

Abstract
In the past decade, the study of mechanisms of cancer immunity has seen a prominent boom, which paralleled the increased amount of research on the clinical efficacy of immune checkpoint blockade in several lethal types of cancers. This conspicuous effort has led to the development of successful immunotherapy treatment strategies, whose medical impact has been recognized by the awarding of 2018 Nobel Prize in Physiology or Medicine to the two pioneers of check point inhibitor research, Tasuku Honjo and James Allison. Despite these promising achievements, the differences in the clinical response rate in different cancer patients and the high risk of toxicity of immune-based therapies represent crucial challenges. More remarkably, the causes responsible for different outcome (success versus failure) in patients with tumor having same histotype and clinical characteristics remain mostly unknown. MicroRNAs (miRNAs), small regulatory non-coding RNA molecules representing the most studied component of the dark matter of the human genome, are involved in the regulation of many pathways of cancer and immune cells. Therefore, understanding the role of miRNAs in controlling cancer immunity is necessary, as it can contribute to reveal mechanisms that can be modulated to improve the success of immune-therapy in cancer patients. Here, we discuss the latest findings on immune pathways regulated by miRNAs in cancer, miRNA-mediated regulation of immune cells in the tumor microenvironment, and miRNAs as potential target for immunotherapies.
Keywords: microRNAs, checkpoint molecules, therapy, cancer
Introduction
In cancer immunity, two opposite outcomes can occur based on the characteristics of tumor microenvironment. At one side, tumor infiltrating immune cells (innate and adaptive immunity) can effectively control tumor progression and eventually eradicate tumor cells. On the other side, tumor cells can develop the ability to escape immunosurveillance and destruction through the process of immunoediting1,2. The discoveries by Drs. Tasuku Honjo and James Allison represent milestones in cancer immunity research. In 1992, Dr. Honjo was the first to identify programmed cell death-1 gene (PDCD1)3 and its importance to regulate immune responses for cancer treatment4. Dr. Allison was the first to determine the anti-tumor efficacy of cytotoxic T lymphocyte antigen-4 (CTLA-4) blockade for treatment of melanoma5. Consequently, there has been an increase of studies that aimed to develop more effective immune responses in many different cancer types. Following these important breakthrough, numerous immune checkpoint blockade-based therapies have been approved by Food and Drug Administration (FDA) for the treatment of a broad range of tumor types6. Specifically, multiple molecules, targeting receptors and ligands expressed on both tumor and immune cells in the tumor microenvironment (TME), have been approved by the FDA starting from 20017. They including CTLA-4 as well as PD-1 receptor and its corresponding ligand (PDL-1). However, the success of these therapies is often reduced, as only a subset of patients usually experiences positive clinical response and many patients suffered from severe toxicity. Therefore, the identification of patients that would have a clinical benefit from immunotherapy is instrumental to reduce unnecessary severe adverse events in patients that do not respond to these therapies. In this regard, the characterization of the TME and immune cell infiltrates can help to identify which components, such as immune cells and soluble signaling factors, are either beneficial or deleterious to patients8,9.
MiRNAs are small (∼ 21 nucleotides) endogenous non-coding RNA molecules involved in post-transcriptional regulation of gene expression. Briefly, gene expression modulation by miRNAs occurs via base pairing of the specific miRNA primary sequence to its corresponding target messenger RNA (mRNA), which can be located either in the 3’ untranslated region (3’UTR) or within the coding sequence. This pairing can lead to either translational repression or cleavage of the mRNA, resulting in the reduced levels of the target protein10,11.
Aberrant expression of miRNAs is often found in cancer12, resulting in dysregulation of expression of genes that control biology of tumor cells13. Since 2011, when hallmarks of cancer were described14 and miRNAs were included as ubiquitous players in the regulation of all cancer hallmarks15, there has been increasing interest in targeting miRNAs as cancer therapy16.
Despite a relevant number of studies, there are no currently approved miRNA-based therapies for cancer treatment. Indeed, the use of miR-34a, tested as therapeutic molecule in a miR-34a restoration clinical trial (MRX34), faced a premature termination due to immune related severe adverse events (clinicaltrials.gov: NCT01829971)17. Despite this initial failure, the RNA-based therapeutics has recently seen a revival18–20. A phase 3 clinical trial using Patisiran, the first RNAi drug approved by the FDA, that specifically inhibits hepatic synthesis of transthyretin in patients with hereditary transthyretin amyloidosis, has been successfully completed21. Moreover, additional clinical trials are undergoing, such as phase 1 study of cobomarsen (MRG-106), evaluating the use a locked nucleic acid (LNA)-based oligonucleotide inhibitor of miR-155 for hematopoietic malignancies including cutaneous T-cell lymphoma (CTCL), mycosis fungoides (MF), chronic lymphocytic leukemia (CLL), diffuse large B-cell lymphoma (DLBCL), and adult T-cell leukemia/lymphoma (ATLL) (ASCO 2018; clinicaltrials.gov: NCT02580552).
Recently, there has also been increasing interest in elucidating the role of miRNAs in the regulation of anti-cancer immune responses and how this could impact the efficacy of different cancer therapeutics22. Indeed, immune responses have both pro- and anti-oncogenic effects14 and functional interactions between immune and cancer cells in TME are very important in determining the course of cancer progression. MiRNAs are involved in mediating and controlling several immune and cancer cell interactions and also important mechanisms of immune responses23. Therefore, it is crucial to carefully assess the specific roles of miRNAs in the TME for the development of effective and safe miRNA-based anti-cancer treatment strategies.
In this review, we discuss the involvement of miRNAs in the regulation of immune cells and potential therapeutic approaches using miRNAs for cancer immunotherapy.
Immune cell pathways regulated by miRNAs in cancer
Monocytes and macrophages
MiRNAs regulate differentiation, activation, and effector functions of cells of innate and adaptive immunity, which have important effects on cancer progression. As an example, in innate immunity, miR-17–5p, miR-20a, and miR-106a regulate monocyte differentiation and maturation by targeting the expression of the transcription factor acute myeloid leukaemia-1 (AML1). Particularly, these three miRNAs are downregulated during monocytopoiesis, resulting in increased levels of AML1, which in turns promotes the transcription of M-CSF receptor (central regulator of monocytic–macrophage differentiation and maturation) and inhibits the expression of miRNA 17–5p–20a–106a24. A comprehensive transcriptional analysis evaluated the association between the expression miRNAs and their transcription factors (TFs) during monocytic differentiation. Several TFs were found to control the expression of miR-21, miR-155, miR-424, and miR-17–92, which regulate monocytic differentiation by controlling MAPK, TGF-β, and JAK-STAT signaling pathway25.
MiRNAs can control also macrophage activation and polarization (M1 and M2). M1 and M2 macrophages have opposite effect on regulating cancer immunity. Particularly, M1 are classically activated by Toll-like receptor (TLR) ligands and IFN-γ, secrete pro-inflammatory cytokines (e.g.: IL-6, IL-12, IL-23, TNF-α), and are involved in T helper 1 (Th1)-mediated immune responses, which control tumor growth or induce tumor elimination. On the other side, M2 macrophages are alternatively activated by IL-4 and IL-13, secrete IL-10 and TGF-β, and mediate anti-inflammatory and pro-tumorigenic responses26. MiR-146a and miR-21 attenuate classical macrophage activation (M1 polarization) by targeting the key adaptor molecules in the TLR/NF-κB pathway. MiR-146a targets IL1R-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6), and miR-21 targets programmed cell death protein 4 (PDCD4). Downregulation of IRAK1 and TRAF6 leads to reduced TLR signaling and attenuated pro-inflammatory cytokine responses controlled by NF-κB. On the other side, miR-155 can promote classical macrophage activation by targeting suppressor of cytokine signaling 1 (SOCS1), which inhibits proinflammatory response, and IL-13RA1, which promotes alternative M2 macrophage polarization. Furthermore, miR-125a/b can increase NF-κB stability by targeting the negative regulator of NF-κB, TNF-α-induced protein 3 (TNFAIP3), promoting NF-κB signaling and M1 response27,28 (Table 1).
Table 1.
Examples of miRNAs involved in immune regulation in cancer.
| miRNA name | Genomic location | Role in cancer immunity | Immune-related targets | Ref. |
|---|---|---|---|---|
| miR-17-5p, miR-20a, miR-106a | 13q31.3 | Inhibition of monocytic differentiation and maturation | AML1 | 24 |
| miR-155 | 21q21.3 | Immune cell activation, proinflammatory responses, monocytic differentiation, Th1, Treg, NK, CTL functions | SOCS1, SMAD2-5, CTLA‐4, SHIP-1,c-Maf | 25,27,32,36,47,49,53,88 |
| miR-424 | Xq26.3 | Activation of monocytic differentiation, anti-tumor immune response | NFI-A, PD-L1 | 25,59 |
| miR-146a | 5q33.3 | Suppression of proinflammatory responses, macrophage polarization, Treg and CTL functions | IRAK1, TRAF6, STAT1 | 27,28,50,55 |
| miR-21 | 17q23.1 | Suppression of NF-κB activation, monocytic differentiation, PGE2-mediated M2 macrophage generation, CTL activation | PDCD4, PTEN, STAT3, DUSP10 | 25,27,48,49,68,110 |
| miR-17-92 | 13q31.3 | Monocytic differentiation, CTL functions, Th1 functions, TFH differentiation | E2F1, AML1, SPHK2, PTEN, TGFβRII, CREB1, RORα | 25,38,39,46,110 |
| miR-23a/27a/24-2 | 19p13.12 | M1 macrophage cytokines | A20 | 69 |
| miR-125a/b | 19q13.41 | Enhance proinflammatory signaling | TNFAIP3 | 27 |
| miR-130a | 11q12.1 | Myeloid suppressor cells regulation | TGFβRII | 72 |
| miR-145 | 5q32 | |||
| miR-29 | 7q32.3 | NK and CTL functions | IFN-γ | 33 |
| miR-150 | 19q13.33 | NK cell maturation | c-Myb | 29,30 |
| miR-181 | 1q32.1 | NK cell maturation | NLK | 29,31 |
| miR30e | 1p34.2 | NK functions | Prf1 | 34 |
| miR-378 | 5q32 | NK functions | GzmB | 34 |
| miR-27a | 19p13.12 | NK functions | Prf1, GzmB | 35 |
| miR-326 | 11q13.4 | Th17 differentiation | Ets-1 | 44 |
| miR-181c | 19p13.12 | Th17 differentiation | Smad7 | 45 |
| miR-23a | 19p13.12 | CTL functions | BLIMP-1 | 90 |
| miR-30b | 8q24.22 | CTL functions | Bcl-6 | 49 |
| miR-15a/16 | 13q14.2 | Treg functions | Foxp3, CTLA4 | 54 |
| miR-34a, | 1p36.22 | Anti-tumor immune response | PD-L1 | 56–58,60 |
| miR-138-5p | 3p21.32 | |||
| miR-200a,b,c | 1p36.33, 12p13.31 |
|||
| miR-513 | Xq27.3 | |||
| miR-138-5p | 3p21.32 | Immune checkpoint regulation | PD1, CTLA-4 | 61,62 |
Natural killer cells
Another important cellular component of the innate immunity includes natural killer (NK) cells, which play critical role in the immune responses to cancer by tumor cell killing and releasing of immunostimulatory cytokines like IFN-γ. Several miRNAs are involved in the regulation of maturation process and effector functions29. The maturation of NK cells is regulated by miR-150. Indeed, in miR-150−/− mice, immature NK cells accumulate in the bone marrow and peripheral organs due to the direct regulation of c-Myb, a miR-150 target, and consequently the downstream molecules c-Myc and Bcl-230. MiR-181 is also involved in the regulation of NK cell development from CD34+ hematopoietic progenitor cells by targeting nemo‐like kinase (NLK), which works an inhibitor of Notch signaling, an important pathway for the development of NK cells31. MiR-155 also regulates effector functions of NK cells. Particularly, high levels of miR-155 enhance the synthesis of IFN-γ by activated NK cells by targeting hematopoietic cell specific 5′ inositol phosphatase (SHIP-1), a potent‐negative regulator of NK‐cell effector functions32. The secretion of IFN-γ is also regulated by miR-29, which is downregulated after NK cell activation, by directly targeting the 3′‐UTR of IFN-γ33. Another important effector function of NK cells is the ability to kill tumor cells (cytotoxic activity) by the release of pore-forming protein perforin (Prf1) and granzyme B (GzmB), which induces pores on plasma membrane and apoptosis of tumor cells, respectively. NK cell cytotoxicity is regulated by miR-30e through targeting perforin; miR-378 through targeting granzyme B; and miR-27a-5p, which induces the downregulation of both perforin and granzyme B34,35.
T helper cells
MiRNAs can also regulate adaptive immunity by controlling the differentiation and functions of different T-cell types. MiR-155 can regulate the polarization of CD4+ T cells (Th1 vs Th2). The knockdown of miR-155 promotes the differentiation of CD4+ T cell towards Th2 phenotype by increasing the levels of its target c-Maf, a potent transactivator of the IL-4 promoter, resulting in enhanced production of Th2 cytokines IL-4, IL-5, and IL-1036. The miR-17–92 cluster (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a) is overexpressed in Th1 cells37 and miR-17 and miR-19b control Th1 responses by promoting proliferation, reducing activation-induced cell death, enhancing IFN-γ production, and suppressing regulatory T-cell (Treg) differentiation. MiR-17 and miR-19b exert their regulatory function by targeting TGFβRII and CREB1 and PTEN, respectively38.
T follicular helper cells (TFH cells) are required for effective humoral immune responses by regulating growth, differentiation, immunoglobulin isotype switching, affinity maturation of B cells, and antibody secretion. TFH cell differentiation is regulated by miR-17–92 cluster by targeting PTEN and the transcription factor RORα, which prevents the expression of genes associated with other T helper subsets, such as Th17 or Th22 cells39.
T helper 17 cells (Th17), named in this way due to the production of IL-17, were found to be associated to tumor tissue and involved in tumor immunity40. Recently, it has been proposed that Th17 cells can promote anti-tumor immune responses, as they negatively correlate with Treg cells and facilitate the recruitment of effector IFN-γ secreting cells, cytotoxic CD8+ T and NK cells, in the same tumor microenvironment41. However, Th17 cells can also have pro-tumorigenic effect by inducing tumor vascularization42 or IL-6 from tumor cells and tumor-associated stromal cells, which leads to STAT3-mediated upregulation of pro-survival and pro-angiogenic genes43. Th17 development is regulated by miRNAs, such as miR-326, which targets the transcription factor Ets-1, a negative regulator on Th17 differentiation44. Th17 cell differentiation is also regulated by miR-181c, which targets Smad7, a negative regulator of TGF-β signaling, resulting in increased TGF-β-induced Smad2/3 signaling followed by inhibition of IL-2 functions (Th17 cell differentiation inhibitor)45.
Cytotoxic T cells
Overexpression of miR-17–92 cluster in CD8+ T-cells enhances interferon IFN-γ production, cytotoxicity, and increases the frequency of cells with memory phenotype by the downregulation of TGFβRII46. MiR-155 is upregulated by T cell receptor (TCR)-mediated activation and it regulates proliferation of CD8+T cells by targeting SOCS147 and the anti-proliferative effect of type I IFN signaling48. MiR-21 and miR-30b also regulate CD8+T cell proliferation by targeting dual specificity phosphatase 10 (DUSP10) and B cell CLL/lymphoma 6 (BCL-6), respectively49. On the other side, miR-146a, which is upregulated by TCR-mediated activation, represses NF-κB signaling by targeting TRAF6 and IRAK150. As we reported above for NK cells, miR-29 can also target mRNA coding for IFN-γ in CD8+T cells, reducing their effector functions33.
T regulatory cells
MiR-155 play an important role also in the regulation Treg cell development and functions. The transcription factor Foxp3, a pivotal lineage specific regulator of Treg cells, binds to the promoter region of the gene BIC, which hosts miR-155 within its sequence51. Deficiency of miR-155 impairs the development of Treg cells resulting in reduced number of thymic and splenic Treg cells52. Furthermore, miR-155 targets the suppressor of cytokine signaling 1 (SOCS1), a negative regulator of IL-2R signaling. Therefore, low levels of miR-155 results in increased levels of SOCS1, determining reduced STAT5 signaling downstream of the IL-2R and accordingly reduced Foxp3 synthesis and diminished proliferative potential53. MiR-15a and miR-16 are expressed at low levels in Treg cells and can target Foxp3 and CTLA4. Their overexpression results in reduced levels of both Foxp3 and CTLA4, resulting in partial reduction of Treg cell-mediated suppression54. MiR-146a is highly expressed in Treg cells and targets STAT1, a key transcription factor necessary for the differentiation of Th1 effector cells. This prevents acquisition of Th1-like properties by Treg cells and enhances effective suppressor function of Treg cells55.
Immune checkpoint molecules
Immune checkpoint molecules are pivotal players in regulating immune responses. MiRNAs regulate the expression of either PD-L1, such as miR-34a-5p56, miR-138–5p57, miR-20058, miR-42459, and miR-51360 or PD-1, such as miR-138–5p61,62 (Table 1). Therefore, expression levels of these miRNAs can affect the engagement between PD-1 receptor and PD-L1 ligand and accordingly modulate T-cell functions63. MiR-138–5p is also implicated in the regulation of another immune checkpoint molecule, the CTLA-462, whose engagement with its receptors (either CD80 or CD86) on antigen presenting cell (APC), such as dendritic cells and macrophages, determines the suppression of effector T-cell activity, whereas on the other hand promoting Treg cells64.
Recently, it has been discovered that highly repetitive primate specific genomic sequences, called pyknons, are transcribed and can harbor binding sites for miRNAs, consequently working as molecular decoy to buffer the levels of target miRNAs, including immune-related miRNAs. Particularly, a specific pyknon transcript, pyknon-90, which is contained inside the primate-specific long non-coding RNA N-BLR and overexpressed in colon cancer, harbors the binding site of miR-200 within its sequence and accordingly can determine the reduction of cellular levels of members of miR-200’s family65. Because miR-200 can target PD-L158, N-BLR has the potential to represent a novel and additional level of regulation of immune check point molecules. Future miRNA-based drugs targeting PD-1/PD-L1 checkpoint can represent a powerful tool to develop more effective anti-tumor immunotherapies. Furthermore, these miRNAs targeting check point molecules can be used as biomarkers to predict the efficacy of PD-1/PD-L1 antibody treatment.
In summary, miRNAs play an important role in regulating several immune cell pathways, controlling differentiation and effector functions of different subsets of immune cells, such as monocytes, macrophages, NK cells, T helper cells, CD8+ T cells, and Treg cells, which are involved in cancer immunity.
MiRNA-mediated regulation of immune cells in the tumor microenvironment
TME presents as an underlying challenge to the efficacy of cancer immunotherapy. A myriad of factors within the TME contribute to an immune-exclusive phenotype including the abundance of immune-regulatory cells, such as myeloid derived suppressor cells (MDSCs), Treg cells, and tumor-associated macrophages (TAMs), via the production of various cytokines, chemokines, and metabolites66. As a result, anti-tumor immunity is blunted due to the inability of tumor infiltrating immune cells, such as cytotoxic T cells and NK cells, from making physical cell-to-cell contact with tumor cells. MiRNAs have been strongly implicated in driving the development and progression of cancer67, making them a likely player in determining the immune contexture of tumor microenvironment. Indeed, recent studies provide evidence of the role that miRNAs play in modulating the tumor immune response by regulating the recruitment, activation, and effector functions of different immune cell types in the TME.
The polarization of TAMs in the TME from a pro-immune M1 phenotype to an immune-regulatory M2 phenotype can be a result of aberrant miRNA regulation in cancer. As discussed above, certain miRNAs responsible for homeostatic regulation of macrophage polarization are differentially expressed in M1 and M2 macrophages. The TME is also rich in various molecules such as TGFβ, vascular endothelial growth factor (VEGF), IL-4, IL-13, or IL-10, and prostaglandin E2 (PGE2) that promote M2 polarization in addition to other pleiotropic immune-suppressive effects. Some miRNAs are able to prevent M2 polarization by such molecules, making those miRNAs translationally significant. For example, LPS and IFN-γ stimulation (M1 polarization stimuli), can increase miR-21 expression, which is able to prevent PGE2-mediated M2 generation by targeting the STAT3 gene68. M1-type stimulation can downregulate the expression of miR-23a/27a/24–2 cluster through the binding of NF-κB to this cluster’s promoter; whereas IL-4 (M2 polarization stimulus) can activate the expression of this cluster through STAT6 singling 69 (Table 1).
Trafficking and homing of immune cells to the TME and into the tumor bed is essential for effective cancer immunotherapy. MiRNAs regulate chemokines responsible for recruitment and infiltration of lymphocytes into the TME. For example, miR-21, which we previously discussed in the context of preventing M2 polarization, also regulates immune cell recruitment. Inhibition of miR-21 resulted in enhanced release of chemokines RANTES (CCL5) and IP-10 (CXCL10) in the breast cancer cell line MCF-7 and accordingly increased lymphocyte migration70 (Figure 1). Like M2 TAMs, the recruitment of MDSCs and Treg cells, which are challenging cells for cancer immunotherapy strategies, can be linked to dysregulation of specific miRNAs in the TME. For example, in a hepatocellular carcinoma model, higher levels of TGFβ suppressed the expression of miR-34a, which increased production of chemokine CCL2, responsible for recruiting Treg cells to the TME71 (Figure 1). Given the pleiotropic roles TGFβ plays in cancer, the regulation of TGFβ signaling is also a unique target of interest. It was recently shown that miR-130a and miR-145 target TGFβ beta receptor II and both miRNAs are downregulated in Gr-1+CD11b+ myeloid cells72 (Table 1). Ectopic expression of miR-130a and miR-145 as well as introduction of mimics benefitted anti-tumor immunity, including reduction of myeloid cells and increase in IFN-γ CD8+ T cells, and limited tumor metastasis72. Given the positive and negative roles miRNAs play in manipulating the TME, further studies on regulation of miRNAs can lead to novel strategies that will synergize with cancer immunotherapy.
Figure 1. The interplay between immune cells and microRNAs in TME.
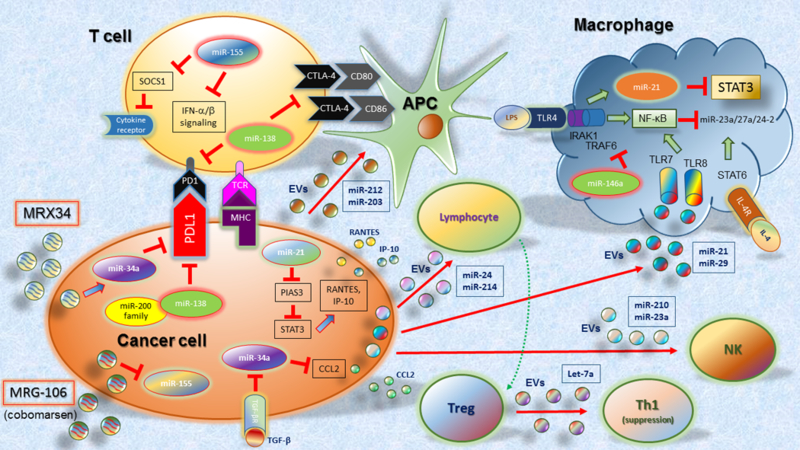
The most significant examples described in the text are included here. Also, the miRNA therapeutic agents MRX34 and MRG-106 (cobomarsen) are included.
Another important aspect to be considered in the regulatory role of miRNAs in the TME is that they can exert their regulatory function also beyond the cells of origin. Indeed, in the TME, miRNAs can be transported inside extracellular vesicles (EVs) and delivered to recipient cells, regulating their biological functions9. This miRNA-mediated cell-to-cell communication represents an active crosstalk involving different cellular components of TME, which include cancer cells, mesenchymal stromal cells (MCS), cancer associated fibroblasts (CAFs), endothelial cells, and immune cells. This intercellular communication is particular important as many miRNAs regulating immune cells are shuttled by exosomes73. In a mouse model of lung cancer, tumor cells can release EVs carrying miR-21 and miR-29, which are delivered to macrophages in the tumor tissue and bind to TLR8 and TLR7, resulting in the activation of the NF-κB pathway (inflammatory response mediated by TNF-α and IL-6)74 (Figure 1). EV miR-212–3p from pancreatic tumor cells (PANC-1) affects immune functions of dendritic cells, inducing immune tolerance75 (Figure 1). EV-mediated delivery of miR-203 from PANC-1 to dendritic cells induces the downregulation of TLR4, TNF-α, and IL-12 levels, resulting in the impairment of the immune response activation76 (Figure 1). MiR-24 released from nasopharyngeal cancer impairs T cell proliferation, their differentiation into Th1 and Th17 cells, and promote the generation of Treg (CD4+CD25high Foxp3+) cells by increasing the levels of pERK, pSTAT1, pSTAT3, and decreasing the levels of pSTAT577 (Figure 1). Tumor EV miR-214 targets PTEN in CD4+ T cells, which promotes the expansion of Treg cells and enhanced immune suppression78 (Figure 1). Treg cells can exert their suppressor function also via EVs. Indeed, let-7d released inside EVs by Treg cells suppresses Th1 cell proliferation and cytokine secretion79 (Figure 1). Tumor EV miRNAs can also regulate effector functions of NK cells. EVs derived from hypoxic tumor cells contain high levels of miR-210 and miR-23a, which impairs NK cell cytotoxicity against different tumor cells in vitro and in vivo80(Figure 1).
In summary, miRNAs play an important role in regulating the immune cell functions also in the TME.
MiRNAs as potential targets for immunotherapies
Traditionally, miRNAs have been used as clinical biomarkers for cancer prognosis, diagnosis, and treatment response9,81. MiRNAs are powerful tumor suppressors and oncogenes with specific functions associated with cancer and have intrinsic ability to affect the efficacy of standard therapies. Moreover, as described above, miRNAs are also important regulators of immune cell functions and immune responses in the tumor microenvironment. With this in mind, targeting miRNAs with miRNA-based therapeutics for cancer has great potential to enhance immunotherapy along with current treatment modalities.
Several strategies are currently in preclinical studies to evaluate the role of miRNA as immunotherapeutic agents. These immunotherapeutic targets are subdivided into two types: miRNA mimics and antagonists. MiRNA mimics traditionally restore miRNAs that have tumor suppressor capabilities, whereas miRNA antagonists act similarly to inhibitors82. Targeting miRNAs can induce multiple effects from increased tumor sensitivity to conventional treatment modalities to directly increasing immunogenicity of the tumor cells themselves.
Over the past decade, miRNA mimics have led the charge for immunotherapy targets. The first and only miRNA-based anti-cancer therapy that reached clinical trial involved the use of mimic of miR-34a, MRX3483,84. MiR-34a was also found to reduce PD-L1 expression in acute myeloid leukemia (AML) by targeting PD-L1 mRNA56. It was shown that miR-34a mimic can also regulate the subtests of immune cells infiltrating tumor tissue. Particularly, it is was demonstrated that in vivo administration of MRX34, besides inducing decreased expression of PD-L1, can also increase tumor infiltration of CD8+ T cells and decrease CD8+PD1+ T cells in a mouse model of NSCLC85. Additionally, combination of MRX34 and radiotherapy (XRT) further enhances CD8+ T cell infiltration, and reduces both the numbers of radiation-induced macrophages and Treg cells85. These results showed the enhanced efficacy of miR-34a mimic to modulate anti-tumor immune responses and control tumor growth in combination with XRT. The ability of miR-34a to potently control immune responses is proved by occurrence of five immune related serious adverse events, which determined the early termination of the phase I clinical trial (on September 2016). Further in depth studies on toxicity and immune related adverse events were conducted in patients with advanced solid tumor. The use of dexamethasone as premedication was found to moderate adverse events and be associated with acceptable safety17.
Additional new potential targets for miRNA mimic therapy are currently being investigated. MiR-124 has the ability to target STAT3, a key pathway regulating immunosuppression in the tumor microenvironment, and administration of miR-124 mimic can increase pro-immunogenic cytokines, such as IFN-γ, TNF-α, and IL-2 in glioma microenvironment, resulting in a potent anti-glioma therapeutic effect86. Similarly, miR-424 acts as suppressor of PD-L1 and CD80 expression. In vivo studies, disruption of PD-L1/PD-1 and CD80/CTLA-4 immune checkpoint signaling and reversion of chemoresistance mediated by restoration of miR-424 result in a synergistic effect, which induces proliferation of functional cytotoxic CD8+T cells, the inhibition of myeloid-derived suppressive cells and regulatory T cells, and increases survival in a mouse model of ovarian carcinoma59.
As we discussed above, miR-138 regulates CTLA-4 and PD-1 expression on CD4+T cells (Table 1), which results in downregulation of Foxp3 expression. In vivo administration of miR-138 mimic induces significant downregulation of CTLA-4, PD-1, and Foxp3 on tumor infiltrating CD4+T cells in a mouse model of glioma, resulting in significant reduction of infiltrating Treg cells62.
Given these preliminary studies, further investigation into several miRNA mimics are warranted, especially with other treatment modalities such as checkpoint inhibitors.
MiRNA inhibitors, as mentioned above, are conventional targets for miRNAs that possess oncogenic potential. These inhibitors have reached clinical trials, with the use of Cobomarsen (MRG105) as anti-miR-155 agent (Figure 1). The inhibition of miR-155 by systemic administration of complementary anti-miR sequences blocks tumor growth and dissemination in vivo87. However, the inhibition of miR-155 by systemic administration may also induce various effects related to immune cell activity, as genetically-induced deficiency of miR-155 in the immune system cells attenuates their functions88. On the other side, the possible negative effects on immune responses induced by systemic administration of miR-155 inhibitor can be circumvented by the overexpression of miR-155 in tumor-specific CD8+ T cells, which resulted in increased efficacy of T-cell–based adoptive therapies89. As another example, miR-23a has been shown to decrease cytotoxicity of effector CD8+ cytotoxic T lymphocytes (CTLs) by targeting the transcription factor BLIMP-1, which regulates effector cell differentiation and their cytotoxic ability. TGF-β released by tumor cells induces the increase of miR-23a levels. Adoptive transfer of CTL cells treated to inhibit miR-23a robustly retards tumor progression in a mouse model of melanoma90. These two examples show that the inhibition of miRNAs that dampen effector functions of immune cells results in a more robust and effective anti-tumor immune responses.
MiRNAs have also been deployed to modulate functional characteristics of chimeric antigen receptor (CAR) T cells in order to increase their efficacy in adoptive cell therapy. Since 1989, when CAR technology was initially established91 and later when anti-CD19 CAR T cell therapy was demonstrated to be effective for the treatment of lymphomas and leukemias92,93, there has been an exponential growth in the research on CAR T cell therapy and discovery of new tumor cell surface targets. This has led to over 100 ongoing clinical trials targeting over 25 different surface molecules in almost every human tissue94. It was recently discovered that CAR T cells specific for epidermal growth factor receptor variant III (EGFRvIII) for the treatment of glioblastoma (GBM) had improved survival and therapeutic efficacy when co-transduced with miR-17–92 and in combination with temozolomide (TMZ)95. This may result from the targeting of TGFβRII and PTEN by miR-17 and miR-19b, respectively, which may increase effector ability. Another example is represented by increased cytotoxicity of CAR T cells specific for HER2 (HER2-CAR T) mediated by the transfection with miR-143 mimic. Overexpression of miR-143 promotes the differentiation of T cells to fully functional memory T cells, decreases apoptosis, and increases T cell cytotoxicity by targeting Glut-1, a regulator of glycolytic metabolism96.
An alternative weapon for anti-tumor immunity strategies is represented by NK cells, which are an important subset of immune cells of the innate immunity. Particularly, they are endowed with highly cytotoxic immune effector function and have potent anti-tumor activity. Furthermore, NK cells can be engineered to generate CAR-NK cells which acquired enhanced specific and effective anti-tumor activity97. Therapeutic manipulation of CAR-NK cells with miRNAs or anti-miRNAs will be of interest and we anticipate the development of such approaches in the near future.
Challenges in miRNA-based therapy
Although it showed great potential, it is surprising to find miRNA-targeted immunotherapeutics have not achieved success in clinical practice as a cancer therapy, yet.
As discussed above, MRX34 proved to have beneficial anti-cancer therapeutic effect in pre-clinical animal model of lung cancer85,98. However, during the trial, several patients developed severe immune-related toxicities, as miR-34a can also regulate immune cell functions85,99,100. A second phase I clinical trial using MRX34 showed that premedication with dexamethasone, a potent anti-inflammatory agent, was required to manage infusion-related adverse events17. Furthermore, it should be also considered the indirect effect of miRNA-based treatment on immune responses. Indeed, it was reported that therapeutic RNA interference (RNAi) molecules have intrinsic immunostimulatory capacity and can activate innate immune response by the activation of TLR eliciting a nonspecific therapeutic effects74. This may increase the risk of misinterpreting the therapeutic effects of miRNA-based therapy or underestimating the possibility of severe immune overreaction101. This is a cautionary tale for miRNA therapeutics and particular attention is required for miRNA-based therapy in order to control the effect on immune cell functions, adverse events, and nonspecific results.
Various strategies for diminishing the adverse effects are envisioned and can include the use of combinatorial miRNA mimic and siRNA therapy102 or combinatorial anti-miRNA and chemotherapy87. These strategies can potentially be used to optimize the dosage for the combination of the single agents and thereby reducing the risk and the levels of treatment-related toxicity103.
Another important aspect than needs to be taken in consideration is the proper delivery methods. Several types of delivery systems have been investigated, which include synthetic nanoparticles (micelles, liposomes, polymers, nanotubes)104 and exosomes, which naturally carry and deliver non-coding RNA105,106. Both systems have pros and cons. The advantage of synthetic nanoparticles is represented by the fact that they can be quickly produced in large and controlled amount and are easily manipulated to increase efficient RNAi molecule loading and cell targeting. However, they have low transfection efficiency in vivo, thus multiple administrations may be required, increasing the risk of immunogenicity and toxicity107. On the other side, exosomes have little or no toxicity and immunogenicity compared with conventional nanocarriers, even after multiple administrations, as they are naturally produced; have increased efficiency in delivery cargo directly into the cytosol; and can be engineered to be loaded with RNAi molecules and express tag on their surface to specifically target tumor cells108. On the other hand, there are not currently available techniques to produce scalable amount, efficiently purify and load exosomes with RNAi molecules to clinical grade standards109.
Conclusions
Ultimately, the role of miRNA as therapeutics against cancer requires deeper study. The potential for several mimics and inhibitors, as seen in their preclinical evidence, is promising, and there is much more to be learned about miRNA involvement in cancer and immunotherapy. Given their potential, there is a strong case to continuing to explore miRNA and their capabilities as immunotherapeutics against cancer.
ACKNOWLEDGEMENT
Dr. Calin is the Felix L. Haas Endowed Professor in Basic Science. Work in Dr. Calin’s laboratory is supported by National Institutes of Health (NIH/NCATS) grant UH3TR00943-01 through the NIH Common Fund, Office of Strategic Coordination (OSC), the NCI grants 1R01 CA182905-01 and 1R01CA222007-01A1, an NIGMS 1R01GM122775-01 grant, a U54 grant #CA096297/CA096300 – UPR/MDACC Partnership for Excellence in Cancer Research 2016 Pilot Project, a Team DOD (CA160445P1) grant, a Ladies Leukemia League grant, a Chronic Lymphocytic Leukemia Moonshot Flagship project, a Sister Institution Network Fund (SINF) 2017 grant, and the Estate of C. G. Johnson, Jr.
REFERENCES
- 1.Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 2015;21(4):687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140(6):883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11(11):3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol 2013;14(12):1212–1218. [DOI] [PubMed] [Google Scholar]
- 5.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271(5256):1734–1736. [DOI] [PubMed] [Google Scholar]
- 6.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 2018;8(9):1069–1086. [DOI] [PubMed] [Google Scholar]
- 7.Van Roosbroeck K, Calin GA. Cancer Hallmarks and MicroRNAs: The Therapeutic Connection. Adv Cancer Res 2017;135:119–149. [DOI] [PubMed] [Google Scholar]
- 8.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12(4):298–306. [DOI] [PubMed] [Google Scholar]
- 9.Anfossi S, Babayan A, Pantel K, Calin GA. Clinical utility of circulating non-coding RNAs - an update. Nat Rev Clin Oncol 2018;15(9):541–563. [DOI] [PubMed] [Google Scholar]
- 10.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116(2):281–297. [DOI] [PubMed] [Google Scholar]
- 11.Paladini L, Fabris L, Bottai G, Raschioni C, Calin GA, Santarpia L. Targeting microRNAs as key modulators of tumor immune response. J Exp Clin Cancer Res 2016;35:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006;6(11):857–866. [DOI] [PubMed] [Google Scholar]
- 13.Munker R, Calin GA. MicroRNA profiling in cancer. Clin Sci (Lond) 2011;121(4):141–158. [DOI] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- 15.Berindan-Neagoe I, Monroig Pdel C, Pasculli B, Calin GA. MicroRNAome genome: a treasure for cancer diagnosis and therapy. CA Cancer J Clin 2014;64(5):311–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer 2011;11(12):849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beg MS, Brenner AJ, Sachdev J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs 2017;35(2):180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov 2013;12(11):847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah MY, Ferrajoli A, Sood AK, Lopez-Berestein G, Calin GA. microRNA Therapeutics in Cancer - An Emerging Concept. EBioMedicine 2016;12:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 2017;16(3):203–222. [DOI] [PubMed] [Google Scholar]
- 21.Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379(1):11–21. [DOI] [PubMed] [Google Scholar]
- 22.Dragomir M, Chen B, Fu X, Calin GA. Key questions about the checkpoint blockade-are microRNAs an answer? Cancer Biol Med 2018;15(2):103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol 2016;16(5):279–294. [DOI] [PubMed] [Google Scholar]
- 24.Fontana L, Pelosi E, Greco P, et al. MicroRNAs 17–5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 2007;9(7):775–787. [DOI] [PubMed] [Google Scholar]
- 25.Schmeier S, MacPherson CR, Essack M, et al. Deciphering the transcriptional circuitry of microRNA genes expressed during human monocytic differentiation. BMC Genomics 2009;10:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer 2017;117(11):1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Squadrito ML, Etzrodt M, De Palma M, Pittet MJ. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol 2013;34(7):350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Self-Fordham JB, Naqvi AR, Uttamani JR, Kulkarni V, Nares S. MicroRNA: Dynamic Regulators of Macrophage Polarization and Plasticity. Front Immunol 2017;8:1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beaulieu AM, Bezman NA, Lee JE, Matloubian M, Sun JC, Lanier LL. MicroRNA function in NK-cell biology. Immunol Rev 2013;253(1):40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bezman NA, Chakraborty T, Bender T, Lanier LL. miR-150 regulates the development of NK and iNKT cells. J Exp Med 2011;208(13):2717–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cichocki F, Felices M, McCullar V, et al. Cutting edge: microRNA-181 promotes human NK cell development by regulating Notch signaling. J Immunol 2011;187(12):6171–6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trotta R, Chen L, Ciarlariello D, et al. miR-155 regulates IFN-gamma production in natural killer cells. Blood 2012;119(15):3478–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma F, Xu S, Liu X, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol 2011;12(9):861–869. [DOI] [PubMed] [Google Scholar]
- 34.Wang P, Gu Y, Zhang Q, et al. Identification of resting and type I IFN-activated human NK cell miRNomes reveals microRNA-378 and microRNA-30e as negative regulators of NK cell cytotoxicity. J Immunol 2012;189(1):211–221. [DOI] [PubMed] [Google Scholar]
- 35.Kim TD, Lee SU, Yun S, et al. Human microRNA-27a* targets Prf1 and GzmB expression to regulate NK-cell cytotoxicity. Blood 2011;118(20):5476–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007;316(5824):608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki K, Kohanbash G, Hoji A, et al. miR-17–92 expression in differentiated T cells - implications for cancer immunotherapy. J Transl Med 2010;8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang S, Li C, Olive V, et al. Molecular dissection of the miR-17–92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood 2011;118(20):5487–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baumjohann D, Kageyama R, Clingan JM, et al. The microRNA cluster miR-17 approximately 92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol 2013;14(8):840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol 2010;10(4):248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kryczek I, Banerjee M, Cheng P, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009;114(6):1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Numasaki M, Fukushi J, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood 2003;101(7):2620–2627. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 2009;206(7):1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol 2009;10(12):1252–1259. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Z, Xue Z, Liu Y, et al. MicroRNA-181c promotes Th17 cell differentiation and mediates experimental autoimmune encephalomyelitis. Brain Behav Immun 2018;70:305–314. [DOI] [PubMed] [Google Scholar]
- 46.Kosaka A, Ohkuri T, Ikeura M, Kohanbash G, Okada H. Transgene-derived overexpression of miR-17–92 in CD8+ T-cells confers enhanced cytotoxic activity. Biochem Biophys Res Commun 2015;458(3):549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dudda JC, Salaun B, Ji Y, et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 2013;38(4):742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gracias DT, Stelekati E, Hope JL, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol 2013;14(6):593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang CC, Zhang QY, Liu Z, Clynes RA, Suciu-Foca N, Vlad G. Downregulation of inflammatory microRNAs by Ig-like transcript 3 is essential for the differentiation of human CD8(+) T suppressor cells. J Immunol 2012;188(7):3042–3052. [DOI] [PubMed] [Google Scholar]
- 50.Boldin MP, Taganov KD, Rao DS, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med 2011;208(6):1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eis PS, Tam W, Sun L, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A 2005;102(10):3627–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol 2009;182(5):2578–2582. [DOI] [PubMed] [Google Scholar]
- 53.Lu LF, Thai TH, Calado DP, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity 2009;30(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X, Robinson SN, Setoyama T, et al. FOXP3 is a direct target of miR15a/16 in umbilical cord blood regulatory T cells. Bone Marrow Transplant 2014;49(6):793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu LF, Boldin MP, Chaudhry A, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010;142(6):914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Li J, Dong K, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal 2015;27(3):443–452. [DOI] [PubMed] [Google Scholar]
- 57.Zhao L, Yu H, Yi S, et al. The tumor suppressor miR-138–5p targets PD-L1 in colorectal cancer. Oncotarget 2016;7(29):45370–45384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen L, Gibbons DL, Goswami S, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 2014;5:5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu S, Tao Z, Hai B, et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun 2016;7:11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gong AY, Zhou R, Hu G, et al. Cryptosporidium parvum induces B7-H1 expression in cholangiocytes by down-regulating microRNA-513. J Infect Dis 2010;201(1):160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smolle MA, Calin HN, Pichler M, Calin GA. Noncoding RNAs and immune checkpoints-clinical implications as cancer therapeutics. FEBS J 2017;284(13):1952–1966. [DOI] [PubMed] [Google Scholar]
- 62.Wei J, Nduom EK, Kong LY, et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol 2016;18(5):639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27(4):450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rigoutsos I, Lee SK, Nam SY, et al. N-BLR, a primate-specific non-coding transcript leads to colorectal cancer invasion and migration. Genome Biol 2017;18(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015;348(6230):74–80. [DOI] [PubMed] [Google Scholar]
- 67.Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol 2014;9:287–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z, Brandt S, Medeiros A, et al. MicroRNA 21 is a homeostatic regulator of macrophage polarization and prevents prostaglandin E2-mediated M2 generation. PLoS One 2015;10(2):e0115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma S, Liu M, Xu Z, et al. A double feedback loop mediated by microRNA-23a/27a/24–2 regulates M1 versus M2 macrophage polarization and thus regulates cancer progression. Oncotarget 2016;7(12):13502–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Z, Han J, Cui Y, Zhou X, Fan K. miRNA-21 inhibition enhances RANTES and IP-10 release in MCF-7 via PIAS3 and STAT3 signalling and causes increased lymphocyte migration. Biochem Biophys Res Commun 2013;439(3):384–389. [DOI] [PubMed] [Google Scholar]
- 71.Yang P, Li QJ, Feng Y, et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012;22(3):291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ishii H, Vodnala SK, Achyut BR, et al. miR-130a and miR-145 reprogram Gr-1(+)CD11b(+) myeloid cells and inhibit tumor metastasis through improved host immunity. Nat Commun 2018;9(1):2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anfossi S, Fu X, Nagvekar R, Calin GA. MicroRNAs, Regulatory Messengers Inside and Outside Cancer Cells. Adv Exp Med Biol 2018;1056:87–108. [DOI] [PubMed] [Google Scholar]
- 74.Fabbri M, Paone A, Calore F, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012;109(31):E2110–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding G, Zhou L, Qian Y, et al. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212–3p. Oncotarget 2015;6(30):29877–29888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol 2014;292(1–2):65–69. [DOI] [PubMed] [Google Scholar]
- 77.Ye SB, Zhang H, Cai TT, et al. Exosomal miR-24–3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J Pathol 2016;240(3):329–340. [DOI] [PubMed] [Google Scholar]
- 78.Yin Y, Cai X, Chen X, et al. Tumor-secreted miR-214 induces regulatory T cells: a major link between immune evasion and tumor growth. Cell Res 2014;24(10):1164–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okoye IS, Coomes SM, Pelly VS, et al. MicroRNA-Containing T-Regulatory-Cell-Derived Exosomes Suppress Pathogenic T Helper 1 Cells. Immunity 2014;41(3):503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berchem G, Noman MZ, Bosseler M, et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. Oncoimmunology 2016;5(4):e1062968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Monroig-Bosque Pdel C, Rivera CA, Calin GA. MicroRNAs in cancer therapeutics: “from the bench to the bedside”. Expert Opin Biol Ther 2015;15(10):1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bader AG, Brown D, Stoudemire J, Lammers P. Developing therapeutic microRNAs for cancer. Gene Ther 2011;18(12):1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bouchie A First microRNA mimic enters clinic. Nat Biotechnol 2013;31(7):577. [DOI] [PubMed] [Google Scholar]
- 84.Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 2012;3:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cortez MA, Ivan C, Valdecanas D, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst 2016;108(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wei J, Wang F, Kong LY, et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res 2013;73(13):3913–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van Roosbroeck K, Fanini F, Setoyama T, et al. Combining Anti-Mir-155 with Chemotherapy for the Treatment of Lung Cancers. Clin Cancer Res 2017;23(11):2891–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mashima R Physiological roles of miR-155. Immunology 2015;145(3):323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ji Y, Wrzesinski C, Yu Z, et al. miR-155 augments CD8+ T-cell antitumor activity in lymphoreplete hosts by enhancing responsiveness to homeostatic gammac cytokines. Proc Natl Acad Sci U S A 2015;112(2):476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin R, Chen L, Chen G, et al. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. J Clin Invest 2014;124(12):5352–5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989;86(24):10024–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012;119(12):2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015;33(6):540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Townsend MH, Shrestha G, Robison RA, O’Neill KL. The expansion of targetable biomarkers for CAR T cell therapy. J Exp Clin Cancer Res 2018;37(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ohno M, Ohkuri T, Kosaka A, et al. Expression of miR-17–92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer 2013;1:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang T, Zhang Z, Li F, et al. miR-143 Regulates Memory T Cell Differentiation by Reprogramming T Cell Metabolism. J Immunol 2018;201(7):2165–2175. [DOI] [PubMed] [Google Scholar]
- 97.Rezvani K, Rouce R, Liu E, Shpall E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol Ther 2017;25(8):1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wiggins JF, Ruffino L, Kelnar K, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res 2010;70(14):5923–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shin J, Xie D, Zhong XP. MicroRNA-34a enhances T cell activation by targeting diacylglycerol kinase zeta. PLoS One 2013;8(10):e77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rao DS, O’Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 2010;33(1):48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Robbins M, Judge A, Ambegia E, et al. Misinterpreting the therapeutic effects of small interfering RNA caused by immune stimulation. Hum Gene Ther 2008;19(10):991–999. [DOI] [PubMed] [Google Scholar]
- 102.Nishimura M, Jung EJ, Shah MY, et al. Therapeutic synergy between microRNA and siRNA in ovarian cancer treatment. Cancer Discov 2013;3(11):1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yin Z, Deng Z, Zhao W, Cao Z. Searching Synergistic Dose Combinations for Anticancer Drugs. Front Pharmacol 2018;9:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tatiparti K, Sau S, Kashaw SK, Iyer AK. siRNA Delivery Strategies: A Comprehensive Review of Recent Developments. Nanomaterials (Basel) 2017;7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bullock MD, Silva AM, Kanlikilicer-Unaldi P, et al. Exosomal Non-Coding RNAs: Diagnostic, Prognostic and Therapeutic Applications in Cancer. Noncoding RNA 2015;1(1):53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays 2011;33(10):737–741. [DOI] [PubMed] [Google Scholar]
- 107.Fischer HC, Chan WC. Nanotoxicity: the growing need for in vivo study. Curr Opin Biotechnol 2007;18(6):565–571. [DOI] [PubMed] [Google Scholar]
- 108.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 2011;29(4):341–345. [DOI] [PubMed] [Google Scholar]
- 109.Yamashita T, Takahashi Y, Takakura Y. Possibility of Exosome-Based Therapeutics and Challenges in Production of Exosomes Eligible for Therapeutic Application. Biol Pharm Bull 2018;41(6):835–842. [DOI] [PubMed] [Google Scholar]
- 110.Liang Y, Pan HF, Ye DQ. microRNAs function in CD8+T cell biology. J Leukoc Biol 2015;97(3):487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]