

Abstract
Background
The leishmaniasis are parasitic diseases caused by protozoans of the genus Leishmania, highly divergent eukaryotes, characterized by unique biological features. To survive in both the mammalian hosts and insect vectors, these pathogens make use of a number of mechanisms, many of which are associated with parasite specific proteases. The metalloprotease GP63, the major Leishmania surface antigen, has been found to have multiple functions required for the parasite’s survival. GP63 is encoded by multiple genes and their copy numbers vary considerably between different species and are increased in those from the subgenus Viannia, including L. braziliensis.
Results
By comparing multiple sequences from Leishmania and related organisms this study sought to characterize paralogs in silico, evaluating their differences and similarities and the implications for the GP63 function. The Leishmania GP63 genes are encoded on chromosomes 10, 28 and 31, with the genes from the latter two chromosomes more related to genes found in insect or plant parasites. Those from chromosome 10 have experienced independent expansions in numbers in Leishmania, especially in L. braziliensis. These could be clustered in three groups associated with different mRNA 3′ untranslated regions as well as distinct C-terminal ends for the encoded proteins, with presumably distinct expression patterns and subcellular localizations. Sequence variations between the chromosome 10 genes were linked to intragenic recombination events, mapped to the external surface of the proteins and predicted to be immunogenic, implying a role against the host immune response.
Conclusions
Our results suggest a greater role for the sequence variation found among the chromosome 10 GP63 genes, possibly related to the pathogenesis of L. braziliensis and closely related species within the mammalian host. They also indicate different functions associated to genes mapped to different chromosomes. For the chromosome 10 genes, variable subcellular localizations were found to be most likely associated with multiple functions and target substrates for this versatile protease.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5465-z) contains supplementary material, which is available to authorized users.
Keywords: Leishmania braziliensis, GP63, Virulence proteins
Background
The leishmaniasis are parasitic infectious diseases caused by flagellated protozoa belonging to the genus Leishmania, family Trypanosomatidae, and which are transmitted by sandflies of the genera Phlebotomus or Lutzomyia. These diseases are found as two major clinical forms, named as cutaneous leishmaniasis (CL) and visceral leishmaniasis (VL), with a global incidence for each in the range of hundreds of thousands of cases per year [1]. Multiple Leishmania species are associated with the leishmaniasis and distinct species, closely related or not, are responsible for the disease in different parts of the world. Those belonging to the subgenus Viannia are restricted to the New World (including L. braziliensis and L. guyanensis), have evolved separately from better known species belonging to the subgenus Leishmania (L. major, L. infantum, L. mexicana and others) and are associated with the mucocutaneous leishmaniasis (MCL), a more aggressive variation of CL [2].
As successful pathogens, the various Leishmania species have developed effective mechanisms to escape the mammalian host immune response and proliferate [3, 4]. Some of these evasion mechanisms are dependent on proteases, which help ensure that the parasites can invade the mammalian tissue, survive, differentiate and multiply [5]. The GP63 protease, also known as leishmanolysin or major surface protease (MSP), was first discovered in 1980 as the major surface antigen of the promastigote form of many species of Leishmania [6]. It was later found to be bound to the cell membrane through a GlycosylPhosphatidylInositol (GPI) anchor and was also identified as an important virulence factor. This is a zinc-dependent metalloproteinase, which belongs to the peptidase family M8 and the metzincin class and includes conserved features such as the motif HEXXHXXGXXH and a pro-peptide located in the protein’s N-terminal region that renders the proenzyme inactive during translation and is removed during its maturation and activation. The GP63 proteins also include an N-terminal signal sequence which directs them to the endoplasmic reticulum and to the Leishmania secretory pathway [7, 8].
GP63 has been found to play multiple roles during Leishmania infection in mammals, starting in the extracellular environment where it acts inactivating the complement cascade, by cleaving C3b into iC3b. This inactivation prevents the formation of the membrane attack complex (MAC), despite allowing the opsonisation of the Leishmania, mediated by iC3b, and facilitating its phagocytosis. GP63 can also facilitate the binding of the parasite to the macrophage through fibronectin receptors, cleaving proteins from the host’s extracellular matrix. Within the macrophages, it also acts to reduce the production of TNF, IL-12 and nitric oxide, which contributes to the protection and survival of the parasite, and provides the Leishmania with a faster entry into the macrophage, through the activation of a host tyrosine phosphatase [7, 9, 10]. GP63 has also been shown to be released through exosomes into the extracellular medium and this may facilitate its uptake by the macrophage even before the internalization of the Leishmania parasite [11]. Lack of GP63 drastically reduces the Leishmania’s ability to establish and maintain an infection, since the hosts are more likely to induce an innate immunity inflammatory response [12]. Within the host cell cytoplasm, GP63 has been shown to cleave the transcriptional factor AP-1, which regulates the production of pro-inflammatory cytokines and nitric oxide by the macrophage [11, 13]. GP63 was also shown to be associated with the inactivation of the mTOR kinase, leading to the inhibition of protein synthesis in the macrophage and providing an ideal environment for the proliferation of the pathogen [14].
Early studies have shown that GP63 is more abundantly expressed in the promastigote stage of the Leishmania life cycle, the proliferative stage within the insect vector. This expression may peak during metacyclogenesis, when the parasite prepares to infect the mammalian hosts, and is subsequently reduced again upon differentiation into amastigotes, the intracellular stage that multiplies within the mammalian macrophages [7, 15, 16]. The abundant GP63 expression in promastigotes indicates relevant functions also in the insect vector, presumably needed for survival and proliferation. Indeed, a potential involvement in the degradation of protein components that would lead to the adhesion of the parasite in the insect gut epithelium has been shown [17, 18]. Due to its wide substrate specificity, GP63 may also perform a nutritional role for the parasite, acting as an endopeptidase [19, 20], or even protect the Leishmania against the insect defences [19].
Concerning the GP63 gene organization, there is a noticeable variation in the number of gene copies encoding these proteins among different Leishmania species. In L, major these genes are present in more than one chromosome and multiple copies have been detected arranged in tandem [21], with the same multi-copy arrangement also found in L. infantum and L. braziliensis [22]. Noteworthy, however, is the substantial increase in the number of gp63 genes reported for L. braziliensis and other species belonging to the subgenus Viannia, when compared with the subgenus Leishmania. This was reported in early studies [23–25] and has been confirmed more recently by results derived from a screening for cosmids harboring multiple GP63 genes from L. braziliensis [26], as well as by genome sequencing data for different Leishmania species [27–29]. No clear biological reasons are known, however, to explain this expansion in the GP63 gene copy number. Here, aiming to contribute further to the understanding of the role of GP63 in Leishmania pathogenesis in general but with a focus on Viannia species, we sought to investigate the GP63 gene expansion further, using a range of in silico tools. We started by better defining the extent of GP63 gene diversity in L. braziliensis, followed by an in-depth analysis of the similarities and differences between different genes from this and related Leishmania species. The GP63 genes were first grouped according to their chromosomal localization followed by phylogenetic comparisons between different trypanosomatid species. Further grouping according to sequence similarities or differences within non-coding and coding elements was also carried out, in order to define putative functional distinctions. Possible mechanisms associated with the gene expansion due to DNA recombination were then investigated and variations in sequence mapped on the GP63 structure and linked with predictions of immunogenic potential. Our results are consistent with a selective expansion of a subset of GP63 genes in L. braziliensis that might be linked to mammalian pathogenesis and might be required for a better protection against the host immune system.
Results
Search for new L. braziliensis GP63 paralogs
The early studies based on hybridization assays [23, 24] had suggested that the total number of GP63 genes found within Leishmania species belonging to the Viannia subgenus is greater than the number of genes available at the TriTrypDB database and identified after the L. braziliensis genome sequencing and annotation. Recent data based on next generation sequencing have also suggested major variations in copy number of GP63 genes between species within the same subgenus, Leishmania or Viannia, that have not yet been included on the annotated genomes [28–30]. Here, to begin to understand the true diversity of the L. braziliensis GP63 genes, we first sought to reevaluate the available L. braziliensis GP63 gene sequences considering that the automatic annotation methods might have missed further genes. We therefore performed a reanalysis of the L. braziliensis genome sequences and searched for possible new GP63 paralogs that might not have been annotated. To do this we performed a search in the L. braziliensis genome using the Hidden Markov Models (HMMs) methodology [31], carried out after a grouping of the entire proteome set from different Leishmania species (described in methods). Nine subsets of GP63 sequences were created using the OrthoMCL tool in order to group these sequences and allow the search to be performed, as shown in Additional file 1: Table S1, with the number of genes in each subset varying in size from 56 to only two. All nine subsets were used to build HMMs and these were then applied for the search of new paralogs in the predicted proteome from L. braziliensis 2904. In general, all HMMs were able to find the GP63 sequences assigned to each subset, however no new paralogs were found during the search. The genome of L. braziliensis strain 2904 deposited on TriTrypDB lists 39 GP63 genes and, in total, the HMMs identified the presence of 38 related sequences. A single gene (LbrM.10.1720) was not recovered using these models and indeed its coding sequence did not provide an alignment with a score high enough to be considered as a GP63. The results of the search for each HMM are summarized in Additional file 2: Table S2 and confirm the gene count number for GP63 genes derived from the L. braziliensis genome sequencing, 38 genes, lower than earlier estimates based on the hybridization studies [23].
Next, we considered that the shot gun nature of the sequencing strategy used for the assembly of the best genome available from a Viannia species, from the L. braziliensis 2904, might have led to the grouping of similar GP63 genes together, causing in turn a reduction in the number of genes found. In order to obtain as many natural GP63 sequences as possible, and therefore have a clearer idea of the true number of genes present in the L. braziliensis genome, we opted to amplify these genes using primers directed to conserved regions of representative genes identified in the genome analysis. The PCR strategy used to amplify the GP63 genes can be seen in Fig. 1, superimposed on a schematic representation of a typical GP63. The scheme highlights conserved elements found on all GP63 sequences, such as the zinc-binding motif, multiple cysteine residues, the GPI anchor site and a nearly universally conserved motif of seven consecutive amino acids we named KDELMAP. Six oligonucleotides annealing to sequences encoding the N-terminal ends of the GP63 sequences were used as 5′ primers, considering the variation previously observed within the N-terminus of the various GP63 genes and in order to maximize the number of genes amplified. As 3′ primers, two sets of two oligonucleotides annealing to the more conserved KDELMAP or the GPI anchor site motifs were alternatively used. Individual PCR reactions were set up with different pairs of oligonucleotides, always with a single 5′ and a single 3′ primer. After amplification, cloning and sequencing a total of 40 different GP63 gene fragments were obtained, with thirty-four of those having sequences different from the ones described in the databases. The new GP63 DNA fragments obtained by PCR are listed in Additional file 3: Table S3, which also includes the set of oligonucleotide primers used to amplify each sequence. In all 31 new gene fragments were found, since some were duplicates (also indicated in the table). The new sequences have all been submitted to GenBank and were compared with the already known GP63 genes from L. braziliensis and these analyses will be discussed further below. They are consistent with a higher copy number for the L. braziliensis GP63 genes than predicted based on the genome sequencing alone and more in agreement with the original estimates based on Southern-blots, although no precise quantification is possible either way. Despite the fact that both the genomic and PCR data used for our analyses are derived from the same L. braziliensis 2904 strain, the question of culturing in different laboratories being responsible for the differences observed regarding gene copy number and identification between the two sets of results can be raised. Nevertheless, considering the limited time frame for the culturing procedures, these could only have any impact on gene or chromosomal duplication events and would not lead to the different gene sequences that were found through PCR and/or DNA sequencing.
Fig. 1.

GP63 general gene features. The scheme highlights conserved elements found on most GP63 sequences, such as the signal peptide, propeptide, zinc-binding motif [8], multiple cysteine residues, the GPI anchor site and a nearly universally conserved motif of seven consecutive amino acids we named KDELMAP. The PCR strategy used to amplify the GP63 genes is also shown with products varying from 1 Kb (up to the KDELMAP region) and 1,6 Kb (finishing on the GPI anchor site)
Genomic analysis of known Leishmania GP63 genes
To clarify the relationship between the multiple GP63 genes in Leishmania, we opted to review their chromosomal organization within the major lineages of pathogenic Leishmania. For L. major, the best studied of the available Leishmania genome sequences, three sets of GP63 genes were found distributed in chromosomes 28 (one gene), 31 (one gene) and 10 (four genes) [21]. Based on the sequences available at the Tritryp database, a similar organization is also observed for L. infantum (summarized in Fig. 2) and L. mexicana (not shown), represented by two and one genes for chromosome 28 in L. infantum and L. mexicana, respectively, and one gene for chromosomes 31 in both species, considering that the L. mexicana chromosome 30 is equivalent to the L. major chromosome 31. Five GP63 genes are also found in chromosome 10 for both species and in agreement with previously reported data for L. infantum [22].
Fig. 2.

Genomic organization of Leishmania GP63 genes. Genomic organization of GP63 genes from Leishmanis sp., showing the general distribution, location and synteny of these genes along chromosomes 10, 28 and 31. a Synteny analysis on chromosome 28 showing the localization of the GP63 gene in L. major and L. infantum and its absence in the equivalent position from the L. braziliensis chromosome 28. b Distribution of GP63 genes in Leishmania sp., chromosome 31. Pseudogenes from the L. braziliensis genome are highlighted in gray. c Expansion of GP63 genes in the L. braziliensis chromosome 10, when compared to L. major and L. infantum, within the same chromosome position, as indicated by the flanking genes on the left (some unrelated genes are also included). The horizontal arrows indicate the transcription sense of the genes and the yellow, black and red colors define the 3’ UTR groups identified for the L. major and L. infantum genes. The diagram for L. infantum does not include the preliminary annotation derived from the recent resequencing of its genome [30]. For the L. braziliensis genes, identical colors group those with similar 3’UTRs, as classified in Table 1
In L. braziliensis, based on the available genomic data for the 2904 strain, major differences in the organization of the GP63 genes are observed when they are compared with those found in species from the Leishmania subgenus. First, no GP63 gene is found on chromosome 28, as highlighted before for other Viannia species [32], despite the presence of orthologues to the same genes flanking the single GP63 sequence from L. major and L. infantum. In contrast, six GP63 genes or gene fragments are found on chromosome 31, again generally flanked by orthologues to the same genes found flanking the GP63 gene found in the L. major and L. infantum chromosome 31. Even more noteworthy, however, are the 33 GP63 genes found clustered on chromosome 10. Again, these are localized to the same region seen harboring the other Leishmania chromosome 10 genes, as confirmed by the presence of neighboring sequences encoding orthologues to those found flanking the L. major and L. infantum GP63 genes from chromosome 10. However, the precise gene organization cannot be properly defined and many of the genes sequenced are assembled in relatively short contigs, as indicated in the scheme from Fig. 2c. Again, this might be due to the high similarity between the gene sequences and the nature of the sequencing strategy which might have prevented a proper assembly of repeated sequences.
The significantly greater number of L. braziliensis GP63 genes from chromosome 10 is supported by our PCR data where primers sets directed to the chromosome 10 genes were able to amplify more genes than the ones originally used for their synthesis. For example, a primer pair designed to amplify the gene LbrM.10.0470 allowed the amplification of eight different gene fragments (G0510B2; G0560B1; G0560B2; G1610B3; G1610B4; G1610B5; G1610B6; G1620B1) and similarly, the primer pair directed to gene LbrM.10.0540 amplified fragments from six different genes (G0510C1; G0510C2; G0540C1; G0560C4; G1640C1; G1640C2). In contrast, two sets of PCR reactions directed to a single GP63 gene from chromosome 31, using the same 5′ primer and two distinct 3′ primers, only led to the amplification of the same gene, LbrM.31.2260 (Additional file 3: Table S3). Indeed, we believe that most of the six GP63 genes annotated from the L. braziliensis chromosome 31 might not exist and in fact are either pseudogenes or derived from genome assembly errors. Only one of those genes (LbrM.31.2260) has GP63-related protein features, such as the propeptide domain (HEXXH), and shares a high similarity (85%) with the L. major and L. infantum chromosome 31 genes. The LbrM.31.2200, LbrM.31.2220, LbrM.31.2230, LbrM.31.2240 and LbrM.31.2250 genes have stop codons in the middle of their sequences and/or in alignments showed identical N-terminal or C-terminal regions to LbrM.31.2260 (data not shown). It is possible that these genes may represent parts of LbrM.31.2260 not properly assembled and this in agreement with our PCR data finding only LbrM.31.2260. Overall these results are consistent with the expansion in the number of GP63 genes in L. braziliensis, and other species belonging to the Viannia subgenus, being mainly directed to the chromosome 10 genes.
GP63 evolutionary analyses
Sequences encoding GP63 related genes are also found in other trypanosomatids and more distantly related kinetoplastids and these include multiple genes from T. brucei, T. cruzi, and others. The number of genes in these parasites is quite variable. T. cruzi has over 150 GP63 genes annotated in the TriTrypDB database, but with many pseudogenes among them. T. brucei and C. fasciculata have a smaller amount with 10 and 18 genes respectively. This multiplicity of GP63 genes along the various kinetoplastids lineages reinforce the multiple roles this protein has, independent of the life cycle of the organism involved. Here, we next sought to assess how the Leishmania GP63 genes are related to those found in more distantly related kinetoplastids and whether some function can be inferred based on which genes are found in each organism. To do this we built a phylogenetic tree comparing the most divergent and representative sequences from the three major sets of Leishmania GP63 genes (from chromosomes 10, 28 and 31) with genes from different Trypanosoma species (T. brucei, T. cruzi and T. theileri) and more distantly related organisms. These included species that parasitize reptiles (L. tarentolae) and plants (Phytomonas sp.), have monoxenous life-cycles in insects (Crithidia fasciculata and Leptomonas pyrrhocoris) and are free living (Bodo saltans). As shown in Fig. 3, the phylogenetic analysis could separate the GP63 genes mapped to the Leishmania chromosome 10 from those genes mapped to chromosomes 28 and 31. We also could observe a clear separation between the Leishmania subgenus based on the genes located on chromosome 10. Noteworthy are the T. cruzi and T. brucei GP63 sequences more closely associated with the GP63 genes from B. saltans and T. theileri. Also, when we observe the clustering of the genes present in chromosomes 28 and 31 from Leishmania, they generally show more proximity to the genes from L. pyrrhocoris, C. fasciculata and Phytomonas sp. Nevertheless, one L. pyrrhocoris and two C. fasciculata genes are more closely related to those from the L. braziliensis chromosome 10.
Fig. 3.

Phylogenetic tree comparing selected Leishmania GP63 paralogs with sequences from more distantly related organisms. The tree demonstrates the proximity between the GP63 genes from the Leishmania chromosomes 28 and 31, with GP63 genes from monoxenes trypanosomatids genes, such as Leptomonas pyrrhocoris and Crithidia fasciculata, as well as, genes from a plant tripanosomatid, like Phytomonas sp. Values for highly supported nodes have been replaced by black and white squares, which represents the Bayesian posterior probabilities and bootstrap support for PhyML, respectively. Bs: Bodo saltans, Tt: Trypanosoma theileri, Tb: Trypanosoma brucei, Tc: Trypanosoma cruzi, Lp: Leptomonas pyrrhocoris, Cf: Crithidia fasciculata, Lb: Leishmania braziliensis M2904, Lmj: Leishmania major, Lmx: Leishmania mexicana, Li: Leishmania infantum, Lt: Leishmania tarentolae
Overall, the gene clusters shown in the tree highlight the higher similarity between the Leishmania sp. genes from chromosomes 28 and 31 with the GP63 genes found in organisms that live in insects only or parasitize plants. For instance, the 38 annotated GP63 genes from Phytomonas are more closely related to the Leishmania chromosome 28 GP63. It is then possible to hypothesize that these genes might me more involved in the insect stage of the parasite life cycle. Genes more closely related to the chromosome 10 GP63 genes can be found in the insect parasites L. pyrrhocoris and C. fasciculate, but in general these genes seem to have suffered a substantial expansion within Leishmania species.
Evaluation of the sequence diversity of the Leishmania GP63 genes from chromosome 10
Considering the expansion of the chromosome 10 GP63 genes in the Leishmania lineage in general, and even more so in L. braziliensis and other Viannia species, we then opted to investigate the origins of their diversity further. To do this we compared the full extent of the chromosome 10 GP63 sequences from relevant Leishmania species using as outgroups selected genes from chromosomes 28 and 31. For this analysis, we also included sequences from L. tarentolae (based on the published genome sequence [33]), where a similar expansion in the chromosome 10 GP63 genes was noticed, with 49 genes found in this chromosome while only one gene was found in chromosome 31 and another in chromosome 33. L. tarentolae is currently classified within the Sauroleishmania subgenus, but it is likely to be more closely related to the Leishmania subgenus than to Viannia [2, 34]. The relevance in including the L. tarentolae sequences is due to the fact that it does not parasitize mammals, only lizards, meaning that any potential role in pathogenesis associated with the chromosome 10 GP63 genes is not dependent on their mammalian hosts. We also included in this analysis the new L. braziliensis sequences generated by us through the PCR approach. The phylogenetic tree shown in Fig. 4 summarizes the results from these analyses based on alignments using the full-length sequences for all proteins (or the full-length PCR fragments). For clarity, only the most divergent representative sequences were used to build this tree, with those very similar or nearly identical to the ones shown purportedly removed from the final figure. In the original analysis all chromosome 10 sequences from the selected species were used but with similar results (not shown). Within each of the three Leishmania subgenera analysed, all GP63 sequences from chromosome 10 are more closely related to sequences from the same or related species than to sequences found in species belonging to the other subgenera. Even within the Leishmania clades, the L. infantum genes (in red) seemed to be more closely related to each other than to their L. major counterparts, although for the two Viannia species (L. braziliensis and L. guyanensis) analysed genes (in green) more closely related between the two species were found. These results are in agreement with independent expansions on the number of the chromosome 10 GP63 sequences in each clade, with major expansions occurring for both Sauroleishmania (in blue) and Viannia species. For the latter species, at least, the start of this expansion may have preceded the split between L. braziliensis and L. guyanensis but has subsequently continued and may be an ongoing process.
Fig. 4.

Phylogeny of Leishmania GP63 paralogs from chromosome 10. Phylogenetic tree comparing multiple chromosome 10 GP63 genes from selected Leishmania species. The tree highlights the separation of the GP63 genes according to the three main Leishmania subgenera (Sauroleishmania, Leishmania and Viannia). Values for highly supported nodes have been replaced by black and white squares, which represents the Bayesian posterior probabilities and bootstrap support for PhyML, respectively. Lb: Leishmania braziliensis M2904, Lmj: Leishmania major, Lmx: Leishmania mexicana, Li: Leishmania infantum, Lt: Leishmania tarentolae, Lg: Leishmania guyanensis
Identification of functional differences between the various Leishmania GP63 genes from chromosome 10
So far not much is known regarding possible functional differences between the various GP63 genes found within any specific trypanosomatid. In Leishmania, with the goal of defining functional differences between multiple GP63, and even prior to the completion of the first Leishmania genomes, early studies investigated the expression pattern of selected genes attempting to identify differences in expression during the parasite life cell cycle [8, 15, 35]. In L. infantum and L. major, three growth stage-specific patterns of expression were observed for the then known GP63 genes, with one gene constitutively expressed, a second gene (or genes) expressed during the log phase of promastigote growth and the third gene expressed only in stationary phase cells and/or amastigotes [15, 25]. Here, by comparing their coding sequences and 3’ UTRs with those from the available genomes, we were able to map those genes within the annotated genome sequences (indicated in Fig. 2): LinJ.10.0510 and LmjF.10.0470 are the constitutively expressed genes (here named Group 1 – colored in black in the figure); LinJ. 10.0490/LinJ.10.0500 and LmjF. 10.0460/LmjF.10.0465 are equivalent to the previously described log phase promastigote genes (Group 2 – highlighted in yellow); LinJ. 10.0520/LinJ.10.0530 from L. infantum and the L. major LmjF.10.0480 gene correspond to the stationary phase/amastigote specific GP63 (Group 3 – colored in red). The Group 1 genes are characterized by unique 3’ UTRs and 3′ intergenic regions absent from the remaining chromosome 10 genes, while the Group 2 and 3 genes share very similar sequences within the first ~ 400 nucleotides of their 3’UTRs, although these subsequently diverge into two distinct patterns that correlate with the two groups (these genes and their groups are highlighted by different colors in the scheme from Fig. 2). We also looked at differences within the coding sequences that could be typical of GP63 genes belonging to any particular group. As previously reported for L. major [25], a clear distinction is observed between the C-terminus of the Group 1 proteins and those from Group 2 and 3. Both L. major and L. infantum Group 1 proteins are characterized by a longer C-terminus enriched in hydrophobic and positively charged residues and lack the typical asparagine required for the GPI anchor. In contrast, the shorter C-terminus from the Group 2 and 3 proteins include the GPI anchor site and end in a stretch of mostly hydrophobic amino acids (Fig. 5).
Fig. 5.

Leishmania major, L. infantum and L. braziliensis C-terminal alignments and groups. Alignment showing the difference in the C-terminuses of the chromosome 10 GP63 sequences having divergent 3’ UTRs. a and b shows the L. major and L. infantum C-terminus sequences, respectively, their groups based on their expression profiles and the predicted GPI anchor site (boxed in green). The motif identified in the constitutively expressed L. infantum group 1 sequence also found in the L. braziliensis group 1 C-terminus is boxed in red, while the motif found in the L. infantum and L. major stage specific genes from Groups 2 and 3 nearly identical to a similar motif from the L. braziliensis group 3 genes is boxed in orange. c Alignment of the C-terminal ends of the different L. braziliensis chromosome 10, with the grouping described in the text and the conserved elements also seen in L. major and L. infantum indicated as for A and B
Next, we attempted to group the L. braziliensis chromosome 10 genes based first on similarities and differences within the putative 3’UTRs. Thirty genes were analyzed based on the sequences available from the reference genome sequence and these were classified into six groups according to their 3’UTR, with the first two groups represented by 8 and 15 genes, respectively, while the remaining groups included only one or two genes (Table 1 – also colored differently in Fig. 2). When these 3’UTRs were compared with the three L. infantum and L. major groups no clear similarities were found with any of the L. braziliensis groups, likely due to the large sequence variation observed between species from the two distinct subgenera. We also looked at the amino acid sequences, looking for features in common for L. braziliensis genes sharing similar UTRs. No association with features such as signal peptide, transmembrane domains and isoelectric points was found, however distinct C-terminus were observed which also separated the L. braziliensis into six groups, with a clear association observed between each 3’UTR group and nearly all of the proteins’ C-terminal ends (Table 1). Since Groups 4, 5 and 6 consists of truncated proteins they will not be considered further here, but for the remaining three groups their C-terminal ends were also compared with those seen to be associated with the L. infantum and L. major groups. A clear association between the two proteins from the L. braziliensis Group 3 and the L. major/L. infantum Groups 2 and 3 can be seen, since they share a nearly identical C-terminus that includes the GPI anchor attachment motif (DGGN [36]). The Group 1 genes from L. braziliensis also share conserved elements with the L. major/L. infantum Group 1, such as the lack of a typical GPI anchor site and the presence of a hydrophilic set of amino acids followed by a hydrophobic region resembling a transmembrane segment. The motif “MRQWRERMTALATVT” found in the L. braziliensis sequences is also very similar to the “MQRWNDRMAGLATAA” motif found in L. infantum LinJ.10.0510 gene. Only the L. braziliensis Group 2 genes then, characterized by a shorter C-terminus missing entirely the GPI anchor site or related hydrophobic sequences, do not seem to have counterparts in L. infantum nor in L. major. Nevertheless, it seems likely that, as observed in L. infantum and L. major, the different groups of L. braziliensis chromosome 10 GP63 genes are also differentially regulated during the parasite growth in culture and this is in agreement with the different 3’UTRs seen associated with each group.
Table 1.
Leishmania braziliensis GP63 3’ UTR and C-terminal end gene groups. Table showing the Gp63 gene groups from chromosome 10 defined according to the 3’ UTR and C-terminuses sequence similarity
| Group | L. braziliensis 3’UTRs | L. braziliensis chromosome 10 GP63 paralogs |
|---|---|---|
| 1 | LbrM.10.0510, LbrM.10.0530, LbrM.10.0560, LbrM.10.0580, LbrM.10.0600, LbrM.10.0610, LbrM.10.1540, LbrM.10.1580 | LbrM.10.0510, LbrM.10.0530, LbrM.10.0550, LbrM.10.0560, LbrM.10.0600, LbrM.10.0610, LbrM.10.1540, LbrM.10.1580 |
| 2 | LbrM.10.0470, LbrM.10.0480, LbrM.10.0500, LbrM.10.0520, LbrM.10.0540, LbrM.10.0570, LbrM.10.1570, LbrM.10.1590, LbrM.10.1610, LbrM.10.1620, LbrM.10.1630, LbrM.10.1640, LbrM.10.1650, LbrM.10.0600, LbrM.10.1680 | LbrM.10.0470, LbrM.10.0500, LbrM.10.0520, LbrM.10.0540, LbrM.10.0570, LbrM.10.1570, LbrM.10.1590, LbrM.10.1610, LbrM.10.1620, LbrM.10.1630, LbrM.10.1640, LbrM.10.1650, LbrM.10.1660, LbrM.10.1680 |
| 3 | LbrM.10.0590, LbrM.10.1690 | LbrM.10.0590, LbrM.10.1690 |
| 4 | LbrM.10.0550, LbrM.10.1720 | LbrM.10.1720 |
| 5 | LbrM.10.0490, LbrM.10.1700 | LbrM.10.0490, LbrM.10.1700 |
| 6 | LbrM.10.1550 | LbrM.10.1550 |
Gene recombination in GP63 sequences from chromosome 10
Through analyzes of the alignments generated in this study, we identified that specific regions of certain GP63 gene sequences were very similar to equivalent regions from other GP63 genes which otherwise were more divergent. For example, certain small motifs generally seen only on the L. braziliensis Group 1 genes were also found in one or more of the group 2 genes and vice-versa, an indication of gene recombination. Indeed, the locus for these genes is reported as having high plasticity [23, 37] and the data from the literature shows that this gene family can be influenced by mosaic or fragmental gene conversion [26, 38]. Here, in order to understand why the expansion of the GP63 genes occurs mainly on chromosome 10, we performed an in-silico search for recombination events targeting these genes so as to better evaluate whether their variability was related to intragenic recombination. The software chosen to find the recombination events (RDP4) uses several tools to determine events such as the likely position of recombination breakpoints and the identity of sequences most closely related to the gene being evaluated. In this study we only considered recombination events that were detected by at least two of the tools tested. Therefore, we decided to perform a gene recombination analysis with all the GP63 genes of L. braziliensis present in databases and the ones generated by us through PCR. We first targeted the chromosome 31 GP63 genes, but no recombination events were detected by the software. In contrast, when the 38 PCR sequences from chromosome 10 were analyzed 30 (or 79%) were reported as recombinant genes. Regarding the database genes, 32 recombination events were found, sometimes with more than one event for the same gene (Fig. 6 and Additional file 4: Table S4). Gene duplication and recombination events then are possibly the major source of the novel GP63 sequences seen in the chromosome 10 from L. braziliensis and closely related sequences.
Fig. 6.

Leishmania braziliensis GP63 recombination events. Representative figure indicating recombination events identified using the RDP4 software. The p-values calculated for the recombination events are indicated as well as the parental and recombinant regions of the chosen gene. The graph shows the gene that underwent a recombination event (in blue) and other two genes that most likely were involved in this recombination, named major parent (in yellow) and minor parent (in purple), identified by pairwise alignment. Despite their definition as major or minor parent, any one of these genes may be involved in the recombination. a Predicted recombination event between two PCR genes, G1620B1 and G0510G4, with the recombinant gene (G0540C1) showing a 100% identity in most of the N-terminus region with G0510G4. b Predicted recombination event between two annotated genes, LbrM10.1630 and LbrM10.0600, showing the putative recombinant gene (LbrM10.0590) with an almost 100% pairwise identity in most of its N-terminal and central regions with the gene LbrM10.0600. A representative GP63 gene structure is shown in the bottom of the figure, discriminating N-terminal, Central and C-terminal regions
Protein structural modeling, mapping of variable regions and B-cell epitope prediction
Based on the crystallized structure of a membrane GP63 from L. major promastigotes, GP63 was identified as a compact protein consisting predominantly of β sheet secondary structure elements divided into three distinct domains (N-terminus, Central domain and C-terminus) and with features typical of the catalytic modules of zinc proteases [39]. After observing the recombination events and sequence variations between the multitudes of L. braziliensis GP63 genes, we decided to investigate where these variations are found along the 3D structure of the protein. Through three-dimensional protein structure predictions, we were able to model the structure of eight divergent GP63 sequences with high modeling scores, as can be seen in Fig. 7. We then mapped on the models the most variable motifs identified by the previous multiple alignments (highlighted in blue in the structures shown in the figure). As can be observed, most of the variable regions were positioned externally on the structures.
Fig. 7.
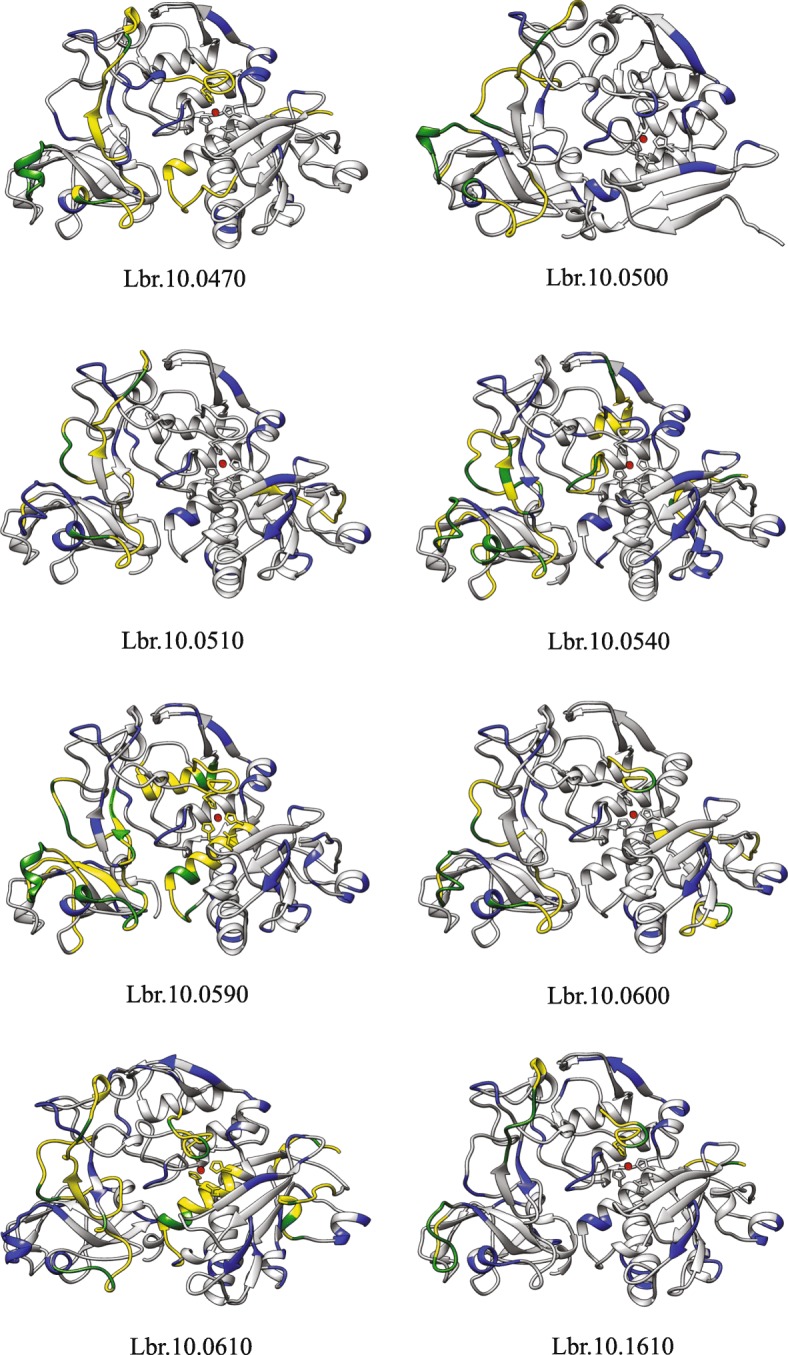
Comparative protein modelling of selected Leishmania braziliensis GP63 paralogs from chromosome 10. Modelled structures of eight representative GP63 paralogs from the L. braziliensis chromosome 10. The segments labelled in blue indicate the variable motifs in each protein, while the segments yellow represent the predicted linear B-cell epitopes and the green markings show variable motifs coinciding with the predicted epitopes
We next sought to evaluate how the various GP63 sequence would be recognized by the B cells from the mammalian immune system and also to predict their ability to induce the production of specific antibodies. Linear and conformational B-cell epitope predictions were carried out using the various chromosome 10 GP63 sequences from L. braziliensis. The linear epitope predictions returned 56 epitopes from the sequences of the modeled proteins. Regarding their localization within the various structures, from the total of 56 epitopes, 41 were mapped to the proteins’ external regions, whereas six were localized internally and nine could not be evaluated due to the comparative nature of the tridimensional structure modeling (Table 2). The modeling is based on a mature GP63 crystallized structure, which lost part of its N-terminal region, during the protein posttranslational modification, which prevented the assessment of epitopes localized to this region. Out of the 41 linear epitopes localized externally, 33 coincide with motifs that display sequence variation, while 8 are found in regions conserved between the different GP63 sequences. Considering only the epitopes that were predicted to localize internally, four coincide with variable sequences while two were associated with conserved regions. As for the prediction of conformational B-cell epitopes, the analysis returned 40 epitopes, with 32 mapped to the proteins’ external region. Twenty-three of those were in motifs with sequence variation, while nine were in conserved regions. Regarding the eight epitopes localized internally, five coincided with variable sequence motifs and three were in conserved motifs. We also identified 14 motifs that were present in both linear and conformational epitope predictions (not shown). As above, most of the epitopes coincided with variable motifs localized externally, as shown in Table 2. Noteworthy also is the fact that a peptide from L. infantum GP63 that has been previously shown to react strongly with sera from dogs infected with visceral leishmaniasis [40] is nearly identical to one of the L. braziliensis B-cell epitopes predicted by our analysis within the Lbr10.0590 polypeptide (not shown). Overall, these results are consistent with variable regions localizing externally and being more capable of inducing an immune response.
Table 2.
Leishmania braziliensis GP63 B-cell epitope prediction. Table showing the distribution of the predicted epitopes along the studied GP63 paralogs
| Total Number of Epitopes | Conserved Region | Sequence Variation Region | Type of Epitopes | Protein Region |
|---|---|---|---|---|
| 41 | 8 | 33 | Linear | External |
| 6 | 2 | 4 | Linear | Internal |
| 32 | 9 | 23 | Conformational | External |
| 8 | 3 | 5 | Conformational | Internal |
Discussion
The in-silico analysis carried out here highlights the strong selective pressure for the expansion in copy number of the chromosome 10 GP63 genes within Leishmania species, and in particular in the Viannia and Sauroleishmania subgenera. The increased number of the chromosome 10 GP63 genes in different Leishmania species evolved independently generating a wide range of paralogs, which display sequence variations and may be generated by recombination. This expansion seems to be an ongoing process that might be related to pathogenesis or defense mechanisms directed to the vertebrate host and the parallel expansion in both Viannia and Sauroleishmania species is something that must be taken into account. Such expansion of multiple genes arranged in tandem, originating from duplication and recombination events, demonstrates the adaptability of Leishmania species to the environment, associated with the evolutionary pressure suffered by the GP63 genes [28]. As a result, the presence of these multi-copy arrays may lead to speciation [41] or indicate the possible need for stage-specific genes [28]. As previously highlighted [42], an expansion in GP63 sequences also occurred independently in other trypanosomatids, such as T. cruzi and T. brucei, and this led to novel GP63 domains which might be associated with species-specific or group-specific functions. It is likely then that this expansion in Leishmania GP63 genes might be related to novel aspects of the pathogenesis of these parasites to the vertebrate hosts, but this still needs to be better defined. When multiple strains from a single species is considered the overall GP63 diversity might be even greater, as recently evaluated [43], and this might be associated with possibly different virulence phenotypes and clinical outcomes for the disease. The recent release of a new L. infantum genome based on data using two distinct methodologies of next generation sequencing, and showing a higher copy number of GP63 genes for this species [30], also highlights the need for better quality genomes in order to properly define the true diversity of these genes for multiple Leishmania and trypanosomatid species.
The expansion observed in the chromosome 10 genes are concentrated in the Group 1 and Group 2 genes and, if we extrapolate the expression for Group 1 based on the data with the L. major and L. infantum genes [15, 25], they are likely to be expressed constitutively with likely functions during the mammalian infection. In contrast, for the Group 3 genes, with only two paralogs, their expression might be restricted to the promastigote stage of the parasite life cycle, therefore with minor or no relevant function in the mammalian host. For all three groups their expression will have to be confirmed but the large sequence variations observed for both Groups 1 and 2 genes, concentrated on potentially immunogenic regions localized on the surface of the GP63 molecules, also imply related expression patterns during the mammalian stage of the Leishmania life cycle. As previously shown in L. major and L. mexicana [44], these expression patterns should be linked to the mRNA 3’UTRs and sorting out the molecular mechanisms associated will be a major endeavor. An important question that emerges regarding the expression of the genes from Groups 1 and 2 is related to the multiple paralogs. Are multiple genes belonging to the same group expressed simultaneously or some are expressed more efficiently than others or alternatively? This will also need to be investigated further.
Another relevant question remaining deals with the functional roles for the distinct groups and how these might be associated with sequence differences between the paralogs. A possible link with the proteins’ subcellular localization is presumed based on the differences in the C-terminus of the subsets identified and the presence or absence of a typical GPI anchor. These differences regarding the presence or absence of GPI anchor sites have been suggested before based on comparisons between the L. major and L. infantum GP63 sequences [8]. Here, the C-terminal Group 2 of L. braziliensis GP63 sequences lacks the GPI anchor signal entirely, which is consistent with proteins that are directly secreted into the extracellular medium, as previously reported for L. mexicana GP63 [45]. This release into the extracellular environment might contribute at the early stages of infection, due to the ability of GP63 to digest the extracellular matrix proteins, facilitating parasite mobility and invasion [46]. Alternatively, these proteins might be selectively transferred to exosomes and later to the macrophages in order to influence its metabolism and promote Leishmania growth [11]. For the L. braziliensis Group 1 proteins, they all share a C-terminus having a likely transmembrane domain with no clear GPI anchor site. Lack of a typical GPI anchor site however, with a more likely transmembrane domain identified, was also seen in the L. major Group 1 gene, which was nevertheless seen to have a GPI anchor [25]. The distinct C-terminal ends nevertheless clearly suggest critical differences in subcellular localization for the distinct GP63 groups, but these need further experimental confirmation.
In early studies performed with L. guyanensis, it was suggested that new GP63 genes may be generated by events of mosaicism through recombination between 5′ and 3’ UTRs and protein coding regions [24], and mosaicism in GP63 sequences was subsequently also found in L. braziliensis genes [26]. The data obtained by us corroborate with other studies investigating GP63 recombination that found it to target mainly the N-terminal and C-terminal regions of the gene [37, 38]. The impact of the GP63 sequence variability in its structure has been investigated in a wider scale, comparing Trypanosoma and Leishmania sequences, and found to be associated with variability in its zinc binding site and presumably activity [42]. In a recent study targeting L. braziliensis GP63 sequences, structural differences have also been found to be associated with sequence variability, implying functional differences, such as during substrate binding, which may affect the interaction with the host [47]. Alternatively, the variability in protein structure could mainly affect recognition by the host immune system and promote infection mainly because the host would need to produce different antibodies to neutralize a single group of proteins. Our results, showing variability concentrated on antigenic regions on the protein’s surface, is in agreement with previously reported data based on L. major and L. infantum sequence analysis where regions of GP63 sequence variation were mapped to the surface of the protein and were associated with immunodominant epitopes [37]. However, more studies are needed to better understand the recognition of different GP63 paralogs by the host immune system.
Overall the data presented here highlights novel and relevant aspects related to the expansion of GP63 genes in L. braziliensis and related Viannia species and raises specific issues regarding the role of GP63 in the parasite pathogenesis during the infection in mammals. It is possible that species belonging to the subgenus Viannia may have added a new level of complexity to GP63 function and this may somehow be related to the capacity of some species to cause the more aggressive mucocutaneous form of the disease. The new questions raised here then, when solved, shall provide novel and relevant knowledge regarding the very unique mechanisms of pathogenesis associated with these parasites.
Conclusions
Our results suggest a greater role for the sequence variation found among the chromosome 10 GP63 genes for the pathogenesis of L. braziliensis and closely related species within the mammalian host. The variation in sequence and the expansion in number of these GP63 genes have occurred independently in different Leishmania lineages, is associated with intragenic recombination events and has a likely role against the host immune response. They also indicate different functions associated to genes mapped to different chromosomes and, for the chromosome 10 genes at least, variable subcellular localizations likely associated with multiple functions and target substrates for this versatile protease.
Methods
Parasites and culture conditions
In this study, we used Leishmania (Viannia) braziliensis (MHOM/BR/75/M2904) in its promastigote form. This is a reference strain from the Evandro Chagas Institute, Belém, Brazil. The cells were cultured at 26 °C in Schneider (Sigma) pH 7.2 supplemented with 20% fetal bovine serum (FBS), antibiotics (Streptomycin / Penicillin 0.1%) and 0.1% Hemin.
PCR, cloning and sequencing
Approximately, 108 L. braziliensis promastigotes were used for total genomic DNA extraction using DNAzol (Invitrogen) and standard procedures. PCR reactions for the amplification of the GP63 sequences were performed using Phusion® High-Fidelity DNA Polymerase (New England Biolabs), following the manufacturer’s protocol and with the oligonucleotides used as primers listed in the Additional file 5: Table S5. After amplification, cloning and sequencing of the PCR products, a nomenclature was created for the newly generated sequences in order to identify from which set of primers they were derived, whether from those encoding the KDELMAP or GPI regions, and defining which annotated GP63 gene it most closely resembles. The newly generated sequences derived from the PCR amplifications were deposited on the GenBank and all accession numbers are listed in Additional file 3: Table S3.
Search for new GP63 paralogs through hidden Markov models
First, the predicted proteomes of the following organisms were downloaded from TritrypDB in August 25, 2014: L. braziliensis strain 2903 [taxid: 1295825], L. braziliensis strain 2904 [taxid: 420245], L. infantum [taxid: 435258], L. major [taxid: 347515], L. donovani [taxid: 981087], L. mexicana [taxid: 929439] and L. tarentolae [taxid: 5689]. GP63 genes were then identified within the downloaded proteomes, considering only genes annotated as GP63, encoding proteins longer than 30 amino acids and with no more than one stop codon per sequence. All of the protein sequences derived from genes that met these inclusion criteria were submitted to the analysis of the OrthoMCL program [48], and grouped according to homology using the Markov Cluster algorithm [49]. Protein sequences from each group were aligned using the MAFFT software (default settings) [50] and the multiple alignments used as input for the hmmbuild, version 3.0, a tool from the HMMER package [51] to build Hidden Markov Models (HMMs). The models were then used with the hmmsearch tool to search for new paralogs within the L. braziliensis strain 2904 proteome. A cutoff of 0.001 for hit significance (e-value < = 0.001) was applied.
GP63 phylogenetic analysis and detection of recombination events
A phylogenetic tree was built with GP63 protein sequences from genes encoded within chromosomes 10, 28 and 31 from diverse Leishmania species and more distantly related organisms. These include the Phytomonas sp. isolate Hart11 [taxid: 134014], Crithidia fasciculata [taxid: 5656], Leptomonas pyrrhocoris [taxid: 157538], Trypanossoma cruzi [taxid: 353153], Trypanossoma brucei [taxid: 185431], Trypanossoma theileri [taxid: 67003] and Bodo saltans [taxid: 75058]. Another tree was made with selected GP63 sequences used in the previous analyses plus the ones obtained by PCR from L. braziliensis as well as GP63 sequences from Leishmania guyanensis [taxid: 5670]. For all trees, the selected sequences were aligned by MAFFT (default settings) and the alignments automatically edited by Trimal [52] to keep just phylogenetically informative sites. ProtTest [53] was then used to predict the best evolutionary model which was subsequently used as a setting to build phylogenetic trees with PhyML, applying the Maximum Likelihood (ML) method [54], and MrBayes, applying the Bayesian method [55, 56]. The branch support for the ML tree was given by non-parametric bootstrap analysis using 1000 replicates. The Bayesian inferred trees were determined by 5,000,000 chains to check for convergence and a 100% burn-in was discarded. The aligned nucleotide sequences from L. braziliensis, obtained from the TriTrypDB database and through PCR, were analyzed for recombination using the RDP4 program [57].
Modelling of GP63 homologs and searches for non-conserved regions
Eight of the most variable paralogs from different L. braziliensis C-terminal groups were chosen for the three-dimensional modelling. The modelling was performed for the amino acid sequences previously obtained from TriTrypDB and applying the SWISS MODEL platform [58]. When the models were completed, their qualities were assessed through Procheck [59]. Specific regions of the protein models were then evaluated using the initial alignment information, highlighting the non-conserved regions which were characterized by amino acid exchanges.
B-cell epitope prediction
Linear B-cell epitope predictions were performed for the protein sequences used in the 3D modeling step. The predictions were carried out using the following programs: AAP12 [60], BCPred12 [61] and BepiPred [62]. Only epitopes predicted by at least two programs, with lengths equal to or greater than 10 amino acids and with scores greater than 0.8 were considered as positive predictions on AAP12 and BCpred12. Epitopes with scores over 0.5 obtained by BepiPred were also included in the analysis.
In addition to the linear prediction, a conformational prediction of epitopes was also performed to evaluate if the protein structures were also able to generate interaction with the immune system. The conformational epitopes were predicted by the CBTOPE web server [63], where only epitopes with more than 10 amino acids and a score above 4 were considered for this study. After the prediction, an assessment was performed to map the localization of all the epitopes on the modeled proteins.
Additional files
Table S1. Subsets of GP63 sequences used to build the Hidden Markov Models (HMMs) (DOCX 13 kb)
Table S2. GP63 genes identified by HMM from the L. braziliensis M2904 proteome. Table showing the number of GP63 genes identified by each HMM after the search for new paralogs within the L. braziliensis M2904 genome sequences. (DOCX 11 kb)
Table S3. Set of oligonucleotides, GP63 sequences obtained by PCR and their respective GenBank accession number. (DOCX 15 kb)
Table S4. Table showing the recombination events found for the L. braziliensis GP63 genes, followed by the programs that detected it and their p-value. Detailed description of the recombination events throughout the L. braziliensis GP63 genes. (XLSX 16 kb)
Table S5. Oligonucleotides used for the PCR reactions. A list of the oligonucleotides used for the PCR reactions in this study. (DOCX 13 kb)
Acknowledgements
We thank members of Dr. O. P. de Melo Neto’s laboratory for helpful discussions and support. The authors thank the Nucleo de Plataformas Tecnológicas (NPT) at the Institute Aggeu Magalhães (FIOCRUZ-PE) for the use of its automatic sequencing facility.
Funding
The Leishmania work in Dr. de Melo Neto’s lab was more recently funded with grants provided by the Brazilian funding agencies FACEPE (APQ-0239-2.02/12 and APQ-1662-2.02/15), CNPq (480899/2013–4, 313934/2013–4 and 401282/2014–7) and CAPES (23038.007656/2011–92). Studentships for the graduate students (ALCN, and ANALMB) were provided by CAPES and FACEPE.
Availability of data and materials
The GP63 sequence datasets analysed in this study are available at TriTrypDB (http://tritrypdb.org/tritrypdb/) and GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under the accession number provided in the Additional file 3: Table S3.
Abbreviations
- CL
Cutaneous Leishmaniasis
- FBS
Fetal Bovine Serum
- GP63
Glycoprotein 63
- GPI
GlycosylPhosphatidylInositol
- HMMs
Hidden Markov Models
- MAC
Membrane Attack Complex
- MCL
Mucocutaneous Leishmaniasis
- MSP
Major Surface Protease
- NPT
Nucleo de Plataformas Tecnológicas
- UTR
Untranslated Region
- VL
Visceral Leishmaniasis
Authors’ contributions
ALCN – experimental and bioinformatic analysis, writing original draft. ANALMB - experimental and bioinformatic analysis. AMR – bioinformatics experimental design. FBM – results discussions. OPMN – conceptualization, supervision, funding acquisition, writing, review and editing. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Artur L. Castro Neto, Email: artur.leonel@gmail.com
Adriana N. A. L. M. Brito, Email: adrineuman@gmail.com
Antonio M. Rezende, Email: antonio.rezende@cpqam.fiocruz.br
Franklin B. Magalhães, Email: franklinmagalhaes@asces.edu.br
Osvaldo P. de Melo Neto, Email: opmn@cpqam.fiocruz.br
References
- 1.Pace D. Leishmaniasis. J Inf Secur. 2014;69(Suppl 1):10–18. doi: 10.1016/j.jinf.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Akhoundi M, Kuhls K, Cannet A, Votýpka J, Marty P, Delaunay P, et al. A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl Trop Dis. 2016;10:1–40. doi: 10.1371/journal.pntd.0004349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaye P, Scott P. Leishmaniasis: complexity at the host–pathogen interface. Nat Rev Microbiol. 2011;9:604–615. doi: 10.1038/nrmicro2608. [DOI] [PubMed] [Google Scholar]
- 4.Arango Duque G, Descoteaux A. Leishmania survival in the macrophage: where the ends justify the means. Curr Opin Microbiol. 2015;26:32–40. doi: 10.1016/j.mib.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Silva-Almeida M, Pereira BA, Ribeiro-Guimarães M, Alves C. Proteinases as virulence factors in Leishmania spp. infection in mammals. Parasit Vectors. 2012;5:160. doi: 10.1186/1756-3305-5-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bordier C. The promastigote surface protease of leishmania. Parasitol Today. 1987;3:151–153. doi: 10.1016/0169-4758(87)90199-2. [DOI] [PubMed] [Google Scholar]
- 7.Olivier M, Atayde VD, Isnard A, Hassani K, Shio MT. Leishmania virulence factors : focus on the metalloprotease GP63. Microbes Infect. 2012;14:1377–1389. doi: 10.1016/j.micinf.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Yao C, Donelson JE, Wilson ME. The major surface protease (MSP or GP63) of Leishmania sp. biosynthesis, regulation of expression, and function. Mol Biochem Parasitol. 2003;132:1–16. doi: 10.1016/S0166-6851(03)00211-1. [DOI] [PubMed] [Google Scholar]
- 9.Gupta G, Oghumu S, Satoskar AR. Mechanisms of immune evasion in Leishmaniasis. Adv Appl Microbiol. 2014;82:155–184. doi: 10.1016/B978-0-12-407679-2.00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Podinovskaia M, Descoteaux A. Leishmania and the macrophage: a multifaceted interaction. Future Microbiol. 2015;10:111–129. doi: 10.2217/fmb.14.103. [DOI] [PubMed] [Google Scholar]
- 11.Atayde VD, Hassani K, da Silva Lira Filho A, Borges AR, Adhikari MC, et al. Leishmania exosomes and other virulence factors: impact on innate immune response and macrophage functions. Cell Immunol. 2016;309:7–18. doi: 10.1016/j.cellimm.2016.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Olivier M, Hassani K. Protease inhibitors as prophylaxis against leishmaniasis: new hope from the major surface protease gp63. Future Med Chem. 2010;2:539–542. doi: 10.4155/fmc.10.17. [DOI] [PubMed] [Google Scholar]
- 13.Contreras I, Gómez MA, Nguyen O, Shio MT, McMaster RW, Olivier M. Leishmania-induced inactivation of the macrophage transcription factor AP-1 is mediated by the parasite metalloprotease GP63. PLoS Pathog. 2010;6:e1001148. doi: 10.1371/journal.ppat.1001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaramillo M, Gomez MA, Larsson O, Shio MT, Topisirovic I, Contreras I, et al. Leishmania repression of host translation through mTOR cleavage is required for parasite survival and infection. Cell Host Microbe. 2011;9:331–341. doi: 10.1016/j.chom.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Ramamoorthy R, Donelson E, Paetz E, Maybodi M, Roberts SC, Wilson ME. Three distinct RNAs for the surface protease gp63 are differentially expressed during development of Leishmania donovani chagasi promastigotes to an infectious form. J Biol Chem. 1992;267:1888–1895. [PubMed] [Google Scholar]
- 16.Schneider P, Rosat JP, Bouvier J, Louis J, Bordier C. Leishmania major: differential regulation of the surface metalloprotease in amastigote and promastigote stages. Exp Parasitol. 1992;75:196–206. doi: 10.1016/0014-4894(92)90179-E. [DOI] [PubMed] [Google Scholar]
- 17.D’Avila-Levy CM, De Almeida Dias F, ACN DM, Martins JL, De Carvalho Santos Lopes AH, ALS DS, et al. Insights into the role of gp63-like proteins in lower trypanosomatids. FEMS Microbiol Lett. 2006;254:149–156. doi: 10.1111/j.1574-6968.2005.00022.x. [DOI] [PubMed] [Google Scholar]
- 18.Pereira FM, Bernardo PS, Dias Junior PFF, Silva BA, Romanos MTV, D’Avila-Levy CM, et al. Differential influence of gp63-like molecules in three distinct Leptomonas species on the adhesion to insect cells. Parasitol Res. 2009;104:347–353. doi: 10.1007/s00436-008-1202-2. [DOI] [PubMed] [Google Scholar]
- 19.D’Avila-Levy CM, Altoé ECF, Uehara LA, Branquinha MH, Santos ALS. Proteins and Proteomics of Leishmania and Trypanosoma. 2014. GP63 function in the interaction of Trypanosomatids with the invertebrate host: facts and prospects; pp. 253–270. [DOI] [PubMed] [Google Scholar]
- 20.Santos ALS, Branquinha MH, D’Avila-Levy CM. The ubiquitous gp63-like metalloprotease from lower trypanosomatids: in the search for a function. An Acad Bras Cienc. 2006;78:687–714. doi: 10.1590/S0001-37652006000400006. [DOI] [PubMed] [Google Scholar]
- 21.Ivens AC, Peacock CS, Worthey EA, Murphy L, Berriman M, Sisk E, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Victoir K, Dujardin JC, de Doncker S, Barker DC, Arevalo J, Hamers R, et al. Plasticity of gp63 gene organization in Leishmania (Viannia) braziliensis and Leishmania (Viannia) peruviana. Parasitology. 1995;111:265–273. doi: 10.1017/S0031182000081828. [DOI] [PubMed] [Google Scholar]
- 24.Steinkraus H, Greer J. DC. S, PJ L. sequence heterogeneity and polymorphic gene arrangements of the Leishmania guyanensis gp63 genes. Mol Biochem Parasitol. 1993;62:173–186. doi: 10.1016/0166-6851(93)90107-9. [DOI] [PubMed] [Google Scholar]
- 25.Voth BR, Kelly BL, Joshi PB, Ivens AC, McMaster WR. Differentially expressed Leishmania major gp63 genes encode cell surface leishmanolysin with distinct signals for glycosylphosphatidylinositol attachment. Mol Biochem Parasitol. 1998;93:31–41. doi: 10.1016/S0166-6851(98)00013-9. [DOI] [PubMed] [Google Scholar]
- 26.Victoir K, Arevalo J, De Doncker S, Barker DC, Laurent T, Godfroid E, et al. Complexity of the major surface protease (msp) gene organization in Leishmania (Viannia) braziliensis: evolutionary and functional implications. Parasitology. 2005;131:207–214. doi: 10.1017/S0031182005007535. [DOI] [PubMed] [Google Scholar]
- 27.Llanes A, Restrepo CM, Del Vecchio G, Anguizola FJ, Lleonart R. The genome of Leishmania panamensis: insights into genomics of the L. (Viannia) subgenus. Sci Rep. 2015;5:8550. doi: 10.1038/srep08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21:2129–2142. doi: 10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coughlan S, Taylor AS, Feane E, Sanders M, Schonian G, Cotton JA, et al. Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus. R Soc Open Sci. 2018;5:172212. doi: 10.1098/rsos.172212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.González-De La Fuente S, Peiró-Pastor R, Rastrojo A, Moreno J, Carrasco-Ramiro F, Requena JM, et al. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci Rep. 2017;7:18050. doi: 10.1038/s41598-017-18374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46:200–204. doi: 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valdivia HO, Scholte LLS, Oliveira G, Gabaldón T, Bartholomeu DC. The Leishmania metaphylome: a comprehensive survey of Leishmania protein phylogenetic relationships. BMC Genomics. 2015;16:887. doi: 10.1186/s12864-015-2091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raymond F, Boisvert S, Roy G, Ritt J-F, Légaré D, Isnard A, et al. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 2012;40:1131–1147. doi: 10.1093/nar/gkr834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maurício IL, Leishmania Taxonomy.In: Brusch F, Gradoni L (Eds.), The Leishmaniases: old neglected tropical diseases. Cham: Springer International Publishing; 2018. p. 15–30.
- 35.Mccoy JJ, Beetham JK, Ochs DE, Donelson JE, Wilson ME. Regulatory sequences and a novel gene in the msp ( GP63 ) gene cluster of Leishmania chagasi. Mol Biochem Parasitol. 1998;95:251–265. doi: 10.1016/S0166-6851(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 36.Schneider P, Ferguson MAJ, McConville MJ, Mehlert A, Homans SW, Bordier C. Structure of the glycosyl-phosphatidylinositol membrane anchor of the Leishmania major promastigote surface protease. J Biol Chem. 1990;265:16955–16964. [PubMed] [Google Scholar]
- 37.Victoir K, Dujardin JC. How to succeed in parasitic life without sex? Asking Leishmania. Trends Parasitol. 2002;18:81–85. doi: 10.1016/S1471-4922(01)02199-7. [DOI] [PubMed] [Google Scholar]
- 38.Mauricio IL, Gaunt MW, Stothard JR, Miles MA. Glycoprotein 63 (gp63) genes show gene conversion and reveal the evolution of Old World Leishmania. Int J Parasitol. 2007;37:565–576. doi: 10.1016/j.ijpara.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 39.Schlagenhauf E, Etges R, Metcalf P. The crystal structure of the Leishmania major surface proteinase leishmanolysin (gp63) Structure. 1998;6:1035–1046. doi: 10.1016/S0969-2126(98)00104-X. [DOI] [PubMed] [Google Scholar]
- 40.Morales G, Carrillo G, Requena JM, Guzman F, Gomez LC, Patarroyo ME, et al. Mapping of the antigenic determinants of the Leishmania infantum gp63 protein recognized by antibodies elicited during canine visceral leishmaniasis. Parasitology. 1997;114:507–516. [PubMed] [Google Scholar]
- 41.Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- 42.Ma L, Chen K, Meng Q, Liu Q. An evolutionary analysis of trypanosomatid GP63 proteases. Parasitol Res. 2011;109:1075–1084. doi: 10.1007/s00436-011-2348-x. [DOI] [PubMed] [Google Scholar]
- 43.Medina LS, Souza BA, Queiroz A, Guimarães LH, Machado PRL, Carvalho EM, et al. The gp63 gene cluster is highly polymorphic in natural Leishmania (Viannia) braziliensis populations, but functional sites are conserved. PLoS One. 2016;11:1–13. doi: 10.1371/journal.pone.0163284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelly BL, Nelson TN, Mcmaster WR. Stage-specific expression in Leishmania conferred by 3′ untranslated regions of L . major leishmanolysin genes (GP63) Mol Biochem Parasitol. 2001;116:101–104. doi: 10.1016/S0166-6851(01)00307-3. [DOI] [PubMed] [Google Scholar]
- 45.Ellis M, Sharma DK, Hilley JD, Coombs GH, Mottram JC. Processing and trafficking of Leishmania mexicana GP63. Analysis using GPI8 mutants deficient in glycosylphosphatidylinositol protein anchoring. J Biol Chem. 2002;277:27968–27974. doi: 10.1074/jbc.M202047200. [DOI] [PubMed] [Google Scholar]
- 46.Mcgwire BS, Chang K, Engman DM. Migration through the extracellular matrix by the parasitic protozoan Leishmania is enhanced by surface metalloprotease GP63. Infect Immun. 2003;71:1008–1010. doi: 10.1128/IAI.71.2.1008-1010.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sutter A, Antunes D, Silva-Almeida M, Costa MG de S, Caffarena ER. Structural insights into leishmanolysins encoded on chromosome 10 of Leishmania (Viannia) braziliensis. Mem Inst Oswaldo Cruz. 2017;112:617–625. doi: 10.1590/0074-02760160522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Stoeckert CJJ, OrthoMCL RDS. Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Enright AJ, Van DS, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katoh K, Misawa K, Kuma K, Miyata TMAFFT. a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eddy S. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 52.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 54.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. SystBiol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 55.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 56.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 57.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1:1–5. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.R a L, J a R, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 60.Chen J, Liu H, Yang J, Chou KC. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids. 2007;33:423–428. doi: 10.1007/s00726-006-0485-9. [DOI] [PubMed] [Google Scholar]
- 61.EL-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B-cell epitopes using string kernels Yasser. J Mol Recognit. 2008;21:243–255. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsen JEP, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ansari HR, Raghava GP. Identification of conformational B-cell epitopes in an antigen from its primary sequence. Immunome Res. 2010;6:6. doi: 10.1186/1745-7580-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Subsets of GP63 sequences used to build the Hidden Markov Models (HMMs) (DOCX 13 kb)
Table S2. GP63 genes identified by HMM from the L. braziliensis M2904 proteome. Table showing the number of GP63 genes identified by each HMM after the search for new paralogs within the L. braziliensis M2904 genome sequences. (DOCX 11 kb)
Table S3. Set of oligonucleotides, GP63 sequences obtained by PCR and their respective GenBank accession number. (DOCX 15 kb)
Table S4. Table showing the recombination events found for the L. braziliensis GP63 genes, followed by the programs that detected it and their p-value. Detailed description of the recombination events throughout the L. braziliensis GP63 genes. (XLSX 16 kb)
Table S5. Oligonucleotides used for the PCR reactions. A list of the oligonucleotides used for the PCR reactions in this study. (DOCX 13 kb)
Data Availability Statement
The GP63 sequence datasets analysed in this study are available at TriTrypDB (http://tritrypdb.org/tritrypdb/) and GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under the accession number provided in the Additional file 3: Table S3.