

Abstract
Most prostate cancer cases remain indolent for long periods of time, but metastatic progression quickly worsens the prognosis and leads to mortality. However, little is known about what promotes the metastasis of prostate cancer and there is a lack of effective prognostic indicators, making it immensely difficult to manage options for treatment or surveillance. Arginyltransferase 1 (Ate1) is the enzyme mediating post-translational protein arginylation, which has recently been identified as a master regulator affecting many cancer-relevant pathways including stress response, cell cycle checkpoints, and cell migration/adhesion. However, the precise role of Ate1 in cancer remains unknown. In this study, we found the occurrence of metastasis of prostate cancer is inversely correlated with the levels of Ate1 protein and mRNA in the primary tumor. We also found that metastatic prostate cancer cell lines have a reduced level of Ate1 protein compared to non-metastatic cell lines, and that a depletion of Ate1 drives prostate cancer cells towards more aggressive pro-metastatic phenotypes without affecting proliferation rates. Furthermore, we demonstrated that a reduction of Ate1 can result from chronic stress, and that shRNA-reduced Ate1 increases cellular resistance to stress, and drives spontaneous and stress-induced genomic mutations. Finally, by using a prostate orthotropic xenograft mouse model, we found that a reduction of Ate1 was sufficient to enhance the metastatic phenotypes of prostate cancer cell line PC-3 in vivo. Our study revealed a novel role of Ate1 in suppressing prostate cancer metastasis, which has a profound significance for establishing metastatic indicators for prostate cancer, and for finding potential treatments to prevent its metastasis.
Introduction:
Prostate cancer (PC) is the second-most diagnosed and the second-most lethal cancer in men in U.S. (1). The mortality from prostate cancer is predominantly caused by metastasis for which effective treatments are limited. From a clinical perspective, it is desirable to definitively treat primary disease in those individuals at high risk of metastasis. However, it remains difficult to discriminate between aggressive and indolent prostate cancer cases. Although significant work has been done to study the driving factors of PC metastasis, our understanding is still incomplete. Thus, making decisions regarding treatment vs. surveillance for primary PC is difficult. For these reasons, a better understanding of PC metastasis would allow for more informed clinical decision making, and lead to new approaches and targets for preventing or treating metastasis.
While the causes of metastasis of any cancer are highly complicated, metastatic occurrence has been linked to several cellular pathways that regulate invasive phenotypes or increase fitness and survival of cancer cells in local and distal tumor microenvironments. These processes include the disruption of normal cell death pathways, and the misregulation of several crucial cytoskeletal pathways governing cell-cell adhesion, cell-matrix interactions, and cell migration, all of which are also associated the accumulation of mutations in the cell (2, 3). Interestingly, all these processes were recently shown to be regulate by post-translational arginylation, although no causal link has been established between arginylation and cancer metastasis (4–7).
Post-translational arginylation is the addition of an extra arginine to the protein, typically on the N-terminus. The best-studied consequences of protein arginylation include changes of the half-life and/or function of the target protein (6, 8). Arginylation preferentially takes place on proteins that are oxidized, misfolded, or otherwise damaged, implying a role for this modification in protein quality control and in stress response (7–9). While the molecular pathways affected by arginylation are still poorly understood, hundreds of proteins have been identified to be arginylated in vivo, indicating a broad impact for this modification (10–12). In mammals including humans, arginylation is solely mediated by Arginyltransferase (Ate1), an enzyme that is highly conserved across bacteria and Eukarya (13). While arginylation is still a poorly-studied phenomenon, the current aggregate understanding of Ate1 implicates it as a master regulator in the cell. Ate1 is ubiquitously expressed in all tissues in animals (14–16). The cellular enzymatic activity of Ate1 in yeast and mammalian cells directly correlates with Ate1 protein level, making Ate1 protein quantification a good proxy for cellular arginylation activity (4,11,15). Previous reports have shown that Ate1 is indispensable for mouse embryogenesis, and that Ate1-knockout fibroblasts have disrupted cell-cell interactions, cell cycle regulation, DNA damage response, and stress response in vitro (4, 6, 17). While many of these phenotypes are commonly related to cancer development, the role of Ate1 in cancer have not been directly examined. Recently, our analyses of publicly available human cancer datasets revealed that Ate1 is reduced in certain types of cancer including prostate cancer. Notably, a further reduction of Ate1 expression was detected in metastatic sites from prostate cancer compared to the primary tumor(17). These observations raised the possibility of Ate1 involvement in metastasis of this disease. However, it is unclear whether the reduction of Ate1 has any causal effects, or if it is mainly correlative for metastatic prostate cancer (18). This study provides evidence that a reduction in Ate1 increases spontaneous and stress-induced mutagenesis, reduces stress-mediated cell death, and promotes cell invasion and migration. Furthermore, by using cell-based and animal-based test models, we found that a reduction of Ate1 is sufficient to drive prostate cancer metastasis
Results:
Reduced Ate1 in primary prostate tumor tissue correlates with current and future metastasis.
In a previous study, we observed that Ate1 is down regulated in remote metastatic sites as compared to primary prostate tumors (17). However, from the standpoint of finding a cause or a potential indicator for metastasis, it is more desirable to determine whether reduced Ate1 in the primary tumor site is predictive of metastatic outcome. Thus, we first examined published, non-hypothesis-driven datasets based on quantitative mRNA sequencing of human prostate cancer samples with different outcomes (19). In the primary prostate tumors of patients with diagnosed metastatic disease, we found significantly reduced Ate1 levels compared to those in patients without metastasis (Fig 1A). To test whether there is a correlation between Ate1 expression in primary tumors and future metastatic outcome, we examined another published dataset of human PC tumors removed surgically (20). Patient outcome data over a five-year post-surgery time frame is available for these samples. Analysis of this dataset revealed that patients who would suffer metastasis of PC within the five-year window have a significantly lower expression level of ATE1 in the primary prostate tumors, compared to those individuals without a metastatic outcome (Fig 1B). To validate the above mRNA-derived results at the protein level, tissue arrays containing human PC samples with or without metastatic outcomes, and individual-matched controls of benign prostate tissues were analyzed. Consistent with the mRNA-based results described above, data from immunostaining showed a significant reduction of Ate1 protein in the primary tumors from individuals with a metastatic outcome, compared to primary tumors from patients who did not exhibit metastases (Fig. 1C). These results suggest that Ate1 may have value as a prognostic indicator of metastasis.
Figure 1: Reduced Ate1 levels are associated with the metastatic outcome of human prostate cancer and increased aggressiveness in prostate cancer cell lines.
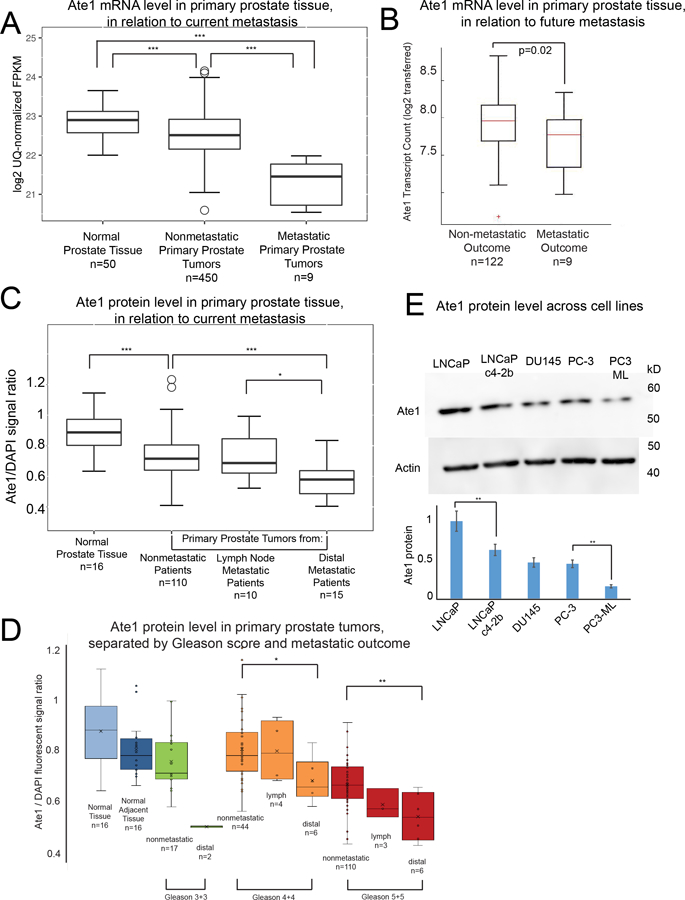
(A) Box and whisker graph showing mRNA level in Fragments per Kilobase of transcript per million mapped reads (FPKM) of Ate1 in primary tumors from patients with metastatic or non-metastatic status in comparison to normal prostate tissue. The data for mRNA were retrieved from the TCGA-PRAD dataset as well as the Beltran dataset (36). The mRNA levels were assessed by RNA sequencing in these databases. See Supp. Fig. S1 for additional information on the normalization protocol. (B) Analysis of dataset of RNA level examined by microarray in excised primary prostate cancer tissue mRNA that had up to 5 years of patient follow-up after time of radical prostatectomy (20). Samples were separated into groups by patient outcome (metastatic or not) to determine the relationship between future metastatic outcome and the Ate1 mRNA levels in the primary tumors. (C) Quantification of Ate1 protein levels on human primary prostate tumor sites by immunohistochemistry, with DAPI staining for loading normalization with DNA. The analysis was performed on human prostate cancer arrays containing primary prostate tumors with either metastatic or non-metastatic outcome, as well as normal (non-diseased) prostate tissues. The samples were separately grouped by the patient outcome (metastatic or non-metastatic) for analysis. (D) Protein level of Ate1 in tissue samples from C were further stratified by Gleason Score and metastatic outcomes. See also Supp. Fig. S2 for representative images. (E) A representative Western blot showing protein levels of Ate1 in multiple human prostate cancer cell lines, including androgen-dependent and indolent cell line LNCaP, its castration-resistant derivative LNCaP c4–2b cells, castration-resistant and intermediate aggressive cell lines DU145 and PC3, and the castration-resistant and highly aggressive PC3-ML, a derivative of PC3. Actin was used as a loading control. The graph shows the quantification from 3 independent repeats, showing a trend of reduced Ate1 correlating with cell line progression and aggressive phenotypes. Statistical significance was assessed with two-tailed Mann-Whitney U test for the human samples, and with the Student’s t-test for cell-based data. * = p<0.05, ** = p<0.01, *** = p<0.001. The monoclonal Ate1 antibody (clone 6F11, Millipore) used in this study has been shown to be highly specific for Western blot and IHC(30, 40). See also Supp. Fig. S3 for additional data demonstrating the specificity of this antibody.
In clinical practice, the malignancy of prostate cancer is often assessed using the morphology-based Gleason score (on a scale of 1–10). The extremely high scores (9 or 10) are more likely to metastasize than lower scores (21). For this reason, we also examined whether there is a correlation between Ate1 and Gleason score and found that the Ate1 level in Gleason score 9/10 group is lower than primary tumors of other scores (Fig 1D, Supp. Fig. S1). Because most patients are diagnosed with lower Gleason scores and lack prognostic indicators of metastasis (22), we next analyzed samples sorted by Gleason score and separated the metastatic and the non-metastatic outcome within each Gleason score. We found that primary tumors with the metastatic outcome consistently showed lower level of Ate1 than the non-metastatic counterpart with the same score. This correlation stays true even among low and intermediate Gleason scores (Fig 1D, Supp. Fig. S1). These data suggest that a reduction of Ate1 in the primary cancer site is correlated with metastasis, and that this relationship is independent of Gleason score. For representative IHC images of histology samples, see Supp. Fig. S2. The Ate1 antibody is known to be highly specific, as further supported by evidence in western blot and immunofluorescence (Supp. Fig. S3).
Ate1 expression inversely correlates with malignancy among human prostate cancer cell lines
Several prostate cancer cell lines have been isolated from human patients to mimic different types and progression stages of prostate cancer in vitro. These cell lines are aimed to represent and reproduce prostate cancer’s clinically significant ability to invade and metastasize, as well as the ability to grow in the absence of exogenous androgens, termed castration resistance (23). To test whether the expression level of Ate1 also correlates with relative malignancy in prostate cancer cell lines, we compared the protein levels of several PC cell lines including LNCaP, LNCaP c4–2b, DU145, PC-3, and PC3-ML (Fig 1E). We found that the non-invasive and androgen-dependent LNCaP cell line has the highest level of Ate1, while the isogenic but castration-resistant and more invasive counterpart LNCaP c4–2b loss about 40% of Ate1. The other castration-resistant and invasive PC cell lines PC-3 and DU145 showed half the Ate1 level compared to LNCaP. Finally, a further reduction of Ate1 was found in PC3-ML cells, a derivative of PC-3 that is more competent in forming distal metastases in mice. This inverse correlation between Ate1 level and the malignancy of PC cell lines is consistent with the observations made in human samples and further supports a possible role of Ate1 in prostate cancer progression.
A reduction of Ate1 protein is seen in cell populations following chronic stress to confer higher stress resistance.
Previous studies demonstrated that fibroblasts quickly upregulate Ate1 in response to high levels of stressors in a matter of hours, and that such an up-regulation is essential for normal stress responses including growth arrest and apoptosis (4). While Ate1 is upregulated in short-term stress, results from this study and previous reports (17) point to a consistent decrease in Ate1 in tumors, which experience prolonged exposure to stressors such as reactive oxygen species (ROS). To examine long-term effects of stress exposure on the cellular level of Ate1, PC-3 and LNCaP cells were stressed for one week with sublethal doses of H2O2, a naturally occurring cellular oxidant commonly associated with oxidative stress or inflammation. We found that Ate1 was reduced by 50–60% in each cell population, and the reduced level of Ate1 in PC-3 remained unchanged for at least 3 days even after the stressor H2O2 was removed (Fig. 2A–B). This result suggests that the level of Ate1 in these cell populations is downregulated in responding to chronic stress.
Figure 2: Ate1 is downregulated following chronic stress.
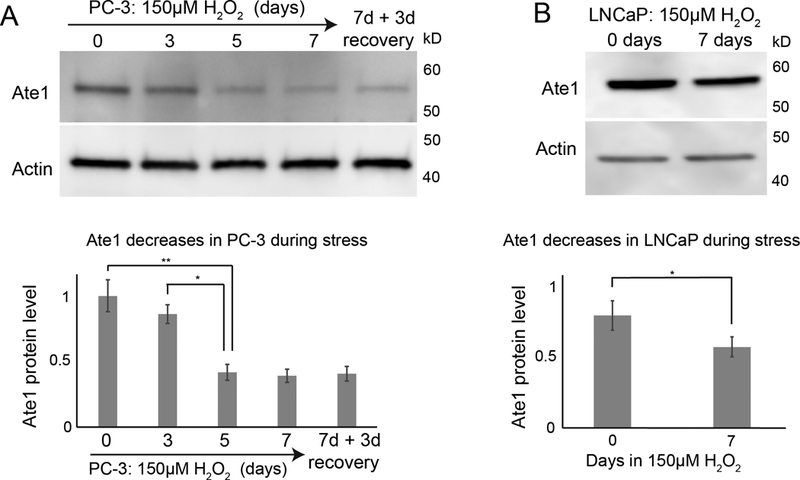
(A) Top panel is a representative Western blot showing the level of Ate1 for PC-3 cells treated with culture media supplemented with 150 μM H2O2 for 7 days then allowed to recover for 3 days in normal media. For matching time points, the “0”-treatment group was treated with H2O through the time course. Actin was used as loading control. Bottom graph shows quantification from three independent repeats. (B) LNCaP cells were treated with 150 μM H2O2 or H2O control for 7 days and Ate1 levels were measured by Western blot. Actin was used as loading control. Bottom graph shows quantification from three independent repeats. Error bars represent Standard Error of Mean (SEM). Statistical significance based on p value from the Student’s t-test: * = p<0.05, ** = p<0.01.
Since Ate1 was shown to be essential for apoptosis during high levels of stress, we hypothesized that the reduction of Ate1 in prostate cancer cells would increase the stress resistance to tumor environment conditions or therapeutic treatments. To test this possibility, we knocked down ATE1 in LNCaP and PC-3 cells with shRNAs targeting ATE1 (sh-ATE1 #1 and #2) or a nonsilencing (sh-NS) control (Fig 3A,B). PC3-ML, an isogenic derivative of PC-3 with higher metastatic potential and a naturally lower level of Ate1, was also used as a positive control. These cells were challenged with increasing concentrations of H2O2. We found that a reduced level of Ate1 is associated with increased cell survival following H2O2 treatment in a dose-dependent manner (Fig 3 C). To test the role of Ate1 in cellular resistance to other types of exogenous and cancer-relevant stress, we next subjected these cells to various doses of Staurosporine (STS), a potent inducer of apoptosis. In both PC-3 and LNCaP cells, the down-regulation of Ate1 increased cell viability in STS treatment (Fig 3D). Finally, we employed gamma irradiation, a common treatment used in radiotherapy, to examine the potential effect of lower Ate1 on resistance to radiation. Seventy-two hours after treatment, the remaining viable cell number was assessed. We found that a reduction of Ate1 corresponded to higher surviving cells (Fig 3E). To further validate this conclusion, we expressed a recombinant GFP-fused Ate1 protein in PC3-ML cells, a highly malignant prostate cancer cell line with reduced Ate1 level and less sensitive to stress compared to its less malignant counterpart PC3. We found that the expression of Ate1-GFP, but not the GFP alone, was able to re-sensitize PC3-ML cells to both STS and gamma irradiation (Figure 4). Together, our data suggests that the reduction of Ate1 in prostate cancer cells leads to cellular resilience when challenged with cancer-relevant stressors, providing low-Ate1 cancer cells with an advantage for survival and proliferation within the tumor environment.
Figure 3: Ate1 reduction in prostate cancer cells promotes stress resistance.
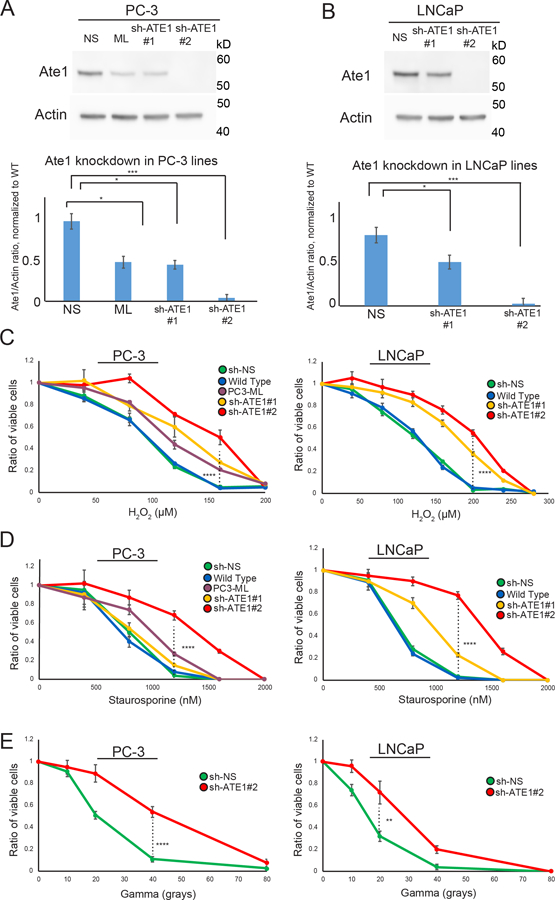
(A) Top panel shows representative Western blot of Ate1 and actin (loading control) for PC-3 cells, which were stably infected with two different shRNAs (#1 and #2) targeting Ate1, or the nonsilencing control. PC3-ML cells, which have naturally lower level of Ate1 compared to the parental PC3 cells, were used as another control. Bottom panel shows quantification from three independent repeats, showing that sh-ATE1#1 has 40–60% efficiency, while #2 has >95% efficiency. For this reason #2 is used for knockdown of Ate1 through this study unless otherwise specified. (B) Similarly as done in (A), Western blots and their corresponding quantifications were presented for LNCaP (C) PC-3 and LNCaP cells either transduced with shRNA against Ate1 or non-silencing control or non-transduced were treated with various doses of H2O2 or Staurosporine for 12 hours, or a single dose of Gamma irradiation followed by 24 hours of recovery, then assessed for cell viability via Calcein AM staining. Graphs showing quantification from three independent experiments. Error bars represent SEM. Statistical significance of group comparison at select data points were assessed with the Student’s t-test: * = p<0.05, *** = p<0.001. See also Supp. Fig. S4 for similar effects of Ate1 reduction on STS and gamma irradiation for human foreskin fibroblasts (HFF), a non-tumorigenic primary cell line.
Figure 4: Ate1 overexpression in prostate cancer cells reduces stress resistance.
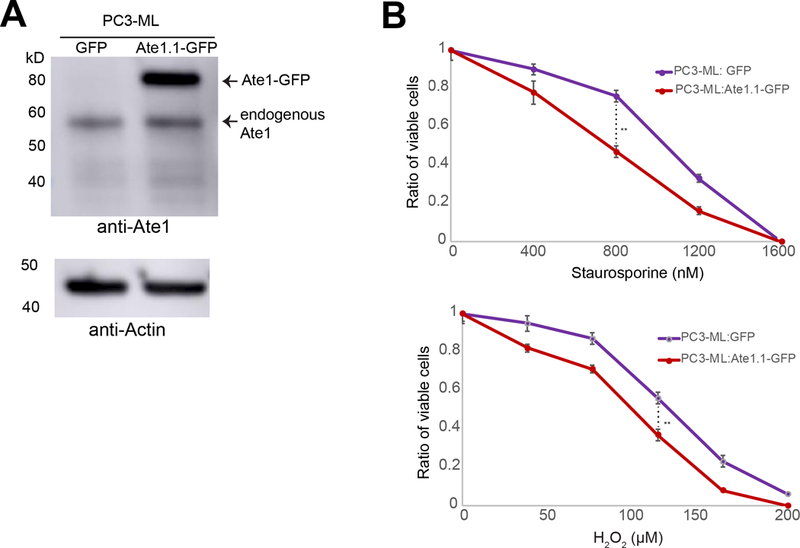
(A) Representative western blot of PC3-ML cells expressing Ate1-GFP. For the construction of recombinant protein, Ate1 is represented by isoform 1 (Ate1.1), the most potent and ubiquitous splice variant of Ate1. It was fused with a C-terminal GFP and cloned into a retroviral vector pBABE-puro for low-level expression in mammalian cells, which is fully functional and not expected to cause cell death as shown by previous reports(4, 30). The expression of GFP alone was used as a control. (B) PC3-ML cells transduced with Ate1.1-GFP or GFP control were treated with various doses of H2O2 or Staurosporine for 12 hours, then assessed for cell viability via Calcein AM staining. Graphs showing quantification from three independent experiments. Error bars represent SEM. Statistical significance of group comparison at select data points were assessed with the Student’s t-test: ** = p<0.01.
Ate1 reduction increases spontaneous and induced mutagenesis
DNA mutations are essential for cancer progression and metastasis. Previous studies showed that ATE1-KO mouse embryonic fibroblasts (MEF) had significant chromosomal aberrations and showed increased mutation frequency following UV irradiations, suggesting a potential role of Ate1 in suppressing the accumulation of mutations (4, 17). To test whether a down-regulation of Ate1 in prostate cancer cells would also lead to higher mutagenic frequencies under DNA-damaging stresses, PC-3 cells with shRNA against ATE1 or a non-targeting control were challenged with sub-lethal mutagenic treatments. These mutagenic conditions include 3mM N-ethyl-N-nitrosourea (ENU), a DNA damage-inducing chemical, or a sublethal 10Gy dose of gamma irradiation, an ionizing and DNA-damaging radiation. Mutation frequencies in the cells were then assessed with the CherryOFF-GFP point mutation reporter as previously described (4). Briefly, a GFP-mCherry dual fluorescent reporter carries a nonsense point mutation on residue TRP98 on mCherry, where only a specific new mutation in that position can restore the red fluorescence, detected with flow cytometry. By using this approach, we found that, compared to the non-targeting control, ATE1-knockdown in PC-3 cells leads to significant increases in mutation frequencies after the treatment of either ENU or gamma irradiation (Figure 5). Additionally, we also observed a smaller but statistically significant increase in mutation frequency in Ate1-reduced cells even without exogenous stress, where endogenous and basal level of stressors may still exit. These results suggest that a down-regulation of Ate1 in cancer cells would allow higher frequencies of mutation, potentially facilitating the acquisition of additional mutations driving metastasis and other malignant phenotypes.
Figure 5: Ate1 reduction increases spontaneous and stress-induced mutagenesis in PC-3 cells.
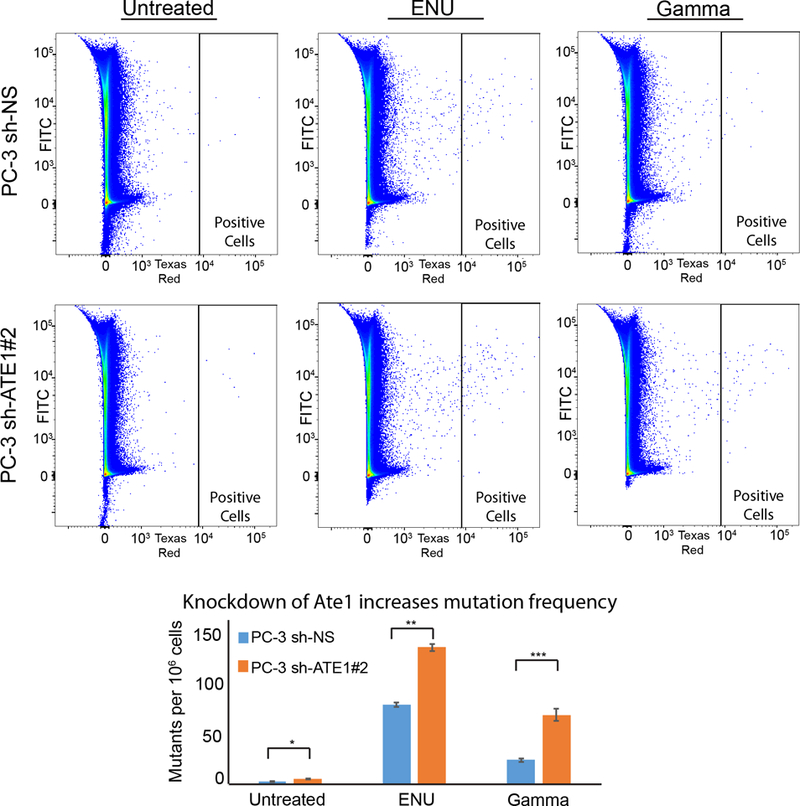
(A) Representative flow diagrams of PC-3 cells carrying CherryOFF-GFP reporter and pLKO.1-puro shRNA targeting Ate1 (shRNA #2) or NS control are shown. Cells were either treated with 3mM ENU for 24 hours, or irradiated with 10Gy of gamma radiation. 72 hours after the initiation of treatments, cells were analyzed by FACS for the acquired mutations that activate the mCherryFP fluorescence (indicated in the window of “Positive Cells”). (B) Quantification of A from three independent repeats shows the changes of mutation rates with the knockdown of Ate1. Error bars represent SEM. Statistical significances of the two-group comparison were assessed with the Student’s t-test: * = p<0.05, ** = p<0.01, *** = p<0.001.
ATE1 reduction drives aggressive prostate cancer cell phenotypes without affecting proliferation rate
To better determine whether the reduction of ATE1 is a driving cause for cancer progression or just a correlative hallmark, we used several established cell-based methods to examine the effects of ATE1-knockdown on prostate cancer cells. Since the readout of most cell-based assays is affected by changes in cell proliferation rates, we first measured whether the cell proliferation rates are affected by the reduction of Ate1. We found that, in non-confluent and non-stressed conditions, shRNA-knockdown of Ate1 has no significant effects on growth rates of either LNCaP or PC-3 cells compared to non-targeting shRNA (Fig 6A).
Figure 6: Ate1 reduction increases soft-agar colony formation, migration, and invasion in prostate cancer cells without affecting proliferation rates.
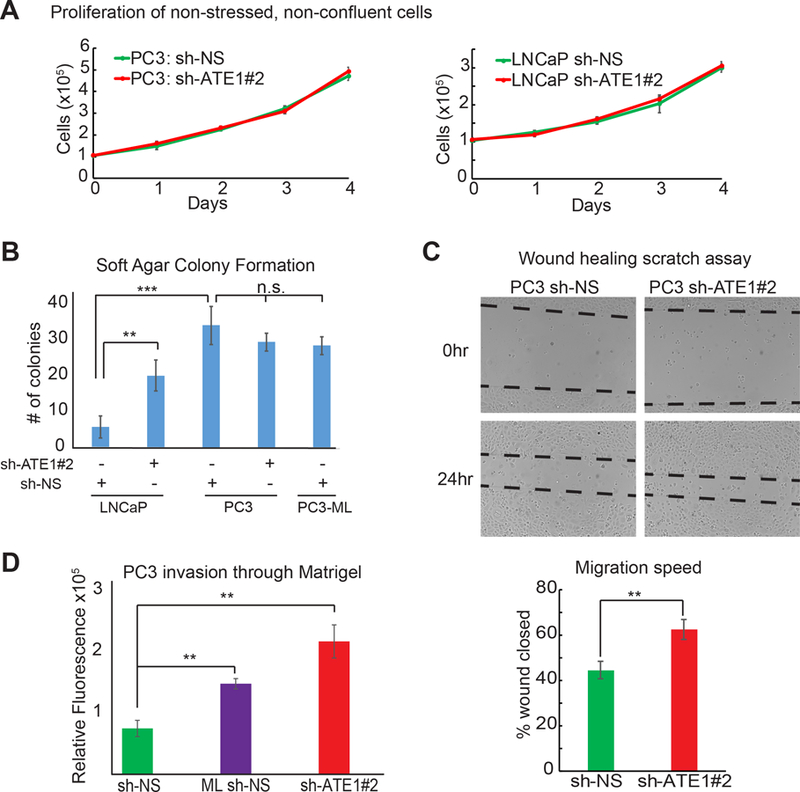
(A) Cell proliferation curves of PC-3 and LNCaP cells expressing shRNA targeting Ate1 (shRNA #2) or NS control are shown. These cells were plated in 10-cm dishes at low density and grown for 4 days without reaching confluency. Viable cells were treated with Calcein AM dye and counted. (B) Graph shows numbers of colonies formed after 2 weeks in soft agar suspensions on 35mm dishes by LNCaP and PC-3 cells expressing sh-ATE1#2 or NS control. Three independent experiments were performed. (C) Left side is the representative photos of PC-3 cells expressing shRNA targeting Ate1 (shRNA #2) or NS control in the wound healing migration assay, before and after 24 hours incubation. The dashed lines indicate edges of the cell wound. Right side is the quantification from three independent experimental repeats. (D) Quantification of cell invasion through Matrigel in Boyden chamber is shown from PC-3 cells expressing shRNA targeting Ate1 (shRNA #2) or NS control. PC3-ML, which is known to be more invasive than PC3, is used as a positive control. The experiment was repeated three times. All error bars represent SEM. Statistical significance based on the Student’s t-test: ** = p<0.01, *** = p<0.001. See also Supp. Fig. S6 for the effects of Ate1 over-expression in PC3-ML on cell migration and Matrigel invasion.
To test whether Ate1 has any effects on the tumorigenic potential of cancer cells, we used soft agar to examine the anchorage-independent colony formation of these cells as previously described (24). We found that a knockdown of Ate1 in LNCaP cells led to significantly more colonies formed by these cells, suggesting that ATE1 is essential for the suppression of this tumorigenic phenotype (Fig 6B). However, the colony formation rate of PC-3 cells or PC3-ML cells were not significantly affected by ATE1 knockdown, likely because these cell lines are highly malignant and already exhibit the maximal level of tumorigenesis readable by this assay.
To test whether ATE1 affects metastasis, we first examined its effects on cell migration by a scratch wound-healing assay on PC-3 cells with Ate1-knockdown or control (25). We found that Ate1-knockdown cells migrated into the wound significantly faster than the control (Fig 6C). We further examined the effect of Ate1 knockdown on invasion of basement membrane by using a Boyden chamber coated with Matrigel as previously described (26). We found that the shRNA-knockdown of Ate1 in PC-3 cells led to a significantly higher invasion rate than the non-targeting control, to a level similar to the positive control of PC3-ML cells, a highly metastatic derivative of PC-3 with naturally down-regulated Ate1 level (Fig 6D). Overall, these data suggest that a reduction of Ate1 in prostate cancer cells can drive progression by provoking anchorage-independent growth, migration, and invasion.
Ate1 reduction drives metastasis of prostate cancer cell in a mouse model
The bioinformatics and cell-based data in this study provide a strong rationale to speculate that the reduction of Ate1 plays a causative role in prostate cancer metastasis. To test this possibility in a prostate-relevant environment in an animal model, we employed prostate orthotropic xenografts on athymic nude mice. PC-3 cells are less competent in forming distal metastases in this test environment, while its derivative PC3-ML cells, which we found to have a naturally lower level of Ate1 than PC-3, are shown to be highly competent to metastasize (27, 28). To test whether a down-regulation of Ate1 would enhance the metastatic potential, PC-3 cells expressing luciferase were stably transduced with shRNA silencing ATE1 (or the non-targeting control). These cells were also transduced with luciferase for monitoring the size and location of primary and any secondary tumors. While no significant difference among the sizes of primary prostate tumors was observed between these cells (Fig 7 A,B), Ate1 knockdown in PC-3 cells drastically increased their frequency of local invasion in seminal vesicles (SV) or testicles, and distal metastasis outside of prostate, SV and testicles (Fig 7C and Supp. Fig. S7). Particularly, the chance of developing a distal metastasis in individual mice by the PC-3 cells with Ate1-knocdown reached a level similar to the positive control of PC3-ML cells, although the number of metastases per mouse was still lower than PC3-ML (Fig 7D). These data suggest that a reduction of Ate1 has a causative effect for both local invasion and distal metastasis in animals.
Figure 7: Ate1 reduction drives metastasis of prostate orthotopic xenograft in mice.
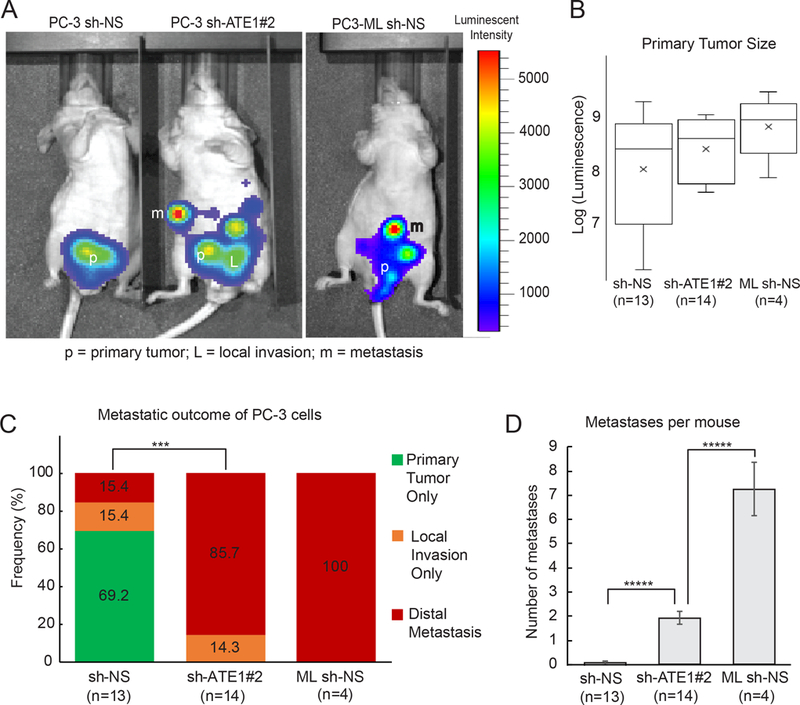
(A) Representative photo of luminescence detection of mouse tumors, 40 days after orthotopic injection of PC-3 and PC3-ML cells into the dorsal prostates of nude mice. The PC3 cells were transduced shRNA targeting Ate1 (shRNA #2) or NS control, and also transduced with luciferase as a tracing marker. PC3-ML, which has been shown to metastasize following orthotopic xenograft (27) was used as a positive control and transduced with NS control shRNA and luciferase. Arrows point to a few remote metastatic sites in these mice. (B) Quantification of primary tumor burden in these mice at the termination of the study was determined by bioluminescent signal count. No statistical significance was found. (C) Percentage of mice containing either only primary tumor, or with local invasion, or distal metastasis by these different cell lines is shown. Statistical significance was assessed by the Fisher’s exact test. *** = p<0.001. (D) Quantification of the average number of distal metastases per mouse. Error bars represent SEM. Statistical significance based on the Student’s t-test: **** = p<0.0001. See also Supp Fig. S7 for representative H&E stained images for the morphology of primary and metastatic tumors, as well as the distribution of the metastatic locations, formed by these above mentioned cells.
Discussion:
The causes of metastasis in prostate cancer are poorly understood. In this study, we found that a down-regulation of Ate1 is commonly detected in advanced stages of prostate cancer, and is correlated with the risk of metastasis. We further showed that Ate1 is down-regulated in prostate cancer cells after chronic stress, and a decrease in Ate1 level is sufficient to increase stress resistance, mutagenesis, anoikis resistance, migration, and invasion at the cellular level. Finally, by using a prostate orthotopic xenograft model, we demonstrated that a reduction of Ate1 in PC-3 cells is sufficient to promote seminal vesicle invasion and distal metastasis of tumor cells. These results suggest Ate1 is required to prevent metastasis of prostate cancer, and that a down-regulation of Ate1 may hold prognostic value for metastasis.
Ate1 is known to be exceptionally conserved through eukaryotic organisms and is also present in a large subset of bacteria. However, our understanding of the physiological relevance of Ate1 is still very limited. Our data suggest several possible pathways for Ate1 to influence the risk of metastasis. As previous studies have shown, Ate1 is essential to regulate normal cell-cell interactions and contact inhibition (17, 29). Ate1 was also shown being essential for stress-induced cell cycle checkpoints and/or apoptosis in mouse fibroblasts (17). Consistently, in this study, similar effects were observed in both human prostate cancer cells (Fig. 3 and 4) and non-tumorigenic Human Foreskin Fibroblasts (Supp. Fig. S4). In particular, while in consistence to previous studies(30), no effects of Ate1 on the growth of prostate cancer cells were observed in non-stressed, non-confluent conditions, a reduction of Ate1 maintains cellular viabilities under chemical stressors or radiation. Therefore a reduced Ate1 would generate growth advantage for cancer cells under the often stressing tumor environments. Furthermore, the present study showed Ate1 reduction increased both spontaneous and exogenous stress-induced mutations in prostate cancer cells, which is another critical driving force for metastasis (31). These effects of Ate1 on stress response, cell cycle checkpoint, cytoskeletal activities, and mutagenesis could explain its role in regulating metastasis (Supp. Fig. S8). In addition to metastasis, Ate1 may also play significant roles in other stages of prostate cancer progression based on the results of this study. For example, we found that the knockdown of Ate1 in LNCaP cells leads to higher colony-formation in soft agar, an indicator for higher tumorigenic potential. This result appears consistent with our other report showing that immortalized MEF cells with Ate1 knocked out spontaneously form tumors in the subcutaneous xenograft assay in mice (17). Furthermore, as seen in the survey of Ate1 expression across cell lines, all tested castration resistant lines showed markedly lower Ate1 expression than androgen-dependent LNCaPs, including the castration-resistant LNCaP-derivative C4–2b. Since AR is a transcription regulator, it is tempting to speculate whether the gene ATE1 is one of its targets. However, when we treated AR-positive LNCaP cells with AR agonist R1881, unlike known AR targets such as FKBP5, the mRNA level of Ate1 was not affected by such a treatment (Supp. Fig. S5). The relationship between AR and Ate1 may require further investigation.
While our data suggest that Ate1 may play a role in suppressing prostate cancer metastasis, it may not act in the manner of a classic metastasis suppressor or tumor suppressor. Ate1 is the only enzyme found in metazoans to mediate posttranslational arginylation. Based on the sequence-based prediction of the N-end rule, which hypothesizes that proteins bearing D, E, N, Q, or C on the 2nd residue are preferable substrates for Ate1, ~25% of any given proteome would be affected by this enzyme. Therefore, Ate1 potentially acts as a global regulator with profound effects on many fundamental cellular pathways. While examination of publicly available datasets from cBioPortal shows that mutations or individual variations of the Ate1 gene are extremely rare in both normal tissues and cancer tissues, changes in its expression level are more common in cancer. This result is consistent with the observations of many other known master regulator genes such as TGF-beta and heat shock proteins, whose expression, rather than mutation, are found to be associated with risk of metastasis (32–34). Furthermore, Ate1 likely affects metastasis in both direct and indirect manners, since it affects several metastasis-related pathways including stress response, cytoskeletal activity, and mutagenesis. Interestingly, a moderate over-expression of recombinant Ate1 resensitizes PC3-ML cells to stress (Figure 4) and also reduced their migration speeds (Supp. Fig. S6), suggesting that these may derived from direct effects of Ate1. However, expression of recombinant Ate1 itself could not reduce the invasive capacity of PC3-ML cells in Matrigel (Supp. Fig. S6). It important to point out that, PC3-ML cells were derived by repeatedly harvesting the low-frequency bone-metastatic colonies from PC3 xenograft in mice. Consistent to the naturally reduced Ate1 level seen in PC3-ML, these cells may contain additional mutations and/or other irreversible adaptations compared to their parental PC-3 cells. If any of these mutations/adaptations are involved in invasion, they would not be reversed by the reintroduction of Ate1. Additional studies would be desirable to elucidate how these different Ate1-depdent effects are involved in the promotion of prostate cancer metastasis.
In this study, by examining publicly available datasets and analyzing tumor tissue arrays of human prostate cancer samples, we found a significant inverse correlation between Ate1 and prostate cancer metastasis. Although many prostate cancers remain indolent for years or even decades, progression to a metastatic state quickly worsens prognosis and survival. However, predicting metastasis in prostate cancer is extremely difficult and cannot be reliably accomplished with current clinically accepted markers including PSA or Gleason scores (22). For example, while Gleason scores of 9–10 may significantly correlate with metastasis, these high score groups are extremely rare among patients at time of diagnosis; most patients are initially diagnosed with intermediate or low scores (35). As this study indicates, lower levels of Ate1 in primary prostate tumors is correlated with risk of future metastatic outcome. Furthermore, as our study found, within the same Gleason score group (including low or intermediate scores), a reduced Ate1 in primary tumor is also correlated with the metastatic outcome. Our results therefore support the potential to use the level of Ate1 as a predictive biomarker for metastasis in prostate cancer patients, although additional work is required to definitively establish this connection. Finally, while the regulatory mechanism for Ate1 expression is still unclear, this study found a sustained suppression of Ate1 expression following chronic treatments of oxidative stressor. While the mechanism of this downregulation is not yet understood, this phenomenon is of great interest because it potentially indicates the involvement of an epigenetic regulation of Ate1 expression, which requires further study to clarify.
Materials and methods
Data Mining
The “TCGA” and “Beltran” datasets were accessed as previously described (19, 36). Briefly, Fragments per Kilobase of transcript per million mapped reads (FPKM) counts of all 500 TCGA prostate-related samples, including normal and tumor samples, were downloaded from GDC Data Portal (https://portal.gdc.cancer.gov). Raw fastq data of RNA-Seq metastatic samples (dbGap phs000909.v.p1) were obtained through authorized access of another database (36). Among 49 metastatic samples in the Beltran dataset, there are 9 samples obtained at prostate site. Read mapping was performed using STAR aligner with GRCh38 reference genome (37), and transcripts in FPKM were quantified using the RSEM tool (38). To normalize across datasets, upper-quartile normalized FPKM (UQ-FPKM) were calculated across TCGA and Beltran’s data sets (Supp. Fig. S1) based on published protocols (39).
Cell Culture
HEK 293T cells (Clone T7) were obtained from ATCC (Manassas, VA, USA). HFF cells were a gift from Dr. John Murray at the University of Indiana. Immortalized MEFs (both WT and ATE1-KO) were a gift from Dr. Anna Kashina at the University of Pennsylvania. These cells were passaged and maintained in DMEM medium supplemented with 10% fetal bovine serum. Human prostate cancer cell lines PC-3, LNCaP, and prostate epithelial line RWPE-1 were obtained from ATCC, while PC3-ML were a gift from Dr. Alessandro Fatatis at Drexel University College of Medicine (Philadelphia, PA, USA) (28) and LNCaP-C4–2B were a gift from Dr. Conor Lynch at Moffitt Cancer Center (Tampa, FL, USA). Human prostate lines were cultured in RPMI medium supplemented with 10% fetal bovine serum. All cells were maintained at 37°C under 5% CO2 in a humidified incubator. Cells were detached using a 0.25% Trypsin solution (Life Technologies, CA, USA). All cells were screened to ensure the lack of mycoplasma and other common pathogens prior to use in mouse experiments.
Western Blot (WB)
Ate1 protein level was assayed using whole-cell protein lysates extracted using RIPA buffer and separated on 4–20% SDS-PAGE gels (Bio-Rad #456–8093) as previously described(4). Antibodies used were anti-Ate1 (Clone 6F11; Millipore #MABS436) and loading control anti-Beta-Actin (Clone AC-15, Sigma #A1978). Pictures were obtained using a GM Amersham Imager 600, and signals were quantified using ImageJ software.
Plasmids and lentiviral construction
Lentiviral vectors expressing shRNA targeting Ate1 (#1 – GCCATGCCTTACGGTGTTTAT (originally designed for mouse Ate1 but also cross-react with human Ate1), #2 - GCCATGCCTTACGGTGTTTAT) or scrambled control (CCTAAGGTTAAGTCGCCCTCG) were purchased from VectorBuilder, coexpressing mCherry as selection marker. pBABE-Ate1.1-GFP was generated as described in our previous work (4). For the fluorescent mutagenesis reporter experiment, these shRNA sequences were inserted into pLKO.1 puro vectors. Puromycin was used at a concentration of 1µg/uL to select pLKO.1 puro-transduced cells. Viral particles were packaged in HEK 293T cells and applied to target cells as previously described (4).
Cell proliferation and viability assays
LNCaP or PC-3 cells (with control or Ate1-targeting shRNA) were seeded at a density of 100,000 cells per 60cm dish. At each day for 4 days, representative plates were trypsinized, resuspended, and counted via slide cell counter (TC-20, Bio-Rad).
To assess viability, LNCaP or PC-3 cells were seeded in 96-well plates at a density of 10,000 cells per well and incubated overnight. Media was replaced with fresh media containing stressor as indicated, and incubated for 12 hours. Cells were washed with PBS, and incubated for 20 minutes at 37°C in PBS containing 1µg/mL Calcein AM (Thermo Fisher #C3100MP). Plates were washed with PBS, dried, and read by Fluorostar Omega platereader at 515/495nm.
Cell irradiation
Cells were treated with gamma irradiation via a Cobalt-60 source monitored by the University of Miami Radiation Control Center. Cells were placed perpendicular to source of radiation and exposed for necessary time according to distance-based calibrations.
Soft agar assay
LNCaP and PC-3 cells (with control or Ate1-targeting shRNA) were seeded in soft agar to evaluate anchorage-independent growth as previously described(24).
Migration and invasion assays
LNCaP or PC-3 cells (with control or Ate1-targeting shRNA) or PC3-ML cells (with pBABE-Ate1.1-GFP or pBABE-GFP) were serum-starved overnight then seeded at a density of 50,000 cells per well onto the top chamber of the BME-coated transwell apparatus (Trevigen #3455–024-K). Media with 10% FBS as a chemoattractant was used in the lower chamber during 48 hour incubation. The invading cells were isolated from the lower chamber and the bottom of the transwell apparatus and quantified via Calcein AM as per manufacturer’s protocol. For scratch migration assays, LNCAP, PC-3, or PC3-ML cells were grown to confluence then wounded with a 200uL pipette tip. Pictures were taken via light microscope at given time points. The percentage of wound close was calculated by comparing the average distance between the migrating border of cells at different time points. Isolated cells within the scratch area were excluded in measurements.
AR stimulation and quantification of mRNA levels
LNCaP cells were washed with PBS and incubated in serum-free media for 1 hour. They were washed again with serum-free media, then incubated for 16 hours in serum-free media containing 0.1nM AR agonist R1881 (or ethanol control). After 16 hours, RNA was extracted from the cells via Direct-zol RNA miniprep kit (Zymo Research) and analyzed on an ABI StepOne Plus qPCR machine with the Taqman gene expression assay (ThermoFisher, Waltham, MA), which provides reagents for measuring the mRNA levels of FKBP5 (assay ID: Hs01561006_m1) and GAPDH (assay ID is Hs99999905_m1). The mRNA level of human ATE1 was measured with these primers:
Forward primer CAGTTCCTAAGCCAGGCAAA
Reverse primer AGCCTGGAAACCCTCAAGTT
Probe: [5′FAM] ACTAATGCAGCAGAACCCAGCTGGA [3′TAMRA]
The mRNA levels of FKBP5, and ATE1 mRNA were normalized to relative GAPDH expression for quantification.
Orthotopic xenographs
PC-3 cells carrying luciferase and shRNA targeting Ate1 or control were prepared by sorting. The cells were injected into the dorsal prostates of 6-week-old athymic nude mice (Envigo, Indianapolis, IN, USA). Mice were randomly assigned to experimental groups under a protocol approved by University of Miami’s Institutional Animal Care and Use Committee. 5×105 PC-3 cells were injected using a repeating syringe dispenser (Hamilton, Reno, NV, USA), yielding a tumor take of 90%. Surgical sites were closed using 9mm wound staples. Investigators were not blinded in this study.
In vivo and Ex vivo imaging
Luciferase-based bioluminescence was imaged as photon flux (photons/s/cm2) on the IVIS Spectrum (Perkin Elmer, Waltham, MA, USA) once weekly starting 7 days post-injection, continuing until termination of experiment at day 40. XenoLight D-Luciferin (Perkin Elmer #122799; 150mg/kg in sterile PBS) was injected intraperitoneally 10 minutes before imaging. At day 40, mice were sacrificed, and prostate and seminal vesicles were excised. These mice were reimaged to analyze distal metastasis signal that exist outside of the prostate, seminal vesicle, and testicles sites. Prostates, seminal vesicles, testicles and any detected distal metastatic sites were harvested and placed into separate wells of a 24-well plate, supplemented with D-Luciferin, and imaged again to confirm the existence of tumor (local or distal). Photon flux was analyzed using Living Image Software (Perkin Elmer).
Histological analysis of tumor samples
Slides containing human prostate tissue sections (Biomax, Derwood, US) was stained with monoclonal rat anti-Ate1 (Clone 6F11; Millipore #MABS436) and DAPI. Optical and fluorescent cell images were obtained using a Zeiss Observer inverted microscope (Zeiss, Germany) and analyzed via Zeiss Zen Pro software.
Tissue masses harvest from the mice, which contain primary or distal tumors as validated by ex vivo imaging with luciferin, were either directly fixed with paraformaldehyde (4% in PBS) or snap-freeze with liquid nitrogen. These tissues were then further processed with paraffin-embedding and sectioning for Hematoxylin and Eosin (H&E) Staining.
Microscopy
Optical and fluorescent imaging of cells were performed on a Zeiss Observer (Jena, Germany) equipped with a series of objectives and Zen Pro software (Carl Zeiss Microscopy, Jena, Germany). Pictures displayed in migration assay were obtained using EC Plan – Neofluar 10x objective lens at 1392×1040 pixel resolution. Histology slides were imaged using a LD Plan – Neofluar 20x objective lens at 1932×1040 pixel resolution and analyzed using Zen Pro software.
Statistical Analysis
Mouse studies were designed with a pre-study power analysis which assumed tumor take with α=0.05, β=0.2, and 20% mortality rate. All healthy pups were included, and post-surgery exclusion was only based on mortality or lack of tumor take. Mice were randomized into experimental groups via a simple online random distribution generator. The investigators were not blinded at any stage.
Cell-based data, which appeared to be normally distributed with similar variances, were analyzed for significance via two-tailed Student’s t-test. Two-group comparisons of continuous outcomes in non-normally distributed human patient samples were carried out using the Mann-Whitney U test. The non-normal distributions of type of metastasis by prostate cancer cell line in the animal-based model were compared by the Fisher’s exact test. Statistical tests were two-tailed and p value p<0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
Cell sorting was performed in the core facility of Sylvester Comprehensive Cancer Center. Ex vivo imaging was performed in the small animal imaging facility at the University of Miami Miller School of medicine. Gamma irradiation was performed in the Radiation Control core facility at the University of Miami Miller School of medicine. Immunostaining of tumor array samples was performed in the histology core facility in the Hussman Institute for Human Genomics at the University of Miami Miller School of medicine. Dr. Priyamvada Rai (University of Miami) provided critical reading for the manuscript. Dr. Yuanfang Guan (University of Michigan) provided technical consultations for the analysis related to Ate1 mRNA levels.
The research presented in this article is funded in part by these grants: DoD (CDMRP), Idea Award, PC140622; DoD (CDMRP), Exploratory Grant, PC141013; NIGMS (NIH), R01GM107333.
Footnotes
Competing Interests
The authors have no competing interests to declare.
References
- 1.Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2016;25(1):16–27. Epub 2015/12/17. [DOI] [PubMed] [Google Scholar]
- 2.Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Seminars in Cancer Biology 2015;35(Supplement):S244–S75. [DOI] [PubMed] [Google Scholar]
- 3.Klein CA. Selection and adaptation during metastatic cancer progression. Nature 2013;501(7467):365–72. Epub 2013/09/21. [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, Birnbaum MD, Patel DM, Morgan WM, Singh J, Barrientos A, et al. Posttranslational arginylation enzyme Ate1 affects DNA mutagenesis by regulating stress response. Cell death & disease 2016;7(9):e2378 Epub 2016/09/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenach PA, Schikora F, Posern G. Inhibition of arginyltransferase 1 induces transcriptional activity of myocardin-related transcription factor A (MRTF-A) and promotes directional migration. The Journal of biological chemistry 2014;289(51):35376–87. Epub 2014/11/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang F, Saha S, Kashina A. Arginylation-dependent regulation of a proteolytic product of talin is essential for cell-cell adhesion. The Journal of cell biology 2012;197(6):819–36. Epub 2012/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang N, Donnelly R, Ingoglia NA. Evidence that oxidized proteins are substrates for N-terminal arginylation. Neurochemical research 1998;23(11):1411–20. Epub 1998/11/14. [DOI] [PubMed] [Google Scholar]
- 8.Varshavsky A The N-end rule pathway and regulation by proteolysis. Protein science : a publication of the Protein Society 2011;20(8):1298–345. Epub 2011/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klemperer NS, Pickart CM. Arsenite inhibits two steps in the ubiquitin-dependent proteolytic pathway. The Journal of biological chemistry 1989;264(32):19245–52. Epub 1989/11/15. [PubMed] [Google Scholar]
- 10.Wong CC, Xu T, Rai R, Bailey AO, Yates JR 3rd, Wolf YI, et al. Global analysis of posttranslational protein arginylation. PLoS biology 2007;5(10):e258 Epub 2007/09/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piatkov KI, Brower CS, Varshavsky A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proceedings of the National Academy of Sciences of the United States of America 2012;109(27):E1839–47. Epub 2012/06/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Decca MB, Bosc C, Luche S, Brugiere S, Job D, Rabilloud T, et al. Protein arginylation in rat brain cytosol: a proteomic analysis. Neurochemical research 2006;31(3):401–9. Epub 2006/05/31. [DOI] [PubMed] [Google Scholar]
- 13.Balzi E, Choder M, Chen WN, Varshavsky A, Goffeau A. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. The Journal of biological chemistry 1990;265(13):7464–71. Epub 1990/05/05. [PubMed] [Google Scholar]
- 14.Kwon YT, Kashina AS, Varshavsky A. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Molecular and cellular biology 1999;19(1):182–93. Epub 1998/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu RG, Brower CS, Wang H, Davydov IV, Sheng J, Zhou J, et al. Arginyltransferase, its specificity, putative substrates, bidirectional promoter, and splicing-derived isoforms. The Journal of biological chemistry 2006;281(43):32559–73. Epub 2006/09/01. [DOI] [PubMed] [Google Scholar]
- 16.Rai R, Mushegian A, Makarova K, Kashina A. Molecular dissection of arginyltransferases guided by similarity to bacterial peptidoglycan synthases. EMBO reports 2006;7(8):800–5. Epub 2006/07/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rai R, Zhang F, Colavita K, Leu NA, Kurosaka S, Kumar A, et al. Arginyltransferase suppresses cell tumorigenic potential and inversely correlates with metastases in human cancers. Oncogene 2016;35(31):4058–68. Epub 2015/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 2008;68(6):1777–85. [DOI] [PubMed] [Google Scholar]
- 19.Weinstein JN, Collisson EA, Mills GB, Shaw KM, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nature genetics 2013;45(10):1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer cell 2010;18(1):11–22. Epub 2010/06/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rusthoven CG, Carlson JA, Waxweiler TV, Yeh N, Raben D, Flaig TW, et al. The prognostic significance of Gleason scores in metastatic prostate cancer. Urologic oncology 2014;32(5):707–13. Epub 2014/03/19. [DOI] [PubMed] [Google Scholar]
- 22.Kuroiwa K, Uchino H, Yokomizo A, Naito S. Impact of reporting rules of biopsy Gleason score for prostate cancer. Journal of clinical pathology 2009;62(3):260–3. Epub 2008/10/28. [DOI] [PubMed] [Google Scholar]
- 23.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nature reviews Cancer 2001;1(1):34–45. Epub 2002/03/20. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki F, Suzuki K, Nikaido O. An improved soft agar method for determining neoplastic transformation in vitro. Journal of tissue culture methods 1983;8(3):109–13. [Google Scholar]
- 25.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature protocols 2007;2(2):329–33. Epub 2007/04/05. [DOI] [PubMed] [Google Scholar]
- 26.Albini A, Benelli R. The chemoinvasion assay: a method to assess tumor and endothelial cell invasion and its modulation. Nature protocols 2007;2(3):504–11. Epub 2007/04/05. [DOI] [PubMed] [Google Scholar]
- 27.Rice MA, Ishteiwy RA, Magani F, Udayakumar T, Reiner T, Yates TJ, et al. The microRNA-23b/-27b cluster suppresses prostate cancer metastasis via Huntingtin-interacting protein 1-related. Oncogene 2016;35(36):4752–61. Epub 2016/02/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, Stearns ME. Isolation and characterization of PC-3 human prostatic tumor sublines which preferentially metastasize to select organs in S.C.I.D. mice. Differentiation; research in biological diversity 1991;48(2):115–25. Epub 1991/11/01. [DOI] [PubMed] [Google Scholar]
- 29.Zhang F, Saha S, Shabalina SA, Kashina A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 2010;329(5998):1534–7. Epub 2010/09/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rai R, Zhang F, Colavita K, Leu NA, Kurosaka S, Kumar A, et al. Arginyltransferase suppresses cell tumorigenic potential and inversely correlates with metastases in human cancers. Oncogene 2015. Epub 2015/12/22. [DOI] [PMC free article] [PubMed]
- 31.Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res 1978;38(9):2651–60. Epub 1978/09/01. [PubMed] [Google Scholar]
- 32.de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-beta signaling in cancer. Journal of the National Cancer Institute 2000;92(17):1388–402. Epub 2000/09/07. [DOI] [PubMed] [Google Scholar]
- 33.Sherman MY, Gabai VL. Hsp70 in cancer: back to the future. Oncogene 2015;34(32):4153–61. Epub 2014/10/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Massague J TGFbeta in Cancer. Cell 2008;134(2):215–30. Epub 2008/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humphrey PA. Gleason grading and prognostic factors in carcinoma of the prostate. Mod Pathol 2004;17(3):292–306. [DOI] [PubMed] [Google Scholar]
- 36.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nature medicine 2016;22(3):298–305. Epub 2016/02/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 2013;29(1):15–21. Epub 2012/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 2011;12:323 Epub 2011/08/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC bioinformatics 2010;11(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurosaka S, Leu NA, Zhang F, Bunte R, Saha S, Wang J, et al. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS genetics 2010;6(3):e1000878 Epub 2010/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.