

Abstract
Thymus-derived regulatory T cells (tTregs) play pivotal roles in immunological self-tolerance and homeostasis. A majority of tTregs are reactive to self-antigens and are constantly exposed to antigenic stimulation. Despite this continuous stimulation, tTreg and conventional T-cell populations remain balanced during homeostasis, but the mechanisms controlling this balance are unknown. We previously reported a form of activation-induced cell death, which is dependent on p53 (p53-induced CD28-dependent T-cell apoptosis, PICA). Under PICA-inducing conditions, tTregs survive while a majority of conventional T cells undergo apoptosis, suggesting there is a survival mechanism that protects tTregs. Here, we report that the expression of RasGRP1 (Ras guanyl-releasing protein 1) is required for PICA, as conventional T cells isolated from RasGRP1-deficient mice become resistant to PICA. After continuous stimulation, tTregs express a substantially lower amount of RasGRP1 compared to conventional T cells. This reduced expression of RasGRP1 is dependent on TGF-β, as addition of TGF-β to conventional T cells reduces RasGRP1 expression. Conversely, RasGRP1 expression in tTregs increases when TGF-β signaling is inhibited. Together, these data show that RasGRP1 expression is repressed in tTregs by TGF-β signaling and suggests that reduced RasGRP1 expression is critical for tTregs to resist apoptosis caused by continuous antigen exposure.
Keywords: Foxp3, RasGRPl, TCR, TGF-β, Tregs
Introduction
Thymus-derived Tregs (tTregs) comprise 5–10% of peripheral blood CD4+ T cells and play a pivotal role in maintaining immune homeostasis [1, 2]. tTregs constitutively express CD25 along with the transcription factor Foxp3 that is essential for Treg functions [3]. Since Tregs suppress the proliferation and activation of other conventional T cells, maintaining the appropriate balance between Treg and conventional T cell populations is critical for effective immune responses and immunological homeostasis.
Activation-induced cell death (AICD) occurs after expansion of antigen-stimulated T cells to reduce the number of these activated T cells [4, 5]. Previously, we have established an in vitro system to study AICD by stimulating T cells with anti-CD3 and CD28 antibodies coated on flat tissue culture plates. Under these conditions, a majority of conventional T cells undergo apoptosis in a p53- dependent manner, while Foxp3+ tTregs are resistant to apoptosis and expand over 7000-fold within 10 days [6]. Since classical AICD is p53 independent [7], we concluded that plate-bound anti- CD3/anti-CD28 antibodies induce apoptosis in a manner distinct from classical AICD and named the process p53-induced CD28- dependent T-cell apoptosis (PICA) [6]. Further analysis revealed that Foxp3+ Tregs require autocrine TGF-β to resist PICA [8]. Conversely, addition of exogenous TGF-β renders conventional T cells resistant to PICA. These data suggest that PICA might play a role in immune regulation by controlling the balance between tTregs and conventional T cells. These data also provide a potential explanation for autoimmunity observed in p53-deficient mice [9].
To elucidate the mechanism that induces PICA, we examined the signaling processes that differ between conventional T cells and tTregs. We identified that expression of RasGRPl is substantially upregulated by antigen stimulation in conventional T cells but remains low in tTregs. We further found that TGF-β is responsible for suppressing RasGRPl expression in tTregs. Additionally, conventional T cells from RasGRPl-deficient mice resist PICA, suggesting that the expression level of RasGRPl plays a critical role in controlling the number of T cells that survive antigen exposure.
Results and discussion
Since CD4+CD25+ Tregs are resistant to PICA, we hypothesized that there are TCR-associated signaling differences between CD4+CD25+ Tregs and conventional T cells that allow for Tregs to resist PICA. To test this hypothesis, we analyzed known signaling molecules that function downstream of TCR stimulation and compared them between CD4+CD25+ Tregs and conventional T cells (Fig. 1).
Figure 1.
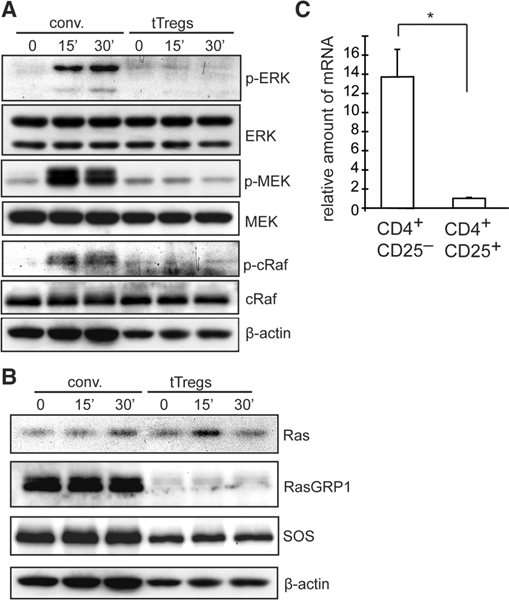
Expression and activation of ERK signaling molecules in conventional T cells and tTregs. Ex vivo expanded CD4+CD25+ tTregs and conventional (conv) CD4+ T cells (CD4+ CD25- cells) isolated from C57BL/6 spleens were stimulated with biotin-conjugated anti-CD3 crosslinked with avidin for 0, 15, or 30 min. (A, B) tTregs and conv cells were analyzed by western blot analysis. Blots are representative of three independent experiments. (C) Real-time PCR assays for expression of Rasgrpl mRNAby conventional (CD4+CD25-) and tTregs (CD4+ CD25+). The relative amount of mRNA was determined using gapdh gene expression as a reference, *p<0.005 Student’s t-test. The data are representative of two independent experiments.
Differences in the Ras/ERK signaling pathway between Tregs and conventional T cells were most evident. While conventional T cells showed a strong upregulation of phosphorylated ERK (indicative of ERK activation) 15–30 min after TCR stimulation, tTregs showed little, if any, upregulation of ERK phosphorylation (Fig. 1A). Compared to conventional T cells, tTregs also showed a substantial decrease in phosphorylation of two molecules upstream of ERK, MEK and c-Raf. These data suggest that the ERK/Ras signaling pathway is uncoupled from TCR stimulation in tTregs.
In T cells, activation of the MAPK/ERK pathway is dependent on two Ras activators, RasGRPl and Sos-1/Sos-2 [10]. Since ERK activation was significantly lower in tTregs than in conventional T cells, we next determined the expression level of RasGRP1 and Sos-1 in tTregs compared to conventional T cells (Fig. 1B and C). We found that tTregs and conventional T cells expressed markedly different levels of RasGRPl (Fig. 1B and C). Compared to conventional T cells, tTregs expressed a much lower level of Ras- GRP1 protein and mRNA. Expression levels of Ras were comparable between conventional T cells and tTregs. Sos-1 expression by tTregs was slightly lower than conventional T cells. Together, the data suggest that tTregs exhibit reduced ERK signaling in part due to lower levels of RasGRP1 expression.
We next addressed the mechanism by which tTregs express lower levels of RasGRPl. Since we used in vitro expanded conventional and regulatory T cells in the experiments described above, we first tested if RasGRPl expression is elevated in conventional T cells by ex vivo expansion (Fig. 2A). Indeed, the expression of RasGRPl did not significantly differ between freshly isolated conventional and regulatory T cells. However, after in vitro stimulation by anti-CD3/CD28 coated beads, expression of RasGRPl increased only in conventional T cells (Fig. 2A). The data suggested that Tregs produce an inhibitory molecule(s) for RasGRPl expression and/or conventional T cells produce a RasGRPl promoting factor(s) during expansion.
Figure 2.
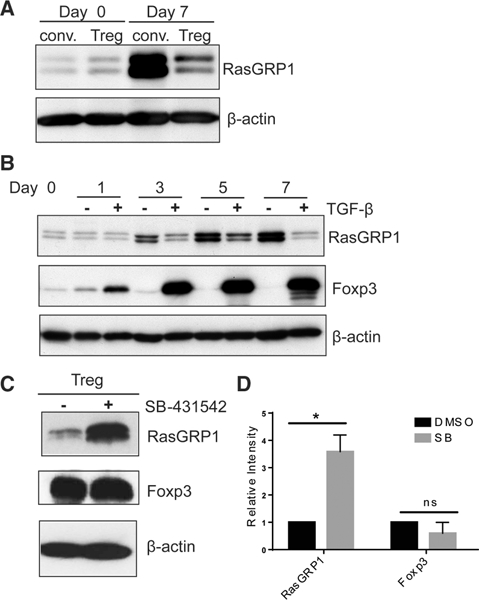
Regulation of RasGRPl expression by TGF-β. (A) CD4+ CD25+ Tregs and CD4+ CD25- conventional T cells were isolated from spleens of C57BL/6 mice and stimulated with anti-CD3/28 coated beads for 7 days in the presence of IL-2. Cells harvested at day 0 and day 7 were lysed in SDS sample buffer for western blot analysis using anti-RasGRPl and anti-p actin antibodies. (B) CD4+CD25- T cells were stimulated with plate-bound anti-CD3 and soluble anti-CD28 antibodies in the presence or absence of TGF-β supplemented with IL-2. After 3 days of stimulation, cells were harvested to remove stimulation and were further cultured in the presence of TGF-β and IL-2 and analyzed by western blot (C) CD4+25+ Tregs were expanded ex vivo for 7 days with anti-CD3 and anti-CD28 coated beads in the presence of IL-2. After 7 days, cells were re-stimulated on anti-CD3 and anti-CD28 coated plates in the presence of IL-2 and a TGF-β type I kinase inhibitor (SB-431542) or DMSO control. Cells were harvested 5 days after stimulation and lysed in SDS sample buffer then western blot analysis was performed using anti-RasGRPl, anti-Foxp3, and anti-p actin antibodies. (A-C) Blots are representative of three independent experiments. (D) Relative intensity of RasGRPl and Foxp3 in SB-43l542 treated Tregs compared to normalized DMSO control. β-actin was used as a loading control. Four spleens were pooled for each independent experiment. Data are shown as mean + SD (n = 3) and are representative of three independent experiments. Student’s t-test, RasGRPl p = 0.016; Foxp3 p = 0.157.
Our previous work showed that Tregs require TGF-β signaling to resist PICA, and that exogenous TGF-β confers PICA resistance to conventional T cells [8]. tTregs express active TGF-β and its receptors [11–13]. Conventional T cells also express TGF-βRI and TGF-βRII, but their expression of active TGF-β ligand is limited due to the lack of TGF-β activation machinery [14–18]. Based on these data, we hypothesized that TGF-β reduces RasGRP1 expression by conventional T cells after activation. Our hypothesis predicted that addition of TGF-β to activated conventional T cells would reduce expression of RasGRP1. When we stimulated CD4+ CD25- conventional T cells by anti-CD3 coated plates with soluble anti-CD28, RasGRPl expression substantially increased after 3 days, and this level of expression was maintained over 7 days (Fig. 2B). In contrast, addition of exogenous TGF-β to conventional T cells resulted in little, if any, increase in RasGRPl expression. The data show that TGF-β is an inhibitor for RasGRPl expression by activated conventional T cells. In accordance with previous data, Tregs and conventional T cells have a basal level of pSMAD2/3 expression without stimulation, and upon stimulation with the addition of exogenous TGF-β, both Tregs and conventional T cells upregulated pSMAD2/3 expression (Supporting Information Fig. 1 and Fig. 2B) [14]. The data suggest that signaling processes downstream of SMAD phosphorylation and/or non-canonical TGF-β signaling are involved in the regulation of RasGRPl expression.
We next tested if the low levels of RasGRPl expression by tTregs require autocrine TGF-β signaling. If autocrine TGF-β is required, then inhibition of TGF-β signaling would increase Ras-GRPl expression. To test this, we re-stimulated ex vivo expanded CD4+CD25+ Tregs from mouse splenocytes with anti-CD3/anti- CD28 coated plates in the presence or absence of a TGF-β type I receptor inhibitor (SB- 43l542). Cells were harvested 5 days after stimulation and the level of RasGRPl expression was determined by western blot (Fig. 2C). Tregs stimulated with the TGF-β receptor signaling inhibitor showed a significant increase in RasGRPl expression compared to cells stimulated with a DMSO control (Fig. 2C and D), suggesting that TGF-β signaling in Tregs is required for maintaining low RasGRPl expression after activation. Inhibition of TGF-β signaling did not significantly reduce expression of Foxp3 by tTregs (Fig. 2D). The data suggest that TGF-β inhibits RasGRPl expression in a manner independent of Foxp3 expression.
RasGRPl has been shown to transduce apoptotic signals in B cells [l9]. Moreover, sustained ERK signaling can promote cell death [20–24]. Therefore, we hypothesized that conventional T cells are susceptible to PICA because of the increase in Ras- GRPl after TCR stimulation, which leads to sustained ERK activation. If downregulation of RasGRPl is important for survival under PICA inducing conditions, then RasGRPl deficient conventional T cells would become resistant to PICA. To test this, we cultured CD4+ CD25- conventional T cells isolated from the spleens of RasGRPl knockout or littermate control mice with plate-bound anti-CD3/anti-CD28 antibody stimulation. As expected, RasGRPl- deficient conventional T cells showed a substantial decrease in the percentage of AnnexinV+ and 7AAD+ cells, and became resistant to PICA, while control cells underwent apoptosis (Fig. 3A, B and Supporting Informaion Fig. 2A). These data show that RasGRPl expression is required for PICA in conventional T cells and suggest that reduced expression of RasGRPl by tTregs is a mechanism by which tTregs resist PICA. Since low expression of RasGRPl in tTregs requires TGF-β signaling, the data illustrate that TGF-β acts as a survival factor in tTregs to resist PICA. Previous work by others demonstrated that TGF-β signaling enhances the ERK signaling pathway by SMAD-dependent and -independent manners in non-lymphoid cells [25–28]. Our finding is therefore unique in that TGF-β signaling suppresses RasGRPl expression in T cells, which subsequently reduces ERK signaling. The mechanism by which TGF-β suppresses RasGRPl expression is currently unknown. Our previous work showed that Foxp3 does not mediate this effect, because TGF-β inhibited PICA in conventional T cells without an increase in Foxp3 expression [8].
Figure 3.
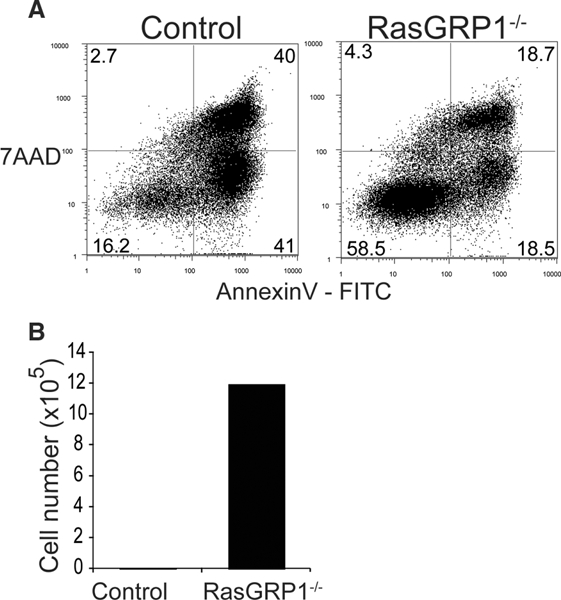
Comparison of PICA between RasGRPl sufficient and deficient conventional CD4+ T cells. Conventional CD4+ T cells (CD4+ CD25- T cells) isolated from the spleens of RasGRPl knockout mice (Rasgrp1−/−) or littermate controls, (Rasgrp1+/+ or +/−) were stimulated with plate- bound anti-CD3/anti-CD28 antibodies supplemented with IL-2. (A, B) Cells were harvested at day 4. Cells were stained with Annexin V (FITC) and 7AAD and analyzed by flow cytometry (A). (B) The total number of live cells was determined by Trypan blue exclusion. (A, B) The data are representative of four independent experiments, where two or three spleens were pooled for each experiment.
The reduction of RasGRPl by TGF-β may be due to several mechanisms, such as the direct effects of SMAD2/3-mediated transcriptional controls. Additionally, our previous work showed that addition of TGF-β to conventional T cells caused activation of Akt and inactivating phosphorylation/degradation of FoxO3a, which may be a mechanism of preventing apoptosis [8]. Others have also found that TGF-β can inactivate FoxOl and FoxO3a in a PI3K-dependent manner [29, 30]. These findings bring up the possibility that TGF-β could control RasGRPl expression in a PI3K-dependent manner.
The TGF-β-mediated mechanisms that control RasGRPl and PICA could also have important clinical implications, especially for chimeric antigen receptor (CAR) T cells, which have been found to undergo activation-induced cell death with repeated antigen exposure [31–33]. PICA could play a significant role in CAR T-cell apoptosis since CD3 and a co-stimulatory domain such as CD28 are co-engaged during CAR T-cell activation, in a manner similar to plate-bound stimulation in PICA. Further studies will need to elucidate the role of TGF-β and RasGRPl in CAR T-cell activation and death.
Concluding remarks
In this study, we unveiled a new insight into a Treg survival mechanism against activation induced cell death. Expression of RasGRPl is reduced in Tregs by TGF-β. RasGRPl-deficient T cells, similar to Tregs, are resistant to p53-dependent CD28-induced apoptosis. The data suggest that TGF-β and TCR signaling cross talk via RasGRPl expression and controls the fate of T cells.
Materials and methods
Mice
C57BL/6 and BALB/c mice were purchased from Jackson Laboratory. Rasgrp1−/− mice were kindly gifted by Dr. JC Stone. Mice were maintained under specific pathogen-free conditions. All procedures were approved and monitored by the Institutional Animal Care and Use Committee of Loyola University Chicago.
Cell culture
Splenocytes were labeled with fluorochrome conjugated anti-CD4 and anti-CD25 antibodies followed by sorting of CD4+CD25+ T cells and CD4+ CD25- T cells using FACSAria (BD Biosciences, Franklin Lakes, NJ). tTregs were expanded in vitro as previously described [6]. Briefly, CD4+ CD25+ T cells were stimulated with immobilized anti-CD3/anti-CD28 antibodies (5 μg/mL each, Biolegend, San Diego, CA, USA) coated on culture dishes in the presence IL-2 (l0 ng/mL, Peprotech, Rocky Hill, NJ, USA). For conventional T cell expansion, CD4+CD25- T cells were stimulated with anti-CD3/anti-CD28 antibodies coated on 4.5 μM polystyrene beads (Polysciences, Inc, Warrington, PA, USA). For the culture with the addition of exogenous TGF-β, CD4+CD25- T cells were stimulated with plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 antibodies (l μg/mL) in the presence of TGF- β (2.5 ng/mL, Peprotech) and IL-2 (l0 ng/mL, Peprotech). After 3 days of stimulation, cells were harvested to remove stimulation and were further cultured in the presence of TGF-β and IL-2. Expanded tTregs or conventional T cells were washed and rested overnight in the presence of IL-2 (l ng/mL) prior to restimulation. For culture with the TGF-β signaling inhibitor, CD4+ 25+ tTregs were expanded with anti-CD3/anti-CD28 antibody coated beads for 7 days in the presence of IL-2. After 7 days, tTregs were restimulated with immobilized anti-CD3/anti-CD28 antibodies (5 μg/mL each, Biolegend) coated on culture dishes with DMSO control or l0 μM SB-43l542 (Sigma, St. Louis, MO, USA) in the presence of IL-2 (l0 ng/mL, Peprotech). For re-stimulation to assess TCR signaling events, expanded cells were stimulated with biotin- conjugated anti-CD3 Ab (5 μg/mL, eBiosciences, San Diego, CA USA) cross-linked by streptavidin (5 μg/mL, EMD Chemicals, Rockland, MA, USA).
Western blot
Cells were lysed either in SDS sample buffer (2% SDS, 125 mM DTT, 10% glycerol, 62.5 mM Tris-HCl, pH 6.8) or RIPA buffer (10 mM Phosphate buffer, 150 mM NaCl, 1% NP40, 0.1% sodium deoxycholate, and 0.1% SDS) containing protease inhibitors and phosphatase inhibitors. Equivalent amounts of proteins adjusted by cell number were loaded in each lane and separated on SDS- PAGE gels. Proteins transferred on PVDF membranes were probed with the following primary antibodies: antibodies specific for phospho-Erk (Thr 202/Tyr204), phospho-Mek1/2 (Ser 217/221), phosphor-cRaf (Ser 259), phospho-smad 2/3 (Ser 423/425) and Mek were from Cell Signaling Technology. Antibodies specific for RasGRP1 and Sos1 were from Santa Cruz Biotechnology (Dallas, TX, USA). The anti-Ras antibody was from BD Biosciences, the anti-Erk Ab was from Millipore (Billerica, MA, USA), the anti-Foxp3 Ab was from eBiosciences, and the anti-p actin anti-body was from Sigma (St. Louis, MO, USA).
Flow cytometry
Harvested cells were stained with 7AAD (BD Biosciences) and AnnexinV (Biolegend) for 15 min at room temperature. Data were collected on BD FACSCanto (BD Biosciences) and analyzed using FlowJo software (FlowJO, Ashland, OR, USA). To assess pSMAD2/3 expression in conventional T cells and Tregs, spleno- cytes were first stained with CD4 and CD25 (Biolegend), followed by permeabilization and pSMAD2/3 intracellular staining with a standard protocol (BD Biosciences). For all flow cytometry and cell sorting, the EJI guidelines were followed.
Supplementary Material
Acknowledgements:
We thank Dr. James Stone for kindly providing Rasgrp1 knockout mice. This research was supported by R01AI055022 and AII100129 (M.I.), and T32 AI007508 (CC).
Abbreviations:
- PICA
p53-induced CD28-dependent T-cell apoptosis
- RasGRPl
Ras guanyl-releasing protein 1
- tTreg
thymus-derived Treg
Footnotes
Conflict of interest: The authors have declared no financial conflicts of interest.
Additional supporting information maybe found online in the Supporting Information section at the end of the article.
References
- 1.Sakaguchi S, Sakaguchi N, Asano M, Itoh M and Toda M, Immunologic self- tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of selftolerance causes various autoimmune diseases. J. Immunol. 1995. 155: 1151–1164. [PubMed] [Google Scholar]
- 2.Levings MK, Sangregorio R and Roncarolo MG, Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J. Exp. Med. 2001. 193: 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hori S, Nomura T and Sakaguchi S, Control of regulatory T cell development by the transcription factor Foxp3. Science 2003. 299:1057–1061. [PubMed] [Google Scholar]
- 4.Smith CA, Williams GT, Kingston R, Jenkinson EJ and Owen JJ, Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature 1989. 337:181–184. [DOI] [PubMed] [Google Scholar]
- 5.Krammer PH, Arnold R and Lavrik IN, Life and death in peripheral T cells. Nat. Reu. Immunol. 2007. 7: 532–542. [DOI] [PubMed] [Google Scholar]
- 6.Singh N, Yamamoto M, Takami M, Seki Y, Takezaki M, Mellor AL and Iwashima M, CD4(+)CD25(+) regulatory T cells resist a novel form of CD28- and Fas-dependent p53-induced T-cell apoptosis. J. Immunol. 2010. 184: 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boehme SA and Lenardo MJ, TCR-mediated death of mature T lymphocytes occurs in the absence of p53. J. Immunol. 1996156: 4075–4078. [PubMed] [Google Scholar]
- 8.Takami M, Love RB and Iwashima M, TGF-beta converts apoptotic stimuli into the signal for Th9 differentiation. J. Immunol. 2012188: 4369–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takatori H, Kawashima H, Suzuki K and Nakajima H, Role of p53 in systemic autoimmune diseases. Crit. Reu. Immunol. 2014. 34: 509–516. [DOI] [PubMed] [Google Scholar]
- 10.Roose JP, Mollenauer M, Ho M, Kurosaki T and Weiss A, Unusual interplay of two types of Ras activators, RasGRP and SOS, establishes sensitive and robust Ras activation in lymphocytes. Mol. Cell. Biol. 2007. 27: 2732–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G et al. , Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003. 198: 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li MO, Sanjabi S and Flavell RA, Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and-independent mechanisms. Immunity 2006. 25: 455–471. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S and Chen W, A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008. 9: 632–640. [DOI] [PubMed] [Google Scholar]
- 14.Tu E, Chia CPZ, Chen W, Zhang D, Park SA, Jin W, Wang D et al. , T cell receptor-regulated TGF-beta type i receptor expression determines T cell quiescence and activation. Immunity 2018. 48: 745–759e746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox FE, Ford HC, Douglas R, Cherian S and Nowell PC, Evidence that TGF-beta can inhibit human T-lymphocyte proliferation through paracrine and autocrine mechanisms. Cell. Immunol. 1993150: 45–58. [DOI] [PubMed] [Google Scholar]
- 16.Gorelik L and Flavell RA, Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000. 12: 171–181. [DOI] [PubMed] [Google Scholar]
- 17.Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limon P, Nakayama M, Leonard WJ et al. , Excessive Th1 responses due to the absence of TGF-beta signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc. Natl. Acad. Sci. U. S. A. 2013110: 6961–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L, Jin H and Li H, GARP: A surface molecule of regulatory T cells that is involved in the regulatory function and TGF-beta releasing. Onco- target 2016. 7: 42826–42836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stang SL, Lopez-Campistrous A, Song X, Dower NA, Blumberg PM, Wender PA and Stone JC, A proapoptotic signaling pathway involving RasGRP, Erk, and Bim in B cells. Exp. Hematol. 2009. 37:122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin P, Poggi MC, Chambard JC, Boulukos KE and Pognonec P, Low dose cadmium poisoning results in sustained ERK phosphorylation andcaspase activation. Biochem. Biophys. Res. Commun. 2006. 350: 803–807. [DOI] [PubMed] [Google Scholar]
- 21.Yang Z, Zhao Y, Yao Y, Li J, Wang W and Wu X, Equol induces mitochondria-dependent apoptosis in human gastric cancer cells via the sustained activation of ERK1/2 pathway. Mol. Cells 2016. 39: 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang TY, Chang GC, Chen KC, Hung HW, Hsu KH, Sheu GT and Hsu SL, Sustained activation of ERK and Cdk2/cyclin-A signaling pathway by pemetrexed leading to S-phase arrest and apoptosis in human non-small cell lung cancerA549 cells. Eur.J. Pharmacol. 2011. 663: 17–26. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Zhang L, Wu S, Teraishi F, Davis JJ, Jacob D and Fang B, Induction of S-phase arrest and p21 overexpression by a small molecule 2[[3-(2,3- dichlorophenoxy)propyl] aminojethanol in correlation with activation of ERK. Oncogene 2004. 23: 4984–4992. [DOI] [PubMed] [Google Scholar]
- 24.Hsu YL, Kuo PL, Lin LT and Lin CC, Asiatic acid, a triterpene, induces apoptosis and cell cycle arrest through activation of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways in human breast cancer cells. J. Pharmacol. Exp. Ther. 2005. 313: 333–344. [DOI] [PubMed] [Google Scholar]
- 25.Hough C, Radu M and Dore JJ, Tgf-beta induced Erk phosphorylation of smad linker region regulates smad signaling. PLoS One 2012. 7: e42513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Principe DR, Diaz AM, Torres C, Mangan RJ, DeCant B, McKinney R, Tsao MS et al. , TGFbeta engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 2017. 36: 4336–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM et al. , TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007. 26: 3957–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulder KM, Role of Ras and Mapks in TGFbeta signaling. Cytokine Growth Factor Reu. 2000. 11: 23–35. [DOI] [PubMed] [Google Scholar]
- 29.Kato M, Yuan H, Xu ZG, Lanting L, Li SL, Wang M, Hu MC et al. , Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol. 2006. 17: 3325–3335. [DOI] [PubMed] [Google Scholar]
- 30.Kurebayashi Y, Baba Y, Minowa A, Nadya NA, Azuma M, Yoshimura A, Koyasu S et al. TGF-beta-induced phosphorylation of Akt and Foxo transcription factors negatively regulates induced regulatory T cell differentiation. Biochem. Biophys. Res. Commun. 2016. 480: 114–119. [DOI] [PubMed] [Google Scholar]
- 31.Tschumi BO, Dumauthioz N, Marti B, Zhang L, Schneider P, Mach JP, Romero P et al. , CART cells are prone to Fas- and DR5-mediated cell death. J. Immunother. Cancer 2018. 6: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID et al. , GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell deathby PD-1 blockade. Mol. Ther. 2016. 24:1135–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L and Alimandi M, CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018. 9: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.