

Summary:
Here we describe a novel pathogenic entity, the activated neutrophil (PMN)-derived exosome. These CD63+/CD66b+ nanovesicles acquire surface-bound neutrophil elastase (NE) during PMN degranulation, NE being oriented in a configuration resistant to αl-antitrypsin (α1AT). These exosomes bind and degrade extracellular matrix (ECM) via the integrin Mac-1 and NE respectively, causing the hallmarks of chronic obstructive pulmonary disease (COPD). Due to both ECM targeting and alAT resistance, exosomal NE is far more potent than free NE.
Importantly, such PMN-derived exosomes exist in clinical specimens from subjects with COPD but not healthy controls, and are capable of transferring a COPD-like phenotype from humans to mice in an NE-driven manner. Similar findings were observed for another neutrophil-driven disease of ECM remodeling (bronchopulmonary dysplasia, BPD). These findings reveal an unappreciated role for exosomes in the pathogenesis of disorders of ECM homeostasis such as COPD and BPD, providing a critical mechanism for proteolytic damage.
Keywords: Neutrophil, Protease, Exosomes, Extracellular Vesicles, Microparticles, Extracellular Matrix, Elastase, ELA-2, Inflammation, Lung disease, COPD, BPD
Graphical Abstract
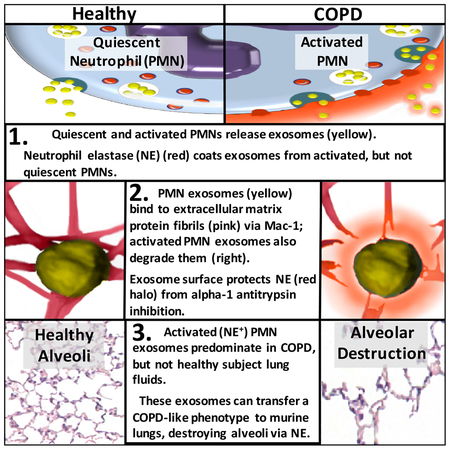
In Brief
In chronic inflammatory lung disease neutrophil-derived pathogenic exosomes bypass the pulmonary antiprotease barrier to promote extracellular matrix destruction
Introduction
Exosomes are extracellular vesicles, <150nm in diameter, released by most if not all cells constitutively and in response to stimuli. Exosomes are considered mediators of cell-cell communication as well as intercellular ferries of a diverse cargo of proteins, lipids, and nucleic acids (Maas et al., 2017). Recent work from ourselves and others has shown that exosomes can also harbor proteases with intact enzymatic activity (Shimoda and Khokha, 2013; Szul et al., 2016).
Proteases derived from PMNs play an important role in certain chronic inflammatory diseases in the lung (Russell et al., 2016). Deranged interplay between the antiprotease barrier and PMN-secreted proteases is implicated in the pathogenesis of several diseases of lung ECM remodeling, including COPD and BPD(Altiok et al., 2006; Richmond et al., 2016; Shapiro et al., 2003; Yasumatsu et al., 2006). COPD, a smoking-associated disease which remains the fourth leading cause of death globally (Raherison and Girodet, 2009), is the classical example of a pulmonary disorder driven by protease-antiprotease imbalance (Gross et al., 1965; Laurell and Eriksson, 1963; Senior et al., 1977). COPD is characterized by inflammatory airway obstruction and emphysema, causing cough, shortness of breath, and respiratory failure (Vogelmeier et al., 2017). Among the protease-antiprotease systems, the PMN-derived serine protease NE and its dominant pulmonary inhibitor α1AT are among the most important in COPD (Shapiro et al., 2003; Sng et al., 2017). NE has a broad substrate specificity and is capable of degrading, among others, two ECM structural proteins which constitute much of the pulmonary interstitial matrix, type I collagen (Kafienah et al., 1998) and elastin. Instillation of NE or certain other proteases into animal airways has been shown to cause emphysema when done in quantities sufficient to overwhelm the anti-protease barrier (Gross et al., 1965; Janoff et al., 1977; Senior et al., 1977). Reduced activity of α1AT is implicated in COPD and genetic deficiency of α1AT begets a form of COPD in humans that occurs spontaneously even in the absence of exposure to tobacco smoke or other risk factors (Laurell and Eriksson, 1963; Stockley, 2014).
Despite this evidence supporting a central role for NE in the development of pulmonary parenchymal distortion, the pulmonary antiprotease barrier imposed by α1AT is robust, and mechanisms by which NE bypasses it to induce ECM destruction remain unclear. We investigated whether NE exists in an exosomal form and whether such exosomes might bypass α1AT and contribute to inflammatory lung disease. This report defines a new exosome population derived from PMNs, which via unfettered NE activity degrades ECM.
Results
Exosomes from Activated PMNs Express Increased Enzymatically Active Surface NE
We examined the exosomes released by PMNs activated with the bacterial formylated peptide (a canonical PMN stimulant) formyl-Methionine-Leucine-Phenylalanine (fMLP), termed ‘activated exosomes’, and those released constitutively from solvent control treated PMNs (‘quiescent exosomes’). Quiescent and activated exosomes were similar in size (~100nm) (Fig. S1A and B), number released per PMN (Fig. S1C), and appearance under electron microscopy (Fig. 1A). Both exosome populations expressed a number of PMN-associated proteins, but these were generally more abundant in activated exosomes (Fig. 1B, S1D), which also contained a generally increased amount of total protein/exosome (Fig S1E). Using fluorescence activated cell sorting (FACS), we found NE was present on the exosome surface by staining for NE after pulldown for the PMN marker CD66b and confirmed that activated exosomes had considerably higher quantities of surface NE compared to quiescent exosomes (Fig. 1C–E) (See Fig. S1F for exosome gating strategy). We determined that the beads bound equal numbers of CD66b+ exosomes from quiescent and activated PMNs (Fig. S1G). However, the mean fluorescence intensity (MFI) for NE was dramatically increased in activated compared to quiescent exosomes (Fig. 1C,D). Nearly all activated exosomes expressed NE on their surface (89.2%), whereas only 1% of quiescent exosomes expressed NE (Fig 1E).
Figure 1. PMN activation leads to release of exosomes with more surface NE than quiescent PMN exosomes.

(A) Electron micrographs of exosomes derived from fMLP stimulated PMNs (activated, right) or DMSO control in PBS (quiescent, left). (B) Unbiased proteomic analysis of exosomes by mass spectroscopy, representative PMN-associated proteins shown. Exosomes (2.5 × 107) derived from PMNs from healthy donor peripheral blood (n=2) were pulled down on anti-CD66b beads, stained with anti-NE AF647, analyzed by flow cytometry (C), representative histogram shown in D) and percentage staining for NE determined (E). Data for C, E depicted as mean +/− SEM. See also figures S1 and S6.
We next investigated whether PMN-derived exosome-associated NE exists in an active and substrate-accessible state on the surface of these exosomes. Using a commercially available BODIPY-tagged elastin (DQ Elastin™) which fluoresces upon cleavage of the elastin molecule, we found that NE exists in an active, substrate-accessible form when associated with activated exosomes. By contrast, quiescent exosomes had little elastolytic capacity (Fig. 2A). These enzymatic results confirmed the elevated levels of NE for activated exosomes determined by Western blot, mass spectroscopy, and FACS (Fig. 1B–D, S1D). We probed the enzymatic properties of exosomal NE using the peptidomimetic colorimetric NE substrate N-methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide (pNA). Again, the activated exosomes were found to have much more activity than quiescent exosomes (Fig. 2B). The exosomal NE was found to have the same Km (140μM) for the NE substrate as the purified protease (Nakajima et al., 1979). The binary difference between NE activity of activated versus quiescent exosomes is consistent with an essentially “all or none” surface distribution and apparent quantity of NE as opposed to the interior of exosomes (Fig. 1D–F).
Figure 2. PMN activation confers increased expression of enzymatically active, α−1 AT resistant NE.

(A) NE activity of activated and quiescent exosomes measured at 5-minute intervals for production of (BODIPY FL) labelled fluorescent elastin fragments from self-quenching BODIPY FL-conjugated bovine neck ligament elastin. (B) NE activity of activated and quiescent exosomes against specific NE peptidomimetic substrate MeOSucAAPVpNA, by pNA generation. (C) 20ng purified NE coincubated with Human NE Inhibitor II, the endogenous NE inhibitor α−1 AT or PBS control, and NE activity by pNA generation measured as in B. (D) NE activity of an equipotent number of activated exosomes (1 × 109) and 20ng of purified NE measured with Human NE Inhibitor II, α1-AT, or PBS control as in B. Single experiment shown in C. and D. displayed separately for clarity. Data shown as mean +/− SEM. See also figure S2.
Exosomal NE is resistant to α1AT inhibition
Exosomal NE and purified NE activity were both largely inhibited by the prototypical, highly specific irreversible small molecular weight active site-targeted NE inhibitor, Human NE Inhibitor II (Fig. 2C, D) (Navia et al., 1989). In contrast, unlike purified NE (Fig. 2C), activated exosomal NE was resistant to inhibition by α1AT (Fig. 2D). Compared to a dose-response for α1AT inhibition of purified NE (Fig. S2A), we ascertained that activated exosomal NE is ~9 times less sensitive to α1AT than solution-phase protease (using exosomes subjected to a single freeze-thaw cycle). We subsequently found that fresh, never frozen activated exosomal NE is virtually completely resistant to α1AT inhibition (See Fig. 3B, Fig. S2B), apparently due to a higher proportion of surface-bound NE, as it is the surface-bound form that is protected from α1-AT inhibition (see below). These experiments demonstrate that quiescent exosomes contain little surface-bound, substrate-accessible NE, but PMN activation leads to exosomes which harbor surface-exposed NE in a position that allows for substrate access yet is resistant to α1-AT.
Figure 3. PMN exosomes load NE extracellularly.

(A) 5 × 109 quiescent exosomes coincubated with purified NE (1μg) or left in PBS, filtered and rinsed in PBS to remove unbound NE. Exosome NE activity measured by pNA. (B)1 × 109 activated exosomes coincubated with protamine sulfate or PBS with or without α1-AT and NE activity measured by pNA. Data from A and B. shown as mean +/− SE of 4 experiments. (C-G) Model of hypothesized mechanism of NE loading upon exosomes. (C) Quiescent PMN releases exosomes (yellow spheres) constitutively as NE (blue shapes) is sequestered intracellularly in primary granules. (D) Activated PMN continues to release exosomes constitutively while degranulating, releasing NE molecules into solution which bind to exosome as it passes through locally elevated halo of degranulated NE. (E) Close-up model of activated exosome, to which multiple NE particles are bound via charge-mediated interactions. (F) Activated exosome in an environment rich in α1-AT (crescents). Steric hindrance impedes α1-AT interaction with exosomal NE, whereas free NE readily complexes with α1-AT molecules in an irreversible fashion. (G) Activated exosome after application of cationic molecules such as protamine sulfate to displace NE from exosome surface, now readily complexed and inactivated by α1-AT. See also figure S3.
When PMNs are activated, their primary granules release NE into solution, some of which binds back to the PMN surface via an ionic interaction with proteoglycans on the cell membrane (Owen, 2008; Owen et al., 1995). To determine whether a similar mechanism might occur with exosomes, quiescent PMN exosomes were treated with purified human NE. The quiescent exosomes then assumed an activated exosome phenotype with surface exposed NE activity (Fig. 3A). Furthermore, we determined that application of the cationic substance protamine sulfate caused activated exosomes to release NE from the exosome surface. Such released NE was completely inactivated by α1-AT present at a dose which had no effect on exosome-bound NE (Fig. 3B). Similar results were obtained with the cationic amino acid l-lysine, but not with the neutral amino acid l-proline (Fig. S2B). These findings are consistent with our hypothesis that NE, which carries a significantly cationic net charge, associates with the exosome surface via high volume but weak ionic interactions. This may be mediated by proteoglycans which bind NE to the cell membrane and have also been observed on the surface of exosomes (Christianson et al., 2013). NE then assumes a catalytically active orientation that sterically hinders α1-AT binding. Collectively, these results also suggest that the exosomes released constitutively by PMNs acquire the majority of surface NE extracellularly when degranulation occurs in the vicinity of exosome release (Fig. 3C–G).
Activated Exosomes Degrade Collagen
We next sought to test whether the activity of exosomal NE extended to type I collagen, the most abundant form of collagen in the lung. This protein can be cleaved by several proteases observed in the proteome of PMN exosomes (Fig. 1B), including NE itself (Kafienah et al., 1998). Type I collagen was visualized by electron microscopy with and without activated and quiescent exosomes at different time points (Fig. 4A–F). Both quiescent and activated PMN exosomes physically associated with collagen fibrils; very few if any could be found in the space between the collagen network (Fig. 4B–C). In contrast, activated (Fig 4E) but not quiescent (Fig 4D) exosomes visibly destroyed the collagen fibrils over time and in the process seemed themselves to disintegrate. The degradation of collagen fibrils by activated exosomes appeared to be localized (Fig. 4E, F), with areas of apparent sparing after the reaction was complete (Fig. 4E), likely representing fibrils that were not in contact with exosomes. This suggests that physical association is important in the exosomes’ capacity to hydrolyze collagen; we hypothesize that this contact focuses the proteolytic process and allows the exosomes to “drill” into the contacted regions of substrate rather than releasing proteolytic enzymes into solution, which would be expected to create a more diffuse reaction.
Figure 4. Exosomes associate with collagen fibrils selectively via Mac-1 and possess NE dependent collagenase activity.

(A) EM image of collagen fibrils after 24 hour-incubation. (B) Collagen fibrils and quiescent exosomes imaged immediately after being combined. (C) Collagen fibrils and activated exosomes imaged immediately after being combined. (D) Collagen fibrils and quiescent exosomes after 24 hour co-incubation. (E) Collagen fibrils imaged after 24 hours of co-incubation with activated exosomes. (F) Closer view of activated exosome with collagen fibril. Arrow denotes area where a collagen fibril appears to be nicked and frayed near exosome. (G) PBS or exosomes of varying quantities coincubated with type I collagen-coated plates. ELISA for exosome associated lactoferrin performed using horse-radish peroxidase-conjugated anti-lactoferrin. (H) ELISA performed as in G. in the presence of MP-9, or RGD peptide at varying concentrations. (I) Activated and quiescent exosomes measured for production of fluorescein isothiocyanate (FITC) from FITC-labelled type I collagen. (J) Activated exosomes with and without Human NE Inhibitor II as well as purified NE measured for type I collagen degradation as in I. See also figure S2.
The specificity of this physical exosome-collagen binding was then tested using an enzyme-linked immunosorbent assay (ELISA) in which we probed type-I collagen coated plates for exosomal lactoferrin after incubating the plates with varying doses of exosomes; a linear increase in exosome-collagen binding was observed with increasing quantities of exosomes (Fig. 4G). We then determined how the exosomes bind to collagen. As tumor exosomes use integrins for tissue targeting (Hoshino et al., 2015), we hypothesized that the leukocyte integrin Mac-1, (composed of the dimerized subunits αM (CD11 b) and β2 (CD18) present in the exosome proteome (Fig 1B)), which binds a wide array of ECM proteins including type I collagen and elastin via the αM-I domain, contributes to the exosome-collagen interaction. To test this, we reproduced the ELISA in the presence of a potent, specific nonapeptide inhibitor (MP-9) of the Mac-1 αM-I domain (Podolnikova et al., 2015) and found the binding to be dose-dependently and completely inhibited (Fig 4H). By contrast, arginine-glycine-aspartate (RGD), a broad integrin inhibitory peptide (D’Souza et al., 1991) unable to bind Mac-1 (Podolnikova et al., 2015; Van Strijp et al., 1993), had no effect even at much higher concentrations than those tested for MP-9 (Fig 4H). These results confirm that exosome binding to collagen fibrils is mediated by Mac-1, specifically via the aM-I domain. In keeping with the relatively broad binding specificity of Mac-1, exosomes were also found to bind elastin (Fig. S2C). Importantly, these findings demonstrate a second mechanism by which proteolytic exosomes exert an outsized degradative capacity in relation to their respective size and protease load. These generate a physical association with the ECM, focusing exosome NE activity in the locus of the proteolytic substrate.
To test the relative contributions of NE and other exosomal proteases to this observed collagenase activity in vitro, we assayed exosomes and purified NE against a FITC-tagged type I collagen substrate (Fig. 4I, J). Purified NE had a robust collagenase activity (Fig. 4J), as previously reported (Kafienah et al., 1998). Similar to our findings with elastin and the synthetic NE substrate, activated exosomes were again found to have a binary difference with quiescent exosomes (Fig. 4I), and the activity ratio between exosome quantity and purified NE was similar to that observed with other NE substrates. Since type I collagen is a substrate for several other PMN-derived proteases, we tested the relative contribution of NE to this activated exosome collagenase activity (using Human NE Inhibitor II) and found the exosomal collagenase activity to be largely blocked (Fig. 4J), to a degree similar to that observed when using the NE specific colorimetric substrate (Fig 2D). Taken together these experiments demonstrate that activated exosomes have the capacity to degrade collagen in a largely NE-dependent manner.
Activated PMN exosomes cause emphysema when administered into murine lungs
The potent ability of these exosomes to bind to and degrade collagen and elastin in vitro coupled with the resistance to α1-AT inhibition suggested that these nanovesicles might disrupt the extracellular matrix in the lungs and cause emphysema. To test this in vivo they were administered intratracheally into mouse airways. In this model, activated but not quiescent PMN exosomes from multiple individual human blood donors caused the hallmarks of COPD, alveolar enlargement, increased airway resistance, and right ventricular hypertrophy (RVH), compared to mice treated with PBS (Fig. 5A–D). This effect was neither strain-nor sex-dependent, occurring in male A/J, as well as female (C57Bl/6) mice (Fig S3A). Alveolar enlargement was observed by 3 days post-administration of activated exosomes and progressed daily, peaking between days 6 and 7 (Fig. 5E). Moreover, alveolar enlargement persisted for at least 3 weeks after a single-dose exosome administration, as would be expected for emphysema (Fig. S3B). Alveolar enlargement was dose-responsive with a minimum dose required for peak effect occurring between 1.67 × 108 and 1.67 × 107 exosomes and significantly reduced alveolar enlargement at a dose of 1.67 × 106 exosomes (Fig. 5F). When exosomes from activated PMNs were treated with Human NE Inhibitor II prior to administration to mouse airways there was a marked reduction in alveolar enlargement (Fig. 5G). Therefore, NE seems to be the dominant driver of the alveolar enlargement seen with activated exosome administration in vivo. To confirm that this alveolar enlargement in mice was associated with breakdown of pulmonary ECM, animals were treated with activated or quiescent exosomes or PBS and bronchoalveolar lavage was performed. The murine bronchoalveolar lavage fluid (BALF) was then measured by mass spectroscopy for quantity of acetyl-proline-glycine-proline (PGP), a collagen fragmentation product associated with ECM turnover and known to be increased in COPD lung secretions (O’Reilly et al., 2009). The PGP levels in BALF of activated exosome-treated but not quiescent exosome-treated animals was significantly increased (Fig. S3C). These findings are consistent with direct alveolar destruction by the exosomes rather than an indirect effect of inflammatory cell influx (which was not observed to any consequential degree; see representative histology and BALF cell counts in Figure 5A and also Fig. S4). This is consistent with the minimal exosome immunogenicity others have noted previously (Zhu et al., 2017). Although the development of emphysema is linked to ECM degradation, derangements of epithelial cell biology and resilience, most notably increased levels of epithelial cell apoptosis (Demedts et al., 2006; Mouded et al., 2009)), have also been associated with COPD pathogenesis. Therefore, we investigated the general effects of activated and quiescent PMN exosomes on human primary airway epithelial cells by performing RNA sequencing and mRNA expression pathway analysis (Ingenuity Pathway Analysis, QIAGEN, Inc.) of cells treated with a quantity of exosomes (3.7 × 105/1.9 cm2 well) equivalent to the number given (per unit area) to animals treated with 1.67 × 108 exosomes, assuming average female A/J mouse pulmonary surface area reported in prior literature (Knust et al., 2009). However, only minor differences in mRNA were observed between activated and quiescent exosome-treated cells, and these differences seem unlikely to account for the alveolar enlargement as these were generally more similar between activated and quiescent exosome-treated cells than PBS-treated cells (Fig. S5), whereas quiescent exosomes did not cause alveolar enlargement. This suggests that, while there may be effects on the cells of exosome-treated animal lungs, the alveolar enlargement is driven primarily by direct hydrolysis of the ECM.
Figure 5. Activated neutrophil exosomes confer a COPD-like phenotype to mice in an NE-dependent fashion and express CD66b and CD63.

(A) Hematoxylin/eosin (H&E) stained photomicrographs of lung tissue of female A/J mice exposed i.t. to PBS,1.67×108 activated or quiescent PMN derived exosomes (n= 3–5 mice per group repeated 8 times with exosomes representing 8 separately tested donors) and sacrificed on day 7. (B) Alveolar size measured as mean linear intercept (Lm). (C) Mice (n=6) exposed to exosomes or PBS control as in A. and pulmonary function testing performed and resistance measured. (D) Weight ratios of right ventricle: left ventricle + septum (RV/LV+S) measured, shown as dot plot with mean indicated by line. (E) Activated exosomes administered as in A, animals sacrificed at days 1–7 with Lm measured as in B. (F) Varying doses of exosomes given as in A., animals sacrificed on day 7, and Lm measured as in B., n ≥ 4 per group. (G) Exosomes from activated PMNs with and without preincubation with Human NE Inhibitor II delivered i.t. into mice as in A. and Lm measured as in B., n ≥ 10 per group. (H) Purified human NE instilled i.t at varying doses, animals sacrificed at day 7, Lm measured as in B., n≥ 4 per group (I) Exosomes from neutrophils stimulated with the CXCR2 ligand PGP or control peptide PGG administered to mice with Lm measured as in B., n ≥ 4 per group. (J) Exosomes delivered to mice as in A., Lm measured as in B., after antibody coated magnetic bead capture and release to purify for CD63, CD66b or MUC4 (negative control), or after bead depletion for CD63 and CD66b, n ≥ 4 per group. Data shown as follows: center line, median; box limits, upper and lower quartiles; whiskers, minimum and maximum values. * p<0.05, ** P<0.01, *** p<0.001. See also figures S3, S4 and S5.
We found that, like activated exosomes, purified human NE can cause doseresponsive alveolar enlargement when introduced into mouse airways (Fig. 5H) as had been previously shown in similar mammalian models (Janoff et al., 1977; Senior et al., 1977). However, the dose required (3333ng/mouse) for modest but significant alveolar enlargement is 10,000 times more than the active site-titrated 0.33ng of NE associated with 1.67 × 107 activated exosomes (Fig. 5F). Thus, NE appears to be far more pathologic in vivo when exosome-associated; this is presumably due both to their resistance to antiprotease activity and their ability to physically associate with ECM proteins.
While we have used fMLP as a model PMN activator, we questioned whether these findings were specific to this compound or reflective of a more general aspect of PMN biology. Consequently we activated PMNs with the CXCR2 ligand Acetyl-Proline-Glycine-Proline (PGP), which contributes to PMN activation in COPD subjects and causes emphysema in mice (Weathington et al., 2006). Figure 5I shows that exosomes from PGP-treated, but not control peptide PGG-treated PMNs, caused alveolar enlargement similar to fMLP-treated PMN derived exosomes. PGP is a biomarker for COPD that is induced by cigarette smoke (CS) and persists in COPD after smoking cessation, propagating chronic PMN-driven inflammation which is now shown to elicit proteolytic exosomes (Gaggar and Weathington, 2016; Weathington et al., 2006).
Pathogenic Activated Exosomes Coexpress CD63 and CD66b
Using a capture and acid pulse-release method from antibody-coated magnetic beads, activated exosomes that cause alveolar enlargement were found to express the PMN-specific and exosome markers CD66b and CD63 respectively but not the airway epithelial marker MUC 4 (Fig. 5J). Furthermore, depletion of the CD66b+ and CD63+ exosome population could each completely prevent the alveolar enlargement (Fig. 5J), demonstrating that the disease-causing exosome is positive for both markers, consistent with their PMN exosomal origin.
Human COPD Lung-Derived CD63+/CD66b+ Exosomes Confer COPD to Mice
We next evaluated whether these activated PMN exosomes exist in lungs of COPD patients. Exosomes were purified from BALF from COPD patients or healthy never smoking controls (NS) (Table S1A). Similar numbers of exosomes of similar size were obtained from COPD compared to NS control BALF (Fig. S6A–D). Figure 6A top panels show representative histology demonstrating marked alveolar enlargement (Fig. 6B) and RVH (Fig. 6C) occurring when BALF exosomes from pooled individuals with COPD, but not pooled BALF exosomes from healthy controls, are administered to mice. It should be noted that the total number of exosomes administered per mouse (2.4 × 109 in 6 doses of 4 × 108) is a pathologically relevant dose. This number of exosomes falls within the range that we observed in 1mL of COPD BALF (mean concentration 2.06 × 109 +/− 0.55/mL), the volume of an adult murine lung. This also falls within the (lower end) range of the exosome number we have previously observed (Szul et al., 2016) (~1 × 109-1 × 1010) in the lung lavage of an adult mouse. When such COPD BALF-derived exosomes were separated into those from current versus former smokers, those from current smokers caused somewhat, albeit non-significantly, increased alveolar enlargement (Fig. 6B). Both caused significant alveolar enlargement compared to never-smoker BALF exosomes (Fig. 6B). A longer exposure time and increased numbers of COPD BALF exosomes (2.4 × 109) were required to observe pathology compared to activated PMN exosomes (Fig. 5F). This likely reflects lesser disease potency due to BALF exosomes being a mixture from different cell sources as compared to those solely of PMN origin. A prominent role for NE in the emphysema-causing activity of the COPD BALF-derived exosomes was confirmed by the ability of Human NE Inhibitor II to markedly blunt the response (Fig. 6A lower left panel, 6B). The COPD BALF-derived exosomes that caused disease were CD66b+, but not MUC4+, as demonstrated by the ability of exosomes captured and released from CD66b, but not MUC4, antibody coated beads to cause alveolar enlargement (Fig. 6D). Interestingly, CD66b capture from COPD BALF enriched for pathogenic exosomes as evidenced by the lower total number (5×108) required to cause alveolar enlargement as compared to those from an unpurified COPD BALF population (2.4×109). This number of CD66b+ BALF-derived exosomes is similar to the number of activated peripheral blood-derived PMN exosomes that cause disease (Fig. 5F). By contrast, CD66b-coated bead capture purification did not enrich for alveolar enlargement activity in exosomes from NS (Fig. 6C). The ability of COPD BALF-derived CD66b+ exosomes to cause alveolar enlargement was completely ablated by treatment with Human NE Inhibitor II (Fig. 6A lower middle and right panels, 6D). It bears noting, however, that Human NE Inhibitor II ablated the alveolar enlargement caused by the mixed population of COPD BALF exosomes by about 70–75%, whereas it completely prevented the alveolar enlargement caused by CD66b+ COPD BALF exosomes. We next tested exosomes from individual COPD and control subject BALF and found that 10/10 COPD subject BALF exosomes caused significant alveolar enlargement (of varying magnitude), whereas mice treated with exosomes from individual control subject BALF had marginal, if any, alveolar enlargement (Fig. 6E). Having demonstrated that COPD BALF exosomes cause alveolar enlargement largely driven by NE present on the CD66b+ population of exosomes, (whereas N/S CD66b+ purified BALF exosomes did not), we next performed pull-down of three COPD and three control N/S BALF exosomes with anti-CD66b beads. To better elucidate the differences in these exosomes, we analyzed an equal number of CD66b+ exosomes from each (Fig. S7E). This showed a dramatically increased staining with an anti-NE antibody via FACS for COPD CD66b+ exosomes in comparison to an equal number of control N/S BALF exosomes (Fig 6F). In fact, almost all (96.1%) CD66b+ exosomes in COPD BALF expressed NE, whereas few (1.3%) of those obtained from NS BALF expressed NE (Fig. 6G). The partial blunting of the pathogenicity of the pool of COPD exosomes by Human NE inhibitor II (Fig 6B) together with the total NE inhibitor II blockage of the purified CD66b+ COPD exosomes (Fig 6D) implies that there might be another, less prominent pathogenic exosome population in COPD BALF which do not express CD66b and operate via a non-NE dependent mechanism.
Fig 6. Human BALF-derived CD63+/CD66b+ exosomes confer a COPD-like phenotype to mice in a NE-dependent manner.

(A) Photomicrographs of H&E stained lung tissue of mice exposed to various exosomes. (B) Exosomes obtained from BALF of healthy never smokers (N/S) (n=10), and subjects with COPD (n=10). Exosomes pooled from BALF of all 10 healthy never smoker subjects (N/S), all 10 pooled COPD subjects with and without Human NE Inhibitor II preincubation, current smoker COPD subjects (n=5) (current smoker COPD”), or former smoker COPD subjects (n=5) (former smoker COPD), administered i.t. (4.0 × 108 exosomes/dose, 6 doses over 12 days) to 8–10 week old mice (n ≥ 15 per group), sacrifice at 14 days and Lms were determined. (C) RV/(LV+S) shown for PBS, N/S, and COPD experiments of panel B. Dot plot displayed with line delineating mean measurement. (D) Exosomes from BALF of pooled healthy N/S subject (n=10) and pooled COPD subjects (n=10) captured on anti-CD66b or anti-MUC4 antibody coated beads and resulting population depleted and/or purified for expression of CD66b or MUC4 used for i.t. mouse exposure; COPD subject BALF purified for CD66b expression (with and without Human NE Inhibitor II) and MUC4 expression, NS subject BALF purified for CD66b expression, and MUC4+ purified exosomes from pooled COPD patient BALF administered to mice i.t. as in (B), n ≥ 4 per group. The dose of exosomes used for these experiments correspond to the entire population of exosomes given in B. after depletion of or purification for the respective marker. Shown for comparison are the Lm of pooled COPD subject and N/S BALF exosome treated animals from experiment in panel B. Representative photomicrographs of these experiments shown in panel A. (E) BALF exosomes from separate individual N/S or COPD subject BALF administered i.t. to mice (n=4 per individual) and Lm measured as in B. Each data point shown represents mean increase of Lm over control (i.e., intra-experimental PBS treated mice) of mice treated with the exosomes from a single N/S or COPD individual subject, shown as mean +/− SEM for the two groups. The standard deviation of control PBS treated mice in these experiments was +/− 0.87μm. (F) Exosomes (2.5 × 107) derived from individual N/S or COPD subjects (n=3/group) were pulled down on anti-CD66b beads, stained with anti-NE AF647, analyzed by flow cytometry and MFI of anti-NE staining of exosomes determined, shown as representative histogram, with quantitative display of mean in inset. (G) Percentage of exosomes that stained for NE analyzed as in F. Data for B. and D. presented as follows: center line, median; box limits, upper and lower quartiles; whiskers, minimum and maximum values. ** P<0.01, *** p<0.001 ****p<0.0001. See also figures S1, S4, S5, and S6 and Table S1.
Pathogenic Exosomes Are Also Present in BPD Lung Secretions
We next tested whether the observation of a population of proteolytic exosomes in the lung capable of transferring disease features from humans to mice was a phenomenon unique to COPD or a more general characteristic of inflammatory lung disease. To investigate this, we purified and pooled exosomes from the aspirated tracheal secretions of intubated neonates with severe BPD (BPD) and gestational age-matched full-term controls (Non-BPD) (n= 5 per group). Donor characteristics are shown in Table S1B. As anesthesia can result in animal loss at this age, these were administered intranasally (i.n.) in 15μL aliquots to neonatal (postpartum day 3) female C57/Bl6 pups at a dose of 4×108 per mouse for three doses (total 1.2 X109 per pup) with sacrifice at postpartum day 14. As shown in Figure 7, mice treated with BPD exosomes, and not non-BPD exosomes, developed the morphological and physiological hallmarks of BPD, namely alveolar hypoplasia (quantified as radial alveolar count (RAC)) (Fig. 7A, B), increased airway resistance (Fig. 7C), and RVH (Fig. 7D). To verify that the alveolar hypoplasia induced by BPD exosomes was mediated by the CD66b+ exosome population, pups (n=4) were also treated i.n. with purified CD66b+ BPD exosomes and found to have the same degree of hypoplasia as that seen with unfractionated BPD exosome exposure (RAC 5.2 +/− 0.37 vs. 5.2 +/− 0.19, respectively). These results complement the COPD results and highlight the generalizability of this pathway in neutrophilic lung disease.
Figure 7. Tracheal aspirate exosomes from BPD subjects confer a BPD-like phenotype to exposed neonatal mice.

(A) Exosomes from tracheal aspirate samples of subjects (n=5/condition) with BPD and non-BPD controls were purified and pooled into BPD and Non-BPD groups. Mice (n=4/group) treated i.n. with 4 × 108 exosomes in 15μL on postnatal days 3, 6, and 12 and sacrificed on postnatal day 14. Lungs stained by H&E for alveolar morphometry and representative histology of treated animals shown. (B) Radial alveolar counts measured for animals treated in A. (C) Mice (n=5) treated i.n. with exosomes as in A., pulmonary function testing was performed, and resistance is shown. (D) RV/(LV+S) of animals treated in A. was measured. See also Table S1.
Discussion
PMNs play a critical role in the innate immune response observed in chronic disease, providing a key pivot between resolution or propagation of organ damage (Richmond et al., 2016). Here we identify PMN-derived, NE-rich exosomes as a new subcellular entity which is a fundamental link between PMN-driven inflammation and tissue damage. Further, we characterize a novel mechanism by which these exosomes become capable of aggressive proteolysis unchecked by antiproteases, and we define a method of ECM-specific targeting. This report seems to provides the first evidence of the capability of a defined non-infectious subcellular entity to recapitulate disease phenotype when transferred from human to mouse. As a result, this investigation reveals an entirely unappreciated aspect of the interplay between inflammation, proteolysis, and matrix remodeling with far-reaching implications for future research.
Given the abundance of antimicrobial proteins observed on these exosomes (including NE itself), it is also likely that these exosomes play a role in innate immune defense against bacterial pathogens. However, in disease, the excessive activity of these PMN exosomes leads to damaged ECM sufficient to trigger alveolar unit loss. This leads to emphysema in the case of COPD and alveolar simplification in the case of BPD. As a consequence, COPD patients with such exosomes might be viewed as functionally α1AT deficient in the alveolar interstitium. It seems likely that such exosomes may contribute to the pulmonary architectural distortion seen in other lung diseases, such as cystic fibrosis, that are associated with excessive PMN-driven inflammation and local elevations of NE.
The resistance to inhibition of exosomal NE may be due to steric hindrance inherent to the position they assume on the exosome membrane, as has been reported to occur when NE is associated with the PMN cell membrane (Owen, 2008; Owen et al., 1995). This seems to be mediated by an ionic interaction between NE molecules and the exosome membrane (Fig. 3). Although the ability of these exosomes to degrade ECM proteins is largely driven by NE, other proteases such as MMP-9 were seen in the activated exosomal proteome (Fig. 1B). In addition to antiprotease resistance, the in vivo potency of exosomal NE is likely enhanced by the physical association of the protease-laden exosome with its substrate, effectively focusing the activity of NE.
PMN microvesicles have been previously shown to diffuse through tissue planes into ECM tissues devoid of PMNs, such as synovial cartilage (Headland et al., 2015). Activated PMN ‘microparticles’ (corresponding to the extracellular vesicles termed ‘exosomes’ in this report) have also been described to disrupt epithelial barriers via degradation of tight junctions (Butin-Israeli et al., 2016). Thus it is probable that activated PMN exosomes ferry NE from areas of PMN activation (e.g., intravascular and alveolar spaces) through cellular planes into adjacent ECM (e.g., alveolar interstitium), allowing PMNs to contribute to tissue remodeling by proxy. This report significantly expands the biological repertoire of the exosome, demonstrating potent biological effects of these particles ex cellula.
Our findings collectively suggest that this phenomenon is driven primarily by direct hydrolysis of the alveolar ECM bed. However, secondary cell effects including apoptosis still must arise, as the cellular components of alveolar units must also be lost for alveolar enlargement to occur. Cellular loss of normal ECM cues is known to provoke apoptosis of overlying pulmonary epithelial cells, a phenomenon called anoikis. Anoikis likely accounts for some or all of the observed increase in apoptotic cells in COPD (Mouded et al., 2009), and the pulmonary ECM in COPD subjects has been recently found to directly contribute to derangements of cellular function (Hedstrom et al., 2018). Such a mechanism would explain the observed enlargement of the alveoli of animals treated with proteolytic exosomes from activated PMNs in the absence of a marked direct effect on airway epithelial cells uncoupled from ECM in vitro. Alternatively, alveolar, in contrast to airway, epithelial cells could be directly affected by proteolytic exosomes and contribute to emphysema.
Quantification of the more destructive exosomal NE rather than solution-phase NE may yield insight into monitoring of disease/therapeutic activity and detecting early disease in persons with COPD. It is possible that previously observed correlations between NE levels and COPD activity and progression (Betsuyaku et al., 2000; Sng et al., 2017) might be strengthened by accounting for the levels of exosome-associated NE. Similarly, the efficacy of NE inhibitors against the exosomal form of NE would appear to be an important consideration in development of therapeutics. Considering that NE inhibition of unfractionated COPD exosomes only partially inhibited alveolar enlargement (Fig. 6B), and since other proteases such as MMP-12 also play prominent roles in COPD (Greenlee et al., 2007; Hautamaki et al., 1997), future studies of nanovesicle association of other proteases seem warranted. Similarly, exosomes of other cell types such as macrophages ought to be examined for matrix-modifying properties in health and disease. Such mechanisms could potentially be involved in a host of other disorders associated with immune cell activation and ECM remodeling (e.g., myocardial infarction, metastatic cancer, and chronic kidney disease). Lastly, these investigations demonstrate feasibility, at least in vitro, of therapeutic strategies that interrupt pathogenic aspects of PMN exosome function (i.e., dissociation of NE from exosomes to render them susceptible to endogenous α−1AT, inhibition of exosomal Mac-1, and inhibition of exosomal NE itself). Parenthetically, protamine sulfate, a compound which caused dissociation of NE from exosomes, is a drug approved for and used commonly in humans. These might be tailored into treatments that ameliorate the presently inexorable progression of COPD and perhaps other diseases such as BPD in which comparable exosome-mediated pathology occurs.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, J. Edwin Blalock, PhD (jeblalock@uabmc.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were purchased from Jackson Laboratories. Experiments were done using female A/J mice (RRID:IMSR_JAX:000646) of 8–12 weeks of age except for BPD exosome and those experiments done on age-matched male A/J mice and female C57/BL6 mice (RRID:IMSR_JAX:000664) as put forth in Fig S4. Mice were housed in the UAB animal housing facility in cages of 5 and for any experiments requiring re-caging this was done upon arrival to the housing facility. Based on our prior experience with murine models of emphysema (Weathington et al., 2006; Wells et al., 2014) a minimum group size of 3 has previously been largely sufficient to demonstrate a difference in alveolar size between conditions. Depending upon the quantity of available exosomes for an experiment, groups of 4 or 5 were used to ensure adequate power and to account for the possibility of animal loss in the course of investigation. Also, key experiments were repeated, including validation of findings with exosomes from PMNs of 8 separate human donors; data from repeat experiments was aggregated, which along with occasional animal loss led to some variation of sample size, as reported in respective figure legends. Animals were assigned to condition by cage (i.e., littermates exposed to same condition) without bias whenever possible. For any conditions requiring animals to be combined from multiple cages, animals were matched for weight and distributed evenly between cages prior to assignment of a given cage to any experimental condition. Mice were conditioned to the housing facility for at least 48 hours prior to any experimentation. All animal experiments were done in compliance with widely accepted ethical standards and were approved by the institutional IACUC under protocols 20254, 20345, and 10075.
Human Subjects
Human Bronchoalveolar Lavage Fluid (BALF) Procurement
Adults were recruited and categorized into cohorts based on the presence or absence of COPD as defined by the GOLD criteria. Demographics, smoking status, smoking duration and intensity as indicated by pack-years were self-reported and corroborated by medical record review. Current smoking status was defined by cigarette smoking within the previous month. Spirometry was performed per ATS/ERS recommendations and the percent of predicted post-bronchodilator forced expiratory volume in 1 second (FEV1) was recorded. BALF was collected during fiberoptic bronchoscopy under conscious sedation. The airways were completely inspected followed by instillation of 200mL of 0.9% sodium chloride and recovered through the bronchoscopic working channel using constant suction. As shown in Table S1A, individual donor BALF exosomes tested in vivo from COPD and N/S subjects (Fig. 6E) were derived from (64% and 20% male, 36% and 80% female subjects, respectively). 9 of these were the same donors used for pooled BALF exosome experiments (Figs. 5, 6) and one additional COPD subject BALF was used to replace a sample that had been depleted already by the pooled exosome experiments. Post-hoc analysis of effect by gender showed no significant difference between male and female subject COPD BALF derived exosomes (mean Lm increase above control 6.81 +/− 2.98μm for males vs. 6.76 +/− 2.29μm for females, P= 0.96). Specimens were allocated to COPD or N/S groups based upon the presence or absence of spirometrie obstruction (post-bronchodilator FEV1:FVC <0.7) as per the Global Diagnosis of Obstructive Lung Disease criteria (Vogelmeier et al., 2017). Because these samples had been pre-collected and housed in our Lung Health Center biorepository, and because of our observed mean exosome return among COPD subjects of 2.06 × 109 +/− 0.55/mL (Figure S6D), corresponding to approximately one mL of BALF exosomes per mouse treated, we based our group sizes upon the amount of BALF available. n=10 samples from n=5 current and n=5 former smokers were identified with at least 10mL of BALF available, which allowed for testing of approximately 100 mice. All procedures were performed with informed consent and were approved by the institutional IRB under protocols F120404001 and IRB-031006001.
Human Neonatal Tracheal Aspirate Isolation
Human BPD and non-BPD control tracheal lavage was harvested from subjects. All samples were obtained with informed consent and approved under UAB institutional protocol IRB-140926006. After pelleting cells and smaller particles by centrifugation (10,000G × 1 hour), tracheal aspirate fluid was ultracentrifuged (150,000G X2 hours) for exosome extraction (n=5 subjects with severe BPD, 5 non-BPD controls). Exosome quantity per subject was similar to our previously published experience (Lal et al., 2018) Exosomes from individual subjects from each group were pooled and resuspended in 200μl PBS to create two pooled groups (BPD and Non-BPD). BPD and non-BPD tracheal aspirate samples were allocated to BPD or non-BPD groups using the physiologic definition of BPD (Lal et al., 2018; Walsh et al., 2004) with sample size of n=5 subjects per group based simply upon the number of BPD specimens available in our repository. Detailed clinical characteristics are shown in Table S2. These were then diluted in PBS so that 4 × 108 exosomes were present in each 15 μl aliquot for dosing. Analysis of gender effect could not be performed due to use of pooled exosome populations.
PMN purification
Whole blood was obtained from healthy volunteers (gender and ages not available) by phlebotomy into heparinized vacuum phlebotomy vials. RBCs were allowed to sediment for 20 minutes at 20°C after 1:1 mixture with 5% de xtran. Supernate was eluted and centrifuged for 7 minutes at 1000G at 20°C. Residua l RBCs were lysed by resuspending pellet in ice-cold 0.2% saline for 30 seconds then rapidly bringing solution to isotonicity with 1.6% saline. Cell pellet was resuspended in filter/sterilized 0.9% saline solution equal to starting volume of whole blood and 10mL of Ficoll gradient solution (GE Healthcare Bio-Sciences) was layered at the bottom of a 50mL centrifuge tube. This was then centrifuged for 40 minutes at 1000G at 4°C. The pellet, now consisting of purified peripheral blood PMNs, was then resuspended in 1mL filter-sterilized phosphate buffered saline (PBS) (pH 7.5 without CaCl2/MgCl2) and set on ice. A 1:10 dilution in Trypan blue dye was performed and cells counted; viability of > 95% was confirmed for all experiments. For stimulation, PMNs were resuspended at a ratio of 1mL of PBS with CaCl2/MgCl2 (+/− activating agents or solvent control) per 5 × 106 cells. Samples were obtained with informed consent and was approved by the UAB institutional under protocols IRB-031006001 and F161121004.
Cultured Cells
Cell lines were obtained with informed consent under UAB institutional protocol IRB-090908003. Primary human airway epithelial (HBE) cells (sourced from the Gregory Fleming Cystic Fibrosis Institute at UAB) were derived from lung explant of a 28 year old nonsmoker potential lung transplant donor without a history of chronic lung disease whose lung was deemed not suitable for transplantation. Briefly, tissues were debrided immediately after surgical resection, washed twice in Minimum Essential Media (Thermo-Fisher, Cat#: 11095–072) with 0.5 mg/ml DTT (Sigma-Aldrich, Cat#: D9779) and 25 U/ml DNAase I (Sigma Aldrich, Cat #: 4716728001), and then placed in dissociation media containing MEM, 2.5 U/mL DNAse I, 100 μg/ml ceftazidime(Sigma-Aldrich, Cat#: C3809), 80 μg/mL tobramycin (Sigma-Aldrich, Cat#:614005),1.25 μg/mL amphotericin B (Sigma-Aldrich, Cat#: A2942), and 4.4 U/mL pronase (Sigma-Aldrich, Cat#: 10165921001) for 24–36 hrs at 4 C. Cells were screened for mycoplasma at least quarterly per institutional standards. Loosened airway epithelial cells were then expanded in Minimum Essential Medium containing BEGM (LONZA, Catalog #: CC-3170) that was exchanged every 24 hrs. Following expansion, first passage cells were grown and terminally differentiated to confluence under polarizing air-liquid interface conditions in 1.9cm2 filters in a 5% CO2 cell culture incubator in Pneumacult-ALI (StemCell Technologies, Cat#: 05001), which was exchanged every 48 hours.
METHODS DETAILS
PMN stimulation
For most experiments 2μM fMLP (Sigma Aldrich) in DMSO (final concentration 0.01%) was used to activate PMNs. Quiescent control PMNs were treated with an 0.01% DMSO in PBS. Cells were incubated for 30 minutes with gentle agitation at 37°C. Acetylated Proline-Glycine-Proline (PGP) and Proline-Glycine-Glycine (PGG) obtained from Bachem were used at 100μg/mL. After PMN activation, tubes were placed briefly on ice then centrifuged at 1000G for 7 minutes at 4°C. Supernate was removed for exosome purification and cell pellet discarded.
Exosome purification
Exosome-containing PMN supernate or BALF was centrifuged at 10,000G at 4°C for 45 minutes and pellet discarded to remove cellular debris, microvesicles and other larger particles. This was repeated a second time for BALF. Resulting supernates were then ultracentrifuged at 150,000G at 4°C for 2 hours and pellet resuspended in filter/sterilized TrisHCl 10mM pH 7.0 for EM experiments and PBS for all other experiments. All solvents were depleted of nano-sized particles by ultracentrifugation prior to pellet resuspension for accuracy of nanotracking analysis. Exosomes were either used fresh without freeze-thaw or, if intended for use more than two weeks after acquisition, were stored at −80°C and subjected to a single freeze-th aw, unless otherwise noted.
Exosome counting
Purity, size, and concentration of all preparations of exosomes was performed upon resuspension using nanotracking analysis on a Nanosight NS300 (Malvern), which counts and describes the size characteristics of the particle population. This was done with Nanosight NTA 3.1 software using a 35μL/min flow rate and read at 25°C on a 488nM laser/filter for increased accuracy of size estimation. All Nanosight readings were performed using PBS or TrisHCl which had been previously depleted of nanoparticle background by ultracentrifugation for increased accuracy.
Murine Intratracheal Administration of Exosomes
Anesthitized mice were administered exosomes or PBS instilled via intratracheal (i.t.) route in 50μL aliquots containing the designated quantity of exosomes set forth in manuscript and/or legend for a given experiment as determined by nanotracking analysis quantification of the individual exosome donation. For some experiments, exosomes were preincubated for 30 minutes with Human NE Inhibitor II (10μM) prior to administration. For experiments using human subject BALF exosomes, i.t. dosing was carried out serially every other day until sacrifice due to the lower disease-causing potency of this population of mixed-cell origin exosomes as described in manuscript. For other experiments, i.t. dosing was carried out once with mouse sacrifice at interval reported in legend, see Fig. 5, 6, S3.
Murine Pulmonary Function Testing
Pulmonary function was measured in mice (treated with PBS or exosomes as per above) as previously published (Nicola et al., 2009), using the flexiVent apparatus (SCIREQ, Montreal, Canada), equipped with Module 1(suitable for mice as small as 10 g and up to adult weights) and FlexiWare 7.6.3 software. Calibration of the flexiVent was done using the tracheal cannula to be used before each experiment. Mice were anesthetized with ketamine/xylazine, and tracheostomy was performed using either a 24G Angiocath for neonatal mice or an 18G blunt needle for adult mice. Measurements were then made with the flexiVent, including perturbations (predefined pressure of volume waveforms) such as forced oscillations, via the tracheostomy using room air in the closed-chest animal at a positive end expiratory pressure at machine default (3cmH2O). The delivered tidal volume was 6 ml/kg, with a respiratory rate of 150/min. Measurements made included total resistance (R; which encompasses Rn, G, and chest wall resistance; chest wall resistance in the mouse is essentially zero), compliance (C), elastance (E), input impedance (Z), airway resistance (Rn or Raw; Newtonian resistance, which is primarily the resistance of the central or upper airways), tissue damping (G), tissue elastance (H), quasistatic compliance (Cst), and hysteresivity (Eta; the ratio of G and H) (Rn, G, and H were calculated by fitting the constant phase model to input impedance). Rn was recorded for each mouse as the average of three separate measurements per mouse. The mice were euthanized at the conclusion of the procedure.
Murine Bronchoalveolar Lavage
A midline incision was performed from the upper abdomen to the mid-cervical region, exposing the lungs and trachea of mouse. An 18 gauge venous catheter was inserted into the cervical trachea and sealed around the cannula with gently tied suture. Bronchoalveolar lavage was then performed by gently instilling 1mL of PBS over a period of 2 minutes per mouse followed by slow retrieval of infused fluid into syringe. C57/Bl6 mice were time bred and litters were combined in order to familiarize dams to surrogacy. Each cage had half of the pups from the two litters combined. Both types of exosomes were administered intranasally to pups, 15 μl corresponding to postpartum days (p)3, p6 and p12. An additional group was administered PBS on these days. At p14, pups were harvested for histology after inflation fixing and pulmonary function tests (SCIREQ FV), as described above. After sacrifice, lungs were prepared as previously described (Wells et al., 2014). In brief, sacrificed mice were immediately dissected by a technician unaware of group assignments to expose thorax and cervical trachea. The hearts were explanted immediately and a technician unaware of group assignments carefully removed right ventricle from the left ventricle and septum by microdissection, then weighed immediately. In parallel, the first technician punctured the cervical trachea and inserted into it an 18g intravenous (IV) catheter with sutures looped around the trachea to provide a seal.1mL of 10% formalin was then instilled at 500μL/second, catheter was removed and suture was tied to hold instilled volume of fluid into lungs. Lungs and trachea were placed en bloc into 10% formalin solution and fixed overnight. They were then sliced with transverse sections and stained with H&E for analysis.
Electron microscopic imaging
97% pure Collagen type I was purchased as PureCol (Advance BioMatrix). 7.5μg of each protein was mixed with 20μL exosome samples suspended in 10mM Tris-HCl, pH 7.0 for images taken of coincubation. Approximately 8μl of exosome suspension was applied to discharge formvar-carbon coated grid. After 2 minutes an excess of liquid was wicked away with a wedge of filter paper. 8μl of stain (1% filtered uranyl acetate) was added onto sample following 15-second incubation. Any excess of stain was then wicked away on the wedge of filter paper and grid was air dried at room temperature. Dry grids were subsequently viewed on a Tecnai Spirit 120kv TEM electron microscope (FEI, Hillsboro, OR). Digital images were taken with an AMT CCD camera (Woburn, MA).
Fluorescent elastin degradation assay
The elastin substrate DQ™ elastin was purchased from Thermo Fisher Scientific, as part of an EnzChek Elastase Assay Kit (E-12056). Along with exosomes, substrate was brought to final concentration of 0.25μg/mL in a total volume of 200μL/well of provided reaction buffer, pH 8.0 in a black 96 well plate. NE activity was measured via fluorescence emission maxima at ~515 nm (BODIPY) using a Spectra Max Gemini fluorescent 96 well plate reader at 5 minute intervals. 1×109 exosomes were used per reaction. Data was read using SoftMax Pro software. Reactions were carried out at 37°C.
Fluorescent collagen degradation assay
A FITC-collagen labeled substrate was purchased from BioVision as part of Collagenase Assay Kit (Fluorometric) (K490). Along with purified human NE (CalBiochem) or exosomes, fluorescent substrate was diluted 1:25 per manufacturer’s instructions and 25μL substrate added in a total volume of 200yL/well of provided assay buffer in a black 96 well plate. Collagen degradation was measured via fluorescence emission maxima at ~520 nm (FITC) using a Spectra Max Gemini fluorescent 96 well plate reader at 5 minute intervals. Data was read using SoftMax Pro software.
Reactions were carried out at 37°C. Inhibition expe riments were performed as above with addition of Human Neutrophil Elastase Inhibitor II (MeOSuc-Ala-Ala-Pro-Ala-CMK, CalBiochem). For these experiments exosomes were coincubated for 30 minutes at 37°C with respective inhibitor prior to addition of substrate. 1×109 exosomes were used per reaction.
Neutrophil elastase assay
The NE substrate N-methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide was purchased from CalBiochem. Along with purified serum-derived human NE (CalBiochem) or exosomes, substrate was brought to final concentration of 0.33mg/mL in a total volume of 150μL/well of PBS pH 7.5 in a 96 well plate. NE activity was measured via absorbance at 410nm wavelength on a Biotek Eon 96 well plate reader at 5 minute intervals. Data was read using Gen 5 Reader 2.03 software. Reactions were carried out at 25°C. Inhibition experiments were performed as a bove with addition of Human Neutrophil Elastase Inhibitor (8μg) or α1-AT (rPeptide) (at a 2:1 molar ratio to active site titrated NE quantity per reaction). For these experiments, protease or exosomes were coincubated for 20 minutes at 25°C with respective inhibitor prior to addition of substrate. For NE loading experiments, quiescent exosomes were co-incubated with purified NE (1 μg) in 50μL PBS, filter/rinsed twice through 50kD filters to remove nonbound NE, then reaction performed with the rinsed exosomes. For NE removal experiments activated exosomes were pre-incubated with 25μM protamine sulfate (CalBiochem),10μM l-lysine (Sigma) or 10μM l-proline (Sigma) for 30 minutes at 25°C prior to filtration. Protamine caused a prominent increase in NE catalytic activity; consequently, curves for NE activity were adjusted to correct for this. 1×109 exosomes were used per reaction except where otherwise stated.
Polyacrylamide Gel
40μL solution containing 1.5 × 1010 exosomes was sonicated at 40Hz in a Branson 2510r-MT sonicator at 20°C for 30 minutes to break apart exosome membranes. 6μL of 10× NuPage reducing agent (Thermo Fisher) was added to sample along with 15μL of 4× NuPage LDS sample buffer. Samples were heated at 100°C for 15 minutes for denaturation. Samples were then added to wells of a 4%−12% Bis-Tris polyacrylamide gel (Thermo Fisher) and run in 1× MOPS running buffer for approximately 50 minutes at 180V. 10μL of Precision Plus Protein All Blue Prestained Protein Standard (Bio-Rad) was run in adjacent well for protein molecular weight comparison. Biosafe Coomasie (Bio-Rad) stain was used for protein visualization per manufacturer’s instructions.
Exosome Western Blots
Exosomes from PMNs of five separate donors were pooled. Exosomes to be compared were matched by number of exosomes per sample along with 2ng of glyceraldehyde 3-phosphate dehydrogenase as exogenous loading control and 1:100 dilution of P3840 Protease Inhibitor Cocktail (Sigma Cat#P3840–1ML). Samples run on SDS-PAGE gel as described above, with the exception of using a 4%−20% mini-Protean TGX gel (Bio Rad). Gel was then transferred to nitrocellulose membrane using Tris-Glycine transfer buffer. Nitrocellulose membranes were then blocked with 5% milk/TBS-T solution for 1 hour at room temperature and incubated with primary antibody overnight at 4C. Membranes were washed 3× with TBS-T ant then incubated 1:5000 dilution with HRP conjugated secondary antibody. Membranes washed 3X as above, and then developed using SuperSignal Chemiluminescent Substrate kit (Thermo Fisher). Antibodies used for detection are as follows: Anti-Lactoferrin polyclonal ab18511(Abcam), Anti-Myeloperoxidase PA1–26402 (Fisher scientific), anti-hELA2 (NE) mAB91671 (Fisher Scientific), and Anti-glyceraldehyde 3-phosphate dehydrogenase AC001 (ABclonal). Glyceraldehyde 3-phosphate dehydrogenase used as loading control was a kind donation from the laboratory of Sang Sang Park which was grown from Escherischia coli BL21 using pET30–2-GAPDH (Addgene catalog #83910) and purified using cobalt resin.
Lung Morphometry
H&E stained lung sections were photographed by a blinded investigator using Infinity 2 photomicrographer. For Lm counts, a 100ym grid was overlaid using the Infinity Analyze 2–1 program. 4 nonconsecutive sections per mouse were analyzed and 3 photomicrographs taken of each section overlaid with 100μm grid. A second investigator (also blinded to groups), counted the number of intersections of alveolar septae with the walls of three predetermined noncontiguous 100μm × 100μm squares of the grid. The number of intercepts for each square were then divided into the 400μm length of its walls to derive a mean linear intercept (Lm). For each mouse the Lm was averaged across each 400μm measurement (total of 36 measurements per lung) for analysis. Lm for control female A/J and C57/BL6 mice were consistent with previous reports(Soutiere et al., 2004). Radial alveolar count was performed as previously described (Lal et al., 2018; Sun et al., 2013). Briefly, a standardized measurement grid was laid over the photomicrographs as per above for Lm, and each alveolar saccule bisected by this line was counted by a blinded observer and normalized to linear length. The results were averaged over three slides from the left lung of each mouse for analysis.
Exosome Immunophenotype Analysis with Antibody-coated Magnetic Beads
Magnetic beads were conjugated to target antibody as described per manufucturer’s protocol in the Dynabeads Antibody Coupling Kit (Invitrogen). Anti-human CD63 coated beads were purchased as pre-conjugated magnetic beads (ThermoFisher Cat#10606D). Antibodies to be conjugated to beads were to the specific human neutrophil marker CD66b/CEACAM 8 (Sigma catalog number SAB4301144–100) and the human epithelial cell marker MUC4 (Abcam catalog number ab60720 RRID:AB_944301). Exosomes were coincubated with antibody conjugated magnetic beads in a shaker for 18 hours at 4°C. Magnetic beads with bound exosomes were held in Eppendorf tubes with a magnet while unbound exosome supernates were removed and captured beads were washed. Elution of captured exosomes from antibody-bead complexes was performed by adding 200μL of 50mM pH 2.5 glycine solution to tube containing beads for 10 minutes, followed by addition of 70 μL 1M pH 7.5 Tris buffer solution to neutralize. Eluted exosomes, now purified for expression of respective markers, were used for further experiments. Both depletion and elution were performed a minimum of two times with fresh beads before each in vivo experiment 97% capture efficiency.
Ion Electrospray Liquid Chromatography Mass Spectroscopy/Mass Spectroscopy
The Coomassie stained bands were excised and destained overnight using a wash in 50% 100mM ammonium bicarbonate/50% acetonitrile. Disulfide bonds were reduced using dithiothreitol (25mM) at 50°C for 30 min foll owed by alkylation of free thiols groups with iodoacetamide (55mM) for 30 min in the dark at room temperature. After removal of excess alkylating agent, the gel pieces were evaporated to dryness prior to reswelling in 100mM ammonium bicarbonate buffer and overnight digestion using mass spectrometry grade trypsin (12.5ng/ml). Tryptic peptides were extracted using solution of 1% formic acid in water and acetonitrile (50/50) and then evaporated to dryness in a Speedvac. Samples were resuspended in 50μL of ddH2O with 0.1% formic acid for mass spectrometry evaluation.
An aliquot (5μl) of each digest was loaded onto a Nano cHiPLC 200μm × 0.5mm ChromXP C18-CL 3μm 120Å reverse-phase trap cartridge (Eksigent, Dublin, CA) at 2μL/min using an Eksigent 415 LC system autosampler. After washing the cartridge for 10 min with 0.1% formic acid in double-distilled water (ddH20), the bound peptides were flushed onto a Nano cHiPLC column (200μm ID × 1cm ChromXP C18-CL 3μm 120Å, Eksigent, Dublin, CA) with a 35 min linear (5–50%) acetonitrile gradient in 0.1% formic acid at 1000nl/min using an Eksigent Nano1D+LC (Dublin, CA). The column was washed with 90% acetonitrile-0.1% formic acid for 5 min and then re-equilibrated with 5% acetonitrile-0.1% formic acid for 10 min. The SCIEX 5600 Triple-Tof mass spectrometer (Sciex, Toronto, Canada) was used to analyze the protein digest. The IonSpray voltage was 2300V and the declustering potential was 80V. Ion spray and curtain gases were set at 10psi and 25psi, respectively. The interface heater temperature was 120°C. Eluted peptides were subject ed to a time-of-flight survey scan from m/z400–1250 to determine the top twenty most intense ions for MS/MS analysis. Product ion time-of-flight scans at 50 ms were carried out to obtain the tandem mass spectra of the selected parent ions over the range from m/z400–1500. Spectra are centroided and de-isotoped by Analyst software, version 1.6 TF (Sciex). A β-galactosidase trypsin digest was used to establish and confirm the mass accuracy of the mass spectrometer.
The tandem mass spectrometry data were processed to provide protein identifications using an in-house Protein Pilot 4.5 search engine (Sciex) using the UniProt Homo sapiens protein database and using a trypsin digestion parameter. Proteins of significance were accepted on the criteria of having at least two peptides detected with a confidence score of 95% or greater using the Paradigm method imbedded in the Protein Pilot software. AcPGP quantity in BALF was measured using a MDS Sciex (Applied Biosystems, Foster City, CA) API-4000 spectrometer equipped with a Shimadzu HPLC (Columbia, MD). HPLC was conducted using a 2.0 × 150 mm Jupiter 4u Proteo column (Phenomenex, Torrance, CA) with A: 0.1% HCOOH and B: MeCN + 0.1% HCOOH: 0 min-0.5 min 5% buffer B/95% buffer A, then increased over 0.5–2.5 min to 100% buffer B/0% buffer A. Background was removed by flushing with 100% isopropanol / 0.1% formic acid. Positive electrospray mass transitions were at 312–112,312–140 M/z for AcPGP. Area under the curve was measured, and AcPGP peptide concentration calculated using a relative standard curve method as previously described (Weathington et al., 2006).
Next-Generation mRNA Sequencing
Each 1.9 cm2 well containing approximately 100,000 human primary airway epithlelial cells was administered either 1μL of PBS, or 1×105 activated or quiescent exosomes (in 1 μL PBS) to the apical surface. After 24 hours, cells were scraped and lysed and total mRNA was harvested with RNEasy kit (Qiagen) per manufacturer’s instructions. mRNA-sequencing was performed on the Illumina NextSeq500 as described by the manufacturer (Illumina Inc., San Diego, CA). Briefly, RNA quality was assessed using the Agilent 2100 Bioanalyzer. RNA with a RNA Integrity Number (RIN) of 7.0 or above was used for sequencing library preparation. RNA passing quality control was converted to a sequencing ready library using the Agilent SureSelect Strand Specific mRNA library kit as per the manufacturer’s instructions (Agilent, Santa Clara, CA). The cDNA libraries were quantitated using qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for Illumina library quantitation (Kapa Biosystems, Woburn, MA) prior to cluster generation. Cluster generation was performed according to the manufacturer’s recommendations for onboard clustering (Illumina, San Diego, CA). We generated between 34–37 million single end 75bp sequencing reads per sample for gene level transcriptional abundance.
mRNA Data Assessment
STAR (version 2.5.3a) was used (parameters used: --outReadsUnmapped Fastx;-- runThread 12; --outSAMtype BAM SortedByCoordinate; --outSAMattributes All;-- readFilesCommands gunzip -c) to align the raw RNA-Seq fastq reads to the human reference genome (GRCh38.p7, Release 25) from Gencode (Dobin et al., 2013). Following alignment, HTSeq-count (version 0.9.1) was used (parameters used: -m union; -r pos; -t exon; -i gene_id; -a 10; -s no; -f bam) to count the number of reads mapping to each gene (Anders et al., 2015). Normalization and differential expression was then applied to the count files using expression was then applied to the count files using the package DESeq2 (version 1.20.0) in R (version 3.4.3) following their vignette (Love et al., 2014). All datasets were then uploaded into the Ingenuity® Pathway Analysis program (version 01–13, QIAGEN) and analyzed both by individual mRNA expression pattern and by average mRNA expression pattern by condition using the Expression Analysis function of the software (Kramer et al., 2014). A heat map with this expression analysis was generated for visual demonstration of the relative upregulation and/or downregulation of cellular processes using the proprietary cellular process denotation/reference database imbedded in the software (Ingenuity Pathway Analysis Knowledge Base). Heat maps of the same data (showing both mean group expression and individual sample expression analyses) were then downloaded with “hierarchical annotation” displayed, in unprocessed form. For clarity, heat maps were then overlaid with thin dashed lines and labelled to delineate boundaries between groups.
FACS Analysis
Streptavidin-conjugated magnetic beads (Exo-Flow exosome purification kit, EXOFLOW700A-1, System Biosciences, Palo Alto, CA) were coupled with biotinylated anti-CD66b antibody (305120, Biolegend, San Diego, CA) according to the manufacturer’s protocol. Subsequently, 2.5 × 107 exosomes (from primary activated or quiescent neutrophil supernatant or subject BALF) were incubated with the conjugated beads, and stained with anti-ELA2 AF647 (IC91671R, R&D Systems, Minneapolis, MN) at 4°C for 2 hours. Samples were washed and acquired on a CytoFLEX Flow Cytometer (Beckman Coulter Life Sciences, Indianapolis,IN) and analysed using FlowJo v10 (FlowJo LLC, Ashland, OR). Results were then normalized to the total protein concentration (measured by Bradford protein assay) in the BALF as a multiple of the lowest reading per experiment.
Type I collagen binding and inhibition.
96-well ELISA assay plates were coated with 5μg type I collagen per well. Exosomes of various amounts were incubated for 30 minutes in the presence of dose dependent MAC peptide, or RGD peptide, or control PBS. Exosomes then incubated in collagen wells for 1 hour at for 1 hour, and washed 3 times with 5%BSA/PBS solution. 4% neutral buffered formalin solution was used to fix, and then the wells were blocked with 5%BSA/PBS solution for 1 hour. 1:1000 dilution of lactoferrin polyclonal antibody added to each well for 1 hour incubation. After 3 washes with 5%BSA/PBS,1:3000 dilution of HRP conjugated secondary antibody was added for 1 hour, followed by additional 3 wash step.1-step Ultra TMB-ELISA substrate was added, and the reaction stopped using 2M sulfuric acid. Absorbance at 450nm was measured to determine amount of exosomes bound to collagen. All incubations performed at room temperature. Absorbances were normalized to collagen only control. Inhibition of binding to type I collagen reported as % inhibition as compared to absorbance of exosome binding without any peptide inhibitor. All experiments performed in triplicate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics were performed using the GraphPad Prism 5.0 or 7.0 software. Two-tailed nonparametric (Mann—Whitney) testing was used for simple significance testing between two groups in which n<4 or with presumed non-Gaussian distribution. Analysis of variance (ANOVA) was performed for multiple comparisons with Kruskal-Wallis post-test for normally distributed data (all groups n=4 or greater). To depict data distribution specifically for Lms, central tendency for all Lm values (except Figure 6E, see below) were presented as median with box limits representing upper and lower interquartile ranges and whiskers representing minimum and maximum values. For figure 6E, in order to save space and synthesize multiple experiments with multiple control groups, data points representing the mean Lm increase above the mean of the respective control of the group (n=4) of animals treated with exosomes from BALF of a single individual were presented by dot plot as a single data point. Data is depicted as mean +/− SEM where denoted as such in the figure legend. To distinguish aggregated RV:(LV+S) measurements from Lm values while still demonstrating data distribution, values are shown as individual data points with line indicating the mean value. Statistical significance was inferred for p values <0.05; p values are presented as: * <0.05, **<0.01, ***<0.001.
DATA AND SOFTWARE AVAILABILITY
Proteomic data
The mass spectroscopy-derived proteomic data for activated and quiescent neutrophil exosomes have been deposited at ExoCarta (www.exocarta.org) under accession number: Vesiclepedia_1398.
Next Generation RNA sequencing data
The exosome-treated human airway epithelial cell mRNA sequencing data have been deposited at the GEO (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GEO: GSE121307.
Supplementary Material
Figure S1. Related to Figures 1 and 6 (A)Representative nanotracking analysis for quiescent (left) and activated (right) PMN derived exosomes. (B) Mean exosome size from four separate experiments comparing quiescent and activated PMN derived exosomes. (C) Exosome count per PMN comparing quiescent and activated PMN derived exosomes of 5 (quiescent) and 7 (activated) donors. (D) Representative Western blot of exosomes (2.5×1010/lane) stained for PMN-associated proteins. Glyceraldehyde 3-phosphate dehydrogenase used as loading control. (E) Coomassie stained polyacrylamide gel of exosomes (1.5×1011/lane) (F) CD66b+ exosomes were pulled-down as described in the method section. The gating strategy for the analysis included a singlets gate (FSC-A vs. FSC-H) and an exosome gate (FSC-A vs SSC-A). (G) Number of exosomes after CD66b pulldown were compared between groups. Data for B. and C. represented as follows: center line, median; box limits, upper and lower quartiles; whiskers, minimum and maximum values. Data for F shown as mean +/− SEM.
Figure S2. Related to Figures 2 and 4 (A). The NE substrate N-methoxysuccinyl-Ala-Ala-Pro-Val pNA at final concentration of 0.33mg/mL was incubated with 20ng recombinant NE previously treated for 30 minutes at 37°C with varying molar ratios of α1-AT. Reactions were carried out at 20°C in 150 μL total volume/well in a 96 well plate. The reaction rate was determined at 10 minutes and plotted against α1-AT ratio. The line indicates the enzymatic rate of NE substrate product generation of the recombinant enzyme. The arrow indicates the enzymatic rate of 1×109 exosomes (corresponding to the uninhibited activity of 20ng NE) incubated at a 2:1 ratio with α1-AT, plotted on the recombinant NE curve for comparison. (B) Activated PMN exosomes (1×109) were measured at 37°C against MeOSuccAAPV-pNA for NE activity after coincubation with 10μM l-lysine, l-proline, +/−15μg of α1-AT and pNA generation at 30 minutes compared to exsosome only positive control. (C) Elastin fibrils alone and with quiescent exosomes were imaged by electron microscopy with negative staining and representative images shown.
Figure S3. Related to Figures 3, 5. (A) Male A/J mice (n= 5 mice per group) and female C57 BL/6 mice (n= 5 mice per group) were administered 1.67×108 exosomes from activated neutrophils or PBS in 50μL via i.t. route. Animals were sacrificed after 7 days. Alveolar size was measured as mean linear intercept (Lm). (B)1.67 × 108 activated exosomes were administered to female A/J mice as in (A) and sacrificed 7,14, or 21 days later, Lm measured as in (A), n ≥ 4 per group. Control Lms for the two strains differed as previously reported (Soutiere et al., 2004) (C) Female A/J mice (n ≥ 6 per group) were treated as in (A) and bronchoalveolar lavage (BAL) was performed. BAL was then analyzed by mass spectroscopy for quantity of the collagen breakdown product AcPGP and results displayed. ** p<0.01, *** p<0.001 Data shown as mean % control pNA release +/− SEM.
Figure S4. Related to Figures 5 and 6. (A) Female A/J mice were treated i.t with 1.67×10 8 exosomes from activated PMNs in 50μL PBS and sacrificed after 1–7 days. PBS and quiescent exosome treated controls were sacrificed at day 7. Quiescent control shown was treated with 1×109 exosomes corresponding to 60× the minimum dose shown to cause disease as shown in Figure 5F. Animals treated i.t with LPS (100μg in 50μL PBS) and sacrificed after 1 day also shown as inflammatory control. Lungs were fixed and stained by H&E, representative images displayed. (B) Mice were treated with activated exosomes, PBS, or LPS as in (A) and bronchoalveolar lavage performed after 1,4 or 7 days, cells counted and stained for differential (n=4 for LPS and 3 for all other groups). Total cell count (B), PMN percentage (C), and absolute PMN count (D) shown.
Figure S5, Related to Figures 5, 6. (A) Primary human HBE cells were treated with exosomes (n=3 wells for activated and quiescent exosomes) or PBS control (n=2) and lysed after 24 hours. Total mRNA was extracted and expression analysis performed for each well. (B) Expression analysis in A. analyzed with all wells from each condition averaged together.
Figure S6. Related to Figures 1 and 6. (A) Representative nanotrackinganalysis of exosomes from BALF of healthy never smokers (B) Representative nanotracking analysis of exosomes from COPD patients. (C) Mean exosome size as determined by nanotracking analysis between exosome populations derived from BALF of healthy never smokers and COPD patients. No significant difference is present. (D) Exosome count per mL of BALF between exosome populations derived from healthy never smokers (n=10) and COPD patients (n=10). No significant difference is present. (E) Number of exosomes after CD66b pulldown were compared between exosomes derived from BALF of N/S control or COPD individuals (n=3 per group). Data for E. depicted as dot plot overlaid on mean +/− SEM.
HIGHLIGHTS.
Exosomes from activated, not quiescent, PMNs harbor surface α1AT insensitive NE
Activated PMN exosomes bind ECM via MAC-1 and degrade ECM via NE
CD66b+/NE+ PMN exosomes cause emphysema and RVH when administered to mice
CD66b+/NE+ PMN exosomes reside in COPD patients and transfer a COPD phenotype to mice
Acknowledgements
We thank Landon Wilson for proteomic analysis, Ralph D. Sanderson for helpful discussions, and Pamela Parker for manuscript preparation.
This study was supported by: R35HL135710, R01HL077783, R01HL114439, R01HL110950 to JEB, R01HL126596 to JEB and AG, R01 HL102371 to AG, R01HL126603 to RT, K08HL123940 to JMW, T32HL105346–07 to DWR and T32 HL105346–05 to KRG; AHA-17SDG32720009 to CL; Veteran’s Affairs: I01BX001756 to AG; Nancy Dunlap Chair to JEB.
The Targeted Metabolomics and Proteomics Laboratory supported in part from the UAB O’Brien Acute Kidney Injury Center (P30 DK079337), the UAB Lung Health Center and the UAB Center for Free Radical Biology and the UAB Heflin Center for Genomic Science supported in part from the Comprehensive Cancer Center Core grant P30 CA031148.
Research reported in this publication was supported by the NHLBI, NIH, and the Family Smoking Prevention and Tobacco Control Act. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Food and Drug Administration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: The authors declare no competing interests.
References:
- Altiok O, Yasumatsu R, Bingol-Karakoc G, Riese RJ, Stahlman MT, Dwyer W, Pierce RA, Bromme D, Weber E, and Cataltepe S (2006). Imbalance between cysteine proteases and inhibitors in a baboon model of bronchopulmonary dysplasia. Am J Respir Crit Care Med 173, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betsuyaku T, Nishimura M, Takeyabu K, Tanino M, Miyamoto K, and Kawakami Y (2000). Decline in FEV(1) in community-based older volunteers with higher levels of neutrophil elastase in bronchoalveolar lavage fluid. Respiration 67, 261–267. [DOI] [PubMed] [Google Scholar]
- Butin-Israeli V, Houser MC, Feng M, Thorp EB, Nusrat A, Parkos CA, and Sumagin R (2016). Deposition of microparticles by neutrophils onto inflamed epithelium: a new mechanism to disrupt epithelial intercellular adhesions and promote transepithelial migration. FASEB J 30, 4007–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, and Belting M (2013). Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A 110, 17380–17385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza SE, Ginsberg MH, and Plow EF (1991). Arginyl-glycyl-aspartic acid (RGD): a cell adhesion motif. Trends Biochem Sci 16, 246–250. [DOI] [PubMed] [Google Scholar]
- Demedts IK, Demoor T, Bracke KR, Joos GF, and Brusselle GG (2006). Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 7, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggar A, and Weathington N (2016). Bioactive extracellular matrix fragments in lung health and disease. J Clin Invest 126, 3176–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee KJ, Werb Z, and Kheradmand F (2007). Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev 87, 69–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross P, Pfitzer EA, Tolker E, Babyak MA, and Kaschak M (1965). Experimental Emphysema: Its Production with Papain in Normal and Silicotic Rats. Arch Environ Health 11, 50–58. [DOI] [PubMed] [Google Scholar]
- Hautamaki RD, Kobayashi DK, Senior RM, and Shapiro SD (1997). Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277, 2002–2004. [DOI] [PubMed] [Google Scholar]
- Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, Gil CD, Nerviani A, Dell’Accio F, Pitzalis C, et al. (2015). Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci Transl Med 7, 315ra190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom U, Hallgren O, Oberg L, DeMicco A, Vaarala O, Westergren-Thorsson G, and Zhou X (2018). Bronchial extracellular matrix from COPD patients induces altered gene expression in repopulated primary human bronchial epithelial cells. Sci Rep 8, 3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoff A, Sloan B, Weinbaum G, Damiano V, Sandhaus RA, Elias J, and Kimbel P (1977). Experimental emphysema induced with purified human neutrophil elastase: tissue localization of the instilled protease. Am Rev Respir Dis 115, 461–478. [DOI] [PubMed] [Google Scholar]
- Kafienah W, Buttle DJ, Burnett D, and Hollander AP (1998). Cleavage of native type I collagen by human neutrophil elastase. Biochem J 330 (Pt 2), 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knust J, Ochs M, Gundersen HJ, and Nyengaard JR (2009). Stereological estimates of alveolar number and size and capillary length and surface area in mice lungs. Anat Rec (Hoboken) 292, 113–122. [DOI] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal CV, Olave N, Travers C, Rezonzew G, Dolma K, Simpson A, Halloran B, Aghai Z, Das P, Sharma N, et al. (2018). Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurell CB, and Eriksson S (1963). The Electrophoretic α;1-Globulin Pattern of Serum in α;1-Antitrypsin Deficiency. Scandinavian Journal of Clinical and Laboratory Investigation 15, 132–140. [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas SLN, Breakefield XO, and Weaver AM (2017). Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol 27, 172–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouded M, Egea EE, Brown MJ, Hanlon SM, Houghton AM, Tsai LW, Ingenito EP, and Shapiro SD (2009). Epithelial cell apoptosis causes acute lung injury masquerading as emphysema. Am J Respir Cell Mol Biol 41, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Powers JC, Ashe BM, and Zimmerman M(1979). Mapping the extended substrate binding site of cathepsin G and human leukocyte elastase. Studies with peptide substrates related to the alpha 1-protease inhibitor reactive site. J Biol Chem 254, 4027–4032. [PubMed] [Google Scholar]
- Navia MA, McKeever BM, Springer JP, Lin TY, Williams HR, Fluder EM, Dorn CP, and Hoogsteen K(1989). Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84-A resolution. Proc Natl Acad Sci U S A 86, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola T, Hagood JS, James Ml., Macewen MW, Williams TA, Hewitt MM, Schwiebert L, Bulger A, Oparil S, Chen YF, et al. (2009). Loss of Thy-1 inhibits alveolar development in the newborn mouse lung. Am J Physiol Lung Cell Mol Physiol 296, L738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly P, Jackson PL, Noerager B, Parker S, Dransfield M, Gaggar A, and Blalock JE (2009). N-alpha-PGP and PGP, potential biomarkers and therapeutic targets for COPD. Respir Res 10, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen CA (2008). Leukocyte cell surface proteinases: regulation of expression, functions, and mechanisms of surface localization. Int J Biochem Cell Biol 40, 1246–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen CA, Campbell MA, Sannes PL, Boukedes SS, and Campbell EJ (1995). Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 131, 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolnikova NP, Podolnikov AV, Haas TA, Lishko VK, and Ugarova TP (2015). Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry 54,1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raherison C, and Girodet PO (2009). Epidemiology of cOpD. Eur Respir Rev 18, 213–221. [DOI] [PubMed] [Google Scholar]
- Richmond BW, Brucker RM, Han W, Du RH, Zhang Y, Cheng DS, Gleaves L, Abdolrasulnia R, Polosukhina D, Clark PE, et al. (2016). Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat Commun 7, 11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW, Gaggar A, and Solomon GM (2016). Neutrophil Fates in Bronchiectasis and Alpha-1 Antitrypsin Deficiency. Ann Am Thorac Soc 13 Suppl 2, S123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior RM, Tegner H, Kuhn C, Ohlsson K, Starcher BC, and Pierce JA (1977). The induction of pulmonary emphysema with human leukocyte elastase. Am Rev Respir Dis 116, 469–475. [DOI] [PubMed] [Google Scholar]
- Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, and Belaaouaj A (2003). Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol 163, 2329–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda M, and Khokha R (2013). Proteolytic factors in exosomes. Proteomics 13, 1624–1636. [DOI] [PubMed] [Google Scholar]
- Sng JJ, Prazakova S, Thomas PS, and Herbert C (2017). MMP-8, MMP-9 and Neutrophil Elastase in Peripheral Blood and Exhaled Breath Condensate in COPD. COPD 14, 238–244. [DOI] [PubMed] [Google Scholar]
- Soutiere SE, Tankersley CG, and Mitzner W (2004). Differences in alveolar size in inbred mouse strains. Respir Physiol Neurobiol 140, 283–291. [DOI] [PubMed] [Google Scholar]
- Stockley RA (2014). Alpha1-antitrypsin review. Clin Chest Med 35, 39–50. [DOI] [PubMed] [Google Scholar]
- Sun H, Choo-Wing R, Fan J, Leng L, Syed MA, Hare AA, Jorgensen WL, Bucala R, and Bhandari V (2013). Small molecular modulation of macrophage migration inhibitory factor in the hyperoxia-induced mouse model of bronchopulmonary dysplasia. Respir Res 14, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szul T, Bratcher PE, Fraser KB, Kong M, Tirouvanziam R, Ingersoll S, Sztul E, Rangarajan S, Blalock JE, Xu X, et al. (2016). Toll-Like Receptor 4 Engagement Mediates Prolyl Endopeptidase Release from Airway Epithelia via Exosomes. Am J Respir Cell Mol Biol 54, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Strijp JA, Russell DG, Tuomanen E, Brown EJ, and Wright SD (1993). Ligand specificity of purified complement receptor type three (CD11b/CD18, alpha m beta 2, Mac-1). Indirect effects of an Arg-Gly-Asp (RGD) sequence. J Immunol 151, 3324–3336. [PubMed] [Google Scholar]
- Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Chen R, Decramer M, Fabbri LM, et al. (2017). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Eur Respir J 49. [DOI] [PubMed] [Google Scholar]
- Walsh MC, Yao Q, Gettner P, Hale E, Collins M, Hensman A, Everette R, Peters N, Miller N, Muran G, et al. (2004). Impact of a physiologic definition on bronchopulmonary dysplasia rates. Pediatrics 114, 1305–1311. [DOI] [PubMed] [Google Scholar]
- Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, and Blalock JE (2006). A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12, 317–323. [DOI] [PubMed] [Google Scholar]
- Wells JM, O’Reilly PJ, Szul T, Sullivan DI, Handley G, Garrett C, McNicholas CM, Roda MA, Miller BE, Tal-Singer R, et al. (2014). An aberrant leukotriene A4 hydrolase-proline-glycine-proline pathway in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 190, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumatsu R, Altiok O, Benarafa C, Yasumatsu C, Bingol-Karakoc G, Remold-O’Donnell E, and Cataltepe S (2006). SERPINB1 upregulation is associated with in vivo complex formation with neutrophil elastase and cathepsin G in a baboon model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 291, L619–627. [DOI] [PubMed] [Google Scholar]
- Zhu X, Badawi M, Pomeroy S, Sutaria DS, Xie Z, Baek A, Jiang J, Elgamal OA, Mo X, Perle K, et al. (2017). Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles 6,1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Related to Figures 1 and 6 (A)Representative nanotracking analysis for quiescent (left) and activated (right) PMN derived exosomes. (B) Mean exosome size from four separate experiments comparing quiescent and activated PMN derived exosomes. (C) Exosome count per PMN comparing quiescent and activated PMN derived exosomes of 5 (quiescent) and 7 (activated) donors. (D) Representative Western blot of exosomes (2.5×1010/lane) stained for PMN-associated proteins. Glyceraldehyde 3-phosphate dehydrogenase used as loading control. (E) Coomassie stained polyacrylamide gel of exosomes (1.5×1011/lane) (F) CD66b+ exosomes were pulled-down as described in the method section. The gating strategy for the analysis included a singlets gate (FSC-A vs. FSC-H) and an exosome gate (FSC-A vs SSC-A). (G) Number of exosomes after CD66b pulldown were compared between groups. Data for B. and C. represented as follows: center line, median; box limits, upper and lower quartiles; whiskers, minimum and maximum values. Data for F shown as mean +/− SEM.
Figure S2. Related to Figures 2 and 4 (A). The NE substrate N-methoxysuccinyl-Ala-Ala-Pro-Val pNA at final concentration of 0.33mg/mL was incubated with 20ng recombinant NE previously treated for 30 minutes at 37°C with varying molar ratios of α1-AT. Reactions were carried out at 20°C in 150 μL total volume/well in a 96 well plate. The reaction rate was determined at 10 minutes and plotted against α1-AT ratio. The line indicates the enzymatic rate of NE substrate product generation of the recombinant enzyme. The arrow indicates the enzymatic rate of 1×109 exosomes (corresponding to the uninhibited activity of 20ng NE) incubated at a 2:1 ratio with α1-AT, plotted on the recombinant NE curve for comparison. (B) Activated PMN exosomes (1×109) were measured at 37°C against MeOSuccAAPV-pNA for NE activity after coincubation with 10μM l-lysine, l-proline, +/−15μg of α1-AT and pNA generation at 30 minutes compared to exsosome only positive control. (C) Elastin fibrils alone and with quiescent exosomes were imaged by electron microscopy with negative staining and representative images shown.
Figure S3. Related to Figures 3, 5. (A) Male A/J mice (n= 5 mice per group) and female C57 BL/6 mice (n= 5 mice per group) were administered 1.67×108 exosomes from activated neutrophils or PBS in 50μL via i.t. route. Animals were sacrificed after 7 days. Alveolar size was measured as mean linear intercept (Lm). (B)1.67 × 108 activated exosomes were administered to female A/J mice as in (A) and sacrificed 7,14, or 21 days later, Lm measured as in (A), n ≥ 4 per group. Control Lms for the two strains differed as previously reported (Soutiere et al., 2004) (C) Female A/J mice (n ≥ 6 per group) were treated as in (A) and bronchoalveolar lavage (BAL) was performed. BAL was then analyzed by mass spectroscopy for quantity of the collagen breakdown product AcPGP and results displayed. ** p<0.01, *** p<0.001 Data shown as mean % control pNA release +/− SEM.
Figure S4. Related to Figures 5 and 6. (A) Female A/J mice were treated i.t with 1.67×10 8 exosomes from activated PMNs in 50μL PBS and sacrificed after 1–7 days. PBS and quiescent exosome treated controls were sacrificed at day 7. Quiescent control shown was treated with 1×109 exosomes corresponding to 60× the minimum dose shown to cause disease as shown in Figure 5F. Animals treated i.t with LPS (100μg in 50μL PBS) and sacrificed after 1 day also shown as inflammatory control. Lungs were fixed and stained by H&E, representative images displayed. (B) Mice were treated with activated exosomes, PBS, or LPS as in (A) and bronchoalveolar lavage performed after 1,4 or 7 days, cells counted and stained for differential (n=4 for LPS and 3 for all other groups). Total cell count (B), PMN percentage (C), and absolute PMN count (D) shown.
Figure S5, Related to Figures 5, 6. (A) Primary human HBE cells were treated with exosomes (n=3 wells for activated and quiescent exosomes) or PBS control (n=2) and lysed after 24 hours. Total mRNA was extracted and expression analysis performed for each well. (B) Expression analysis in A. analyzed with all wells from each condition averaged together.
Figure S6. Related to Figures 1 and 6. (A) Representative nanotrackinganalysis of exosomes from BALF of healthy never smokers (B) Representative nanotracking analysis of exosomes from COPD patients. (C) Mean exosome size as determined by nanotracking analysis between exosome populations derived from BALF of healthy never smokers and COPD patients. No significant difference is present. (D) Exosome count per mL of BALF between exosome populations derived from healthy never smokers (n=10) and COPD patients (n=10). No significant difference is present. (E) Number of exosomes after CD66b pulldown were compared between exosomes derived from BALF of N/S control or COPD individuals (n=3 per group). Data for E. depicted as dot plot overlaid on mean +/− SEM.
Data Availability Statement
Proteomic data
The mass spectroscopy-derived proteomic data for activated and quiescent neutrophil exosomes have been deposited at ExoCarta (www.exocarta.org) under accession number: Vesiclepedia_1398.
Next Generation RNA sequencing data
The exosome-treated human airway epithelial cell mRNA sequencing data have been deposited at the GEO (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GEO: GSE121307.