

Abstract
Background
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) has previously been demonstrated to play a pro-inflammatory role in allergic airways disease and COPD through the upregulation of the E3 ubiquitin ligase MID1 and the subsequent deactivation of protein phosphatase 2A (PP2A).
Methods
Biopsies were taken from eight IPF patients presenting to the Second Affiliated Hospital of Jilin University, China between January 2013 and February 2014 with control samples obtained from resected lung cancers. Serum TRAIL, MID1 protein and PP2A activity in biopsies, and patients’ lung function were measured. Wild type and TRAIL deficient Tnfsf10−/− BALB/c mice were administered bleomycin to induce fibrosis and some groups were treated with the FTY720 analogue AAL(s) to activate PP2A. Mouse fibroblasts were treated with recombinant TRAIL and fibrotic responses were assessed.
Results
TRAIL in serum and MID1 protein levels in biopsies from IPF patients were increased compared to controls. MID1 levels were inversely associated while PP2A activity levels correlated with DLco. Tnfsf10−/− and mice treated with the PP2A activator AAL(s) were largely protected against bleomycin-induced reductions in lung function and fibrotic changes. Addition of recombinant TRAIL to mouse fibroblasts in-vitro increased collagen production which was reversed by PP2A activation with AAL(s).
Conclusion
TRAIL signalling through MID1 deactivates PP2A and promotes fibrosis with corresponding lung function decline. This may provide novel therapeutic targets for IPF.
Keywords: TRAIL , MID1, PP2A , Fibrosis, E3 ubiquitin ligase
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common form of the idiopathic interstitial lung disorders accounting for 50–60% of cases [1]. There is marked heterogeneity in the histopathological findings, radiological profiles, treatment responsiveness and the rate of disease progression across individuals diagnosed with IPF. This is thought to be due to the complexity of the interplay between environmental and endogenous factors and the multiplicity of co-activated pathogenic pathways that have been demonstrated to underpin this disease [2].
IPF consistently clinically manifests as chronic and progressive exertional dyspnoea and dry cough of gradual onset as a result of restrictive ventilation impairment and respiratory failure [1]. Current medical interventions for IPF are either supportive therapy (oxygen therapy) or lung transplantation, and few effective pharmacological therapies are available for its treatment. IPF has a poor prognosis with a median survival of 3 years and a 5 year survival rate of 45% [3]. The traditional concept of IPF pathogenesis was that the initial event is an inflammatory process characterised by infiltration of inflammatory cells such as macrophages, lymphocytes and neutrophils in the alveoli (alveolitis). Inflammation was believed to lead to recruitment of fibroblasts and myofibroblasts and finally to collagen formation and irreversible fibrosis. However, clinical trials of drugs inhibiting an underpinning inflammatory process such as Etanercept, a TNF-α antagonist, have consistently failed to show any efficacy in slowing disease progression [4]. In contrast, antifibrotic drugs such as nintedanib and pirfenidone have been demonstrated to slow disease progression and have recently been approved for use in treatment of IPF in the USA and Europe [5, 6]. Thus, the current consensus is that IPF is a primary epithelial and fibroblastic disease, and pro-inflammatory and inflammatory mediators are involved in the epithelial-dependent fibrotic pathway.
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is a member of the tumour necrosis factor family that is principally expressed by airway epithelial cells [7, 8], and leukocytes including monocytes [9], macrophages [10], lymphocytes [11] and neutrophils [12]. It is best described for its role in inducing apoptosis in a variety of transformed cells [13–15]; however, we have recently described the fibrogenic role played by TRAIL signalling in mouse models of allergic and neonatal infection-induced airways disease and in eosinophilic esophagitis [16–18]. We have also shown that TRAIL induces the E3 ubiquitin ligase midline-1 (Mid1) to inhibit the activation status of the protein phosphatase (PP)2A by interacting with the α4 regulatory subunit that is required for the ubiquitin-specific modification and proteasome-mediated degradation of its catalytic subunit PP2Ac [19–21]. PP2A is the most abundant cellular protein phosphatase. Reduced activity due to Mid1 upregulation resulted in hyperphosphylation of mitogen-activated protein kinases (MAPKs) and inhibitor of κBα (IκBα) protein, thereby promoting p38 MAPK, c-Jun N-terminal kinase (JNK) and nuclear factor-κB (NF-κB) activity [22–26].
Here we investigate lung tissue derived from IPF patients for the pro-inflammatory fibrogenic upregulation of MID1 and PP2A. We also investigate the role of TRAIL-Mid1-PP2A signalling in the rodent fulminant-induced bleomycin model of pulmonary fibrosis and explore its relevance to collagen production by lung fibroblasts - thought to underpin much of the disease phenotype in human disease.
Methods
Lung biopsies were collected from eight IPF patients from the Department of Respiratory and Critical Care Medicine, the Second Affiliated Hospital of Jilin University between January 2013 and February 2014. A clinical diagnosis of IPF was identified according to histological confirmation of usual interstitial pneumonia (UIP) or the presence of a UIP pattern on high-resolution computed tomography (HRCT). A total of 21 candidates without known ILD (eg. environmental factors, occupational exposure, connective tissue diseases, drug injury) underwent blood tests for screening of connective tissues diseases (including; ANCA, ANA, rheumatic blood test, complement C3,C4) and upon negative results underwent HRCT. Six patients were categorized as definite UIP and 4 as not UIP from the HRCT and did not undergo transbronchoscopic biopsy. The remaining 11 patients classified as possible UIP according to the ATS/ERS/JRS/ALAT statement criteria underwent transbronchosopic biopsy of which eight had histologically confirmed UIP. The ATS/ERS/JRS/ALAT guidelines state that “the sensitivity and specificity of this approach (transbronchosopic biopsy) for the diagnosis for UIP pattern is unknown” and therefore not always suitable for the diagnosis of IPF. However, in each of the eight cases here the diagnosis of IPF was made on the basis of all features present (clinical parameters, CT images, histology). Importantly, control tissues were from tissues distal to resected lung cancer in patients with no clinical evidence of IPF or other fibrotic lung disease. Written informed consent was obtained from individual subjects, and all the studies were approved by the Medical Ethics Committee of the Second Affiliated Hospital of Jilin University, Changchun, Jilin, P.R. China.
Mice
Wild-type (WT) and TRAIL deficient (Tnfsf10−/−) male BALB/c mice (6–14 weeks-old) were obtained from the Australian Bioresources facility (Moss Vale, Australia). Animals were housed with ad libitum access to food and water with a 12-h light-and-dark cycle.
Bleomycin-induced pulmonary fibrosis
Mice were anaesthetised with Alfaxan by tail vein injection, then 50 μl solution containing bleomycin (2 mg/kg, Sigma-Aldrich, USA) was intratrachealy administered. Control mice were administered saline instead. All procedures were performed in a sterile environment [27]. In order to activate PP2A, mice received the nonphosphorylatable FTY720 analog, (S)-2-amino-4-(4-heptyloxyphenyl)-2-methylbutan-1-ol (AAL(s)) intranasaly starting at day − 1 and daily throughout the model with bleomycin administration at day 0. Mice were sacrificed 1, 4, 8, or 21 days after bleomycin instillation via pentobarbitone overdose, and one lung was taken for histology and one was homogenised for protein quantification.
Lung capacity
Vital capacity was assessed under Ketamine and Xylazine induced anaesthesia, as the volume of air that entered the lungs when the airway pressure was increased from 2 to 30 cmH2O by the ventilator (PVs-P Flexivent manoeuvre). Compliance was calculated as the measured change in volume divided by this applied pressure change. Three inflations were performed and averaged per mouse [27, 28].
Histological preparations
Lungs were collected in 10% neutral buffered formalin solution for 24 h before being transferred to 70% ethanol. Tissue was then embedded in paraffin and stained with either hematoxylin and eosin (eosinophils), periodic acid Schiff (PAS positive mucus producing epithelial cells), Masson’s Trichrome (Collagen), toluidine blue (mast cells) or processed further for immunohistochemistry. Lungs across experimental groups were all processed as a batch for either histologic staining or the immunostaining protocol.
Collagen area enumeration
The area of peribronchial trichrome staining in a paraffin-embedded lung was outlined and quantified using a light microscope (Olympus) attached to an image-analysis system (Image-Pro Plus 6; Media Cybernetics, Maryland, USA). Results are expressed as the area of trichrome staining per micrometre length of basement membrane of major airways. At least 10 counts were made from each mouse as previously described [17, 29].
Terminal dUTP nick end labelling (TUNEL) assay
Longitudinal sections of the left single-lobe lung were stained with TUNEL assay kits (Promega, Sydney, Australia) according to the manufacturer’s instructions. Apoptosis in lung parenchyma was assessed by enumerating the numbers of TUNEL positive and negative cells in 20 randomised, 100mm2 fields by light microscopy [28].
Quantitative RT-PCR
We performed quantitative RT-PCR with SYBR Green (Invitrogen, Sydney, Australia) on a Realcycler (Eppendorf, Sydney, Australia) with the following cycling conditions: 2 min at 50 °C followed by 2 min at 95 °C. Amplification was recorded during 40 cycles of 15 s at 95 °C followed by 45 s at 60 °C. We quantified mRNA copy number using cDNA standards for all genes of interest. We normalised expression to the housekeeping gene Hprt with all primer sequences previously described [22].
Elisa
Concentrations of human MID1 (Cusabio, USA) and PP2A activity (R&D systems, USA) in homogenized lung biopsies and TRAIL (R&D systems, USA) in serum were determined using a sandwich ELISA according to the manufacturer’s instructions [30, 31].
Primary mouse fibroblast culture
Lung tissue was collected under sterile conditions and cut into approximately 1 × 1 mm tissue fragments. They were washed three times with PBS and then spread on the bottom of 6-well plates in complete DMEM with 10% (vol/vol) FCS. The media was changed every 2–3 days. Cell monolayers formed around tissue blocks in 1–2 weeks. Tissue fragments were removed using sterile forceps if more than 60% of the plate was covered by attached fibroblasts. When fibroblasts reached > 90% confluence, 0.25% trypsin was used to lift the cells, which were then removed in two culture flasks for passage (1:2). Fibroblasts were passaged 3 times with this method. We used passage 4 and cultured them until 80% confluence, serum-starved them for 24 h and incubated them with recombinant (r)TRAIL (0 ng/ml-100 ng/ml) for 24 h in serum-free DMEM.
Statistical analysis
Statistical significance was analysed using Student’s t-test or Mann-Whitney test as appropriate. All graphed data are expressed as mean ± SEM. Correlation analysis was done using a Spearman test. All data was analysed using GraphPad Prism 7 (La Jolla, CA).
Results
IPF diagnosis
Patients with a diagnosis of IPF as expected had a reduced percentage predicted carbon monoxide diffusing capacity (% pred DLco) and forced vital capacity (% pred FVC) (Table 1). Controls had normal DLCO and FVC. Transbronchial biopsy sections for IPF patients had distorted lung architecture and obliterated alveolar structure with hyperplastic pneumocytes, inflammation and fibroblast proliferation (Fig. 1).
Table 1.
Participant demographics for the control and IPF populations included in the study
| Control | IPF | p value | |
|---|---|---|---|
| Gender (male/ total) | 5/8 | 5/8 | |
| Age (min-max) | 56 (45–62) | 65 (50–83) | 0.08 |
| dLCo (25th–75th) | 92.27 (89.2–96.53) | 43.85 (28.73–53.73) | < 0.001 |
| FVC (25th – 75th) | 79.85 (78.5–80.8) | 44.2 (28.75–51.5) | < 0.001 |
| Smokers | 2/8 | 3/8 |
Fig. 1.
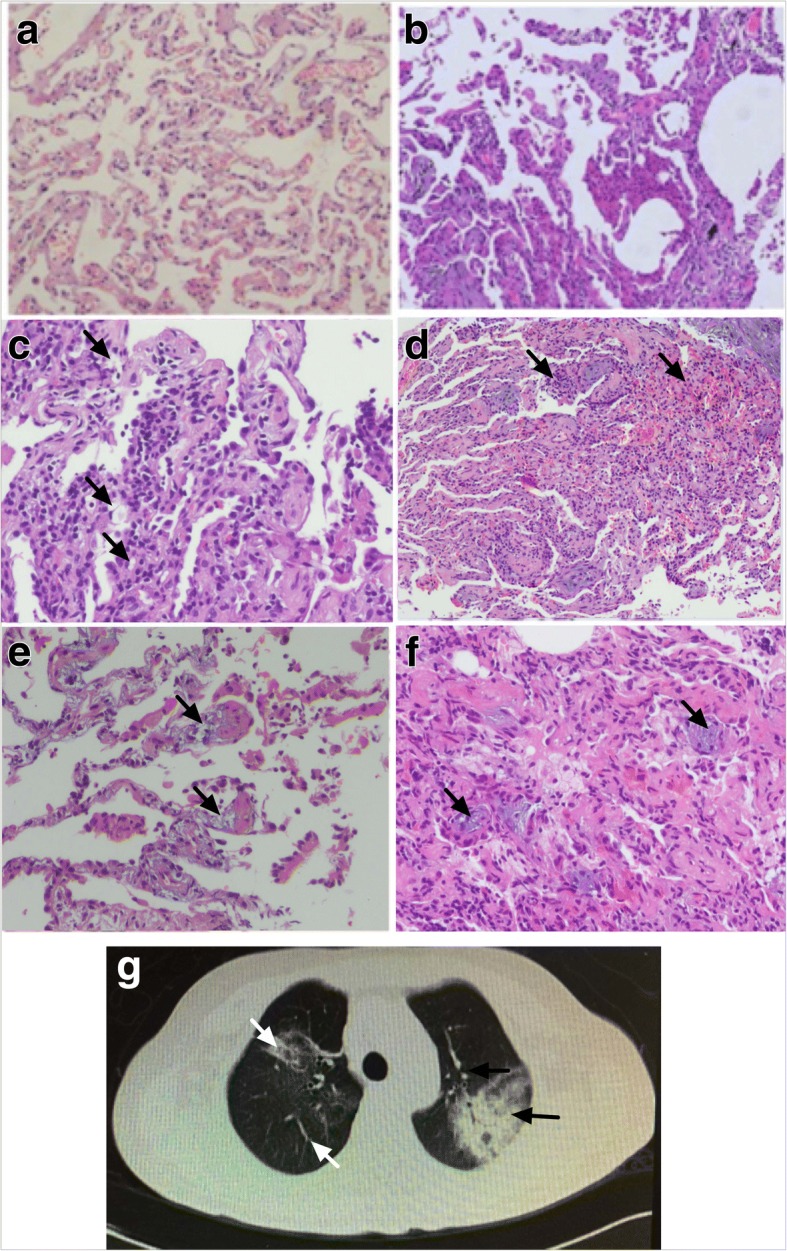
Lung and alveolar structure is disrupted, and inflammatory responses occur in IPF. Representative images of haematoxylin and eosin stained transbronchial biopsy sections at 200x magnification with arrows highlighting described features: Normal lung architecture from control patients (a), distortion of the normal lung architecture and obliteration of alveolar architecture consistent with IPF (b), hyperplasic pneumocytes (c), inflammatory infiltrates (d), and fibroblastic proliferation (e, f). Representative high-sresolution computerised-tomography image showing areas of air-space consolidation, ground glass opacities, small nodular opacities and bronchial wall thickening and dilatation (g)
TRAIL in serum and MID1 protein in biopsies are increased in IPF
We assessed protein levels of TRAIL in the serum and MID1 and phosphatase activity of PP2A in homogenised lung specimens compared to controls. We found that TRAIL and MID1 protein levels were significantly upregulated in IPF patients (Fig. 2a, b). As expected, IPF patients had a reduced percentage predicted carbon monoxide diffusing capacity (% pred DLco) (Table 1). Notably MID1 protein and PP2A activity levels in lung biopsies from IPF patients correlated with DLco (Fig. 2e, f).
Fig. 2.

Serum from IPF patients had elevated levels of TRAIL (a) and lung biopsies elevated MID1 (b) protein, which corresponded with a trend towards decreased activity of PP2A (c) when compared to those from control subjects. The percentage predicted diffusing lung capacity for carbon monoxide (DLco) did not have a strong inverse association with serum TRAIL (d) but was inversely associated with MID1 protein (e) and correlated with the levels of PP2A activity (f) in lung biopsies from IPF patients. n = 8, * p < 0.05
Elevated Mid1 and reduced PP2A activity occurs in bleomycin-induced pulmonary fibrosis, and the absence of TRAIL or activation of PP2A reverses these effects and improve lung function
We then assessed a mouse model of lung fibrosis induced by bleomycin. In support of the human data WT BALB/c mice with bleomycin-induced lung fibrosis displayed a marked upregulation of Mid1 and a downregulation of PP2A activity in their lungs (Fig. 3a, b). Furthermore, WT mice treated with the synthetic PP2A activator AAL(s) and Tnfsf10−/− mice were protected from the upregulation of Mid1 and the deactivation of PP2A in response to bleomycin exposure (Fig. 3a, b). WT mice treated with AAL(s) and Tnfsf10−/− mice were also protected from the bleomycin-induced decline in lung function as shown by an unaffected vital capacity (VC) and peak compliance in response to bleomycin exposure (Fig. 3c, d).
Fig. 3.

Wild-type mice treated with the PP2A activator AAL(s) and Tnfsf10−/− mice were protected from increases in Mid1 mRNA, and the loss of PP2A activity and lung function induced by bleomycin. Mid-1 mRNA was downregulated in AAL(s) treated WT mice and Tnfsf10−/− mice exposed to bleomycin (a) while PP2A activity was increased (b). The vital capacity (VC) and pressure at the peak of compliance (Cpk) were both decreased by bleomycin 21 days post exposure but Tnfsf10−/− mice or those treated with AAL(s) were protected (c-d). n = 5–8, * p < 0.05
Pro-fibrotic factor mRNA levels are increased in bleomycin-induced pulmonary fibrosis in mice, and the absence of TRAIL or activation of PP2A reverses these effects
We next investigated the expression of pro-fibrotic factors and found that bleomycin-exposed WT mice had elevated levels of collagen-α2(I), transforming growth factor β1 (TGFβ), matrix metalloproteinase (MMP) 9 and C-C motif chemokine ligand (CCL) 2 mRNA. AAL(s) treated WT mice and Tnfsf10−/− mice were protected from the increases in these pro-fibrotic mediators (Fig. 4).
Fig. 4.

Wild type mice treated with AAL(s) and Tnfsf10−/− mice had reduced expression of collagen-α2(I) (a), TGFβ (b), mmp9 (c) and ccl2 (d) in their lungs 21 days after bleomycin exposure. n = 7–12, * p < 0.05
Collagen deposition and apoptosis levels are increased in bleomycin-induced pulmonary fibrosis in mice, and the absence of TRAIL and activation of PP2A reverses these effects
We then investigated the levels of collagen deposition and apoptosis. Histological investigation of Masson’s trichrome stained transverse lung sections demonstrated that bleomycin induced increases in collagen deposition around the airways compared to saline treated controls (Fig. 5a). AAL(s) treated WT mice and Tnfsf10−/− mice had reduced or inhibited collagen deposition, respectively. TUNEL staining also revealed that bleomycin induced increases in the percentage of TUNEL positive cells that peaked after one day but was still elevated after 21 days (Fig. 5b, c). AAL(s) treated WT mice and Tnfsf10−/− mice have reduced levels of TUNEL positive staining.
Fig. 5.

AAL(s) treated wild type mice and Tnfsf10−/− mice were protected from bleomycin-induced collagen deposition and apoptosis. Collagen deposition in Masson’s trichrome stained lung sections (a). Images at 200x magnification of representative sections from Masson’s trichrome for (b) Wild Type saline and (c) bleomycin treated (d) wildtype bleomycin treated with AAL(s), (e) TRAIL−/− bleomycin treated and (f) TRAIL −/− saline. Apoptosis in terms of the percentage of TUNEL positive cells in lung sections (g), which peaked at one day post bleomycin but was still significantly elevated after day 21(h). n = 6, * p < 0.05
Proliferation and release of Mid1 and collagen-α2(I) mRNA were increased in mouse lung fibroblasts treated with rTRAIL
Next, we isolated fibroblasts from mouse lungs and cultured them with increasing concentrations of rTRAIL in-vitro. Over 24 h we found that proliferation of fibroblasts increased in the presence of TRAIL in a dose-dependent manner peaking at a dose of 1 ng/ml (data not shown). This dose was sufficient to induce the expression of Mid1 and collagen-α2(I) in fibroblast cultures, while collagen expression but not Mid1 was inhibited by addition of AAL(s) (Fig. 6 a, b).
Fig. 6.

rTRAIL stimulated Mid1 and collagen- α2(I) production in isolated primary mouse fibroblasts. Mid1 (a) and collagen-α2(I) (b) expression per copy of HPRT in cultured fibroblasts. n = 3 independent experiments * p < 0.05
Discussion
We have previously demonstrated that TRAIL signalling through the ubiquitin ligase Mid1 and dysregulation of PP2A activity plays a key role in lung fibrosis in house dust mite and ovalbumin-induced allergic airways disease models [17, 22, 32]. The pro-inflammatory TRAIL pathway also is required for fibrosis in Aspergillus fumigatus and ovalbumin-induced models of eosinophilic oesophagitis and cigarette smoke induced models of COPD [16, 28, 33].
Here we show that TRAIL and MID1 are associated with pathogenesis and remodelling in IPF. TRAIL in patient serum and MID1 in the lungs of patients with active IPF are both upregulated (Fig. 2a, b). MID1 protein had an inverse correlation to lung function DLco in these IPF patients (Fig. 2e) while levels of PP2A activity, the effector of the pathway, were directly correlated (Fig. 2f). McGrath et al., showed lower levels of TRAIL in the serum of IPF patients, though they did not examine signalling downstream of TRAIL [12] . More recent studies have shown elevated levels of TRAIL in airway epithelial cells isolated from active regions within IPF lungs [34] which is in line with our findings of increased MID1 and decreased PP2A activity in lung biopsies. Furthermore, post-hoc analysis of Schiller et al.’s recent proteomic profile including eleven IPF patient biopsies vs three healthy controls demonstrates MID1 to be significantly upregulated [35] (supplementary table s1 p = 0.012).
Our study expands on the previous studies by showing MID1 protein upregulation in IPF lung tissue and characterising the downstream signalling pathways affected by it in the form of reduced phosphatase activity of PP2A. Of particular interest, is the association of PP2A activity levels with a decline in DLco. This suggests that there may be a causal relationship to IPF pathogenesis supporting potential modulation of activity or the negative regulator E3 ubiquitin ligase MID1 as a therapeutic target. In support of this are our observations that AAL(s) treated WT mice and Tnfsf10−/− mice were protected from bleomycin-induced fibrosis. However, these data contrast with the previous McGrath et al., study that demonstrated an increase in bleomycin-induced fibrosis in Tnfsf10−/− mice [12]. McGrath et al., used female mice on a C57BL/6 background while we used male mice on a BALB/c background suggesting the mouse strain or gender may exert a significant influence on the bleomycin model in the absence of TRAIL. This is of interest as IPF is biased towards males though the reason for this is unknown [36]. We have also found that female Tnfsf10−/− mice on a BALB/c background to spontaneously develop lung fibrosis associated with the small airways later in life [28], though these changes themselves may be due to alterations in the microbiome with repeated unfiltered room air exposures in the models used [28, 37]. Together, this suggests that there may be gender specific roles for either inflammatory or apoptotic TRAIL signalling that also play a role in lung fibrosis and is worthy of further investigation. It also highlights that experimental data from murine models need to be interpreted in the context of disease observed in patients where TRAIL and its downstream signalling pathway is augmented.
The complexity of the gender and strain influences observed in in-vivo models lead us to follow a more reductionist approach and conduct in-vitro cell culture experiments. This approach was previously employed to demonstrate that MID1-PP2A signalling is induced in primary human epithelial cells in the presence of recombinant TRAIL [22]. In fibrotic lung models TRAIL has been shown to be broadly expressed by multiple cells types including epithelial cells, endothelial cells, fibroblasts and alveolar macrophages [12, 17, 22, 28, 38]. Here we have demonstrated in cultured primary fibroblasts recombinant TRAIL directly induced both proliferation and the production of collagen in a dose dependent manner (Fig. 6). Furthermore, AAL(s) inhibited collagen expression without affecting Mid1 expression in-vitro. In contrast AAL(s) treated WT mice showed reduced Mid1 expression. This supports our proposal that PP2A is downstream of Mid1 on a cellular level while the effect of PP2A activation on Mid1 expression in-vivo occurs because of a blunted inflammatory and pro-fibrotic lung response involving a complex interaction between multiple cell types that cannot be replicated in isolated cells in-vitro. Thus, this combination of ex-vivo and in-vivo studies add further mechanistic support to a role for Mid1 in the promotion of fibrosis.
The key role for MID1 in fibrosis suggests this pathway may be suitable as a therapeutic target. The FTY720 analogue Fingolimod is undergoing phase 2 and phase 3 clinical trials involving patients with relapsing and relapsing-remittent multiple sclerosis [39], and was generally well tolerated at the approved dose [40]. However, FTY720 in its phosphorylated form is also a sphingosine 1-phospate receptor modulator, which inhibits lymphocyte migration and results in lymphopenia. Therefore, non-phosphorylatable FTY720 analogues such as AAL(s) and small molecules targeting MID1 may be more suitable for clinical trials in IPF patients, should any be available in the future.
A limitation of our study was that the diagnosis of IPF in these participants was reliant on transbronchial biopsies which is not in accordance with current clinical guidelines. To address the unknown specificity of our approach we excluded the possibility of IPF participants being incorrectly classified as controls by using control tissues distal to resected lung cancer in patients with no clinical evidence of IPF or other fibrotic lung disease. In regard to the unknown specificity of using transbronchial biopsies for the diagnosis of IPF our approach could have resulted -if anything- in a smaller difference in MID1 between IPF patients and controls in our study. However, despite this limitation, we found a significant difference in serum TRAIL, tissue MID1 and a trend in PP2A between groups which is suggestive of consistent and robust data. Though further studies utilising tissue from a population with clinical diagnosis according to the most recent Fleischner Society White Paper [41] could now be warranted to precisely identify which patients would most benefit from novel therapeutics targeting MID1.
Conclusions
TRAIL signalling through MID1 deactivates PP2A and promotes fibrosis with corresponding lung-function decline. This may provide novel therapeutic targets for IPF.
Acknowledgements
We would like to thank M. Smyth, Peter MacCallum Cancer Centre and J. Peschon, Amgen for providing Tnfsf10-/- mice and the China Scholarship Council for a travel Scholarship to J Li. We appreciate technical assistance from H. MacDonald, J. Grehan and staff from the HMRI Bioresources facility, staff from Department of Respiratory and Critical Care Medicine, the Second Affiliated Hospital of Jilin University.
Funding
This study was supported by the National Health and Medical Research Council (NH&MRC 1011153) (J.M.) and the International Cooperation Program of Jilin Province (No. 20140414066GH). M.R.S and P.H.M were supported by National Health and Medical Research Council Fellowships. Funding bodies played no role in the design of the study nor collection, analysis, or interpretation of data nor in writing the manuscript.
Availability of data and materials
All materials used are commercially available from the suppliers identified in the methods section. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
AC conceptualised, designed and performed the experiments, analysed data and wrote the manuscript. JL performed experiments, analysed data and edited the manuscript. HDT and JCM synthesized AAL(S) for use as an activator of PP2A and developed the dosing regimen. APdeS, MRS and PMH interpreted data and edited the manuscript. XL collected clinical materials and biopsies and performed clinical experiments with JZ providing supervision and interpretation of clinical data. JM coordinated, designed and supervised experiments, interpreted the data and edited the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Written informed consent was obtained from individual subjects, and all the studies were approved by the Medical Ethics Committee of the Second Affiliated Hospital of Jilin University, Changchun, Jilin, P.R. China.
The Animal Care and Ethics Committee of the University of Newcastle, Australia approved all animal experiments, which were conducted and reported in accordance with the ARRIVE guidelines.
Consent for publication
Not applicable to this publication
Competing interests
A.C. is currently acting as an Associate Editor for BMC Pulmonary Medicine. J.M. is currently acting as a Section Editor for BMC Pulmonary Medicine. The authors have no other competing interests to declare.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Adam M. Collison, Email: Adam.Collison@Newcastle.edu.au
Junyao Li, Email: lijunyao191@126.com.
Ana Pereira de Siqueira, Email: Ana.Pereiradesiqueira@newcastle.edu.au.
Xuejiao Lv, Email: lvxuejiao0311@126.com.
Hamish D. Toop, Email: toophamish@gmail.com
Jonathan C. Morris, Email: Jonathan.Morris@unsw.edu.au
Malcolm R. Starkey, Email: Malcolm.Starkey@newcastle.edu.au
Philip M. Hansbro, Email: Philip.Hansbro@newcastle.edu.au
Jie Zhang, Email: doctorzhangj@sina.com.
Joerg Mattes, Phone: 61-2-40420209, Email: Joerg.Mattes@Newcastle.edu.au.
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daccord C, Maher TM. Recent advances in understanding idiopathic pulmonary fibrosis. F1000Res. 2016. 10.12688/f1000research.8209.1. eCollection 2016. [DOI] [PMC free article] [PubMed]
- 3.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61(11):980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raghu G, Brown KK, Costabel U, Cottin V, du Bois RM, Lasky JA, Thomeer M, Utz JP, Khandker RK, McDermott L, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178(9):948–955. doi: 10.1164/rccm.200709-1446OC. [DOI] [PubMed] [Google Scholar]
- 5.King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 6.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 7.Weckmann M, Collison A, Simpson JL, Kopp MV, Wark PAB, Smyth MJ, Yagita H, Matthaei KI, Hansbro N, Whitehead B, et al. Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nat Med. 2007;13(11):1308–1315. doi: 10.1038/nm1660. [DOI] [PubMed] [Google Scholar]
- 8.Singhera GK, Chan TS, Cheng JY, Vitalis TZ, Hamann KJ, Dorscheid DR. Apoptosis of viral-infected airway epithelial cells limit viral production and is altered by corticosteroid exposure. Respir Res. 2006;7:78. doi: 10.1186/1465-9921-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halaas O, Vik R, Ashkenazi A, Espevik T. Lipopolysaccharide induces expression of APO2 ligand/TRAIL in human monocytes and macrophages. Scand J Immunol. 2000;51:244–250. doi: 10.1046/j.1365-3083.2000.00671.x. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, Wang D, Liu D, Liu M, Ge Y, Jiang M, Liu Y, Zheng D. Tumor necrosis factor-related apoptosis-inducing ligand induces the expression of proinflammatory cytokines in macrophages and re-educates tumor-associated macrophages to an antitumor phenotype. Mol Biol Cell. 2015;26(18):3178–3189. doi: 10.1091/mbc.e15-04-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arbour N, Rastikerdar E, McCrea E, Lapierre Y, Dorr J, Bar-Or A, Antel JP. Upregulation of TRAIL expression on human T lymphocytes by interferon b and glatiramer acetate. Mult Scler. 2005;11:652–657. doi: 10.1191/1352458505ms1222oa. [DOI] [PubMed] [Google Scholar]
- 12.McGrath EE, Laurie A, Marriott HM, Mercer P, Cross SS, Arnold N, Singleton V, Thompson AA, Walmsley SR, Renshaw SA, Sabroe I, Chambers RC, Dockrell DH, Whyte MK. Deficiency of tumour necrosis factor-related apoptosis-inducing ligand exacerbates lung injury and fibrosis. Throax. 2012;67:796–803. doi: 10.1136/thoraxjnl-2011-200863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collison A, Foster PS, Mattes J. Emerging role of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) as a key regulator of inflammatory responses. Clin Exp Pharmacol Physiol. 2009;36(11):1049–1053. doi: 10.1111/j.1440-1681.2009.05258.x. [DOI] [PubMed] [Google Scholar]
- 14.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5(2):157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 15.Pan G, O'Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 16.Collison AM, Sokulsky LA, Sherrill JD, Nightingale S, Hatchwell L, Talley NJ, Walker MM, Rothenberg ME, Mattes J. TNF-related apoptosis-inducing ligand (TRAIL) regulates midline-1, thymic stromal lymphopoietin, inflammation, and remodeling in experimental eosinophilic esophagitis. J Allergy Clin Immunol. 2015;136(4):971–982. doi: 10.1016/j.jaci.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collison A, Li J, Pereira de Siqueira A, Zhang J, Toop HD, Morris JC, Foster PS, Mattes J. Tumor necrosis factor-related apoptosis-inducing ligand regulates hallmark features of airways remodeling in allergic airways disease. Am J Respir Cell Mol Biol. 2014;51(1):86–93. doi: 10.1165/rcmb.2013-0490OC. [DOI] [PubMed] [Google Scholar]
- 18.Starkey MR, Nguyen DH, Essilfie AT, KimRY HM, Collison AM, Yagita H, Foster PS, Horvat JC, Mattes J, Hansbro PM. Tumor necrosis factor-related apoptosis-inducing ligand translates neonatal. Mucosal Immunol. 2013;18(10):65. doi: 10.1038/mi.2013.65. [DOI] [PubMed] [Google Scholar]
- 19.McConnell JL, Watkins GR, Soss SE, Franz HS, McCorvey LR, Spiller BW, Chazin WJ, Wadzinski BE. Alpha4 is a ubiquitin-binding protein that regulates protein serine/threonine phosphatase 2A ubiquitination. Biochemistry. 2010;49(8):1713–1718. doi: 10.1021/bi901837h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S. MID1, mutation in Opitz syndrome, encodes an ubiquitin ligase that targets phophatase 2A for degradation. Nat Genet. 2001;29:287–294. doi: 10.1038/ng762. [DOI] [PubMed] [Google Scholar]
- 21.Watkins GR, Wang N, Mazalouskas MD, Gomez RJ, Guthrie CR, Kraemer BC, Schweiger S, Spiller BW, Wadzinski BE. Monoubiquitination promotes calpain cleavage of the protein phosphatase 2A (PP2A) regulatory subunit alpha4, altering PP2A stability and microtubule-associated protein phosphorylation. J Biol Chem. 2012;287(29):24207–24215. doi: 10.1074/jbc.M112.368613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collison A, Hatchwell L, Verrills N, Wark PA, de Siqueira AP, Tooze M, Carpenter H, Don AS, Morris JC, Zimmermann N, et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat Med. 2013;19(2):232–237. doi: 10.1038/nm.3049. [DOI] [PubMed] [Google Scholar]
- 23.Sim AT, Ludowyke RI, Verrills NM. Mast cell function: regulation of degranulation by serine/threonine phosphatases. Pharmacol Ther. 2006;112(2):425–439. doi: 10.1016/j.pharmthera.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Cornell TT, Hinkovska-Galcheva V, Sun L, Cai Q, Hershenson MB, Vanway S, Shanley TP. Ceramide-dependent PP2A regulation of TNFalpha-induced IL-8 production in respiratory epithelial cells. Am J Physiol. 2009;296(5):L849–L856. doi: 10.1152/ajplung.90516.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shanley TP, Vasi N, Denenberg A, Wong HR. The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol. 2001;166(2):966–972. doi: 10.4049/jimmunol.166.2.966. [DOI] [PubMed] [Google Scholar]
- 26.Miskolci V, Castro-Alcaraz S, Nguyen P, NVancura A, Davidson D, Vancurova I. Okadaic acid induces sustained activation of NFkB and degradation of the nuclear IkBa in human neutrophils. Arch Biochem Biophys. 2003;417:44–52. doi: 10.1016/S0003-9861(03)00336-9. [DOI] [PubMed] [Google Scholar]
- 27.Liu G, Cooley MA, Jarnicki AG, Hsu AC, Nair PM, Haw TJ, Fricker M, Gellatly SL, Kim RY, Inman MD et al. Fibulin-1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI Insight. 2016;1(9):e86380. 10.1172/jci.insight.86380. [DOI] [PMC free article] [PubMed]
- 28.Haw TJ, Starkey MR, Nair PM, Pavlidis S, Liu G, Nguyen DH, Hsu AC, Hanish I, Kim RY, Collison AM, et al. A pathogenic role for tumor necrosis factor-related apoptosis-inducing ligand in chronic obstructive pulmonary disease. Mucosal Immunol. 2015. [DOI] [PubMed]
- 29.Broide DH. Allergen-induced peribronchial fibrosis and mucus production mediated by I B kinase -dependent genes in airway epithelium. Proc Natl Acad Sci. 2005;102(49):17723–17728. doi: 10.1073/pnas.0509235102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francois Denizot RL. Rapid colorimetric assay for cell growth and survival, modifications to the tetrazolium dy procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 31.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 32.Girkin JL, Hatchwell LM, Collison AM, Starkey MR, Hansbro PM, Yagita H, Foster PS, Mattes J. TRAIL signaling is proinflammatory and proviral in a murine model of rhinovirus 1B infection. Am J Physiol Lung Cell Mol Physiol. 2017;312(1):L89–L99. doi: 10.1152/ajplung.00200.2016. [DOI] [PubMed] [Google Scholar]
- 33.Sokulsky LA, Collison AM, Nightingale S, Le Fevre A, Percival E, Starkey MR, Hansbro PM, Foster PS, Mattes J. TRAIL deficiency and PP2A activation with salmeterol ameliorates egg allergen driven eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol. 2016;00151:02016. doi: 10.1152/ajpgi.00151.2016. [DOI] [PubMed] [Google Scholar]
- 34.Akram KM, Lomas NJ, Forsyth NR, Spiteri MA. Alveolar epithelial cells in idiopahtic pulmonary fibrosis display upregulation of TRAIL, DR4 and DR5 expression with simultaneous preferential over-expression of pro-apoptotic marker p53. Int J Clin Exp Pathol. 2014;7(2):552–564. [PMC free article] [PubMed] [Google Scholar]
- 35.Schiller HB, Mayr CH, Leuschner G, Strunz M, Staab-Weiinitz C, Preisendorfer S, Eckes B, Moinzadeh P, Krieg T, Schwartz DA, Hatz RA, Behr J, Mann M, Eickelberg O. Deep proteome progiling reveals common prevalence of MZB1-positive plasma B cells in human lung and skin fibrosis. AJRCCM. 2017;196(10):1238–1240. doi: 10.1164/rccm.201611-2263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol. 2013;5:483–492. doi: 10.2147/CLEP.S54815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, Hansbro PM. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- 38.Starkey MR, Nguyen DH, Essilfie AT, Kim RY, Hatchwell LM, Collison AM, Yagita H, Foster PS, Horvat JC, Mattes J, et al. Tumor necrosis factor-related apoptosis-inducing ligand translates neonatal respiratory infection into chronic lung disease. Mucosal Immunol. 2014;7(3):478–488. doi: 10.1038/mi.2013.65. [DOI] [PubMed] [Google Scholar]
- 39.Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen JA, Chun J. Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69(5):759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- 41.Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, Goldin JG, Hansell DM, Inoue Y, Johkoh T, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner society white paper. Lancet Respir Med. 2018;6(2):138–153. doi: 10.1016/S2213-2600(17)30433-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All materials used are commercially available from the suppliers identified in the methods section. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.