

Abstract
The diploid D-genome lineage of the Triticum/Aegilops complex has an evolutionary history involving genomic contributions from ancient A- and B/S-genome species. We explored here the possible cytonuclear evolutionary responses to this history of hybridization. Phylogenetic analysis of chloroplast DNAs indicates that the D-genome lineage has a maternal origin of the A-genome or some other closely allied lineage. Analyses of the nuclear genome in the D-genome species Aegilops tauschii indicate that accompanying and/or following this ancient hybridization, there has been biased maintenance of maternal A-genome ancestry in nuclear genes encoding cytonuclear enzyme complexes (CECs). Our study provides insights into mechanisms of cytonuclear coevolution accompanying the evolution and eventual stabilization of homoploid hybrid species. We suggest that this coevolutionary process includes likely rapid fixation of A-genome CEC orthologs as well as biased retention of A-genome nucleotides in CEC homologs following population level recombination during the initial generations.
Keywords: cytonuclear coevolution, homoploid hybrid speciation (HHS), genes encoding cytonuclear enzyme complexes (CECs), Triticum/Aegilops complex
Introduction
Hybridization is an important process in plant evolution, often leading to speciation via genome doubling or at the homoploid level (Soltis and Soltis 2009; Abbott et al. 2013; Soltis et al. 2014; Yakimowski and Rieseberg 2014). During homoploid hybrid speciation (HHS), the early stages often involve sterility or other fitness barriers that need to be overcome by natural selection for genomically and phenotypically new species (Rieseberg et al. 1995; Coyne and Orr 2004; Abbott et al. 2010). Historical evidence of this process has emerged from genetic and genomic analyses of nuclear genes and from discordance between organellar and nuclear markers (Arnold et al. 1988; Rieseberg 1991; Wendel et al. 1991; Dowling and Secor 1997; Hermansen et al. 2011). The prevalence of HHS in plant evolution is underscored by the increasing frequency with which such discordance and hybrid ancestries are revealed, as summarized in recent reviews (Gross and Rieseberg 2005; Yakimowski and Rieseberg 2014; Nieto et al. 2017; Folk et al. 2018). The most extensive and detailed studies involve hybrid species of Helianthus (Rieseberg 1991; Gross et al. 2003; Rieseberg et al. 2003; Gross and Rieseberg 2005), Iris (Anderson and Hubricht 1938; Anderson 1949; Arnold 1992, 1994, 1997), Senecio (Abbott et al. 2000; James et al. 2005; Abbott et al. 2009), and Heuchera (Folk et al. 2017).
One of the consequences of HHS is mosaicism of the nuclear genome, in which the genome of the derived homoploid hybrid contains a blend of genes and genomic segments from its progenitor lineages (Rieseberg 1991; Arnold 1997; Gross et al. 2003; Abbott et al. 2009; Schumer et al. 2014). A representative recent example concerns the D-genome species in the Triticum/Aegilops complex, which apparently was derived from complex hybridizations involving ancient A- and B/S-genome species as parents (Marcussen et al. 2014; Sandve et al. 2015; Li et al. 2015a, 2015b; El Baidouri et al. 2017). Phylogenomic analyses initially revealed that the relationships among A-genome species (T. monococcum, T. urartu, A-subgenome of T. aestivum), B/S-genome species (Ae. speltoides), and D-genome species (Aegilops tauschii) varied among nuclear genes, with topologies A (B, D) and B (A, D) being similar in quantity (overall genomic admixture ratio of A- and B/S-genomes as 1:1), both being more frequent than D (A, B) (Marcussen et al. 2014; Li et al. 2015b). In addition, phylogenomic investigations of chloroplast genomes and the evolutionary dynamics of gene-based transposable elements (TEs) and homoeoSNPs also support the homoploid hybrid origin of the ancestor of the bread wheat D-genome, but with a more complex nature (Sandve et al. 2015; Li et al. 2015a, 2015b; El Baidouri et al. 2017).
As is the case with allopolyploid evolution (Gong et al. 2012, 2014; Sehrish et al. 2015; Sharbrough et al. 2017), stabilization of homoploid populations derived from interspecific hybridization is likely to involve epistatic selection to overcome negative fitness consequences resulting from merger of two differentiated nuclear genomes in the cytoplasm of only one of the two progenitor genomes. The molecular mechanisms involved in these potential nuclear-cytoplasmic disruptions are not well understood, even though this cytonuclear incompatibility is a well-known aspect of hybridization (Levin 2003; Fishman and Willis 2006; Bomblies and Weigel 2007; Burton et al. 2013; Sloan 2015).
The vast majority of cytonuclear enzyme complexes (hereafter abbreviated as CECs) is derived from nuclear genes that encode proteins that are targeted to the organelles(Rand et al. 2004; Millar et al. 2005; Woodson and Chory 2008; Van Wijk and Baginsky 2011). A subset of these organellar protein complexes are assembled from multiple subunits encoded by both the nuclear and organellar (mitochondrial and plastid) genomes, and so are cytonuclear co-encoded enzyme complexes (CCECs). Both categories provide the opportunity to look for the evolutionary footprints of cytonuclear adjustments to disruptions accompanying genome merger and/or genome doubling (Bock et al. 2014; Sloan et al. 2014; Weng et al. 2016). Our prior work using allopolyploids and the exemplar CCEC enzyme Rubisco (1, 5-bisphosphate carboxylase/oxygenase) showed that paternal nuclear rbcS genes (encoding small subunits of Rubisco, SSUs) were altered, presumably via gene conversion, to be maternal-like, and that gene expression was biased in the same direction (Gong et al. 2012, 2014). To the best of our knowledge, these types of evolutionary processes have not been studied in the context of HHS, nor has this approach been extended to the whole-genome level.
In this paper, we present the results of a global analysis of cytonuclear coevolution in Ae. tauschii, a species with compelling evidence of bi or multiparental ancestry (Marcussen et al. 2014; Sandve et al. 2015; Li et al. 2015a, 2015b). We confirmed a previously inferred derivation in Ae. tauschii of organelles from a taxon resembling the modern A-genome species. Using predictions of protein subcellular localization, we also characterized the composition of nuclear genes with respect to their ancestral parentage, in an effort to address whether CECs in Ae. tauschii have a biased heritage and/or if they have experienced gene conversion in the course of evolution. We show that D-genome CECs in Ae. tauschii are indeed biased in their genome-diagnostic SNPs towards the maternal, A-genome parent, whereas nuclear genes as a whole do not show this bias. These data represent the first evidence bearing on possible genome-wide epistatic selection favoring retention of maternal CEC homologs and nucleotides during hybrid speciation.
Results
Phylogenetic Analysis of Chloroplast Genes Indicates a Shared A-genome Cytoplasmic Ancestry with Ae. tauschii
To investigate the cytonuclear coevolution following HHS, it is necessary to determine the maternal origin of the cytoplasmic organelles. Toward this end, we phylogenetically analyzed cpDNA gene orthologs in representative species of the D-genome lineage (including species of D-, M-, and S*-genome groups) and representative species of A- and S-genome groups in the Triticum/Aegilops complex (T. aestivum is known to have B- or S-cpDNA from its tetraploid parent, T. turgidum, and was categorized into S-genome group). Our analysis used only the chloroplast genes rather than whole chloroplast genomes adopted in a previous study (Li et al. 2015b), to explore whether potentially noisy hypervariable plastid intergenic regions could impact phylogenetic inference.
As shown in figure 1 and supplementary figure 1, Supplementary Material online, relative to the S-genome groups, the concatenated chloroplast genes of the D-genome lineage phylogenetically align with those from the A-genome group in both Neighbor-Joining (NJ) and Maximum Likelihood (ML) trees. We note that the overall topology of the cpDNA genes is identical to that obtained using whole cpDNA genomes (Li et al. 2015b), thus confirming this earlier result. Given the strict maternal inheritance of both chloroplast and mitochondria in wheat (Greiner et al. 2015), we infer that the D-genome lineage harbors organelles that are closely related to those of the A-genome, and thus likely obtained these genomes through ancient hybridization.
Fig. 1.

Neighbor-Joining (NJ) tree of species in the D-genome lineage of Triticum/Aegilops complex and the outgroup species (Hordeum vulgare) inferred from phylogenetic analysis of concatenated chloroplast gene orthologs. Representative species in A- and B/S-genome groups (A-genome group: T. monococcum and T. urartu and B/S-genome group: T. aestivum and Ae. speltoides) and D-genome lineage (D-genome group: Ae. cylindrica and Ae. tauschii; M-genome group: Ae. geniculata; S*-genome group: Ae. bicornis, Ae. longissima, Ae. searsii, and Ae. sharonensis) are included and shown as in Li et al. (2015b) (colored bars and names). Bootstrap values are shown at nodes. The right panel summarizes the chloroplast phylogeny of the A- and B/S-genomes (red and green lines, respectively) in the context of homoploid hybridization events between ancient A- and B/S-genome species. The scale bar represents substitutions and indels per nucleotide position. See text for additional explanation.
Nuclear Gene Homologs Predicted to Encode Proteins Assembled into CECs
To characterize the profile of nuclear genes encoding the components of CECs, we employed TargetP and LOCALIZER (Emanuelsson et al. 2007; Sperschneider et al. 2017) to predict the subcellular localization of nuclear genes in genome assemblies of representative species in Triticum/Aegilops complex. Nuclear CEC genes predicted to encode proteins targeted to organelles were clustered into homolog groups using OrthoFinder. This was done for the diploid D-genome species Ae. tauschii (2D), the A-genome species T. urartu (2A), and the B/S-genome species Ae. speltoides (2B). In addition, we included homoeologs from the allopolyploid wheats, specifically the A- and B/S-subgenomes within both tetraploid T. turgidum and hexaploid T. aestivum (denoted as 4A, 4B, 6A, and 6B, respectively), which might additionally diagnose B-genome parental SNPs involved in ancient hybridization events (fig. 2 and table 1).
Fig. 2.
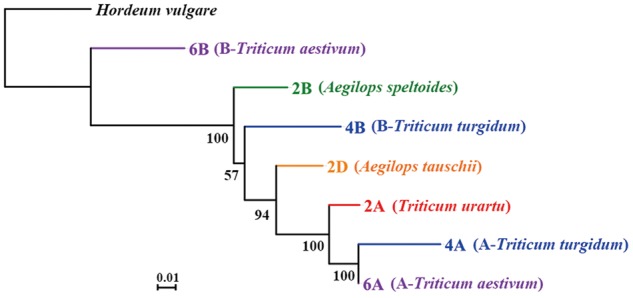
Neighbor-Joining (NJ) tree based on concatenated gene homologs encoding cytonuclear enzyme complexes (CECs) in representative species and subgenomes of the Triticum/Aegilops complex and the outgroup species (Hordeum vulgare). Bootstrap values are shown at each node. In addition to the diploids (2A-T. urartu, 2B-Ae. speltoides, and 2D-Ae. tauschii) and the outgroup species (H. vulgare), the gene homologs of A- and B/S-subgenomes of the tetraploid (T. turgidum, denoted as 4A and 4B, in blue), and hexaploid wheat (T. aestivum, denoted as 6A and 6B, in purple) are included. The scale bar represents substitutions and indels per nucleotide position.
Table 1.
Gene Homolog Groups for Nuclear Genes Encoding Cytonuclear Enzyme Complexes (CECs) in Representative Species and Subgenomes in the Triticum/Aegilops Complex.
| 2D | 2A | 2B | 4A | 4B | 6A | 6B | |
|---|---|---|---|---|---|---|---|
| Nuclear genes encoding CECs | 2,216 | 4,362 | 2,821 | 2,870 | 2,867 | 3,261 | 3,233 |
| Nuclear genes encoding CECs categorized in homolog groups | 2,216 | 4,362 | 2,820 | 2,795 | 2,800 | 3,261 | 3,233 |
| Categorization percentage | 100.00% | 100.00% | 99.96% | 97.39% | 97.66% | 100.00% | 100.00% |
| Number of homolog groupsa | 2,038 | 4,138 | 2,494 | 2,612 | 2,592 | 2,850 | 2,769 |
Note.—Nuclear genes encoding putative CECs in the diploid species (2A-T. urartu, 2B-Ae. speltoides, and 2D-Ae. tauschii) and the A- and B/S-subgenomes within the tetraploid (T. turgidum, denoted as 4A and 4B, respectively) and hexaploid wheats (T. aestivum, denoted as 6A and 6B, respectively) were predicted by TargetP and LOCOLIZER, which were categorized into homolog groups via OrthoFinder.
All gene groups identified are included, including those lacking corresponding homologous groups in some species and/or subgenomes.
Depending on the taxon and genome, between 2,216 and 4,362 gene homologs were predicted to encode proteins targeted to mitochondria and plastids (the first row, table 1). Of note, relatively conserved percentages of nuclear genes encoding CECs (97.39–100.00%) were identified in syntenic regions of respective diploid species (2A and 2B) and subgenomes of tetraploid and hexaploid species (4A, 4B, 6A, and 6B) (the second and third rows in table 1). We suspect that the observed discrepancies among taxa and genomes in putative CEC gene numbers categorized into homolog groups (the second through fourth rows, table 1; supplementary fig. 2, Supplementary Material online) reflects differences in genome assembly and annotation quality as well as gene models being incorrectly collapsed in some cases. Additionally, variation in nuclear CEC predictions may reflect differential gene family expansion or contraction among species. To minimize noise and error in our predictions for subsequent evolutionary analyses, we selected the most conserved CEC gene homologs (n = 150) that were predicted in all seven taxa and genomes (supplementary table 1, Supplementary Material online).
Notably, the homologs of well-known nuclear genes encoding proteins vital for organellar function in plants, such as Rubisco (rbcS), ATP synthase (beta subunit), and the enzymes in TCA cycle (e.g., isocitrate dehydrogenase subunit), were captured in our TargetP and LOCALIZER prediction (supplementary table 1, Supplementary Material online). On the basis of further validation by cropPAL, of the 20 proteins with a predicted subcellular localization, 4 were annotated as being nuclear or cytoplasmic, and the other 16 confirmed the software-based predictions. We infer that our predicted CEC gene set is indeed highly enriched for organellar proteins, notwithstanding the imperfect information regarding the subcellular localizations of the proteins as well as the prediction software.
Concatenated and Consensus Gene Trees Reveal Biased Retention of A-genome Ancestry in the D-genome Species Ae. tauschii
Given that the D-genome lineage has a shared A-genome chloroplast DNA ancestry, we explored the possibility that D-genome nuclear genes are biased in their ancestral retention of nuclear genes from its two progenitor genomes (A- and B/S-). To test this, nuclear gene homologs encoding predicted CECs in the study species were input into phylogenic analyses. To simplify phylogenic inference, for the groups that include multiple homologs in any genome, we sorted and paired homologs in terms of their hierarchical similarity, which were then input into phylogenetic analyses.
For the putative nuclear CEC genes as predicted above (supplementary table 1, Supplementary Material online), NJ and ML trees were built based on the concatenated supergene alignments. Both analyses showed that nuclear genes encoding putative CECs in 2D are phylogenetically sister to their A-genome homologs (2A, 4A, and 6A), and that this D + A group is derived relative to the paraphyletic B-genomes (2B, 4B, and 6B) (fig. 2 and supplementary fig. 3, Supplementary Material online). Despite the paraphyly of the B-genomes, this phylogenetic topology is mostly consistent with that based on chloroplast genes (fig. 1). Considering the intrinsic limitation of phylogenetic reconstruction based on concatenation methods (e.g., possible variance among genes with respect to substitution processes and rates, Gadagkar et al. 2005) and the relatively low bootstrap value connecting 4B to the A- and D-genome clades (bootstrap value as 57 and 62 in NJ and ML tree, respectively, fig. 2 and supplementary fig. 3, Supplementary Material online), we also inferred the phylogenies separately for each gene using Bayesian methods, and constructed a consensus phylogenetic tree by integrating all single gene trees (fig. 3). In line with the foregoing topology based upon the concatenated alignment, most genes encoding putative CECs in Ae. tauschii display closer phylogenetic relationships with diploid A genomes or polyploid A subgenomes (2A, 4A, and 6A) than they do with diploid B-genomes or polyploid B subgenomes (2B, 4B, and 6B) (fig. 3).
Fig. 3.

Superimposed ultrametric gene trees in a consensus plot representing phylogenic relationship among gene homologs encoding cytonuclear enzyme complexes (CECs) in representative species and subgenomes of the Triticum/Aegilops complex and the outgroup species (Hordeum vulgare). In addition to the diploids (2A-T. urartu, 2B-Ae. speltoides, and 2D-Ae. tauschii) and the outgroup species (H. vulgare), the gene homologs of A- and B/S-subgenomes of the tetraploid (T. turgidum, denoted as 4A and 4B, respectively) and hexaploid wheat (T. aestivum, denoted as 6A and 6B, respectively) are included. Among those 150 nuclear gene homolog pairs encoding CECs, 82 and 55 nuclear D-genome homologs exhibit closer phylogenetic relationships to A- and B/S-genomes/subgenomes, respectively.
To test the statistical significance of this apparently biased maintenance of A-genome ancestry in Ae. tauschii, we compared the putative CEC genes to background whole-genome genes (background genes included the putative CEC genes, table 2). To accomplish this, we tabulated genome-diagnostic SNPs/indels (from the A- and B/S-genome) in gene homologs of Ae. tauschii (supplementary table 2, Supplementary Material online). This was inferred by inspection of the SNP/indels composition at homologous nucleotide positions of aligned gene homologs for the species studied (supplementary table 2, Supplementary Material online). A typical case of this analysis is shown for rbcS homologs (encoding small subunits of Rubisco, SSUs) in figure 4a, which illustrates biased retention of A-genome SNPs/indels. Overall, for nuclear genes encoding putative CECs, the number of A-genome diagnostic SNPs/indels was higher than the number of B/S-genome diagnostic SNPs/indels (17,502 A-genome SNPs/indels vs. 16,541 B/S-genome SNPs/indels, table 2). This bias in composition for nuclear genes that putatively encode CECs was statistically significant (Parametric Fisher’s Exact test and binomial test, P value <0.01, table 2). In addition to the mosaic biased retention of A-genome SNPs/indels in Ae. tauschii as shown for the rbcS gene of figure 4a, some extreme cases of complete or near-complete loss of B-genome SNPs/indels (loss of B-allele) were also detected in genes encoding putative CECs (supplementary table 3, Supplementary Material online and fig. 4b).
Table 2.
A- and B/S-genome Ancestry in Ae. tauschii as Reflected and Quantified by the Number of A- and B/S-genome Diagnostic SNPs/Indels for Nuclear Genes Encoding CECs, and Compared with All Nuclear Genes as a Control for Systematic Biases.
| Genome-diagnostic SNPs/indels | Number of SNPs/Indels |
|
|---|---|---|
| Nuclear Genes Encoding CECs | Whole-genomic Genesb | |
| A-genome SNPs/indels | 17,502c,d | 1,547,018c,d |
| B/S-genome SNPs/indels | 16,541c,d | 1,519,036c,d |
| Ambiguous SNPs/indels with undetermined genomic origina | 36,070c | 6,922,851c |
Ambiguous SNPs/indels could result from autapomorphic evolution of SNPs/indels following speciation and/or hybridization, or from segregating ancestral polymorphism, or from multiple mutations at a site that obscures history.
Background whole-genomic genes includes the putative predicted nuclear CEC genes.
Denotes numbers utilized in Fisher’s Exact test, with the numbers of SNPs/indels identified in nuclear genes encoding CECs and background whole-genomic genes as observed and expected counts, respectively.
Denotes respective numbers utilized in Binomial test, with the null hypothesis being that the probability of having A-genome SNPs/indels is equal to that of having B/S-genome SNPs/indels in nuclear genes encoding CECs. The expected success rate is estimated as 0.505, which was calculated as 1,547,018/(1,547,018 + 1,519,036).
Fig. 4.

Exemplary CEC gene homologs representing the mosaic biased retention of A-genome ancestry and the complete loss of B-genome allele in Aegilops tauschii. Panels (a) and (b) illustrate the SNPs and indels of nuclear rbcS3 homologs encoding the SSUs (small subunits) of Rubisco and homologs encoding F-box only protein 7-like in representative species and subgenomes of the Triticum/Aegilops complex, respectively. In addition to the diploid species (designated as 2A-T. urartu, 2B-Ae. speltoides, and 2D-Ae. tauschii), the gene homologs of A- and B/S-subgenomes of the tetraploid (T. turgidum, denoted as 4A and 4B, in blue) and hexaploid wheat (T. aestivum, denoted as 6A and 6B, in purple) are shown. Within the sequence alignment, A- and B/S-genome diagnostic SNPs and indels are denoted in red and green circles, respectively. Autapomorphic D-genome specific SNPs and indels are represented by dark black dots. Nucleotide positions are noted above the sequence alignment.
Collectively, the phylogenetic results combined with the statistical analyses of shared, genome-diagnostic SNPs/indels support an interpretation that genes encoding putative CECs in Ae. tauschii have experienced biased retention of nuclear genes and the genomic SNPs/indels from one of its two progenitor genomes, specifically the same genome as that of the maternal organelle donor.
Discussion
Hybrid speciation can arise either through HHS or via allopolyploidy (Soltis and Soltis 2009). It is well-established that the former is much rarer than the latter (Soltis and Soltis 2009; Kay et al. 2011), although many additional cases of hybrid speciation are being discovered (Folk et al. 2018) with the increasing application of genomic tools to phylogenetic analyses (Folk et al. 2018). Potentially reduced fitness in the early generations, or “hybrid breakdown,” is a challenge that needs to be surmounted for successful establishment of a newly formed taxon (Rieseberg et al. 1995; Coyne and Orr 2004; Soltis and Soltis 2009; Abbott et al. 2010; Kay et al. 2011; Abbott et al. 2013). The mechanisms underlying the eventual stabilization of hybrid derivatives is thus of considerable interest (Soltis and Soltis 2009; Abbott et al. 2010; Schumer et al. 2014; Nieto et al. 2017). Given the commonly observed cytonuclear dimension of hybrid dysfunction (Levin 2003; Fishman and Willis 2006; Bomblies and Weigel 2007; Burton et al. 2013; Sloan 2015), a promising avenue of investigation is to explore the association between cytonuclear genomic interactions with hybrid breakdown in early-generation natural and artificial hybrids (Burton et al. 2013; Sehrish et al. 2015; Sharbrough et al. 2017; Wang et al. 2017). In addition, clues into the targets of epistatic selection may derive from the analysis of the inherent genic incompatibilities that may follow the merger of two nuclear genomes in the cytoplasmic background of only one of the two parents (Sharbrough et al. 2017).
Here, we characterized, for Ae. tauschii, one of the possible outcomes of cytonuclear conflict, namely, biased retention of nuclear ancestry from the maternal rather than paternal progenitor genome. Using a global analysis of nuclear genes, we demonstrate that there indeed exists such a bias, and that it is more profound for nuclear CEC genes than for the genome as a whole. This result is suggestive of cytonuclear selection for enhanced function, although we recognize that functional studies are lacking to prove this for any specific putative CEC. A promising future direction in this respect is to conduct functional studies in experimental systems involving reciprocal crosses. Additionally, in older stabilized natural hybrid species such as Ae. tauschii, insights may emerge from “mix and match” transgenic replacement experiments of native putative CEC genes with those from the alternative progenitor parent. The genes we tabulate here represent a list of candidates that might be suitable for functional validation via reciprocal transgenic experiments.
The HHS origin of the D-genome lineage in the Triticum/Aegilops complex featured multiple rounds of hybridizations into an ancient D-genome progenitor, as has been ascertained by phylogenetic inferences using both plastid and nuclear genes (Marcussen et al. 2014; Sandve et al. 2015; Li et al. 2015a, 2015b), and through investigation of TE insertions and SNP mutation dynamics (El Baidouri et al. 2017). As reported earlier (Li et al. 2015b) and confirmed here, the most recent maternal parent of Ae. tauschii in this complex evolutionary history had a plastid genome similar to modern-day A-genome diploids. The question arises as to how selection might operate to reduce cytonuclear conflict and hence lead to biased retention of maternal gene copies/ancestry during hybrid speciation. After initial hybridization, at least two scenarios may be envisioned: 1) As suggested by the cases of retention of only A-genome CEC SNPs/indels (supplementary table 3, Supplementary Material online and fig. 4b), it seems likely that maternal orthologs encoding putative CECs were fixed early during the homoploid hybridization process either through directional selection to optimize cytonuclear function, or passively through drift and fixation of unrecombined A alleles; and 2) As evidenced by genes that contain a mix of SNPs from both progenitor lineages (fig. 4a), some CECs likely originated following multiple recombination events between paternal and maternal haplotypes—we note that under this scenario, it may be that selection still favored A-genome SNPs in protein domains that differed between the parents and that lead to differences in cytonuclear function. These two scenarios are not mutually exclusive, and it seems probable that both were operative during the critical establishment phase of the newly recombined lineage now represented by Ae. tauschii. It may be possible to design experiments to evaluate the relative importance of these phenomena across generations, using fast-cycling synthetic hybrid populations of Arabidopsis or other species.
Materials and Methods
Data Collection
Chloroplast genomes from the Triticum/Aegilops complex completed by Gornicki et al. (2014) and Middleton et al. (2014) were downloaded from NCBI. Species names and respective accession numbers are as follows: Aegilops bicornis (KJ614417), Ae. cylindrica (KF534489), Ae. geniculate (KF534490), Ae. longissima (KJ614416), Ae. searsii (KJ614415), Ae. sharonensis (KJ614419), Ae. speltoides (JQ740834), Ae. tauschii (JQ754651), T. monococcum (KC912690), T. urartu (KC912693), T. aestivum (KC912694), Hordeum vulgare (KC912687), and Secale cereal (KC912691).
Genomic assemblies and respective gene annotations of T. urartu (Ling et al. 2018) and T. aestivum (International Wheat Genome Sequencing C 2014) were retrieved from plant Ensemble (http://plants.ensemble.org; last accessed June 2018). The genomes of Ae. tauschii (Luo et al. 2017), Ae. speltoides, and T. turgidum ssp. dicoccoides (Avni et al. 2017), were downloaded from IWGSC (International Wheat Genome Sequencing Consortium).
Construction of Chloroplast Phylogenetic Trees
All chloroplast gene orthologs in the Triticum/Aegilops complex were identified and grouped using OrthoFinder (Emms and Kelly 2015) and default parameter settings. The MAFFT tool was employed to align the chloroplast genes of different species into the same ortholog group (Katoh and Standley 2013). Resulting genes from each species were concatenated into a supergene alignment. Both NJ and ML trees were constructed from this alignment using MEGA 6.0 (Tamura et al. 2013) under the Jukes–Cantor substitution model using other default settings. Bootstrap evaluation of support for each node resolved.
Inference of Genomic Ancestry of Nuclear Genes Encoding Cytonuclear Enzyme Complexes (CECs)
CECs are organellar proteins with subunits encoded by nuclear rather than organellar genomes. CEC subunits are targeted to cytoplasmic organelles after cytoplasmic translation (Millar et al. 2005; van Wijk and Baginsky 2011). Putative CEC genes in the Triticum/Aegilops complex were identified using the prediction software packages, TargetP and LOCALIZER with default settings. Protein descriptions and subcellular localizations for the CEC genes in A. tauschii were curated from the online UniProt database (https://www.uniprot.org/; last accessed April 2018) and cropPAL (http://crop-pal.org/; last accessed April 2018) (Hooper et al. 2016).
The taxa used in this analysis were the D-genome Ae. tauschii, the A-genome T. urartu, the B/S-genome Ae. speltoides, and the A- and B/S-subgenomes within both tetraploid T. turgidum ssp. dicoccoides (A and B/S genome) and hexaploid T. aestivum (A and B/S genome) and outgroup H. vulgare. Respective gene homologs were categorized into groups based on their homology using OrthoFinder under default parameter settings. As for the groups enclosing multiple gene copies within species, we utilized custom python scripts to sort and pair the homologs in each species or subgenome in terms of their hierarchical similarity.
The genomic ancestry of D-genome nuclear genes encoding putative CECs after HHS was initially inferred based on their overall phylogenetic clustering pattern relative to their homologs in diploid and polyploid A- and B/S-species and subgenomes. The first phylogenic analysis was performed using concatenation, as described above for the chloroplast genes. Homologs within each group were aligned using MAFFT and further concatenated into a supergene alignment. Both rooted NJ and ML trees were also constructed based on this supergene alignment using MEGA 6.0 (Tamura et al. 2013) under Jukes–Cantor substitution model with bootstrap evaluation, as illustrated using Figtree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/; last accessed June 2018). A second phylogenetic inference was based on the consensus phylogenetic tree. Each individual Bayesian tree was constructed based on aligned homologs within each group by Markov Chain Montel Carlo (MCMC) methods integrated into the program BEAST (Metropolis et al. 1953; Drummond et al. 2012), in which we adopted the HKY nucleotide substitution model, a Relaxed Clock Log Normal model, and a Calibrated Yule tree-prior model with other parameters set as default settings. All individual phylogenetic trees were integrated into a consensus tree using the LogCombiner v2.4.8 module incorporated into the BEAST software.
Statistical Significance of Biased Maintained A-genomic Ancestry in D-genome Nuclear Genes Encoding CECs
To evaluate whether any observed bias in the maintenance of genomic ancestry in D-genome nuclear putative CEC genes was statistically significant, we quantified the number of genic SNPs/indels in homologs contributed by the A- and B/S-genome species, respectively. These genome-diagnostic SNPs/indels in each D-genome homolog were inferred by comparison with respective homologs in the diploid species and the subgenomes of the polyploids studied (SNPs/indels diagnostic of A- or B/S-genomic origin). Accordingly, A- and B/S-genome ancestries were quantified as the number of A- and B/S-genome SNPs/indels for nuclear genes encoding putative CECs compared with the same calculation conducted for background whole-genomic genes (including nuclear CEC genes). Statistical significance of the difference between CEC and all genes was tested based on Fisher’s Exact test and binomial test (details described in table 2 footnote). Because this strategy involves both diploids and the subgenomes of the polyploids, it effectively addresses possible systematic biases and/or different ages of ancestry.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This study was supported by the National Key Research and Development Program of China (2016YFD0101004), the National Natural Science Foundation of China (31500176), the Recruitment Program of Global Youth Experts, the Program of Changbai Mountain Scholar, the Fundamental Research Fund for the Central Universities (2412017BJ005), and the National Science Foundation Plant Genome Program (to J.F.W.).
References
- Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJ, Bierne N, Boughman J, Brelsford A, Buerkle CA, Buggs R.. 2013. Hybridization and speciation. J Evol Biol. 262:229–246. [DOI] [PubMed] [Google Scholar]
- Abbott RJ, Brennan AC, James JK, Forbes DG, Hegarty MJ, Hiscock SJ.. 2009. Recent hybrid origin and invasion of the British Isles by a self-incompatible species, Oxford ragwort (Senecio squalidus l., Asteraceae). Biol Invas. 115:1145. [Google Scholar]
- Abbott RJ, Hegarty MJ, Hiscock SJ, Brennan AC.. 2010. Homoploid hybrid speciation in action. Taxon 59:1375–1386. [Google Scholar]
- Abbott RJ, James JK, Irwin JA, Comes HP.. 2000. Hybrid origin of the Oxford ragwort, Senecio squalidus L. Watsonia 23:123–138. [Google Scholar]
- Anderson E, Hubricht L.. 1938. Hybridization in Tradescantia. III. The evidence for introgressive hybridization. Am J Bot. 256:396–402. [Google Scholar]
- Anderson E. 1949. Introgressive hybridization. New York: John Wiley and Sons. [Google Scholar]
- Arnold ML. 1992. Natural hybridization as an evolutionary process. Annu Rev Ecol Syst. 231:237–261. [Google Scholar]
- Arnold ML. 1994. Natural hybridization and Louisiana irises. BioScience 443:141–147. [Google Scholar]
- Arnold J, Asmussen MA, Avise JC.. 1988. An epistatic mating system model can produce permanent cytonuclear disequilibria in a hybrid zone. Proc Natl Acad Sci U S A. 856:1893.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold ML. 1997. Natural hybridization and evolution. New York: Oxford Univ. Press. [Google Scholar]
- Avni R, Nave M, Barad O, Baruch K, Twardziok SO, Gundlach H, Hale I, Mascher M, Spannagl M, Wiebe K.. 2017. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 3576346:93–97. [DOI] [PubMed] [Google Scholar]
- Bock DG, Andrew RL, Rieseberg LH.. 2014. On the adaptive value of cytoplasmic genomes in plants. Mol Ecol. 2320:4899–4911. [DOI] [PubMed] [Google Scholar]
- Bomblies K, Weigel D.. 2007. Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat Rev Genet. 85:382.. [DOI] [PubMed] [Google Scholar]
- Burton RS, Pereira RJ, Barreto FS.. 2013. Cytonuclear genomic interactions and hybrid breakdown. Annu Rev Ecol Evol Syst. 441:281–302. [Google Scholar]
- Coyne JA, Orr HA.. 2004. Speciation. Sunderland (MA: ): Sinauer Associates; 545 pp. [Google Scholar]
- Dowling TE, Secor CL.. 1997. The role of hybridization and introgression in the diversification of animals. Annu Rev Ecol Syst. 281:593–619. [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A.. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 298:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Baidouri M, Murat F, Veyssiere M, Molinier M, Flores R, Burlot L, Alaux M, Quesneville H, Pont C, Salse J.. 2017. Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol. 2133:1477–1486. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, Von Heijne G, Nielsen H.. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 24:953–971. [DOI] [PubMed] [Google Scholar]
- Emms DM, Kelly S.. 2015. Orthofinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman L, Willis JH.. 2006. A cytonuclear incompatibility causes anther sterility in Mimulus hybrids. Evolution 607:1372–1381. [DOI] [PubMed] [Google Scholar]
- Folk RA, Soltis PS, Soltis DE, Guralnick R.. 2018. New prospects in the detection and comparative analysis of hybridization in the tree of life. Am J Bot. 1053:364–375. [DOI] [PubMed] [Google Scholar]
- Folk RA, Mandel JR, Freudenstein JV.. 2017. Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of Angiosperms. Syst Biol. 66:320–337. [DOI] [PubMed] [Google Scholar]
- Gadagkar R, Rosenberg S, Kumar S.. 2005. Inferring species phylogenies from multiple genes: concatenated sequence tree versus consensus gene tree. J Exp Zool B: Mol Dev Evol. 304B1:64–74. [DOI] [PubMed] [Google Scholar]
- Gong L, Olson M, Wendel JF.. 2014. Cytonuclear evolution of Rubisco in four allopolyploid lineages. Mol Biol Evol. 3110:2624–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Salmon A, Yoo MJ, Grupp KK, Wang Z, Paterson AH, Wendel JF.. 2012. The cytonuclear dimension of allopolyploid evolution: an example from cotton using Rubisco. Mol Biol Evol. 2910:3023–3036. [DOI] [PubMed] [Google Scholar]
- Gornicki P, Zhu H, Wang J, Challa GS, Zhang Z, Gill BS, Li W. 2014. The chloroplast view of the evolution of polyploid wheat. New Phytol 204:704–714. [DOI] [PubMed] [Google Scholar]
- Greiner S, Sobanski J, Bock R.. 2015. Why are most organelle genomes transmitted maternally? Bioessays 371:80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross BL, Rieseberg LH.. 2005. The ecological genetics of homoploid hybrid speciation. J Hered. 963:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross BL, Schwarzbach AE, Rieseberg LH.. 2003. Origin(s) of the diploid hybrid species Helianthus deserticola (Asteraceae). Am J Bot. 9012:1708–1719. [DOI] [PubMed] [Google Scholar]
- Hermansen JS, Saether SA, Elgvin TO, Borge T, Hjelle E, Saetre G-P.. 2011. Hybrid speciation in sparrows I: Phenotypic intermediacy, genetic admixture and barriers to gene flow. Mol Ecol. 2018:3812–3822. [DOI] [PubMed] [Google Scholar]
- Hooper CM, Castleden IR, Aryamanesh N, Jacoby RP, Millar AH.. 2016. Finding the subcellular location of barley, wheat, rice and maize proteins: the compendium of crop proteins with annotated locations (croppal). Plant Cell Physiol. 571:e9–e9. [DOI] [PubMed] [Google Scholar]
- International Wheat Genome Sequencing C. 2014. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788. [DOI] [PubMed] [Google Scholar]
- James JK, Abbott RJ, Soltis P.. 2005. Recent, allopatric, homoploid hybrid speciation: the origin of Senecio squalidus (Asteraceae) in the British Isles from a hybrid zone on Mount Etna, Sicily. Evolution 5912:2533–2547. [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 304:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay KM, Ward KL, Watt LR, Schemske DW.. 2011. Plant speciation In: Harrison SP, Rajakaruna N, editors. Serpentine: the evolution and ecology of a model system. Berkeley (CA: ): Univ. of California Press. [Google Scholar]
- Levin DA. 2003. The cytoplasmic factor in plant speciation. Syst Bot. 28:5–11. [Google Scholar]
- Li L-F, Liu B, Olsen KM, Wendel JF.. 2015a. Multiple rounds of ancient and recent hybridizations have occurred within the Aegilops-Triticum complex. New Phytol. 2081:11–12. [DOI] [PubMed] [Google Scholar]
- Li L-F, Liu B, Olsen KM, Wendel JF.. 2015b. A re-evaluation of the homoploid hybrid origin of Aegilops tauschii, the donor of the wheat D-subgenome. New Phytol. 2081:4–8. [DOI] [PubMed] [Google Scholar]
- Ling H-Q, Ma B, Shi X, Liu H, Dong L, Sun H, Cao Y, Gao Q, Zheng S, Li Y, et al. 2018. Genome sequence of the progenitor of wheat a subgenome Triticum urartu. Nature 5577705:424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M-C, Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, Huo N, Zhu T, Wang L, Wang Y, et al. 2017. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 5517681:498.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcussen T, Sandve SR, Heier L, Spannagl M, Pfeifer M, Jakobsen KS, Wulff Brande BH, Steuernagel B, Mayer K, Olsen OA.. 2014. Ancient hybridizations among the ancestral genomes of bread wheat. Science 3456194:1250092. [DOI] [PubMed] [Google Scholar]
- Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E.. 1953. Equation of state calculations by fast computing machines. J Chem Phys. 216:1087–1092. [Google Scholar]
- Middleton CP, Senerchia N, Stein N, Akhunov ED, Keller B, Wicker T, Kilian B. 2014. Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PLoS One 9:e85761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar AH, Heazlewood JL, Kristensen BK, Braun H-P, Møller IM.. 2005. The plant mitochondrial proteome. Trends Plant Sci. 101:36–43. [DOI] [PubMed] [Google Scholar]
- Nieto FG, Alvarez I, Fuertes-Aguilar J, Heuertz M, Marques I, Moharrek F, Pineiro R, Riina R, Rossello JA, Soltis PS, et al. 2017. Is homoploid hybrid speciation that rare? An empiricist's view. Heredity (Edinb). 1186:513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Haney RA, Fry AJ.. 2004. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol Evol. 1912:645–653. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. 1991. Homoploid reticulate evolution in Helianthus (Asteraceae): evidence from ribosomal genes. Am J Bot. 789:1218–1237. [Google Scholar]
- Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C.. 2003. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 3015637:1211.. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Van Fossen C, Desrochers AM.. 1995. Hybrid speciation accompanied by genomic reorganization in wild sunflowers. Nature 3756529:313. [Google Scholar]
- Sandve SR, Marcussen T, Mayer K, Jakobsen KS, Heier L, Steuernagel B, Wulff BBH, Olsen OA.. 2015. Chloroplast phylogeny of Triticum/Aegilops species is not incongruent with an ancient homoploid hybrid origin of the ancestor of the bread wheat D-genome. New Phytol. 2081:9–10. [DOI] [PubMed] [Google Scholar]
- Schumer M, Rosenthal GG, Andolfatto P.. 2014. How common is homoploid hybrid speciation? Evolution 686:1553–1560. [DOI] [PubMed] [Google Scholar]
- Sehrish T, Symonds VV, Soltis DE, Soltis PS, Tate JA.. 2015. Cytonuclear coordination is not immediate upon allopolyploid formation in Tragopogon miscellus (Asteraceae) allopolyploids. PLoS One 1012:e0144339.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharbrough J, Conover JL, Tate JA, Wendel JF, Sloan DB.. 2017. Cytonuclear responses to genome doubling. Am J Bot. 1049:1277–1280. [DOI] [PubMed] [Google Scholar]
- Sloan DB. 2015. Using plants to elucidate the mechanisms of cytonuclear co-evolution. New Phytol. 2053:1040–1046. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Triant DA, Wu M, Taylor DR.. 2014. Cytonuclear interactions and relaxed selection accelerate sequence evolution in organelle ribosomes. Mol Biol Evol. 313:673–682. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Segovia Salcedo MC, Jordon Thaden I, Majure L, Miles NM, Mavrodiev EV, Mei W, Cortez MB, Soltis PS, Gitzendanner MA.. 2014. Are polyploids really evolutionary dead-ends (again)? A critical reappraisal of Mayrose et al. (2011). New Phytol. 2024:1105–1117. [DOI] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE.. 2009. The role of hybridization in plant speciation. Annu Rev Plant Biol. 60:561–588. [DOI] [PubMed] [Google Scholar]
- Sperschneider J, Catanzariti AM, Deboer K, Petre B, Gardiner DM, Singh KB, Dodds PN, Taylor JM.. 2017. Localizer: subcellular localization prediction of both plant and effector proteins in the plant cell. Sci Rep. 7:44598.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.. 2013. Mega6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 3012:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wijk KJ, Baginsky S.. 2011. Plastid proteomics in higher plants: current state and future goals. Plant Physiol. 1554:1578.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Dong Q, Li X, Yuliang A, Yu Y, Li N, Liu B, Gong L.. 2017. Cytonuclear variation of Rubisco in synthesized rice hybrids and allotetraploids. Plant Genome. 103:1–11. [DOI] [PubMed] [Google Scholar]
- Wendel JF, Stewart JM, Rettig JH.. 1991. Molecular evidence for homoploid reticulate evolution among Australian species of Gossypium. Evolution 453:694–711. [DOI] [PubMed] [Google Scholar]
- Weng M-L, Ruhlman TA, Jansen RK.. 2016. Plastid–nuclear interaction and accelerated coevolution in plastid ribosomal genes in Geraniaceae. Genome Biol Evol. 86:1824–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson JD, Chory J.. 2008. Coordination of gene expression between organellar and nuclear genomes. Nat Rev Genet. 95:383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakimowski SB, Rieseberg LH.. 2014. The role of homoploid hybridization in evolution: a century of studies synthesizing genetics and ecology. Am J Bot. 1018:1247–1258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.