

SUMMARY
S-adenosylmethionine (SAM) is an essential metabolite that acts as a cofactor for most methylation events in the cell. The N6-methyladenosine (m6A) methyltransferase METTL16 controls SAM homeostasis by regulating the abundance of SAM synthetase MAT2A mRNA in response to changing intracellular SAM levels. Here we present crystal structures of METTL16 in complex with MAT2A RNA hairpins to uncover critical molecular mechanisms underlying the regulated activity of METTL16. The METTL16-RNA complex structures reveal atomic details of RNA substrates that drive productive methylation by METTL16. In addition, we identify a polypeptide loop in METTL16 near the SAM binding site with an autoregulatory role. We show that mutations that enhance or repress METTL16 activity in vitro correlate with changes in MAT2A mRNA levels in cells. Thus, we demonstrate the structural basis for the specific activity of METTL16 and further suggest the molecular mechanisms by which METTL16 efficiency is tuned to regulate SAM homeostasis.
In Brief
Doxtader et al. determine crystal structures of the N6-methyladenosine methyltransferase METTL16 in complex with RNA substrates. METTL16 can be regulated by the conformation of the RNA substrate and by an autoregulatory loop near the SAM binding pocket. Modulating the catalytic activity of METTL16 changes MAT2A mRNA levels in cells.
Graphical Abstract
INTRODUCTION
S-adenosylmethionine (SAM) is an essential metabolite that acts as the high-energy methyl donor for most methylation events of DNA, RNA, and protein, which are important for proper gene regulation (Janke et al., 2015; Kaelin and McKnight, 2013; Mentch and Locasale, 2016; Su et al., 2016). SAM is also utilized in the biosynthesis of polyamines and lipids (Walsh et al., 2018). Intracellular SAM levels are tightly controlled, and dysregulation has been linked to abnormalities in stem cell maintenance, cell proliferation, immunity, and aging (Brunet and Rando, 2017; Ducker and Rabinowitz, 2017; Shiraki et al., 2014). Generating SAM from methionine and ATP requires methionine adenosyltransferase (MAT) enzymes (Sakata et al., 1993). In mammals, SAM homeostasis can be maintained via modulating the major SAM synthetase, MAT2A, through a negative feedback mechanism (Martínez-Chantar et al., 2003). Recent studies show that fluctuations of intracellular SAM concentration result in altered levels of MAT2A mRNA, due to differential activity of the essential RNA methyltransferase METTL16 on the 3′ untranslated region (3′ UTR) (Pendleton et al., 2017; Shima et al., 2017).
As the most prevalent internal modification observed for mRNAs, controlled m6A modification plays an important role in pluripotency, cell differentiation, and cell signaling, such as circadian rhythm (Meyer and Jaffrey, 2017; Roundtree et al., 2017). Specific methylation of an RNA can affect its processing, translation, splicing, and stability. Most m6A marks on mRNAs are written by the methyltransferase complex METTL3/METTL14, targets of which share a consensus sequence of DRACH (Dominissini et al., 2012; Linder et al., 2015; Meyer et al., 2012). However, little else is known about how the poly-peptide complex specifically recognizes its RNA targets for methylation (Meyer and Jaffrey, 2017).
In contrast to the METTL3/METTL14 complex, METTL16 has a distinct set of targets for m6A modification, including the 3′ UTR of MAT2A mRNA and the U6 snRNA (Pendleton et al., 2017; Shima et al., 2017; Warda et al., 2017). Although a longer conserved sequence of UACAGAGAA, with a neighboring GU motif, has been proposed as the consensus sequence for METTL16, little is known about METTL16 specificity due to the limited number of validated direct targets. METTL16 can alter both the splicing and stability of MAT2A mRNA by directly binding hairpin structures in the 3′ UTR and adjusting the dwell time and methylation efficiency in response to changes in intracellular SAM concentration (Pendleton et al., 2017; Shima et al., 2017). Therefore, METTL16 plays a key role in a feedback mechanism that controls SAM homeostasis. How METTL16 is tuned to regulate SAM biosynthesis has remained an outstanding question in understanding maintenance of intracellular SAM levels.
Here we show the molecular mechanisms underlying the activity of METTL16 on the MAT2A 3′ UTR. Crystal structures of METTL16 in complex with cognate RNA stem-loops from the MAT2A 3′ UTR provide a physical model to elucidate how an RNA m6A writer recognizes its substrate. We identify RNA structural features that influence methylation efficiency and show that each wild-type hairpin manifests intrinsic propensity for modification, encoded by its own sequence. Moreover, we determine that a key polypeptide loop near the SAM binding site can autoregulate METTL16 activity. Finally, we show that activating and inhibitory mutations of METTL16 can change the methyltransferase activity at a given intracellular SAM concentration, resulting in different MAT2A mRNA levels in cells. Together, our findings provide a mechanistic model for the specificity and modulation of METTL16, an important SAM homeostasis factor.
RESULTS
Structure of METTL16 in Complex with the MAT2A 3′ UTR Hairpin 1
To investigate METTL16 function on MAT2A mRNA, we reconstituted the methyltransferase activity in vitro using the first hairpin (hp1) of the MAT2A 3′ UTR (Figures 1A and 1B) (Pendleton et al., 2017; Shima et al., 2017). Size-exclusion chromatography coupled with multi-angle static light scattering experiments indicate that wild-type, full-length METTL16 is monomeric in solution, suggesting that certain mechanisms are likely to differ from the METTL3/METTL14 heterodimer (Figure 1C) (Śledź and Jinek, 2016; Wang et al., 2016a, 2016b). We determined that the isolated methyltransferase domain (MTD16, residues 1–310) is capable of catalyzing adenosine methylation as efficiently as the full-length METTL16 construct in vitro (Figures S1A, S1B, and S1C). Moreover, truncating the hairpin structure to ~30 nucleotides does not diminish the ability of the RNA to be specifically modified by METTL16 (Figures S1C and S1D). By stabilizing the RNA stem region through removing mismatches, we were able to purify and crystallize stable complexes of METTL16 bound to the optimized hp1 oligonucleotide (hp1x) (Figures S1B and S1C). We determined and refined the complex structure using data that extend to a minimum Bragg spacing of 1.7 Å (Figure 1D and Table S1), to yield a three-dimensional atomic model of a functional m6A writer in complex with substrate RNA.
Figure 1. Structure of METTL16 Bound to MAT2A Hairpin 1.

(A) Domain organization of human METTL16. Crystallization construct (MTD16) is indicated as a blue bar underneath.
(B) Organization of the retained intron isoform of MAT2A mRNA. Exons are indicated with blocks. Hairpins 1–6 (hp1–hp6) within the 3′ UTR are shown as schematics.
(C) SEC-MALS profile for full-length METTL16. The left axis is the absorbance at 280 nm (blue peak), and the right axis is the measured molecular weight from the scattering data (green line) at each elution volume. The theoretical molecular weight for full-length monomeric METTL16 is 63 kDa, and the average measured molecular weight from MALS data is 58 kDa.
(D) Overall structure of MTD16-hp1x in cartoon representation. Protein is shown in blue, RNA in orange, and “clamp” loops interacting with the RNA in cyan.
(E) Surface representation colored by the vacuum electrostatic potential of MTD16. RNA is in orange cartoon representation.
(F) Close-up view of the catalytic pocket of MTD16, with key side chains shown as blue sticks. Catalytic motif, NPPF (184–187), is labeled with black boxes. Target adenosine for methylation (Ade17) is shown as orange sticks.
(G) The catalytic pocket of MTD16-hp1x colored as in (F), aligned with apo MTD16 (PDB: 6B91) shown as pink cartoon. RMSD is 0.4 Å for 181 Cα atoms. The catalytic motif is labeled with black boxes. Conformational difference for Phe187 and Phe188 is indicated with black arrows.
(H) The catalytic pocket of MTD16-hp1x colored as in (D), aligned with MTD3 (PDB: 5K7M) shown as green cartoon. RMSD is 4.5 Å over 104 residues. The catalytic motifs for MTD16 (NPPF, 184–187) and MTD3 (DPPW, 395–398) are shown as sticks. Phe188 from MTD16 and Tyr406 from MTD3 are also shown as sticks. Residues are labeled with corresponding colored boxes and text.
See also Figure S1.
The global architecture of the METTL16-hp1x complex exhibits a 1:1 stoichiometry, where the methyltransferase utilizes three long polypeptide segments (highlighted in cyan) to clamp the RNA hairpin structure from various angles, resulting in extensive intermolecular contact (Figures 1D and 1E). In particular, the longest prong of the “clamp” (residues 189–213) is stably folded into a rigid yet extended conformation, but is mostly missing due to disorder in the structures of METTL16 without RNA (PDB: 2H00; Ruszkowska et al., 2018). The target adenine base is captured in a deep hydrophobic pocket near the proposed catalytic motif, Asn-Pro-Pro-Phe (residues 184–187, NPPF) (Figure 1F). Although the METTL16-hp1x complex crystallized in the presence of SAH, no electron density was observed for the cofactor. Comparing the catalytic pocket conformation of the MTD16-RNA complex with structures of other states without RNA—apo or with SAH bound—suggests an induced fit mechanism, where Phe187 and Phe188 swing around to interact with the adenine to be modified (Figures 1G and S1E). Thus, forming a complex with the substrate RNA involves specific folding of various regions of METTL16.
METTL16 differs significantly from METTL3/METTL14 in primary sequence (<13% identity for the catalytic domain of METTL3), stoichiometry, and substrate RNA consensus sequence. Nevertheless, structural alignment of MTD16 with MTD3 reveals a similar global architecture, with a different permutation of secondary structures as is common in the MTase superfamily (Bujnicki et al., 2002; Iyer et al., 2016) (Figures S1F). Superimposition of the catalytic cavities shows that the catalytic motif of MTD3 (DPPW) aligns with MTD16 (NPPF) for the backbone atoms, and the side chains are likely to adopt slightly different conformations upon binding RNA (Figure 1H). The importance of Tyr406 in METTL3 has been established biochemically (Śledź and Jinek, 2016; Wang et al., 2016a), and it may serve a role similar to Phe188 of METTL16, to contribute to a snug, hydrophobic pocket for the adenine ring (Figure 1H). Given the structural similarity between METTL16 and METTL3, we also used the MTD16/RNA complex structure to model a potential RNA substrate bound to MTD3 (Figure S1G). The extended loops lining the catalytic cavity (purple) of METTL3 may serve an analogous function to the “clamp” of METTL16, to contact the RNA substrate. However, the stem-loop structure of hp1x clashes substantially with METTL14, suggesting that the structural features of METTL3 substrates would be significantly different (Figure S1H). Structural requirements for METTL3 substrates, if any, are not obvious through biochemical studies (Liu et al., 2014, 2015; Roost et al., 2015; Zhou et al., 2016), but the ability of METTL3/METTL14 to readily modify linear oligonucleotides is congruent with its flatter, more open RNA binding surface as compared to METTL16. Furthermore, while the crystallized portion of METTL16 is as active as the full-length polypeptide, the catalytic domains of METTL3/METTL14 also require the N-terminal CCCH motifs of METTL3 for full activity (Śledź and Jinek, 2016; Wang et al., 2016a). It is likely that the additional domain is required for proper positioning of the RNA substrate within the catalytic cavity of METTL3/METTL14, whereas the extended loops of METTL16 are sufficient to orient its RNA substrate for m6A modification. Therefore, while the catalytic activities of METTL3 and METTL16 are likely to share a similar structural basis, substrate specificity diverges dramatically due to the location of the extended loops and the presence of other RNA-binding domains.
Substrate RNA Loop Sequence Is Necessary for m6A Writing Activity
An atomic model of the interactions between METTL16 and RNA substrate provides an explanation for the conserved consensus motif among known targets, UAC(m)AGAGAA (Parker et al., 2011; Pendleton et al., 2017). The RNA conformation in the complex structure can be divided into three different regions: loop, transition, and stem (Figures 2A and S2A). The crystal structure of hp1x also shows that the predicted secondary structure of the MAT2A hairpins that includes a “duckbill” shape was inaccurate (Parker et al., 2011; Pendleton et al., 2017) (Figure S2B). The RNA loop region contains the 5′ half of the motif, UAC(m)AG (14–18), where the bases splay out to maximize sequence-specific contacts with the protein (Figures 2B and 2C). Several hydrogen bonds define both the identity and orientation of the bases of Ura14 and Cyt16. Although Ade15 does not make obvious hydrogen bonds with MTD16, the base is sandwiched between the rings of Phe46 and Cyt16, and the Watson-Crick edge is tightly packed to recognize the shape of the adenine base (Figure 2B). Gua18 flips inward to make several contacts with various amino acids as well as with Gua11 (Figure 2C). Moreover, Gua18 stacks atop the stem axis and serves as a cap for the RNA transition region. Base substitutions in the loop region cause marked reduction of in vitro methylation activity (Figure 2D). While the point mutations modestly affect affinity (Figure S2C), the enzymatic assays are performed with excess RNA (10 times apparent KD). Thus, the overall effect of the mutations is primarily at a step after complex formation. In summary, the structure of METTL16 with cognate RNA reveals how the loop sequence contributes to the conformation necessary for productive catalysis.
Figure 2. Specific RNA Loop Sequence Required for METTL16 Activity.

(A) Overall conformation of hp1x in the complex structure with MTD16. Nucleotides are numbered from 5′ to 3′. Target adenosine for methylation is indicated with a red star. Nucleotides in the conserved consensus motif of METTL16 have boxed labels.
(B and C) Detailed interactions in the RNA loop region. RNA nucleotides are shown as sticks, color-coded by base identity as in (A). Nucleotides within the consensus sequence are outlined with boxes: UAC (14–16) (B) and G18 (C). Protein residues are shown as gray cartoon and side-chain sticks, labeled with black text. Hydrogen bonds are marked with dashed lines.
(D) In vitro methylation activity of METTL16 on wild-type hp1 and hp1 with the indicated loop mutations. Methylated Ade is in red with an asterisk. Mutated base is boldfaced and underlined. Bars correspond to amounts of tritium incorporated into methylated RNA substrates shown as disintegrations per minute (DPM). Data are shown as mean DPM ± SD from three replicates.
(E–G) Detailed interactions in the transition region of hp1x. Each panel contains a pair of bases, where (E) contains the pair closest to the loop region, (F) is the next layer of the transition region, and (G) is the final pair, closest to the stem region. RNA bases are shown as sticks, color-coded as in (A). Nucleotides within the consensus sequence are outlined with black boxes. Polypeptide is shown as gray cartoon, with side chains represented with sticks. The ordered water molecule is shown as a red sphere. Hydrogen bonds are shown as dashed lines.
(H) In vitro methylation activity of METTL16 on the indicated RNA substrates shown as schematics underneath. Data are shown as mean DPM ± SD from three replicates.
See also Figure S2.
To compare how substrate RNA mutations affect METTL3/METTL14 activity, we measured in vitro methylation activity as well as binding affinity of the complex. Using MALAT-1 as the RNA substrate, we introduced transition mutations to each position of the consensus sequence “GGACU” (Liu et al., 2015). Our results show that the METTL3/METTL14 complex also has little tolerance for transition mutations of the GGACU consensus sequence at every position except for the final position for methyltransferase activity, while RNA binding affinity remains similar (Figures S2D and S2E). The slight variance from the DRACH consensus sequence determined from genome-wide sequencing experiments may be due to the fact that we are only using a minimal METTL3/METTL14 writer complex, and the other members of the complex may allow METTL3/METTL14 to modify a wider range of substrates. In short, for both METTL16 and METTL3/METTL14, the RNA sequence surrounding the methylated adenosine is important for the methylation efficiency, likely to support a particular conformation that affects a step after complex formation.
Transition and Stem Structures of METTL16 RNA Substrates
MTD16 also makes various direct contacts with the transition region of the RNA substrate, which consists of three unusual base pairs (Figures 2E–2G). The 3′ half of the consensus sequence, AGAA (19–22), and a conserved GU (8–9) motif on the 5′ side are critical to support the conformation of the transition region as observed in the crystal structure. The layer closest to the loop region is the Ura9-Ade19 base pair, where the glycosidic bonds are oriented in trans (Figure 2E). The next pair is an unusual interaction between the bases of Gua20 and Ade21, where an ordered water molecule mediates their contact (Figure 2F). The position of Gua20 base is further stabilized through additional hydrogen bonds to the backbone amide protons of Arg200 and Asn201 (Figure 2F). The final transition pair includes Ade22 and Gua8, which interact using their Hoogsteen and sugar edges, respectively (Figure 2G), as observed in certain RNA helix ends (Elgavish et al., 2001). The base of Gua8 is supported on the Hoogsteen edge by the guanidino group of Arg200. Thus, Arg200, located at the tip of one of the prongs of the RNA binding clamp, seems to be a critical residue to support the observed conformation of the transition region, given the extensive interactions it makes with two of the three RNA transition layers.
Although no direct contacts are observed between METTL16 and the stem region, the double-stranded RNA is likely to enable the observed transition structure. Removing the stem region or mutating it to become single-stranded obliterates the in vitro methylation activity and complex assembly (Figures 2H and S2F). To test the general role of the structural context of the RNA substrate for METTL16 activity, we used another known substrate, the U6 snRNA (Pendleton et al., 2017; Warda et al., 2017). Using only the consensus region of the U6 snRNA after removing the double-stranded RNA is similarly detrimental to the methylation reaction (Pendleton et al., 2017; Warda et al., 2017) (Figures S2G and S2H). Therefore, METTL16 recognizes a specific RNA sequence, as well as the structural context of the consensus motif.
RNA Loop/Transition Features Tune METTL16 Efficiency
The 3′ UTR of MAT2A mRNA includes six hairpins that have been proposed to contribute differently to METTL16-mediated regulation in cells via distinct mechanisms (Pendleton et al., 2017; Shima et al., 2017). Using individual hairpins and recombinant enzyme, we show that the six hairpins also manifest different propensities for methylation by METTL16 in vitro (Figures 3A and S3A). Aligning the sequences of the six stem-loops reveals two divergent features within the conserved loop/transition regions: (1) a linker sequence of variable sequence and length between the two blocks of conservation and (2) a G-to-A transition in hp5 (Figure 3B). While hp1 contains the largest linker sequence, hp6 has one of the shortest, differing by two nucleotides. METTL16 can methylate hp1 more efficiently than hp6, and swapping the stem regions does not change the trend (Figures 3C and 3D). The affinity of the two hairpins and the chimeras are similar, suggesting that the length of the linker region can modulate the catalysis or turnover efficiency of the enzyme (Figure S3B). Thus, while the stem region of the RNA is essential for methylation by METTL16, the loop/transition region modulates the methylation efficiency of an RNA substrate.
Figure 3. RNA Loop/Transition Features Tune METTL16 Efficiency.

(A) In vitro methylation activity of METTL16 on the indicated RNA substrates. Data are shown as mean DPM ± SD from three replicates.
(B) Sequence alignment of the human MAT2A 3′ UTR RNA hairpins. Conserved sequences are highlighted in yellow. The key variable regions in the loop and transition areas are indicated with red boxes.
(C) Schematics of the RNA substrates used in (D). Hp1 sequences are shown in orange, and hp6 sequences are shown in gray.
(D) In vitro methylation activity of METTL16 on the loop/stem chimeras of hp1 and hp6 illustrated with secondary structure schematics in (C). Data are shown as mean DPM ± SD from three replicates.
(E) Overall structure of MTD16-hp6 shown as gray cartoon, superimposed on the MTD16-hp1x structure colored as in Figure 1D. RMSD = 0.1 Å for 216 Cα atoms. Longer RNA linker in hp1 versus hp6 is indicated with a black arrow.
(F) Close-up view of the structural differences in hp1x RNA backbone (orange) compared to hp6 RNA backbone (gray). R200 and N39 are shown as sticks in blue (MTD16-hp1) and gray (MTD16-hp6) and labeled with black text. The additional two bases in hp1x linker are labeled with black text.
(G) In vitro methylation activity of METTL16 on hp1 and hp6. Wild type (WT) METTL16 is compared to full-length METTL16 containing the indicated point mutations. Data are shown as mean DPM ± SD from three replicates.
(H) In vitro methylation activity of wild type METTL16 on the wild type hairpins is compared to the effect of RNA mutations at the G-to-A position. Data are shown as mean DPM ± SD from three replicates.
(I) In vitro methylation activity of METTL16, wild type or containing R200Q mutation, on hp5. Data are shown as mean DPM ± SD from three replicates.
See also Figure S3.
To investigate the molecular basis for how METTL16 can accommodate different sizes of the loop/transition regions, we determined a crystal structure of the catalytic domain in complex with hp6, at 3.0 Å resolution (Figure 3E and Table S1). Structural alignment shows that the polypeptide conformations are highly similar (Cα RMSD of ~0.1 Å), with the exception of a short stretch that interacts with the additional nucleotides (12–13) of hp1 (Figure 3E). METTL16 “pinches off” the longer RNA linker region when in complex with hp1, while the rest of the nucleotides superimpose reasonably to the analogous positions between hp1 and hp6 (Figures S3C and S3D). Nevertheless, conformational deviations can be observed in both of the loop and transition regions (Figure 3F). To dissect the contribution of each region, we tested how mutations of key interacting residues (Asn39 near the loop and Arg200 for the transition) affect enzymatic activity (Figure 3G). Perturbing the interaction between Asn39 and the hp1 loop region does not alter methylation activity significantly. However, mutating Arg200 near the RNA transition region (R200Q) markedly increases m6A modification efficiency. We chose to mutate Arg200 to a glutamine because the particular mutation has also been reported in large intestinal cancer patients, possibly implicating METTL16 in disease (Giannakis et al., 2016; Muzny et al., 2012). Neither mutation significantly affects the overall affinity of the protein-RNA complex (Figure S3E). Therefore, structural changes near the RNA transition region are likely to affect the methylation efficiency of METTL16 at a step after initial binding of the substrate RNA.
The RNA transition region also contains a notable variation for the fifth hairpin (hp5), where a Gua is substituted with Ade (G-to-A) (Figure 3B). The sequence divergence is conserved in the penultimate hairpin of most vertebrate SAM synthetase 3′ UTR sequences. Moreover, hp5 is also the most efficiently methylated among the six human hairpins (Figure 3A). To test whether the G-to-A variation is the source of methylation propensity differences among the hairpins, we compared how it affects different hairpins. When we introduce the G-to-A mutation into hp1 or hp6, we observe a ~3-fold increase in the in vitro methylation activity of METTL16 (Figure 3H). Reverting hp5 to the consensus sequence (A-to-G) has the opposite effect, reducing methylation significantly. The altered Gua is equivalent to Gua20 of hp1, which makes direct contact with Arg200 (Figure 2F). Since G-to-A and R200Q mutations are both in the transition region and increase METTL16 activity in vitro, we combined the mutations to test whether the effect is additive. Wild-type METTL16 and the R200Q mutant manifest similar activity on wild-type hp5, suggesting that the activating effects of the R200Q mutation and the G-to-A swap are likely to share a common mechanism (Figure 3I). Gel shift assays show minor changes in the apparent protein-RNA affinity, suggesting that the increased methylation rates are at a step after binding (Figures S3F and S3G). Together, our data suggest that the intrinsic propensity for each wild-type RNA to be modified is encoded into the nucleotide sequence and that the transition region of the stem-loop is a key region to tune the methylation efficiency.
Autoregulatory K-Loop of METTL16 Blocks the SAM Binding Pocket
In both structures of METTL16-RNA complexes, an ordered polypeptide loop containing a key lysine residue (K-loop, residues 163–167) occludes the SAM binding site (Figures 4A–4C, S4A, and S4B). Extensive hydrophobic interactions anchor the K-loop to the core methyltransferase domain, resulting in the side chain of Lys163 to occupy the cofactor binding pocket. In contrast, in structures without RNA, the K-loop has a different conformation, where Lys163 is solvent-exposed and other hydrophobic side chains such as Met167 swing away from the domain (PDB: 6B92; Ruszkowska et al., 2018) (Figure 4B). To first validate the location of the SAM (and SAH) binding site, we tested four mutations that would interfere with cofactor binding. All three alanine substitutions (N184A, R82A, and E133A) as well as a mutation identified in large intestinal cancer patients (G110C, curated study by COSMIC COSU646) abrogate in vitro methyltransferase activity (Figure 4D). However, alanine substitutions that would destabilize the observed K-loop conformation in the METTL16-RNA complex—K163A or M167A—dramatically activate methylation, while having little effect on RNA affinity (Figures 4D and S4C). At saturating levels of substrate RNA, we monitored methylation reactions over different SAM concentrations. Destabilizing mutations within the K-loop transform METTL16 to become hyperactive, requiring lower concentrations of SAM to have the same level of in vitro methylation activity (Figure 4E). Therefore, a polypeptide loop in METTL16 near the SAM binding pocket acts as an intramolecular autoregulatory switch and likely dampens METTL16 activity during SAM binding and/or catalytic turnover. To compare the inhibitory effect of the K-loop to potential interference by SAH, we tested the METTL16 activity in the presence of various amounts of SAH. In contrast to the intact K-loop, the presence of SAH does not significantly decrease METTL16 activity, even at 10-fold molar excess to SAM (Figure S4D). To determine whether the K-loop mutations increase METTL16 activity through a common mechanism as the RNA transition region mutations discussed above, we measured how the methylation activity changes for hp5 that contains a native G-to-A variation (Figure 4F). We observe that the fold activation caused by both of the K-loop mutations is similar for hp1 and hp5, indicating that the two regulatory mechanisms are additive. Thus, modulation of METTL16 activity by the RNA transition region (e.g., R200Q or G-to-A) is independent of the auto-regulatory effect of the K-loop. Therefore, our structural and biochemical data have uncovered the K-loop as an independent, auto-regulatory switch that can dramatically affect the methyltransferase efficiency of METTL16.
Figure 4. Autoregulatory K-Loop of METTL16 Blocks the SAM Binding Pocket.

(A) Superimposition of MTD16-hp1x (as shown in Figure 1D) onto the structure of MTD16-SAH complex (PDB: 6B92) shown as pink cartoon representation. RMSD is 0.3 Å for 180 Cα atoms. SAH are shown as magenta sticks and labeled with a black arrow. K-loop region is indicated with a black dashed circle.
(B) Close-up view of the detailed interactions of the K-loop with the SAM binding pocket. Residues used in mutagenesis studies are shown in stick representation (blue, MTD16-hp1x; pink, MTD16-SAH). Black arrows highlight conformational differences.
(C) 2Fobs - Fcalc map contoured at 1.0 σ for MTD16-hp1x is shown for a portion of the K-loop (blue sticks).
(D) In vitro methylation activity of wild-type or mutant METTL16 on hp1. Data are shown as mean DPM ± SD from three replicates.
(E) In vitro methylation activity over a range of SAM concentrations, with wild-type or mutant METTL16, for hp1. Data are shown as mean DPM ± SD from three replicates.
(F) In vitro methylation activity of wild-type or mutant METTL16 relative to the wild-type activity on the indicated RNA substrates. Data are shown as the ratio of the means, and error is ± the relative SD.
See also Figure S4.
Modulating METTL16 Activity Alters MAT2A mRNA Levels
Through structure-guided mutagenesis studies, we reveal various mutations that can significantly inhibit or enhance METTL16 activity in vitro. To determine how the observed changes in enzymatic efficiency affect METTL16 function in intact cells, we introduced the METTL16 mutations and monitored its function as a “SAM sensor.” When SAM is limiting, METTL16 has slower turnover and thus dwells longer on hp1, which induces splicing of an otherwise retained intron of MAT2A (Pendleton et al., 2017). In addition, when SAM is abundant, increased m6A modification on the hairpins in the MAT2A 3′ UTR promotes degradation of MAT2A mRNA (Pendleton et al., 2017; Shima et al., 2017). To investigate how METTL16 mutations affect MAT2A mRNA levels, we used a previously characterized β-globin reporter that includes the MAT2A retained intron, flanking exons, and 3′ UTR and coexpressed various mutants of METTL16 (Pendleton et al., 2017) (Figures 5A, S5A, and S5B). Consistent with both mechanisms of action, the catalytically inactive but RNA-binding-competent N184A mutation of METTL16 stimulates increased MAT2A mRNA abundance relative to wild-type, even under high SAM conditions. In contrast, the SAM-hypersensitive K163A mutation causes reduced MAT2A mRNA levels compared to wild-type METTL16, even at low levels of intracellular SAM (Figures 5B and 5C). Furthermore, R200Q, another mutation near the RNA transition region that enhances METTL16 methyltransferase activity, also dramatically reduces mRNA levels, throughout a range of intra-cellular SAM concentrations (Figures 5D and 5E). In addition, we analyzed the effects of the METTL16 regulatory mutations on MAT2A mRNA splicing. In cells transfected with catalytically inactive METTL16, MAT2A is nearly completely spliced regardless of the levels of SAM, whereas hyperactive METTL16 mutations result in higher levels of the retained intron isoform, even at low levels of SAM (Figures S5C–S5F). Together, our in vitro and in vivo results show that certain mutations near the RNA transition region or the K-loop can activate METTL16 enzymatic efficiency, which can have a direct impact on the steady-state levels of MAT2A mRNA. Such modes of regulation over METTL16 activity are presumably evolutionarily selected to maintain appropriate physiological SAM levels.
Figure 5. Modulating METTL16 Activity Alters MAT2A mRNA Levels.

(A) Schematic of the reporter construct used in cell-based assays.
(B) Representative northern blot of β-globin (MAT2A mRNA) in cells transfected with the reporter (A) and wild-type or mutant FLAG-METTL16, from cells grown over a range of methionine concentrations. Images were cropped from the same gel at the same exposure. GAPDH northern blot is shown below as a loading control.
(C) Quantification of relative mRNA normalized to GAPDH, expressed relative to WT at 1,000 μM methionine from three replicates of (B). Data are shown as the mean ± SD. p values are indicated with asterisks over bars that were compared using unpaired Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001.
(D) Representative northern blot of β-globin (MAT2A mRNA) in cells transfected with the reporter (A) and wild-type or mutant FLAG-METTL16, from cells grown over a range of methionine concentrations. Images were cropped from the same gel at the same exposure. GAPDH northern blot is shown below as a loading control.
(E) Quantification of relative mRNA normalized to GAPDH, expressed relative to WT at 1,000 μM methionine from three replicates of (D). Data are shown as the mean ± SD. p values are indicated with asterisks over bars that were compared using unpaired Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figure S5.
DISCUSSION
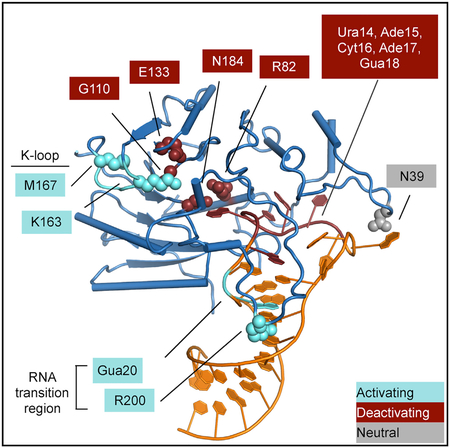
Our structural and biochemical data allow us to present a mechanistic model for METTL16 activity, specificity, and regulation (Figure 6). METTL16 plays a vital role in SAM homeostasis, by controlling the steady-state levels of the SAM synthetase, MAT2A, by modulating both splicing and stability of the mRNA. Through a three-dimensional view of the complex of METTL16 and MAT2A 3′ UTR hairpins at atomic resolution, we reveal how the methyltransferase domain recognizes the cognate RNAs to mark them with m6A. In addition to the catalytic activity, the C-terminal half of METTL16, including the vertebrate conserved regions, plays a role in recruiting factors to the mRNA to promote splicing (Pendleton et al., 2017). Thus, specific binding, affinity, dwell time, and catalytic efficiency of METTL16 toward RNA are all important elements to maintain SAM homeostasis (Figures 6A and 6B). Our results show that RNA affinity does not always correlate with methylation efficiency, underscoring the importance of distinguishing the two roles of METTL16. Genome-wide studies have reported many sites that METTL16 can bind (Brown et al., 2016; Pendleton et al., 2017; Warda et al., 2017), but given the sequence and structural requirements of METTL16 that we identify in this study, the targets that are methylated efficiently may be significantly fewer. METTL16-hp1 interactions are important for splicing, while the other hairpins primarily participate in m6A-dependent degradation. Our findings of how different hairpins exhibit variable propensities for binding and modification will be important to understand how the six hairpins are utilized to integrate multiple input signals to output the proper amount of steady-state MAT2A mRNA and thus SAM production.
Figure 6. Regulatory Mechanisms of METTL16.

(A) In an autoinhibited state, the METTL16 K-loop occludes the SAM binding pocket. This results in less methylation of the MAT2A 3′ UTR, increased splicing, and more stability for the transcript. The increased abundance of MAT2A mRNA results in an increase of SAM biosynthesis.
(B) METTL16 can be activated when the K-loop changes conformation to allow for SAM binding. This results in higher methylation efficiency of MAT2A mRNA, less splicing, and more degradation. The decreased abundance of MAT2A mRNA results in reduced SAM biosynthesis.
(C) Mutations tested in this study are marked by effect on in vitro methylation activity: activating (cyan), inhibitory (red), or unaffected (gray). Protein mutations are shown as spheres, and RNA mutations are indicated with outlined boxes.
METTL16 efficiency is linked to different rates of SAM biosynthesis. In this study we have identified various protein and RNA mutations that decrease or increase the methyltransferase activity of METTL16 (Figure 6C). The deactivating mutations are near the ribose and methionine moieties of SAM, as well as the RNA loop region. We also reveal at least two independent mechanisms to activate the efficiency of METTL16, both of which can directly affect MAT2A mRNA levels in cells. First, mutations in the substrate or the enzyme near the RNA transition region can increase methyltransferase activity. Second, observing a closed conformation of the K-loop occluding the SAM binding site in the crystal structures led to the finding that METTL16 can exhibit an autoinhibitory conformation. The K-loop is closed in the structures of the protein-RNA complexes but is open in both apo and SAH-bound structures (Ruszkowska et al., 2018; PDB: 2H00). It is possible that RNA binding and closure of the K-loop region are coupled. Based on the structures, how the K-loop might rearrange upon RNA binding is not obvious. Although the enhancement effects of the RNA transition region and the K-loop are additive, whether the RNA binding event and the K-loop conformational switch—and thus SAM binding—are truly independent will need a more thorough investigation. Moreover, even though the structures clearly provide a molecular explanation for how the K-loop might interfere with SAM binding, what is less obvious is the enhancement of enzymatic activity when the RNA transition region is disturbed. Some possible reasons for increased methylation include: (1) a faster release of the methylated RNA, (2) an alternative conformation with the mutation that directly stabilizes the transition state, (3) an allosteric effect to make SAM binding or the chemical reaction more favorable, and (4) an allosteric effect to make SAH release more efficient. This study will provide the groundwork for further investigations to distinguish these possibilities. The R200Q mutation near the transition region is found in certain cancer patients, and our model for METTL16 function will also be useful to determine the mode of action for other disease-associated mutations.
Although METTL16 and METTL3/METTL14 are both m6A writers, they are distinct in important ways. Insight from MTD16-RNA complex structures suggests that RNA binding likely induces rearrangement and folding of the catalytic cavity in both enzymes. However, the interaction of each catalytic MTD with the RNA substrate is confined by the unique architectural context. The catalytic domain of METTL16 is monomeric and manifests full activity without the C-terminal region. In contrast, METTL3/METTL14 is an obligate heterodimer, and the catalytic domains require additional help from the CCCH zinc-binding motifs of METTL3 in order to bind and methylate RNA substrates. METTL16 prefers an RNA substrate that contains a double-stranded RNA adjacent to the consensus sequence, while METTL3/METTL14 does not require such structural context (Liu et al., 2014). Such differences could be due to the wider, pre-folded cavity on METTL3/METTL14 (Śledź and Jinek, 2016; Wang et al., 2016a, 2016b), in contrast to METTL16, which folds upon binding RNA to form a deep concave pocket within the catalytic domain. Lastly, autoinhibition via the K-loop may be unique to METTL16. In METTL3, the loop that is analogous to the K-loop based on structural superimposition does not undergo a significant conformational change upon cofactor binding and also does not contain a lysine residue within the loop. Therefore, METTL16 and METTL3/METTL14 diverge in both how they recognize substrates and how they are regulated.
Tunable METTL16 activity allows for tight control of an essential metabolite, SAM. Other SAM sensors, such as SAMTOR, link metabolism to cellular function (Gu et al., 2017), but METTL16 is unique in that it couples SAM sensing with SAM biosynthesis. During this feedback process, METTL16 activity can be modulated in various ways, through sequence and structural changes of both substrate RNA and protein. Controlling enzymatic activity through conformational changes or allostery has been observed in other methyltransferases, such as METTL3, the DNA cytosine methyltransferase complex DNMT3A/3L, and the m7G cap methyltransferase RNMT (Guo et al., 2015; Varshney et al., 2016; Wang et al., 2016a). Many of these examples involve another factor, such as METTL14, histone H3 tail, and RAM, respectively. Our data reveal multiple mechanisms to attenuate or activate METTL16, which might also be triggered by other factor(s) and/or modification(s) that act on either METTL16 or the substrate RNA.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yunsun Nam (yunsun.nam@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
293A-TOA cells were cultured at 37°C in DMEM supplemented with 2 mM L-glutamate, penicillin-streptomycin and 10% FBS (Millipore-Sigma). Methionine-free DMEM (Thermo Fisher 21013024) was supplemented with 1 mM sodium pyruvate, 0.4 mM L-cysteine (Millipore-Sigma) and Tet-Free FBS (Atlanta Biologicals) was used in place of FBS. L-methionine (Millipore-Sigma) was added to Met-free media to obtain the concentration specified.
Expression of recombinant proteins
Specified constructs were transformed into Rosetta (DE3) pLysS cells (Novagen). Target proteins were expressed in cultures grown in autoinduction media at 18°C overnight (Studier, 2005).
METHOD DETAILS
Plasmids
METTL16 constructs for expression in E. coli were cloned into the pET21 vector (Novagen) by amplifying fully spliced cDNA (Pendleton et al., 2017) using the primers indicated in Table S2. For expression of METTL16 constructs in human cells, mutagenesis was done with the primers indicated in Table S2 using the wild-type FLAG tagged construct.(Pendleton et al., 2017).
Protein purification
Cultures were harvested and sonicated in lysis buffer (50 mM Bis-Tris [pH 7.0], 1 M NaCl, 10% glycerol, 1 mM dithiothreitol (DTT) and supplemented with protease inhibitors). Hexahistidine-tagged METTL16 was further purified by affinity for Ni-NTA (QIAGEN) and the tag was removed by digestion with Tobacco Etch Virus (TEV) protease. Target proteins were further purified by ion-exchange chromatography and followed by gel filtration chromatography. All recombinant METTL16 constructs were analyzed by SDS-PAGE, and UV spectroscopy. Proteins were then combined with synthesized RNA constructs (Millipore-Sigma) and assembled complexes were further purified by gel-filtration. The final complex in buffer containing 20 mM Tris (pH 7.8), 50 mM NaCl and 5mM DTT was also supplemented with 1.5 mM SAH (Millipore-Sigma) for crystallization experiments.
Crystallization and data collection
The crystals of the MTD16-hp1x complex were obtained using the hanging-drop, vapor-diffusion method by mixing 1 μL protein-RNA complex (10 mg/mL) with 1 μL reservoir solution containing 0.1 M Tris (pH 8.5), 20% glycerol, and 19% polyethylene glycol (PEG) 10000 and incubating at room temperature. The complex crystals with hp6 were generated by mixing 1 μL protein-RNA complex (10 mg/mL) with 1 μL reservoir solution containing 0.95 M sodium citrate. Se-SAD and native datasets were collected at APS-19-ID at wavelengths of 0.97942 Å and 0.97924 Å, respectively.
Structure determination and refinement
Data were indexed, integrated, and scaled by the program HKL3000 (Minor et al., 2006). The initial phases for SeMet-MTD16-hp6 were determined by single-wavelength anomalous dispersion (SAD) using Phenix.Autosol, and density modification and automatic modeling were performed by Phenix.Autobuild (Adams et al., 2010). Model building was finalized manually using COOT (Emsley and Cowtan, 2004). The structure of MTD16-hp1x was determined by molecular replacement using the MTD16 structure from MTD16-hp6 as a searching model, with the program PHASER (McCoy et al., 2007). All the models were refined using Phenix.refine (Zwart et al., 2008). The PROCHECK program was used to check the quality of the final model, which shows good stereochemistry according to the Ramachandran plot (Laskowski et al., 1993). All structure figures were generated by PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC. Software used in this project was curated by SBGrid (Morin et al., 2013).
In vitro methylation assay
The in vitro methylation assay was carried out in triplicate with a 15 μL reaction mixture containing: 50 mM Tris (pH 8.5), 66 mM NaCl, 0.01% Triton-X, 1 mM DTT, 50 μM ZnCl2, 0.2 U/μL RNasin, and 1% glycerol, 300 nM METTL16 and 1 μM RNA substrate (Millipore-Sigma for all oligonucleotides, extended hp1 and FL U6 were in vitro transcribed) (Walker et al., 2003; Wang et al., 2016a). In vitro methylation assays with METTL3/METTL14 were performed in the same buffer, but with 100 nM of the heterodimer and 5 μM RNA substrate. 3H-SAM (Perkin Elmer) was used at 1 μM when held constant in in vitro methylation reactions. In the SAM titration experiment, 3H-SAM was mixed at a 1:3.3 molar ratio with cold SAM (Millipore-Sigma) and added at the concentrations indicated (Figure 4E). In the SAH competition in vitro methylation assay, SAH (Millipore-Sigma) and 3H-SAM were added at the indicated concentrations (Figure S4D). Each reaction was incubated at 37°C for 2 hr. Half of the reaction mixture was blotted on Hybond N+ nylon membranes (GE Healthcare Amersham) and crosslinked with UV (254 nm). The membranes were washed with reaction buffer, deionized water, and 95% ethanol, in that order, and then subjected to liquid-scintillation counting using the TriCarb 2010 TR Scintillation Counter (Perkin Elmer). Levels of RNA with the incorporated 3H-methyl group are shown as disintegrations per minute (DPM). Data are shown as mean ± SD from three replicates.
Electrophoretic Mobility Shift Assays
RNAs were radiolabeled with ATP[γ−32P] using T4 polynucleotide kinase, and incubated with METTL16 protein constructs in a buffer containing 50 mM Tris (pH 8.5), 66 mM NaCl, 5 6mM DTT, 50 μM ZnCl2, 10 ng/μL yeast tRNA and 10% glycerol. Protein-RNA complexes were resolved by 8% Tris-Glycine native PAGE and apparent affinities were determined by the concentration of protein at which half of the RNA signal had shifted to the complex species (Ryder et al., 2008).
Transfection
293A-TOA cells (Sahin et al., 2010) at ~80% confluence were transfected using Transit-293 (Mirus) following manufacturer’s protocol. Cells were transfected with 100 ng of b globin reporter (Pendleton et al., 2017), 100 ng FLAG-METTL16 and 700 ng pcDNA3 per well.
Methionine depletion
Eighteen hours post-transfection, cells were washed twice with Dulbeccos’ Phosphate Buffer Saline (PBS) with calcium chloride and magnesium chloride (Millipore-Sigma). Media was replaced with methionine-free media supplemented with the indicated concentration of L-methionine (Figures 5C, 5E, S5D, and S5F). Cells were harvested 6 hr later with TRI-Reagent.
Northern blotting
Standard northern blot protocols were followed. The template for the b-globin probe was made by digesting plasmid βΔ1,2(B-A) (Conrad et al., 2006) with NcoI and in vitro transcription was performed using SP6 polymerase in the presence of radiolabeled α- 32P-UTP.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of the cell-based experiments can be found in the corresponding figure legends. Northern blots were quantified with Imagequant 5.2 and each experiment was performed with at least three biological replicates. Unpaired Student’s t tests were used to test significance * p < 0.05, ** p < 0.01, *** p < 0.001. (Pendleton et al., 2017)
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the structure factors and coordinates for MTD16-hp1x and MTD16-hp6 complexes reported in this paper are Protein Data Bank: 6DU4, 6DU5.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-FLAG M2 antibody | Millipore Sigma | Cat#F3165; RRID:AB_259529 |
| Mouse anti-Actin | Abcam | Cat#ab6276; RRID:AB_2223210 |
| Goat-anti mouse IgG IRDye 800CW | LI-COR Biosciences | Cat#92632210; RRID:AB_621842 |
| Bacterial and Virus Strains | ||
| Rosetta (DE3) pLysS | Novagen | Cat# 70956 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| H3 S-adenosylmethionine | Perkin Elmer | Cat# NET155H001MC |
| S-adenosylmethionine chloride dihydrochloride | Miliipore-Sigma | Cat# A7007 |
| S-adenosylhomocysteine | Millipore-Sigma | Cat#A9384 |
| Selenomethionine | Acros Organics | Cat# 3211-76-5 |
| P32-adenosine triphosphate | Perkin Elmer | Cat# NEG035C001MC |
| METTL16 FL WT | This paper | N/A |
| METTL16MTD (1–310) | This paper | N/A |
| FL METTL16 R200Q | This paper | N/A |
| FL METTL16 N39A | This paper | N/A |
| FL METTL16 N184A | This paper | N/A |
| FL METTL16 R82A | This paper | N/A |
| FL METTL16 E133A | This paper | N/A |
| FL METTL16 G110C | This paper | N/A |
| FL METTL16 K163A | This paper | N/A |
| FL METTL16 M167A | This paper | N/A |
| FL METTL3/METTL14 WT | Wang et al., 2016a | N/A |
| Deposited Data | ||
| MTD16-hp1x structure | This paper | PDB: 6DU4 |
| MTD16-hp6 structure | This paper | PDB: 6DU5 |
| Experimental Models: Cell Lines | ||
| Human 293A-TOA | Sahin et al., 2010 | N/A |
| Oligonucleotides | ||
| RNA sequences | Table S2 | N/A |
| Primers for protein and RNA constructs | Table S2 | N/A |
| Recombinant DNA | ||
| Plasmid: FL (1–562) METTL16 WT pET21 | This paper | N/A |
| Plasmid: METTL16 MTD (1–310) WT pET21 | This paper | N/A |
| Plasmid FL METTL16 R200Q pET21 | This paper | N/A |
| Plasmid FL METTL16 N39A pET21 | This paper | N/A |
| Plasmid FL METTL16 N184A pET21 | This paper | N/A |
| Plasmid FL METTL16 R82A pET21 | This paper | N/A |
| Plasmid FL METTL16 E133A pET21 | This paper | N/A |
| Plasmid FL METTL16 G110C pET21 | This paper | N/A |
| Plasmid FL METTL16 K163A pET21 | This paper | N/A |
| Plasmid FL METTL16 M167A pET21 | This paper | N/A |
| Plasmid FL METTL3/METTL14 pET21 | Wang et al., 2016a | N/A |
| Plasmid β MAT-WT | Pendleton et al., 2017 | N/A |
| Plasmid pRZ | Walker et al., 2003 | N/A |
| Plasmid pRZ extended hp1 | This paper | N/A |
| Plasmid pRZ FL U6 | This paper | N/A |
| Plasmid pET21 | Novagen | N/A |
| Plasmids FLAG-METTL16 WT pcDNA3 | Pendleton et al., 2017 | N/A |
| Plasmid FLAG-METTL16 pcDNA3 K163A | This paper | N/A |
| Plasmid FLAG-METTL16 pcDNA3 N184A | This paper | N/A |
| Plasmid FLAG-METTL16 pcDNA3 R200Q | This paper | N/A |
| Plasmid βΔ1, 2 (B-A) (β-globin probe) | Conrad et al., 2006 | N/A |
| Software and Algorithms | ||
| ImageQuant 5.2 | GE Healthcare Life Sciences | RRID:SCR_014246 |
| Phenix.Autosol | Adams et al., 2010 | RRID:SCR_014224; http://www.phenix-online.org/ |
| COOT | Emsley and Cowtan, 2004 | RRID:SCR_014222; https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHASER | McCoy et al., 2007 | RRID:SCR_014219; http://www.phenix-online.org/ |
| Phenix.refine | Zwart et al., 2008 | RRID:SCR_014224; http://www.phenix-online.org/ |
| PROCHECK | Laskowski et al., 1993 | https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ |
| PyMol Molecular Graphics System 1.8 | Schrodinger, LLC | RRID:SCR_000305; https://pymol.org/2/ |
Highlights.
METTL16 recognizes substrate RNA sequence and structure for methylation
Methylation activity of METTL16 can be modulated by the substrate RNA conformation
METTL16 is autoregulated through its SAM binding pocket
Mutations in the catalytic domain of METTL16 regulate MAT2A mRNA levels in cells
ACKNOWLEDGMENTS
We thank support from the Cecil H. and Ida Green Center Training Program in Reproductive Biology Sciences Research and also members of the Structural Biology Laboratory at UT Southwestern for help with data collection. Y.N. is a Southwestern Medical Foundation Scholar in Biomedical Research (Endowed Scholar Program at UT Southwestern), a Pew Scholar (27339), and a Packard Fellow (2013–39275). This study was supported by grants from the NIH NIGMS (R01GM122960 to Y.N., 2T32GM008297 to K.A.D., and 5T32GM007062 to A.M.S.), American Heart Association (17POST33410713 to P.W.), the Welch Foundation (I-1851–20170325 to Y.N. and I-1915–20170325 to N.K.C.), Cancer Prevention Research Institute of Texas (R1221), and American Cancer Society/Harold C. Simmons Comprehensive Cancer Center (ACS-IRG-02-196 to Y.N. and RSG-14-064-01-RMC to N.K.C.). The use of SBC 19ID beamline at Advanced Photon Source is supported by United States Department of Energy contract DE-AC02–06CH11357.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and two tables and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.07.025.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Kinzig CG, DeGregorio SJ, and Steitz JA (2016). Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 113, 14013–14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, and Rando TA (2017). Interaction between epigenetic and metabolism in aging stem cells. Curr. Opin. Cell Biol 45, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujnicki JM, Feder M, Radlinska M, and Blumenthal RM (2002). Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J. Mol. Evol 55, 431–444. [DOI] [PubMed] [Google Scholar]
- Conrad NK, Mili S, Marshall EL, Shu M-DD, and Steitz JA (2006). Identification of a rapid mammalian deadenylation-dependent decay pathway and its inhibition by a viral RNA element. Mol. Cell 24, 943–953. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-Carbon Metabolism in Health and Disease. Cell Metab. 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgavish T, Cannone JJ, Lee JC, Harvey SC, and Gutell RR (2001). A:A and A:G base-pairs at the ends of 16 S and 23 S rRNA helices. J. Mol. Biol 310, 735–753. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. (2016). Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 15, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, Scaria SM, Harper JW, Gygi SP, and Sabatini DM (2017). SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wang L, Li J, Ding Z, Xiao J, Yin X, He S, Shi P, Dong L, Li G, et al. (2015). Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517, 640–644. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Zhang D, and Aravind L (2016). Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. BioEssays 38, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke R, Dodson AE, and Rine J (2015). Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol 31, 473–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG Jr., and McKnight SL (2013). Influence of metabolism on epigenetics and disease. Cell 153, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, and Thornton JM (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst 26, 283–291. [Google Scholar]
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, and Jaffrey SR (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Chantar ML, Latasa MU, Varela-Rey M, Lu SC, García-Trevijano ER, Mato JMM, and Avila MAA (2003). L-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells: role of S-adenosylmethionine. J. Biol. Chem 278, 19885–19890. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, and Locasale JW (2016). One-carbon metabolism and epigenetics: understanding the specificity. Ann. N Y Acad. Sci 1363, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, and Jaffrey SR (2017). Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol 33, 319–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, and Sliz P (2013). Collaboration gets the most out of software. eLife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF, et al. ; Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BJ, Moltke I, Roth A, Washietl S, Wen J, Kellis M, Breaker R, and Pedersen JS (2011). New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 21, 1929–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, and Conrad NK (2017). The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 169, 824–835.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roost C, Lynch SR, Batista PJ, Qu K, Chang HY, and Kool ET (2015). Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc 137, 2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, and He C (2017). Dynamic RNA Modifications in Gene Expression Regulation. Cell 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruszkowska A, Ruszkowski M, Dauter Z, and Brown JA (2018). Structural insights into the RNA methyltransferase domain of METTL16. Sci. Rep 8, 5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder SP, Recht MI, and Williamson JR (2008). Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol. Biol 488, 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin BB, Patel D, and Conrad NK (2010). Kaposi’s sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 6, e1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata SF, Shelly LL, Ruppert S, Schutz G, and Chou JY (1993). Cloning and expression of murine S-adenosylmethionine synthetase. J. Biol. Chem 268, 13978–13986. [PubMed] [Google Scholar]
- Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, Kumagai S, Ochiai K, Suzuki T, and Igarashi K (2017). S-Adenosylmethionine Synthesis Is Regulated by Selective N6-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep. 21, 3354–3363. [DOI] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, and Kume S (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794. [DOI] [PubMed] [Google Scholar]
- Śledź P, and Jinek M (2016). Structural insights into the molecular mechanism of the m(6)A writer complex. eLife 5, e18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Su X, Wellen KE, and Rabinowitz JD (2016). Metabolic control of methylation and acetylation. Curr. Opin. Chem. Biol 30, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney D, Petit A-P, Bueren-Calabuig JA, Jansen C, Fletcher DA, Peggie M, Weidlich S, Scullion P, Pisliakov AV, and Cowling VH (2016). Molecular basis of RNA guanine-7 methyltransferase (RNMT) activation by RAM. Nucleic Acids Res. 44, 10423–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SC, Avis JM, and Conn GL (2003). General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 31, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT, Tu BP, and Tang Y (2018). Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism. Chem. Rev 118, 1460–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, and Nam Y (2016a). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 63, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. (2016b). Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578. [DOI] [PubMed] [Google Scholar]
- Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, Sloan KE, and Bohnsack MT (2017). Human METTL16 is a N6-methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou KI, Parisien M, Dai Q, Liu N, Diatchenko L, Sachleben JR, and Pan T (2016). N(6)-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. J. Mol. Biol 428 (5 Pt A), 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung L-W, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, et al. (2008). Automated structure solution with the PHENIX suite. Methods Mol. Biol 426, 419–435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the structure factors and coordinates for MTD16-hp1x and MTD16-hp6 complexes reported in this paper are Protein Data Bank: 6DU4, 6DU5.