

Summary
Early embryogenesis is accompanied by reductive cell divisions requiring that subcellular structures adapt to a range of cell sizes. The interphase nucleus and mitotic spindle scale with cell size through both physical and biochemical mechanisms, but control systems that coordinately scale intracellular structures are unknown. We show that the nuclear transport receptor importin α is modified by palmitoylation, which targets it to the plasma membrane and modulates its binding to nuclear localization signal (NLS)-containing proteins that regulate nuclear and spindle size in Xenopus egg extracts. Reconstitution of importin α targeting to the outer boundary of extract droplets mimicking cell-like compartments recapitulated scaling relationships observed during embryogenesis, which were altered by inhibitors that shift levels of importin α palmitoylation. Modulation of importin α palmitoylation in human cells similarly affected nuclear and spindle size. These experiments identify importin α as a conserved surface area-to-volume sensor that scales intracellular structures to cell size.
Graphical Abstract
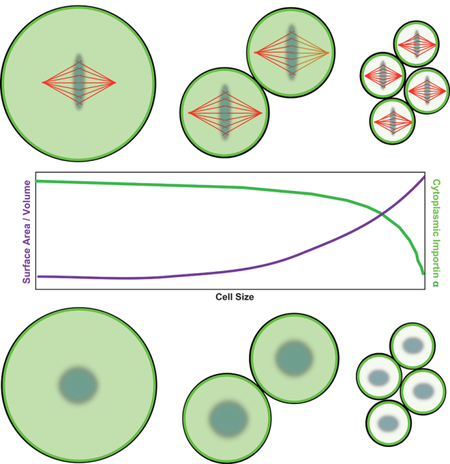
INTRODUCTION
Cell size varies widely among different organisms and cell types, and especially during early embryonic development of many animal species, when rapid divisions in the absence of growth decrease cell volume dramatically. Correct intracellular scaling is crucial for cell function, architecture, and division, but whether and how organelles and subcellular structures are coordinately scaled is poorly understood. One unifying mechanism could be the physical effect of cell volume, which shows a strong correlation with both spindle and nuclear size (Crowder et al., 2015; Vuković et al., 2016). Furthermore, microfluidic encapsulation of cytoplasmic extracts prepared from Xenopus eggs revealed volume-dependent scaling of spindles and nuclei (Good et al., 2013; Hara and Merten, 2015; Hazel et al., 2013). However, size relationships in cell-like compartments did not fully recapitulate those observed in vivo, and experiments with embryo extracts showed that in addition to changes in volume, changes in cytoplasm composition during development also decrease spindle and nuclear size (Levy and Heald, 2010; Wilbur and Heald, 2013). A common biochemical mechanism appears to involve importin α, a highly conserved and abundant nuclear transport factor that binds nuclear localization sequence (NLS)-containing proteins (Miyamoto et al., 2016). Cytoplasmic levels of importin α decrease during early Xenopus development, which directly affects import of cargos including the nuclear lamins, structural proteins required for nuclear growth (Levy and Heald, 2010; Vuković et al., 2016). Cytoplasmic importin α also acts to inhibit NLS-containing spindle assembly factors (Forbes et al., 2015). One such factor is the microtubule depolymerizing kinesin kif2a, which is liberated from importin α in smaller cells of the embryo where it acts to decrease spindle size (Wilbur and Heald, 2013). Intriguingly, concomitant with the decrease in cytoplasmic importin α levels, an increase in its plasma membrane staining was observed (Wilbur and Heald, 2013).
We therefore set out to test the hypothesis that importin α partitioning to the plasma membrane acts as a cell surface area-to-volume sensor that coordinately scales intracellular structures to cell size. Our experiments reveal a previously unidentified post-translational lipid modification of importin α that mediates its membrane association and controls spindle and nuclear size in Xenopus embryos and human cells, and in artificial cell-like compartments. We propose that by modulating the activity of a wide range of NLS-containing cargos, importin α acts as a global sensor that links cell size to fundamental aspects of cellular physiology and organization.
RESULTS
Palmitoylation Drives Importin α Plasma Membrane Association
Membrane association of importin α was previously implicated in nuclear envelope assembly and was shown to be regulated by phosphorylation (Hachet et al., 2004). To identify the cellular membranes that importin α binds and its targeting mechanism, we fractionated Xenopus egg extract on a sucrose gradient and probed for importin α by Western blot. Importin α was found in the cytoplasm and also co-fractionated with membranes containing the plasma membrane-associated protein 1-catenin (Figure 1A). When added to egg extract, exogenous GFP-tagged importin α was similarly enriched in the plasma membrane fraction, but not the fractions reported to contain ER and Golgi membranes (Figure S1A) (Hill et al., 2005). As described previously (Wilbur and Heald, 2013), the fraction of membrane-associated importin α increased during development (Figure 1B). Lipid modifications of plasma membrane proteins such as Ras promote their membrane insertion (Eisenberg et al., 2013), and sequence analysis identified cysteines at positions 230 and 454 of importin α as potential sites for lipid modification by palmitoylation. A biochemical assay for cysteine palmitoylation showed that recombinant importin α, in addition to Ras, is subject to this modification in egg extract (Figure S1B). However, palmitoylation can also occur on serine residues, and mass spectrometry analysis of recombinant importin α incubated in egg extract revealed palmitoylation of S154 and S490 (data not shown). An importin α mutant in which the residues C230, C454, S154 and S490 were all changed to alanine, termed importin α-NP, was no longer palmitoylated (Figure 1C).
Figure 1. Palmitoylation drives importin α plasma membrane association.

(A) Immunoblot of Xenopus eggs fractionated over a sucrose gradient and probed with importin α and beta-catenin antibodies. Importin α is found in the cytoplasm and also cofractionates with beta-catenin as a marker for plasma membrane mostly in the heavy membrane fraction.
(B) Immunoblot of cytoplasm and membrane fractions isolated from stage 3 and stage 8 embryos and probed with importin α and beta-catenin antibodies. A greater fraction of membrane-associated importin α is observed at stage 8.
(C) Blot of immunoprecipitated recombinant wild-type and mutant importin α proteins (2S: S154A, S490A; NP: S154A, S490A C230A, C454A) retrieved from Xenopus egg extract and probed with importin α antibodies or streptavidin to quantify incorporation of biotin-labeled palmitate (Martin, 2013).
(D) Blot of immunoprecipitated recombinant importin α retrieved from Xenopus egg extract following a 1 hour incubation with DMSO solvent, 10 μM palmostatin, or 1 μM Wnt-C59 and probed with importin α antibodies and streptavidin to quantify incorporation of biotin-labeled palmitate (Martin, 2013).
(E) Fluorescence images of GFP-tagged wild-type or NP mutant importin α added to Xenopus egg extract, co-stained with FM 4–64X to visualize plasma membrane lipid derived vesicles, in the presence of DMSO control or drugs that alter palmitoylation. Two vesicles are shown for each condition. GFP-importin α wt localization to vesicles was enhanced by Palmostatin and inhibited by Wnt-C59, whereas GFP-importin α NP did not co-localize with plasma membrane lipid vesicles under any condition. Scale bar, 10 μm.
Both integral and peripheral membrane proteins can be modified by palmitoylation, which may be permanent or transient and can affect protein trafficking or stability (Linder and Deschenes, 2007). A large family of palmotoyl-acyl transferase (PAT) enzymes, and two acyl protein thioesterases (APT1 and APT2) have been identified that mediate cytosolic palmitoylation and depalmitoylation, respectively (Lin and Conibear, 2015). The inhibitor Wnt-C59 targets the predominant serine PAT, PORCN (Proffitt et al., 2013), while the drug palmostatin B potently inhibits APT1 and depalmitoylation of Ras in cells (Dekker et al., 2010). We observed that addition of Wnt-C59 decreased the fraction of palmitoylated importin α, and conversely, addition of palmostatin to egg extract increased the fraction of palmitoylated importin α significantly (Figure 1D). By fluorescence microscopy, a dye marking the outer leaflet of the plasma membrane, FM 4–64X, stained a subset of vesicles in the egg extract (Xiong et al., 2009). Palmostatin enhanced and Wnt-C59 inhibited the localization of GFP-tagged importin α to FM4–64X-positive vesicles, while GFP-importin α-NP did not colocalize with the plasma membrane vesicles (Figure 1E). Thus, the nuclear import factor importin α is palmitoylated, and its dynamic association with plasma membrane vesicles can be modulated by addition of inhibitors to egg extract.
Altering Levels of Importin α Palmitoylation Modulates Spindle and Nuclear Size
To test the effects of increased importin α palmitoylation and membrane association on spindles and nuclei, we added palmostatin to reactions containing sperm chromosomes and egg extract arrested in either mitosis or interphase (Figure 2A, B). A significant decrease in the size of both structures indicated that by sequestering importin α to extract membranes, hyper-palmitoylation impairs its cytosolic functions in nuclear import and spindle assembly. Remarkably, these defects were rescued by the addition of the non-palmitoylated mutant importin α-NP, demonstrating that altering the palmitoylation dynamics of other proteins is not consequential for these processes. Conversely, Wnt-C59 treatment increased both spindle length and nuclear diameter (Figure 2C, D), suggesting that inhibition of importin α membrane association enhances spindle and nuclear growth.
Figure 2. Palmitoylation status of importin α modulates both spindle and nuclear size.

(A) Fluorescence images and quantification of spindle lengths in metaphase-arrested egg extract reactions containing 1 μM recombinant wild-type or NP mutant importin α in the presence of DMSO or 10 μM palmostatin. Importin α NP reverted the decrease in spindle length caused by importin α hyperpalmitoylation. Mean ± SD, 64 spindles from 3 extracts. ** = p < 0.0005.
(B) Fluorescence images of DNA staining and quantification of nuclear diameter in interphase egg extract reactions containing 1 μM recombinant wild-type or NP mutant importin α in the presence of DMSO or 10 μM palmostatin. Importin α NP reverted the decrease in nuclear diameter caused by importin hyperpalmitoylation. Mean ± SD, 78 nuclei from 3 extracts. ** = p < 0.0005.
(C) Fluorescence images and quantification of spindle lengths in metaphase-arrested egg extract reactions containing DMSO or 1 μM Wnt-C59, which reduced importin α palmitoylation and increased spindle length. Mean ± SD, 67 spindles from 3 extracts. * = p < 0.005.
(D) Fluorescence images of DNA staining and quantification of nuclear diameter in interphase egg extract reactions containing 1 μM Wnt-C59, which reduced importin α palmitoylation and decreased nuclear size. Mean ± SD, 319 nuclei from 3 extracts. ** = p < 0.0005.
The Palmitoylation Status of Importin α Regulates its Binding to NLS-Containing Cargos
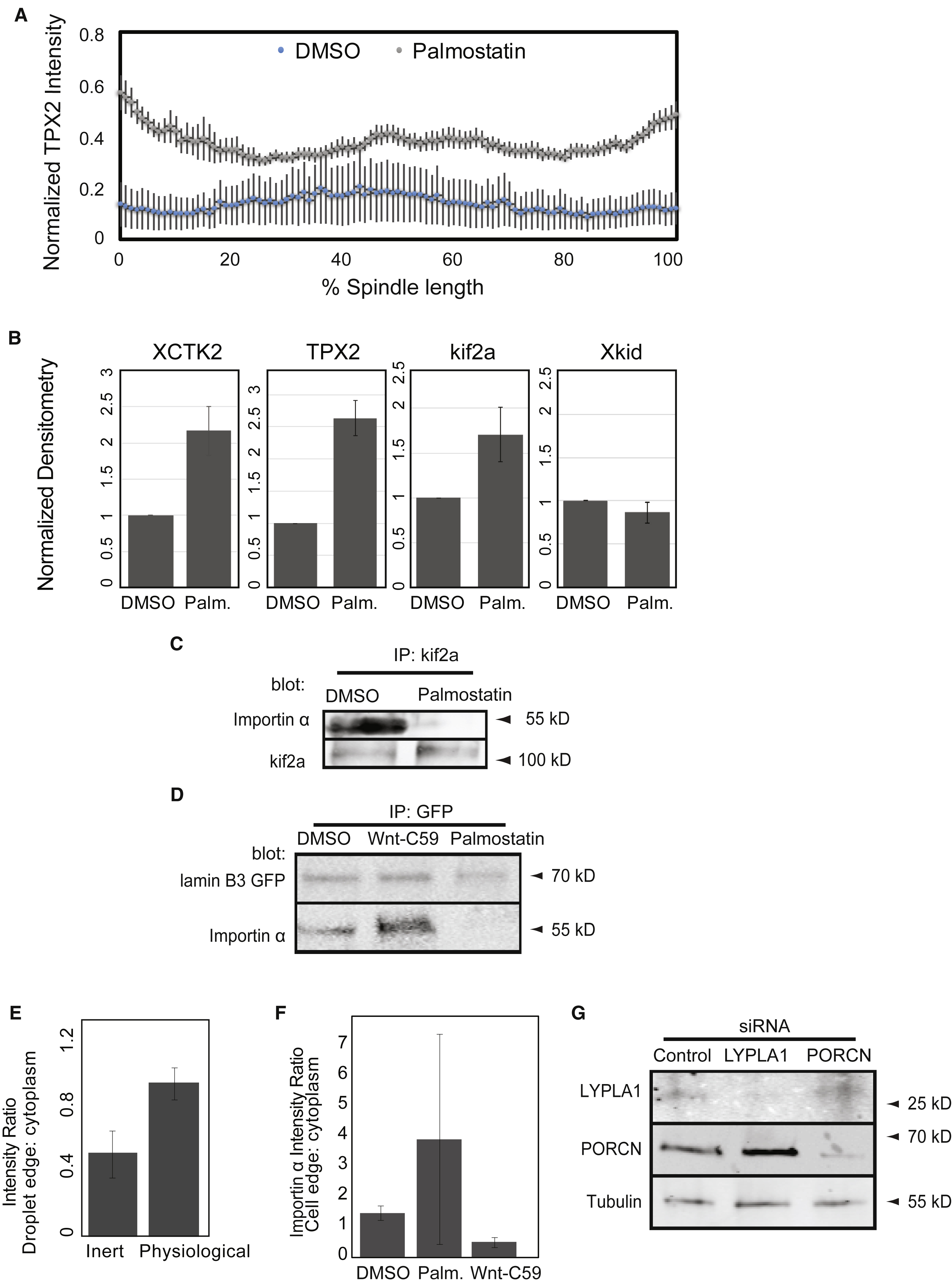
Intriguingly, a highly conserved asparagine residue of importin α crucial for NLS binding is adjacent to cysteine 230, suggesting that modification by palmitoylation at this site could interfere with cargo binding (Leung et al., 2003). To investigate how importin α partitioning to membranes affected cargo localization and transport, we examined kif2a, the microtubule depolymerizing kinesin that functions to scale the spindle during development (Wilbur and Heald, 2013), as well as lamin B3, which is limiting for nuclear growth in egg extracts (Levy and Heald, 2010). Addition of palmostatin to spindles pre-formed in egg extract caused enrichment of kif2a on spindle microtubules within 30 minutes as spindle size decreased, which was reversed by addition of importin α-NP (Figure 3A), suggesting that importin α hyper-palmitoylation impaired its binding to NLS-containing spindle assembly factors. Likewise, import of embryonic lamin B3 into the nucleus was decreased by palmostatin treatment, which reduced nuclear size and was reversed by addition of importin α-NP (Figure 3B). Mis-regulation of importin α cargos upon hyper-palmitoylation could also be monitored biochemically, as a number of spindle proteins containing NLS sequences, including kif2a, TPX2, and XCTK2, were enriched in microtubule pellets in the presence of palmostatin (Figure 3A and Figure S2A, B). Furthermore, palmostatin treatment reduced the levels of importin α-associated kif2a by co-immunoprecipitation (Figure S2C). Similarly, association of GFP-lamin B3 with importin α was reduced by palmostatin and stimulated by Wnt-C59 (Figure S2D). Thus, dynamic palmitoylation of importin α impacts its ability to bind cargos that impact both spindle and nuclear size.
Figure 3. Importin α palmitoylation regulates its binding to NLS-containing cargos.

(A) Fluorescence images and quantification of kif2a association with spindle microtubules in metaphase-arrested egg extract reactions containing 1 μM recombinant wild-type or NP mutant importin α in the presence of DMSO or 10 μM palmostatin. Importin α NP reverts the increase in kif2a localization caused by importin α hyperpalmitoylation. Middle panel: Line scan quantification of fluorescence intensity across the length of 35 spindles normalized for length. Quantification shows mean ± SD from two extracts, ** = p < 0.0005. Right panel: Immunoblot of microtubules pelleted from metaphase-arrested egg extract reactions containing DMSO or 10 μM palmostatin probed with kif2a and tubulin antibodies. Hyperpalmitoylation of importin α enhances kif2a association with microtubules, mean ± SD from 2 experiments, p <0.005.
(B) Fluorescence images and quantification of nuclear lamin staining in interphase egg extract reactions containing 1 μM recombinant wild-type or NP mutant importin α in the presence of DMSO or 10 μM palmostatin. Importin α-NP reverts the decrease in nuclear accumulation caused by importin hyperpalmitoylation. Right panel: Quantification of the mean intensity of lamin B1 staining in 277 nuclei from 3 extracts, mean ± SD from two extracts, ** = p < 0.0005. Scale bars, 10 μm.
Casein Kinase II-Dependent Phosphorylation Regulates Importin α Palmitoylation and Activity
It was shown previously that membrane association of importin α is reduced following its phosphorylation by casein kinase II, and that a phosphomimetic mutant of importin α in which residues T311, T322, T402, T405, S406, and T409 were mutated to glutamic acid, termed importin α-E, did not associate with membranes in Xenopus egg extracts (Hachet et al., 2004). Consistent with these findings, we observed that importin α-E was not palmitoylated on cysteine residues (Figure 4A), and that GFP importin α-E did not associate with plasma membrane vesicles, even upon palmostatin treatment (Figure 4B). To investigate the impact of CKII-mediated phosphorylation of importin α on subcellular scaling, we added the CKII inhibitor Quinalizarin (Qz) to reactions containing sperm chromosomes and egg extract arrested in either interphase or mitosis (Figure 4C, D). Both nuclear and spindle size decreased upon Qz treatment, and these effects were reversed by addition of importin α-NP, although spindle assembly was aberrant. These results indicate that CKII phosphorylation of importin α regulates its palmitoylation and function in subcellular size control and suggest that other CKII substrates also contribute to spindle morphology.
Figure 4. Casein kinase II-dependent phosphorylation regulates importin α palmitoylation and spindle and nuclear size.

(A) Blot of immunoprecipitated recombinant mcherry-SNAP-tagged wild-type and phosphomimetic mutant importin α proteins retrieved from Xenopus egg extract probed with streptavidin following Acl-biotin exchange chemistry (Wan et al., 2007). Hydroxylamine (HA) cleaves palmitate from cysteine residues to reveal a free thiol that reacts with HPDP-biotin. HA was omitted as a negative control.
(B) Fluorescence images of GFP-tagged wild-type importin α or importin α E added to Xenopus egg extract co-stained with FM 4–64X to visualize plasma membrane lipid derived vesicles, in the presence of DMSO control or Palmostatin. Like importin α-NP, importin α-E did not localize to vesicles under any condition. Scale bar, 10 μm.
(C) Fluorescence images of metaphase spindles and interphase nuclei in egg extract reactions containing DMSO or 50 μM Quinalizarin (Qz), an inhibitor of casein kinase II (CKII). Size increases were reversed by addition of importin α-NP, although spindle assembly was aberrant.
(D) Quantification of nuclear areas in part C. Mean ± SD, 122 nuclei from 3 extracts. * = p < .05, *** = p < .0005.
Importin α Partitioning-Dependent Subcellular Scaling Occurs in Microfluidic Droplets
Given that cytoplasmic levels of importin α contribute to spindle and nuclear size control (Levy and Heald, 2010; Wilbur and Heald, 2013), and that palmitoylation and membrane association of importin α alters its cargo binding and transport functions, we wondered whether partitioning of importin α between the cytoplasm and outer cell membrane contributes to intracellular scaling. Such a mechanism could link nuclear and spindle size to the ratio of cell membrane area to cytoplasmic volume as it changes during development. To test whether importin α can act as a surface area/volume sensor, we took advantage of a microfluidic system in which Xenopus extract cytoplasm is encapsulated within cell-like compartments of defined sizes and reconstitutes volume-dependent spindle scaling (Good et al., 2013). We compared droplets formed with the polyethylene glycol (PEG)-ylated stearate previously used to form the artificial lipid boundary, which inserted a long inert PEG “head group” between the lipid and cytoplasm of droplets, with droplets formed using a crude bovine liver lipid extract that closely mimics the physiological sterol and lipid components of the Xenopus egg plasma membrane (Hill et al., 2005). GFP-or RFP-tagged importin α localized to the crude lipid boundary, but not to the artificial, inert boundary (Figure 5A, C and Figure S2E). Comparing droplet diameters between 25 μm and 225 μm, spindles and nuclei were significantly smaller in lipid droplets formed using crude lipids than in the droplets containing inert PEGylated surfactant, and importantly were of similar size to the spindles and nuclei found in cells of the corresponding diameter in Xenopus embryos (Figure 5B, D). These size effects were abrogated by addition of GFP-importin α-NP, indicating that importin α localization to the outer membrane drives physiological scaling.
Figure 5. Compartment size, membrane composition, and importin α partitioning modulate spindle and nuclear size.

(A) Left panel: Schematic of spindles assembled in droplets encapsulated within a passivated boundary (inert membrane) or physiological membrane lipids in the presence of 1 μM GFP-tagged wild-type or NP mutant importin α. Right panel: Fluorescence images showing spindles and wild-type importin α localization in droplets of similar sizes formed within either inert or physiological lipid boundaries. The physiological lipid boundary increases the ratio of importin α at the droplet periphery compared to the interior, see Fig. S2E for quantification.
(B) Plots of spindle length at varying cell and droplets diameters. While both types of droplets demonstrated size-dependent spindle scaling, spindles formed in droplets bounded by physiological lipids were smaller, and showed similar scaling properties compared to spindles of corresponding embryo cell sizes in vivo (p < 0.005). Right panel: Addition of the non-palmitoylatable mutant, but not wild-type importin α, reverted the spindle scaling regime to that of the inert boundary (p < 0.05).
(C) Left panel: Schematic of nuclei assembled in droplets encapsulated within a passivated boundary (inert membrane) or physiological membrane lipids in the presence of 1 μM RFP-tagged wild-type or NP mutant importin α. Right panel: Fluorescence images showing nuclei and wild-type importin α localization in droplets of similar sizes formed within either inert or physiological lipid boundaries.
(D) Plots of nuclear diameter at varying cell and droplets diameters. While both types of droplets demonstrated droplet size-dependent nuclear scaling, nuclei formed in droplets bounded by physiological lipids were smaller than those formed in inert boundary droplets, and showed similar scaling properties compared to nuclei of corresponding embryo cell sizes in vivo (p < 0.005). Right panel: Addition of the non-palmitoylatable mutant, but not wild-type importin α, reverted the nuclear scaling regime to that of the inert boundary (p < 0.05). P-values indicate statistical difference between y-intercepts of regression lines from 3 extracts, calculated using an analysis of covariance. Scale bars, 10 μm.
Importin α Palmitoylation Regulates Nuclear Scaling in the Xenopus Embryo
To model whether importin α could act as a sensor of the cell surface area/volume ratio in vivo, we used cell size measurements at different stages of Xenopus laevis development and the reported importin α concentration in the embryo (5 μM) (Levy and Heald, 2010; Wilbur and Heald, 2013) to calculate the maximum amount of importin α that could localize to the plasma membrane during early development, and the degree of cytoplasmic depletion due to outer membrane partitioning (Figure 6A). At early stages, when the surface area to volume ratio is relatively low, at most only 1% of importin α could localize to the plasma membrane. This is in contrast to stage 8 cells, which have a comparatively higher surface area to volume ratio that could allow nearly 50% of total cellular importin α to localize to the plasma membrane. This calculation is supported by previous observations that while total importin α concentration remains constant at 5 μM throughout development, cytoplasmic levels decrease almost 50% by stage 8 (Levy and Heald, 2010). To test this model in vivo, we injected either palmostatin or Wnt-C59 into fertilized X. laevis eggs, which enhanced and inhibited importin α membrane association, respectively (Figure 6B). Few mitotic cells were observed under these conditions, but nevertheless embryos underwent cleavage divisions, enabling measurement of nuclei in cells with diameters ranging from 25 to 300 μm. Remarkably, nuclear scaling was impaired by both drugs, with palmostatin-injected embryos forming nuclei of significantly smaller sizes than in control embryos, while nuclei remained large in the Wnt-C59-injected embryos over the entire range of cell sizes (Figure 6C). To test the specificity of palmostatin for modulating importin α-dependent scaling, we co-injected either wild type importin α or the non-palmitoylated mutant. Importin α-NP injection itself increased nuclear size in the embryo, and this increase was not significantly reversed by co-injection of palmostatin (Figure 6D). These results indicate that importin α palmitoylation and partitioning drives nuclear size scaling in vivo.
Figure 6. Palmitoylation levels of importin α modulate nuclear scaling in vivo.

(A) Schematic showing the known values for cell size and total importin α concentration during X. laevis development and the predicted cytoplasmic concentrations of importin α at various stages during development. Right panel: Graph of the predicted decrease in cytoplasmic importin α concentrations due to its progressive sequestration at the plasma membrane as the surface area/volume ratio increases during embryogenesis.
(B) Left panel: Fluorescence images of importin α localization and histone H2B to label nuclei in embryos at stage 7 following injection of DMSO, palmostatin or Wnt-C59 into the zygote. Palmostatin increased, and Wnt-C59 decreased the ratio of importin α at the cell periphery compared to the cell interior see Figure S2E for quantification. Right panel: Mean intensity ratio of importin α at the cell membrane compared to the cell center in embryos at stage 7 that had been injected with DMSO, palmostatin, or Wnt-C59. Mean ± SD from 30 cells, p < 0.005. Scale bar, 10 μm.
(C) Plot of nuclear diameters at different cell diameters following drug or vehicle injection. Palmostatin treatment led to earlier onset of nuclear scaling, while Wnt-C59 inhibited nuclear scaling. (p < 0.05). P-values indicate statistical difference between y-intercepts of regression lines from 3 experiments, calculated using an analysis of covariance.
(D) Plot of nuclear diameters at different cell diameters upon co-injection of palmostatin with wild type or non-palmitoylated importin α. Importin α-wt did not reverse the nuclear size decrease, while importin α-NP increased nuclear size and abrogated the effects of palmostatin. (p < .05). P-values indicate statistical difference between y-intercepts of regression lines from 3 experiments, calculated using an analysis of covariance.
Importin α Palmitoylation-Dependent Size Control Also Occurs in Human Cells
Three of the four palmitoylated residues identified in the major importin α isoform found in Xenopus eggs and embryos are conserved in the human homolog KPNA2/importin α1. We therefore tested whether altering palmitoylation status and plasma membrane association of importin α in human cells acted similarly to control nuclear and spindle size. Following inhibitor treatment of human retinal pigment epithelial 1 (RPE-1) cells for 12 hours, we observed that palmostatin enhanced importin α plasma membrane localization, while Wnt-C59 decreased importin α membrane staining (Figure 7A, B, S2F). Consistent with the Xenopus extract and embryo results, palmostatin treatment decreased spindle and nuclear size while Wnt-C59 treatment had the opposite effect. As an independent assay for the effects of drug treatment, we transfected siRNAs against the major predicted targets of palmostatin and Wnt-C59, PORCN and LYPLA1 (APT1 in Xenopus), respectively, reducing their levels to 10% of control siRNA (Figure S2G). Depletion of the enzyme that drives palmitoylation increased nuclear and spindle size, while depletion of the enzyme that removes palmitate decreased nuclear and spindle size (Figure 7C, D). Together these results suggest that importin α palmitoylation is a conserved mechanism for regulating spindle and nuclear size in Xenopus and human cells.
Figure 7. Palmitoylation modulates importin α localization, nuclear and spindle size in human cells.

(A) Fluorescence images of importin α localization and quantification of spindle lengths in RPE-1 cells treated with either DMSO, 50 μM palmostatin or 10 μM Wnt-C59 for 12 hours. Palmostatin treatment increased importin α localization to the plasma membrane and decreased spindle size while Wnt-C59 treatment had the opposite effect. Mean ± SD, 199 cells from 3 experiments. ** = p < 0.0005.
(B) Fluorescence images of DNA and importin α staining and quantification of nuclear area of RPE-1 cells treated with either DMSO, 50 μM palmostatin or 10 μM Wnt-C59 for 12 hours. Palmostatin treatment increased importin α localization to the plasma membrane and decreased nuclear size while Wnt-C59 treatment had the opposite effect. Mean ± SD, 213 cells from 3 experiments. ** = p < 0.0005.
(C) Fluorescence images of metaphase spindles and quantification of spindle lengths in HCT 293 cells 3 days after transfection of either scrambled siRNA or siRNAs targeted to LYPLA1 and PORCN. LYPLA1 knockdown decreased spindle size while PORCN knockdown had the opposite effect. Mean ± SD, 168 cells from 3 experiments. *** = p < 0.0005.
(D) Fluorescence images of metaphase spindles and quantification of nuclear area in HCT 293 cells 3 days after transfection of either scrambled siRNA or siRNAs targeted to LYPLA1 and PORCN. LYPLA1 knockdown decreased nuclear area while PORCN knockdown had the opposite effect. Mean ± SD, 87 cells from 3 experiments. * = p < 0.05.
DISCUSSION
Our results support a mechanism by which palmitoylation-dependent importin α membrane association regulates mitotic spindle and nuclear scaling during Xenopus embryogenesis by providing a measure of the cell surface area-to-volume ratio. Previous studies of spindle and nuclear scaling have focused on either volume-dependent or biochemical differences separately (Good et al., 2013; Levy and Heald, 2010; Wilbur and Heald, 2013). We have identified a pathway that links these two modes via the progressive cytoplasmic depletion of importin α by its exponential sequestration to the plasma membrane during reductive cell divisions. These findings support a conceptual model of embryogenesis whereby physical parameters such as the nucleo-cytoplasmic ratio are linked biochemically to the regulation of cellular processes, for example zygotic genome activation at the mid-blastula transition (Jukam et al., 2017).
Our data indicate that the palmitoylation state of importin α is in dynamic equilibrium. Increasing palmitoylation levels of importin α by addition of palmostatin affected spindle and nuclear size in cell-free Xenopus egg extracts, and cytoplasmic staining of GFP-importin α could be observed upon palmostatin treatment of extract droplets and embryos. These results indicate that importin α can be sequested to cytoplasmic membrane pools in addition to the plasma membrane. Interestingly, by inhibiting importin α CKII is required to maintain this equilibrium. Importin α palmitoylation sites, as well as CKII phosphorylation sites (Hachet et al., 2004) are conserved to varying degrees among the 7 human importin α isoforms (data not shown). How phosphorylation at these sites acts to block palmitoylation, and where and when this regulation occurs, remain open questions.
Given the diverse functions of importin α in gene regulation, cell physiology and differentiation (Miyamoto et al., 2016), it will be interesting to investigate how its cell size-dependent partitioning contributes to cellular function, fate, and homeostasis in a variety of contexts. For example, cell surface area to volume ratio could affect the distribution of transcription factors between the cytoplasm and nucleus by regulating the availability of importin α for transport. In addition to its transport functions, importin α is also thought to participate in gene regulation inside the nucleus, and to modulate signaling at the plasma membrane (Miyamoto et al., 2016). Future studies will elucidate how palmitoylation of importins impacts membrane association and activities of NLS-containing cargos. Finally, importin α levels are elevated in a number of cancers and this is associated with malignant transformation (Christiansen and Dyrskjøt, 2013). Additional investigation promises to reveal new connections between cell size, importin α function, and fundamental biological mechanisms.
STAR METHODS
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Rabbit polyclonal anti-importin α | Levy and Heald, 2010 | N/A |
| Rabbit polyclonal anti-kif2a | Abcam | ab37005 |
| Rabbit polyclonal anti-TPX2 | This paper | N/A |
| Monoclonal anti-GFP | Clontech | Cat#632380 |
| Rabbit anti-XCTK2 | Walczak et al, 1997 | N/A |
| Rabbit anti-Xkid | Funabiki et al, 2000 | N/A |
| Goat anti-LYPLA1 | Abcam | ab77640 |
| Monoclonal anti-GFP | Clontech | Cat#632380 |
| Rabbit anti-PORCN | Abcam | ab105543 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Palmostatin B | Millipore Sigma | Cat#178501–5MG |
| Wnt C-59 | Cayman Chemical | Cat#1243243–89-1 |
| Quinalizarin | Sigma-Aldrich | Q2763 |
| Experimental Models: Cell Lines | ||
| Human: hTERT RPE-1 | UC Berkeley Cell Culture Facility | N/A |
| Human: HCT 116 | UC Berkeley Cell Culture Facility | N/A |
| Human: HEK-293 | UC Berkeley Cell Culture Facility | N/A |
| Experimental Models: Organisms/Strains | ||
| Xenopus laevis | NXR | |
| Oligonucleotides | ||
| siRNA: LYPLA1 | Origene | Cat#SR307076 |
| siRNA:PORCN | Origene | Cat#SR324902 |
| Recombinant DNA | ||
| Plasmid: Importin1α GFP | This paper | N/A |
| Plasmid: Importin α RFP | This paper | N/A |
| Plasmid: Importin α GFP NP | This paper | N/A |
| Plasmid: Importin α GFP E | This paper | N/A |
| Plasmid: Pet20 (a,b,c) | EMD Millipore | Cat#69739 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rebecca Heald (bheald@berkeley.edu).
Experimental Model and Subject Details
Cell culture
RPE-1, HCT 293 and HCT 116 cells were cultured as described previously (Kiyomitsu and Cheeseman, 2012).
Animal Models
All animals were maintained in accordance with standards established by the Institutational Care and Use Committee at the University of California, Berkeley.
Method Details
Xenopus egg extracts, spindle and nuclear assembly reactions, and drug treatments
Cytostatic factor-arrested (CSF) egg extracts were prepared from freshly laid eggs of X. laevis and used for spindle assembly reactions as described (Hannak and Heald, 2006; Maresca and Heald, 2006). Briefly, eggs in metaphase of meiosis II were collected, dejellied and fractionated by centrifugation. The cytoplasmic layer was isolated, supplemented with 10 μg/ml each of the protease inhibitors leupeptin, pepstatin and chymostatin (LPC), 20 μM cytochlasin D, and a creatine phosphate and ATP energy regeneration mix, and stored on ice for up to 6 hours. Typical spindle reactions contained 25 μl CSF extract, X. laevis sperm at a final concentration of 500–10,000 per μl, 2 μM rhodamine-labeled porcine brain tubulin, and 2 μM Hoechst dye. Nuclear assembly reactions were performed as described(Levy and Heald, 2010). Final concentrations of 10 μM Palmostatin or 1 μM Wnt-C59 dissolved in DMSO (EMD Millipore) were added to extracts for one hour. CKII inhibitor Quinalizarin (Qz) (Sigma-Aldrich) was added to extracts at a final concentration of 50 μM.
Fractionation of egg membranes
Between 400 and 1,000 X. laevis eggs were homogenized in oocyte homogenization buffer (OHB; 250 sucrose, 5 MgCl2, 10 HEPES, pH 7.4; 10 μl/egg) containing LPC (10 μg/ml) using 15 strokes of a Teflon-coated pestle in a glass Dounce homogenizer. Homogenates were centrifuged at 500 g for 5 min, and the supernatant was saved. The pellet was resuspended in the same volume of OHB and re-homogenized as above before centrifugation at 500 g for 5 min. The second supernatant was combined with the first and contained crude membranes (~3 ml), which were overlaid on top of a discontinuous sucrose gradient composed of 50% sucrose/OHB (6 ml) and 20% sucrose/OHB (6 ml), and centrifuged at 30,000 g for 1 hour in a swinging bucket rotor (Beckman SW41). Two whitish bands of membranes were visible, the lower, heavier membrane bands at the interface between 20 and 50% sucrose and the light membranes ~1 cm from the top.
Immunofluorescence of spindle and nuclei spin-downs
Importin α antisera were raised against full-length Xenopus importin α (Levy and Heald, 2010). Anti-TPX2 (unpublished) antibody was used at 1:500. Anti-kif2a (abcam) was used at 1:5,000. Immunofluorescence analysis was performed as previously described (Loughlin et al., 2011). Briefly, spindles or nuclei in extract were fixed with 3.7% formaldehyde, sedimented through a 40% glycerol cushion onto coverslips, post-fixed in 100% methanol, and blocked with PBS + 1% BSA. Coverslips were incubated to equilibrium with 1:1000 dilution of primary antibody unless noted, washed extensively, and incubated with a 1:1000 dilution of secondary antibody (Alexa-labeled anti-rabbit or anti-mouse; Invitrogen). Images were obtained on an epifluorescence microscope (BX51; Olympus) under 40X magnification (0.75 NA; UPlanFl N; Olympus) with TRITC, DAPI, and FITC filters (Chroma Technology Corp.) by an Orca-ER cooled CCD camera (Hamamatsu Photonics). Immunofluorescence quantification was performed by manually collecting 25-pixel-width linescans in ImageJ, locally correcting for background intensity if it was more that 10 intensity units above the camera offset. Data was then normalized to 100% spindle length, and intensity was averaged within each 1% length bin. Depending on the length of the spindle, ~2 to 6 pixels were averaged per length bin. Spindles from each condition were averaged under these normalized spindle length conditions with error propagation and intensity plotted as a function of 0–100% spindle length. Immunofluorescence intensity ratios of importin α in embryos and droplets were determined by calculating the integrated densities of a 6-pixel-width line along the cell/droplet periphery compared to the same line shifted ~20 pixels into the cytoplasm of the cell. Spindle lengths and nuclei diameter and area were measured and analyzed using ImageJ/Fiji.
Protein purification
Wild-type importin α was used as a template to introduce cysteine to alanine or serine to alanine mutations by Quikchange mutagenesis (Stratagene/Agilent Technologies, Santa Clara CA, USA). All proteins were cloned into pSNAP-T7 vector and purified as described for his-tagged proteins (Qiagen).
Immunoprecipitation, immunoblotting, and MAP isolation
Immunoprecipitation was performed by cross-linking 0.25 μg/μl of antibody to 20 μl of Dyna beads (Thermo Fisher Scientific). Beads were incubated at 4°C for 90 minutes with 50 μl of egg extract. Beads were washed twice with phosphate-buffered saline (PBS) buffer and twice with PBS + 0.5 M NaCl, before they were resuspended in Laemmli sample buffer and subjected to Western blot analysis. Antibodies used for western blotting and immunoprecipitations include anti-GFP JL8 monoclonal (Clontech), anti-importin α, kif2a, and TPX2 as mentioned above, anti-XCTK2 (gift from C. Walczak, anti-xkid (gift from H. Funabiki, Rockefeller University), anti-LYPLA1 (abcam), and anti-PORCN (abcam). Immunoblotting utilized 1:1000 dilution of all antibodies or antisera except TPX2 which was used at 1:500 dilution and was performed as described (Loughlin et al., 2011). Microtubule associated proteins were isolated from Xenopus extract as described (Budde et al., 2005) and quantified by Western blot. Blots were scanned with an Odyssey Infarer Imaging System (LI-COR Biosciences). Alternatively, samples were processed further for detection of palmitoylation.
Detection of palmitoylation
Palmitoylated residues were detected as described previously by Acyl-Biotin Exchange (ABE) (Wan et al., 2007) and by metabolic labeling with alkyne palmitic acid followed by click chemistry (Martin, 2013). Briefly, ABE chemistry is based on the substitution of biotin for palmitoyl modifications through a sequence of three chemical steps: unmodified cysteine thiols are blocked with NEM; palmitoylation thioesters are cleaved by hydroxylamine (HA) (+NH2OH); and the newly exposed cysteinyl thiols are marked with thiol-specific biotinylating reagent (HPDP-biotin in our experiments). Immunoprecipitated importin α was resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mm EDTA) containing 1% SDS, 2% Triton X-100, 0.5% NP-40, protease inhibitors, 10 mM TCEP (Sigma), and 10 mm NEM (Thermo Scientific). After 1 μM HA treatment for 1 hour, the beads were washed in lysis buffer 3 times and incubated with HPDP-biotin for 1 hour. HA was omitted as a negative control for each of the reactions. After washing 3 more times in lysis buffer, the precipitates were eluted with SDS loading buffer, subjected to SDS-PAGE, and blotted with importin α antibodies and 800 nm-labeled streptavidin (Licor). In the second method, metabolic labeling of extract with alkyne-palmitate was performed for 2 hours followed by immunoprecipitation and click chemistry as described (Martin, 2013). Briefly, alk-16–labeled extract was mixed 1:1 with 0.1 mM triethanolamine (TEA) buffer. 10 μl 10% (w/v) SDS in H2O was added as well as CuAAC reactants at final concentrations of 100 μM az-biotin 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 100 100 μM tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) and 1 mM CuSO4·5H2O for a final volume of 50 μl. CuAAC reactions were allowed to proceed for 1 hour at room temperature, and then washed with PBS. The samples were then resuspended in SDS loading buffer, subjected to SDS-PAGE, and blotted importin α antisera and 800 nm-labeled streptavidin (Licor).
Physiological membrane preparation
Total bovine liver extracts (Avanti Polar Lipids, Alabaster, Alabama) were used to form physiological membrane droplets. To purify the lipids, the extract (shipped in chloroform) was mixed with 3.75 ml of chloroform/methanol (1:2) per 1 ml of liver extract. After vortexing and incubating 5 minutes, 1.25 ml of chloroform was mixed and incubated for one minute, followed by 1.25 ml of water, mixing and incubating another minute before centrifugation. The lower phase was collected with a Pasteur pipette and evaporated under a nitrogen stream before desiccating under vacuum overnight. The resulting lipids were resuspended in squalene to a lipid concentration of 50 mg/ml.
Droplet formation by emulsification
Droplets of various sizes containing either spindles or nuclei and different lipid boundaries were formed by adding 2 μl of the extract reaction mixture on ice to 50 μl of the appropriate surfactant/lipid mixture. Cithrol DPHS resuspended in squalene at 50 mg/ml served as the surfactant to form inert membrane droplets, while total bovine liver extracts (preparation detailed above) were used as the physiological membrane surfactant. The extract/surfactant mixture was then pipetted up and down with a 20 μl pipette tip set to maximum volume approximately 10–20 times, depending on the desired size range of droplets desired. The emulsified mixture was then added to a custom-built chamber composed of vacuum grease (Dow Corning) on a cover slip and imaged as above. All droplet experiments were performed in this manner with the exception of the first of each of the three experiments, which were performed in microfabricated chambers as outlined below.
Microfabrication
Microfluidic devices were fabricated as described (Good et al., 2013). Briefly, a ~25 μm thick film of SU-8 2025 was deposited on a four-inch diameter silicon substrate, soft-baked, and exposed through a mask using a UV contact aligner. After a second bake, the wafers were developed in SU-8 developer and silanized under vacuum prior to use. PDMS (Sylgard elastomer 184) was mixed 10:1 (base to curing agent by weight) and poured onto the wafer, degassed and cured at 65°C overnight. The cured elastomer was then removed from the wafer and inlet and outlet holes (~0.5 mm diameter and ~3 mm diameter, respectively) were punched using stainless steel tubing. The devices were bonded to microscope slides following a ~20s exposure to oxygen plasma and stored at ~65°C until a few hours prior to use. On the day used, devices were filled with Aquapel rain repellent glass treatment, flushed with a gentle stream of nitrogen gas, and dried in an oven for ~4 hours to remove any residual solvent.
Microfluidic extract encapsulation
The encapsulation was performed as described (Good et al., 2013). Briefly, using a T-junction microfluidic device (oil/surfactant in one channel, extracts in second channel), spindle reactions were encapsulated in droplets of varying size. The process was monitored under a stereo microscope in a cold room, with all equipment and reagents equilibrated to 4°C. Approximately 10 μl of spindle or nuclear assembly reaction was loaded into a short length of PTFE tubing mounted on a Hamilton syringe. After flushing devices with the oil/surfactant mixture (Squalene containing either 50 mg/ml Cithrol DPHS or 50 mg/ml bovine liver physiological lipids), extract was introduced, using flow rates for the two phases selected to achieve particular droplet volumes. As droplets were produced, they accumulated in a ~3 mm-diameter open reservoir, from which they were transferred by gentle pipetting to form a monolayer in separate custom-built imaging chambers, surrounded by oil. These chambers were brought to 22°C to initiate spindle or nuclear assembly, and imaged 45 minutes later.
Importin α membrane partitioning calculations
Importin α membrane partitioning (Figure 5A) was calculated using a set of parameters based on the measured importin α concentrations in X. laevis (Levy and Heald, 2010), the average predicted footprint of a single importin α molecular at the membrane (Erickson, 2009), the average Xenopus embryo cell sizes of throughout early development (Wühr et al., 2008), and the predicted surface area and volumes of the cells assuming they are spherical in shape. Using these values, the predicted cytoplasmic levels of importin α as a percent of the total throughout development as a function of changing cell size, assuming as many molecules of importin α titrate to the membrane as space will allow, is given by the following formulas:
IR = reduced cytoplasmic importin α concentration percent
IT = number of total importin α molecules
•IS = number of importin α molecules at the surface
[k] = total cellular concentration of importin α
V = cell volume
N = Avogadro’s number
S = cell surface area
h = molecular footprint of importin α
r = cell radius
IP = cytoplasmic percent of importin α concentration
In vitro fertilization
X. laevis males were injected with 500 U of human chorionic gonadotropin hormone (hCG) 12–24 hours before dissection and testes were stored at 4°C in 1X MMR (100 mM NaCl, 1.8 mM KCl, 2 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES-NaOH pH 7.6) for 1–2 weeks. X. laevis females were primed with 100 U of pregnant mare serum gonadotropin (PMSG, National Hormone and Peptide Program, Torrance, CA) at least 48 hours before use and boosted with 500 U of hCG 14 hours before the experiment. Females were squeezed gently to deposit eggs onto petri dishes coated with 1.5% agarose in 1/10X MMR. 1/3 of a X. laevis testes was added to 1 ml of ddH2O and homogenized using scissors and a pestle. Any liquid in the petri dishes was removed and the eggs were fertilized with 500 μl of sperm solution per dish. Eggs were swirled in the solution to individualize eggs as much as possible and incubated for 10 min. Dishes were flooded with 1/10X MMR, swirled and incubated for 10–20 minutes. Jelly coats were removed with a 2% cysteine solution (in ddH2O-NaOH, pH 7.8). After extensive washing (>4X) with 1/10X MMR, embryos were incubated at 23°C. At stage 2–3, fertilized embryos were sorted and placed in fresh 1/10X MMR in new petri dishes coated with 1.5% agarose in 1/10X MMR.
Embryo microinjection
DMSO, 1 mM palmostatin or 100 μM Wnt-C59 were loaded into a needle pulled from a 1 mm glass capillary tube (TW100F-4, World Precision Instruments) using a P-87 Micropipette Puller (Sutter Instrument). Embryos were placed in a mesh-bottomed dish with 1/10X MMR 3% Ficoll and microinjected in both blastomeres with 1 nl of the appropriate solution using a Picospritzer III microinjection system (Parker) equipped with a MM-3 micromanipulator (Narishige). Injected embryos were transferred to a new dish and incubated at 23°C in 1/10X MMR 3% Ficoll until stages 6–9, then processed for immunostaining.
Embryo whole mount immunofluorescence
At desired stages, embryos were fixed for 1–3 hours using MAD fixative (2 parts of methanol (Thermo Fisher Scientific), 2 parts of acetone (Thermo Fisher Scientific), 1 part of DMSO). After fixation, embryos were dehydrated in methanol and stored at −20°C. Following gradual rehydration in 0.5X SSC (1X SSC: 150 25mM NaCl, 15 mM Na citrate, pH 7.0), embryos were bleached with 1–2% H2O2 (Thermo Fisher Scientific) in 0.5X SSC containing 5% formamide for 2–3 hours under light, then washed in PBT, a PBS solution containing 0.1% Triton X-100 (Thermo Fisher Scientific) and 2 mg/ml bovine serum albumin (BSA). Embryos were blocked in PBT supplemented with 10% goat serum (Gibco – Thermo Fisher Scientific) and 5% DMSO for 1–3 hours and incubated overnight at 4°C in PBT sup plemented with 10% goat serum and the primary antibodies: 1:500 rabbit anti-histone H2B (ab1791; Abcam), 1:500 rabbit anti-importin α (gift from Karsten Weis). Embryos were then washed 4 X 2 hours in PBT and incubated overnight in PBT supplemented with goat anti-rabbit secondary antibodies coupled either to Alexa Fluor 488 or 568 (Invitrogen – Thermo Fisher Scientific). Embryos were then washed 4 X 2 hours in PBT and gradually dehydrated in methanol. Embryos were finally cleared in Murray’s clearing medium (2 parts of Benzyl Benzoate, 1 part of Benzyl Alcohol). For confocal microscopy, embryos were placed in a chamber made using a flat nylon washer (Grainger) attached with nail polish (Sally Hansen) to a slide and covered by a coverslip, or in a chamber made of silicon grease (Beckman Coulter) between slide and coverslip, and filled with Murray’s clearing medium.
Confocal microscopy and nuclear size quantification
Confocal microscopy was performed on a Zeiss LSM 780 NLO AxioExaminer using the Zeiss Zen Software. For imaging of histone H3 and importin α, embryos were imaged using a Plan-Apochromat 20x/1.0 Water objective and laser power of 12%, on multiple 1024×1024 px planes spaced 1 μm in Z. Images are mean averages of 2 scans with a depth of 16 bits. Pinhole size was always chosen to correspond to 1 airy unit. Nuclear diameter at stages 6, 7, 8 and 9 was measured using histone H3 fluorescence signal segmentation in Fiji and utilizing the line tool. Cell diameter was measured at the central plane of the nucleus.
Cell culture, drug treatments, immunofluorescence, RNAi, and microscopy
RPE-1, HCT 293 and HCT 116 cells were cultured as described previously (Kiyomitsu and Cheeseman, 2012). Cells were treated with DMSO, 50 μM palmostatin, 10 μM Wnt-C59, or 5 μM Qz for 12 hours. For live-cell imaging, cells were cultured with 50 ng/ml Hoechst33342 for 10 minutes prior to observation. RNAi experiments were carried out with 27mer siRNA duplexes corresponding to regions of PORCN and LYPLA1. The oligonucleotides (Origene) were transfected at an amount of 100 pmol per 6 well plate into HCT 293 cells using the Lipofectamine 2000 system (Thermo Fisher) in accordance with the supplier’s recommendations. Cells were fixed with methanol for 3 minutes followed by 3 washes in PBS 0.3% triton X 100 for DNA (Hoeschst3342) and microtubule (1:2000 E7 anti-beta tubulin antibody) staining. For importin α staining (1:1000 antibody gift from Karsten Weis), cells were fixed with 3% paraformaldehyde and 2% sucrose. Images were acquired on an Andor/Nikon spinning disk confocal. For both fixed and live cells, 10 Z sections were acquired in 1 μm steps using a Nikon Plan-fluor 40x/0.75 NA objective. Distance measurements and mean fluorescent intensities were analyzed using ImageJ/Fiji software.
Quantification and Statistical Analysis
All experiments were performed in biological triplicate unless otherwise indicated. The “N” for each experiment can be found in the figure legends and represents independently generated samples for in vitro experiments, cells for live cell experiments, and Xenopus embryos for all in vivo experiments. Bar graphs present the mean ± SD; p values were generated for these by two-tailed Student’s t test. Throughout the figures, asterisks indicate the significance of the p value: *p < 0.05; **p < 0.005; ***p < 0.0005. A significant result was defined as p < 0.05.
Supplementary Material
(A) Left: Sucrose gradient of egg membranes to which recombinant GFP-importin α was added. Right: Fractions illuminated with blue light reveal that exogenous importin α co-fractionates at a much higher concentration with the plasma membrane-containing than the heavier membrane fraction (Hill et al., 2005).
(B) Blot of immunoprecpitated Ras and importin α subjected to ABE chemistry to detect palmitoylation. Omission of 1 μM hydroxylamine (HA) serves as a negative control.
{kind=link}
(A) Line scan quantification of TPX2 immunofluorescence intensity with normalized spindle lengths upon addition of DMSO or 10 μM palmostatin.
(B) Quantification of microtubule pelleting upon addition of DMSO or 10 μM palmostatin to spindle assembly reactions in egg extacts. Palmostatin increased the fraction of XCTK2, TPX2 and kif2a bound to microtubules, but did not affect chromokinesin Xkid.
(C) Immunoprecipitation of kif2a and immunoblot of importin α following addition of DMSO or 10 μM palmostatin to egg extract. Palmostatin decreased the amount of importin α associated with kif2a.
(D) Immunoprecipitation of GFP-lamin B3 and immunoblot of importin α following treatment of extracts with DMSO, 50 μM palmostatin or 10 μM Wnt-C59. Palmostatin decreased the amount of importin α associated with the nuclear lamin, while Wnt-C59 increased co-precipitating importin α.
(E) Mean intensity ratio of importin α at the edge compared to the center in extract droplets encapsulated using synthetic or physiological lipids. Mean ± SD from 18 droplets, p < 0.005.
(F) Mean intensity ratio of importin α at the cell membrane compared to the cell center in RPE-1 cells that have been treated with DMSO, palmostatin, or Wnt-C59. Mean ± SD from 30 cells, p < 0.05.
(G) Blot of HEK-293 cells transfected with either control, LYPLA1 or PORCN siRNA.
{kind=link}
Highlights.
The nuclear transport factor importin α is lipid-modified by palmitoylation
Phosphorylation by casein kinase II blocks importin α palmitoylation
Palmitoylated importin α binds to the membrane, reducing its cytoplasmic levels
Cytoplasmic levels of importin α coordinately scale spindle and nuclear size
A modified form of the protein importin α regulates the scaling of intracellular structures concomitant with cell size.
ACKNOWLEDGMENTS
We thank Dr. Romain Gibeaux for advice on figure preparation, other members of the Heald lab, present and past, for support and fruitful discussions, and Stephanie Malinich for sequence alignments and analysis. We thank Claire Walczak for kif2a antibodies, and Dan Levy and Thomas Surrey for advice and comments on the manuscript. CB was supported by a fellowship from the American Cancer Society (grant #127637-PF-15-004-01-CCG). RH was supported by NIH MIRA grant R35 GM118183 and the Flora Lamson Hewlett Chair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bird SL, Heald R, and Weis K (2013). RanGTP and CLASP1 cooperate to position the mitotic spindle. Mol. Biol. Cell 24, 2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde PP, Desai A, and Heald R (2005). Analysis of microtubule polymerization in vitro and during the cell cycle in Xenopus egg extracts. Methods 38, 29–34. [DOI] [PubMed] [Google Scholar]
- Chang C-C, Huang T-L, Shimamoto Y, Tsai S-Y, and Hsia K-C (2017). Regulation of mitotic spindle assembly factor NuMA by Importin-β. J. Cell Biol 216, 3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen A, and Dyrskjøt L (2013). The functional role of the novel biomarker karyopherin α 2 (KPNA2) in cancer. Cancer Lett 331, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder ME, Strzelecka M, Wilbur JD, Good MC, von Dassow G, and Heald R (2015). A comparative analysis of spindle morphometrics across metazoans. Curr. Biol 25, 1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Schölermann B, et al. (2010). Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol 6, 449–456. [DOI] [PubMed] [Google Scholar]
- Eisenberg S, Laude AJ, Beckett AJ, Mageean CJ, Aran V, Hernandez-Valladares M, Henis YI, and Prior IA (2013). The role of palmitoylation in regulating Ras localization and function. Biochem. Soc. Trans 41, 79–83. [DOI] [PubMed] [Google Scholar]
- Erickson HP (2009). Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes DJ, Travesa A, Nord MS, and Bernis C (2015). Nuclear transport factors: global regulation of mitosis. Curr. Opin. Cell Biol 35, 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good MC, Vahey MD, Skandarajah A, Fletcher DA, and Heald R (2013). Cytoplasmic volume modulates spindle size during embryogenesis. Science 342, 856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachet V, Köcher T, Wilm M, and Mattaj IW (2004). Importin α associates with membranes and participates in nuclear envelope assembly in vitro. EMBO J 23, 1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannak E, and Heald R (2006). Investigating mitotic spindle assembly and function in vitro using Xenopus laevis egg extracts. Nat. Protoc 1, 2305–2314. [DOI] [PubMed] [Google Scholar]
- Hara Y, and Merten CA (2015). Dynein-Based Accumulation of Membranes Regulates Nuclear Expansion in Xenopus laevis Egg Extracts. Dev. Cell 33, 562–575. [DOI] [PubMed] [Google Scholar]
- Hazel J, Krutkramelis K, Mooney P, Tomschik M, Gerow K, Oakey J, and Gatlin JC (2013). Changes in cytoplasmic volume are sufficient to drive spindle scaling. Science 342, 853–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Southern NM, MacIver B, Potter E, Apodaca G, Smith CP, and Zeidel ML (2005). Isolation and characterization of the Xenopus oocyte plasma membrane: a new method for studying activity of water and solute transporters. Am. J. Physiol. Renal Physiol 289, F217–24. [DOI] [PubMed] [Google Scholar]
- Jukam D, Shariati SAM, and Skotheim JM (2017). Zygotic Genome Activation in Vertebrates. Dev. Cell 42, 316–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyomitsu T, and Cheeseman IM (2012). Chromosome-and spindle-pole-derived signals generate an intrinsic code for spindle position and orientation. Nat. Cell Biol 14, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak S, and Gönczy P (2013). Mechanisms of spindle positioning: cortical force generators in the limelight. Curr. Opin. Cell Biol 25, 741–748. [DOI] [PubMed] [Google Scholar]
- Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, and Corbett AH (2007). Classical Nuclear Localization Signals: Definition, Function, and Interaction with Importin α. J. Biol. Chem 282, 5101–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SW, Harreman MT, Hodel MR, Hodel AE, and Corbett AH (2003). Dissection of the Karyopherin a Nuclear Localization Signal (NLS)-binding Groove. J. Biol. Chem 278, 41947–41953. [DOI] [PubMed] [Google Scholar]
- Levy DL, and Heald R (2010). Nuclear Size Is Regulated by Importin α and Ntf2 in Xenopus. Cell 143, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DTS, and Conibear E (2015). ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 4, e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder ME, and Deschenes RJ (2007). Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol 8, 74–84. [DOI] [PubMed] [Google Scholar]
- Loughlin R, Wilbur JD, McNally FJ, Nédélec FJ, and Heald R (2011). Katanin Contributes to Interspecies Spindle Length Scaling in Xenopus. Cell 147, 1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca TJ, and Heald R (2006). Methods for studying spindle assembly and chromosome condensation in Xenopus egg extracts. Methods Mol. Biol 322, 459–474. [DOI] [PubMed] [Google Scholar]
- Martin BR (2013). Chemical approaches for profiling dynamic palmitoylation. Biochem. Soc. Trans 41, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Yamada K, and Yoneda Y (2016). Importin α: a key molecule in nuclear transport and non-transport functions. J. Biochem 160, 69–75. [DOI] [PubMed] [Google Scholar]
- di Pietro F, Echard A, and Morin X (2016). Regulation of mitotic spindle orientation: an integrated view. EMBO Rep 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proffitt KD, Madan B, Ke Z, Pendharkar V, Ding L, Lee MA, Hannoush RN, and Virshup DM (2013). Pharmacological Inhibition of the Wnt Acyltransferase PORCN Prevents Growth of WNT-Driven Mammary Cancer. Cancer Res 73, 502–507. [DOI] [PubMed] [Google Scholar]
- Riggs B, Bergman ZJ, and Heald R (2012). Altering membrane topology with Sar1 does not impair spindle assembly in Xenopus egg extracts. Cytoskeleton (Hoboken) 69, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuković LD, Jevtić P, Edens LJ, and Levy DL (2016). New Insights into Mechanisms and Functions of Nuclear Size Regulation. In International Review of Cell and Molecular Biology, pp. 1–59. [DOI] [PubMed]
- Wan J, Roth AF, Bailey AO, and Davis NG (2007). Palmitoylated proteins: purification and identification. Nat. Protoc 2, 1573–1584. [DOI] [PubMed] [Google Scholar]
- Wilbur JD, and Heald R (2013). Mitotic spindle scaling during Xenopus development by kif2a and importin α. Elife 2, e00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wühr M, Chen Y, Dumont S, Groen AC, Needleman DJ, Salic A, and Mitchison TJ (2008). Evidence for an upper limit to mitotic spindle length. Curr. Biol 18, 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J-P, Mahalingham B, Alonso JL, Borrelli LA, Rui X, Anand S, Hyman BT, Rysiok T, Müller-Pompalla D, Goodman SL, et al. (2009). Crystal structure of the complete integrin aV13 ectodomain plus an a/1 transmembrane fragment. J. Cell Biol 186, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Left: Sucrose gradient of egg membranes to which recombinant GFP-importin α was added. Right: Fractions illuminated with blue light reveal that exogenous importin α co-fractionates at a much higher concentration with the plasma membrane-containing than the heavier membrane fraction (Hill et al., 2005).
(B) Blot of immunoprecpitated Ras and importin α subjected to ABE chemistry to detect palmitoylation. Omission of 1 μM hydroxylamine (HA) serves as a negative control.
(A) Line scan quantification of TPX2 immunofluorescence intensity with normalized spindle lengths upon addition of DMSO or 10 μM palmostatin.
(B) Quantification of microtubule pelleting upon addition of DMSO or 10 μM palmostatin to spindle assembly reactions in egg extacts. Palmostatin increased the fraction of XCTK2, TPX2 and kif2a bound to microtubules, but did not affect chromokinesin Xkid.
(C) Immunoprecipitation of kif2a and immunoblot of importin α following addition of DMSO or 10 μM palmostatin to egg extract. Palmostatin decreased the amount of importin α associated with kif2a.
(D) Immunoprecipitation of GFP-lamin B3 and immunoblot of importin α following treatment of extracts with DMSO, 50 μM palmostatin or 10 μM Wnt-C59. Palmostatin decreased the amount of importin α associated with the nuclear lamin, while Wnt-C59 increased co-precipitating importin α.
(E) Mean intensity ratio of importin α at the edge compared to the center in extract droplets encapsulated using synthetic or physiological lipids. Mean ± SD from 18 droplets, p < 0.005.
(F) Mean intensity ratio of importin α at the cell membrane compared to the cell center in RPE-1 cells that have been treated with DMSO, palmostatin, or Wnt-C59. Mean ± SD from 30 cells, p < 0.05.
(G) Blot of HEK-293 cells transfected with either control, LYPLA1 or PORCN siRNA.