

Abstract
The Suppressor of T cell receptor Signaling (Sts) proteins, particularly Sts-1, have recently emerged as potential immunostimulatory targets for drug development. Genetic inactivation of the Sts proteins dramatically increases host survival of systemic infection and leads to improved pathogen clearance. The protein tyrosine phosphatase (PTP) activity of these proteins arises from a C-terminal 2-histidine phosphatase (HP) domain. To identify new inhibitors of the HP activity of Sts-1 we miniaturized a phosphatase assay to 1,536-well format and conducted a 20,580 compound screen. Among the hits were two classes of structurally related compounds, tetracycline variants and sulfonated azo dyes. These hits had low micromolar to nanomolar IC50 values. Orthogonal screening confirmed the validity of these inhibitors and demonstrated that both act competitively on Sts-1 phosphatase activity. When tested on other PTPs, PTP1B and SHP1, the tetracycline variant, doxycycline, and the sulfonated azo dye, Congo red, are selective inhibitors of Sts-1HP with selectivity indices ranging from 19 to as high as 200. The planar polyaromatic moieties present in both classes of compounds suggested a common binding mode. Mutation of either tryptophan 494 or tyrosine 596, located near the active site of the protein, reduced the Ki of the inhibitors from 3- to 18-fold, indicating that these residues may help promote binding of substrates with aromatic groups. This work provides new insights into substrate selectivity mechanisms and describes two classes of compounds that can serve as probes of function or as a basis for future drug discovery.
Keywords: Sts, TULA, UBASH3, tetracycline, PTP
Graphical Abstract
The T cell Receptor (TCR) complex recognizes antigens and is responsible for activating T cells. The TCR signaling pathway is highly regulated, both positively and negatively, by tyrosine kinases and protein tyrosine phosphatases (PTPs) (1–3). Two members of the Suppressor of TCR Signaling (Sts) family of proteins, Sts-1 and Sts-2, act as negative regulators of signaling pathways downstream of the TCR (4, 5). Functional inactivation of the Sts proteins (Sts−/−) in mice leads to profound resistance to systemic infection by the fungal pathogen Candida albicans, and the bacterial pathogen Francisella tularensis (6–9). This resistance phenotype is characterized by enhanced survival, rapid clearance of the pathogen and altered inflammatory response. Systemic C. albicans infections, with close to 50,000 cases per year in the United States alone, have high mortality rates that have not declined in the last 20 years (10–12). F. tularensis, which is the causative agent of tularemia (rabbit fever), is one of the most infectious pathogens known and is classified as a category A bioterrorism agent (13, 14). Untreated, patients with tularemia have a fatality rate of 50 to 60% (15). The striking resistance phenotype and enhanced survival rates of Sts−/− mice suggest that therapeutic inactivation of Sts function generates a unique immune response that helps to reduce pathogen burden and prevent the accompanying destructive inflammation. As such, the pharmacological inhibition of Sts activity presents a potential opportunity to generate immuno-stimulatory therapies, to be used in combination with existing standard-of-care antibiotics or antifungals, as a means to treat deadly pathogen infections.
Sts-1 and Sts-2, which share 40% sequence identify, are multi-domain proteins composed of an N-terminal ubiquitin association (UBA) domain, a Src-homology 3 (SH3) domain, and a C-terminal histidine phosphatase (HP) domain. The UBA and SH3 domains are believed to mediate protein-protein interactions and may be involved in spatiotemporal regulation of the Sts catalytic function (16–18). The HP domain of these proteins are homologous to the phosphoglycerate mutase (PGM) family, a subgroup of the histidine phosphatase superfamily (5, 19, 20). The HP domain of the Sts proteins, like other members of the PGM family, employs a 2-step catalytic mechanism that involves nucleophilic attack by one of the two conserved histidine residues and formation of a phosphor-histidine intermediate (19, 21–24). While the Sts proteins are known to catalyze the dephosphorylation of phosphotyrosines on protein substrates, they are structurally, functionally and mechanistically distinct from canonical protein tyrosine phosphatases (PTPs) such as PTP1B and SHP1 (5, 20). An established substrate of both Sts-1 and Sts-2, in vivo, is the kinase Zap-70 (4, 5, 25). For this substrate, and all others measured, Sts-1 has substantially greater catalytic activity than Sts-2. Similarly, Sts-1 appears to play the predominant role in the observed resistance phenotype (8).
The striking resistance to pathogen infection observed in Sts-deficient mice presents an obvious opportunity for pharmacological interventions. Despite the potential as an immunostimulatory target, little has been reported regarding drug discovery or development. X-ray crystal structures, and complementary biochemical data, suggest that Sts-1 has a well-defined and seemingly druggable active site that is structurally unlike other PTPs or PGMs (19, 20). To date, however, no specific inhibitors of either Sts-1 or Sts-2 have been reported. These proteins can be inhibited with the general competitive inhibitor, orthovanadate, and the SHP-2 inhibitor PHPS1 (4-[2-[1,5-dihydro-3-(4-nitrophenyl)-5-oxo-1-phenyl-4H-pyrazol-4-ylidene]hydrazinyl]-benzenesulfonic acid) inhibits Sts-1 with a Ki of 1 μM (20, 26, 27).
To identify small molecule inhibitors of Sts-1 for use as functional probes or as a basis for further drug development we miniaturized an established phosphatase assay and conducted a 20,000 compound high throughput screen of the Sts-1 phosphatase domain Sts-1HP. The screen yielded 51 active compounds (IC50 < 10 μM) that were inactive in an enzyme-minus counter-screen. Within these hits were several groups of compounds with similar structural scaffolds. Among these, tetracycline analogs and sulfonate-containing azo dyes, were the largest two groups, with more than 10 and 5 variants present in the screening results, respectively. Both compound classes were determined to be competitive inhibitors of Sts-1HP that selectively inhibit this phosphatase, when compared to the PTPs SHP1 and PTP1B. Based on the conserved planar, aromatic structures of these inhibitors we speculated that aromatic residues on the protein were likely involved in critical protein-inhibitor interactions. Consistent with this hypothesis, mutation of either a tyrosine or tryptophan residue near the active site of Sts-1 reduces the effectiveness of these inhibitors without altering the kinetic parameters of the enzyme. Taken together, these data confirm the druggability of the Sts-1 phosphatase active site and provide the first examples of selective Sts-1 inhibitors. The results from the screen, as well as the initial structure-activity and mechanism of action data, provide a foundation for future drug development efforts.
RESULTS AND DISCUSSION
Assay design, miniaturization and testing:
The Sts proteins, particularly Sts-1, have recently emerged as potential targets for the treatment of deadly pathogen infections (6–9). An altered immune response and increased fungal or bacterial clearance in Sts knockout mice generates a profound resistance phenotype (6–8, 28, 29). Small molecules that can potently and selectively inhibit Sts-1 phosphatase activity would be extremely valuable, both as probes of function and as a foundation for drug development. We set out to conduct a high-throughput screen with the goal of identifying one or more class of compounds that inhibit Sts-1 phosphatase activity, and do so selectively when compared to canonical PTPs. As previously reported, the isolated HP domain of Sts-1 displays similar kinetics as the intact protein (20). Thus, for ease of protein expression and purification, we conducted the screen with Sts-1HP. After testing several established phosphatase assays, we settled on the 3-O-methylfluorescein phosphate (OMFP) based fluorescence assay to screen for inhibitors of Sts-1HP. Sts-1HP generates the fluorescent product, 3-O-methylfluorescien, with reasonable kinetics (Km of 190 μM, kcat of 786 s−1) (20). We were able to successfully miniaturize this assay to 1,536-well plate format and confirmed that it behaved well, yielding satisfactory statistics. To test the assay performance in this format, we first conducted a 1,280 compound pilot screen, in triplicate, using the bioactive small molecules in the Library of Pharmacologically Active Compounds (Sigma Aldrich). As expected, the assay ran well and we initially identified 10 active compounds for a 0.86% hit rate (Fig. 1). Among these hits, listed in Table 1, were several known PTP inhibitors including aurintricarboxylic acid, morin, dephostatin and 3,4-methyldephostatin (30–34). These results confirmed that our assay could be used in high-throughput to effectively screen for inhibitors of the Sts-1 phosphatase activity.
Figure 1.

Pilot screen. The % response (A) for the 1,280 LOPAC library compounds, conducted in triplicate, is shown. The high control is plotted in red, the low control is plotted in green, the PHPS1 standard is plotted in magenta, and the samples are plotted in black. Compounds that had a response greater than three times the standard deviation (31.8% response in this case) were selected as active. The potent Sts-1 inhibitor, PHPS1, which has an IC50 of 17.6 μM (B), was used as a positive control. A correlation plot (C) of the compounds screened in duplicate plates is shown (R2 = 0.933). The hit cutoffs are designated by red lines.
Table 1.
Active compounds from LOPAC pilot
| Name | Molecular Weight (g/moL) | Averaged Max. Response (%) | StdDev. Max. Response |
|---|---|---|---|
| Aurintricarboxylic Acid | 422.34 | 89.95 | 2.30 |
| 6-Hydroxy-DL-DOPA | 213.19 | 64.86 | 1.15 |
| Reactive Blue 2 | 774.16 | 58.99 | 2.49 |
| L-α-methyl DOPA | 211.21 | 57.45 | 2.91 |
| Methyl-3,4-Dephostatin | 168.15 | 46.07 | 3.57 |
| Doxycycline | 444.43 | 45.03 | 1.60 |
| Morin | 302.24 | 44.94 | 4.06 |
| Hispidin | 246.22 | 44.44 | 2.78 |
| Dephostatin | 168.15 | 43.47 | 4.40 |
| Minocycline | 456.49 | 32.59 | 1.69 |
20,580 compound screen:
Having established the efficacy of our approach, we conducted a screen of 20,580 compounds. The general workflow from assay optimization through to completion of our screen is shown in Fig. 2A. These compounds were a subset of the Scripps Drug Discovery Library (SDDL) selected to maximize structural diversity in the screen. The performance of the assay was excellent, with an average Z’ of 0.83 ± 0.03 and an average S:B of 3.76 ± 0.26 (n=17 plates) (Fig. 2C). The assay yielded 115 active compounds (0.56% hit rate), using a standard cut-off (3×SD of average response; 23.12%) (Fig 2B). After performing a counterscreen (Z’ of 0.84 ± 0.03 and an average S:B of 5.44 ± 0.08) to eliminate false positives, 98 commercially available compounds were advanced. A 10-point dose response titration, using a 3-fold dilution series, was used to establish IC50 values for these compounds. After completing the titrations and additional counterscreen, 51 active compounds, defined as having an IC50 < 10 μM in the primary OMFP assay and inactive (IC50>10 μM) in the counterscreen, were identified.
Figure 2.

20,580 compound high-throughput screen. The 20K screen, summarized in (A), yielded 115 initial hits using a standard cutoff (23.12% response, in this case) (B). After retesting and counterscreens, 51 active compounds (with IC50 < 10 μM) were identified and advanced for further testing. All of the assays performed well, yielding reasonable Z´ and signal-to-background values (C).
Major inhibitor classes identified in screen:
To investigate initial SAR in the data, we mined the screening results for obvious structural classes of compounds. While several classes of compounds were identified, two groups in particular represented nearly 30% of the total number of actives. One large class of hits, which contained 12 variants in the primary screen, was the tetracycline derivatives. The structures of the six best tetracycline compounds from the HTS are shown in Figure 3. These compounds displayed IC50 values that ranged from low micromolar to high nanomolar (Table 2). Another large, structurally similar, class of compounds that was identified was the sulfonated azo dyes (Fig. 4). These azo dyes also showed low to sub-micromolar IC50 values in the primary screen (Table 3). Additional compound classes included a small set of 3,4-dihydroxyphenylalanine (L-dopa) variants, and a number of 6-hydroxyuracil (barbituric acid) derivatives.
Figure 3.

Tetracycline derivatives identified in the screen. Shown are the structures and names of the top tetracycline active compounds (IC50 < 10 μM) selected in the screening campaign.
Table 2.
Active Tetracycline derivatives identified in 20K compound screen
| Name | Mol. Weight (g/mol) | Primary IC50 (μM) | Counterscreen IC50 (μM) | Promiscuity Indexa | Ki (μM) |
|---|---|---|---|---|---|
| Methacycline | 478.9 | 0.67 | >14.5 | 6 of 73 | 326 ± 35 |
| Meclocycline | 476.9 | 1.8 | >14.5 | 3 of 67 | NDb |
| Tetracycline | 444.4 | 1.9 | >14.5 | 3 of 73 | 199 ± 20 |
| Oxytetracyline | 460.4 | 3.6 | >14.5 | 4 of 67 | ND |
| Doxycycline | 444.4 | 4.1 | >14.5 | 5 of 73 | 99 ± 19 |
| Demeclocycline | 464.9 | 4.7 | >14.5 | 1 of 67 | ND |
The promiscuity index is the number of times this compound appeared as a hit in a screen compared to the number of total screens conducted that contained this compound – a lower number is desirable. This metric measures promiscuity reflected in all screens carried out by the Scripps Research Molecular Screening Center. Additional promiscuity measures can be seen in Tables S1 and S2.
ND: Not determined
Figure 4.

Sulfonated azo dyes. Shown are the structures and names of the top sulfonated azo dye variants that were identified as active compounds (IC50 < 10 μM) in the screening campaign
Table 3.
Active sulfonated azo dyes identified in 20K compound screen
| Name | Mol. Weight (g/mol) | Primary IC50 (μM) | Counterscreen IC50 (μM) | Promiscuity Indexa | Ki (μM) |
|---|---|---|---|---|---|
| Congo Red | 696.7 | 0.92 | >14.5 | 11 of 73 | 0.079 ± 0.02 |
| Chicago Sky Blue | 992.8 | 1.0 | >14.5 | 8 of 67 | NDb |
| NF-279 | 1401.1 | 2.4 | >29 | 5 of 75 | ND |
| Evans Blue | 960.8 | 3.1 | 23.6 | 19 of 82 | 0.39 ± 0.11 |
| PPNDS | 694.4 | 6.9 | >29 | 3 of 75 | ND |
The promiscuity index is the number of times this compound appeared as a hit in a screen compared to the number of total screens conducted that contained this compound – a lower number is desirable. This metric measures promiscuity reflected in all screens carried out by the Scripps Research Molecular Screening Center. Additional promiscuity measures can be seen in Tables S1 and S2.
ND: Not determined
We decided to further investigate the tetracyclines as Sts-1 phosphatase inhibitors. Despite the low promiscuity (a measure of how often a compound shows up in screens) observed in our screening data, some tetracycline derivatives appear frequently in HTS assays reported in PubChem (Table S1). To confirm that these inhibitors were specifically inhibiting the Sts phosphatase activity and not interfering with our assay, we determined the inhibition constants of a subset of these compounds with an orthogonal approach. Using an absorbance-based assay, which relies on detection of p-nitrophenol, we calculated the Ki values for the inhibition of Sts-1HP by methacycline, tetracycline and doxycycline (Table 2). While the results show the same trend as the primary screen IC50 data, the values reflect a significantly lower level of inhibition. Further analysis of the protein-inhibitor interactions (see below) suggests that these tetracycline derivatives may be binding peripherally at the active site. This may allow partial access of the smaller p-nitrophenyl phosphate (PNPP) substrate while more effectively blocking access of the larger OMFP substrate. This would explain why the titration data, which employed the OMFP substrate, shows a much higher level of inhibition. As Sts-1 is a protein tyrosine phosphatase and the native substrate is, as such, a protein molecule, the IC50 values determined using the OMFP assay are likely more indicative of the physiologically relevant response to these inhibitors.
Interestingly, while tetracycline use as antimicrobial agents is well known, additional clinical phenotypes have been reported. One example is the antifungal effect of tetracycline analogs when used alone or combined with antifungal agents (35–38). Several mechanisms have been proposed to explain these observations including enhancement of calcium release, downregulation of efflux pump protein expression and effects on biofilm formation (37, 39, 40). Considering the profound resistance to C. albicans infection and the enhanced fungal clearance conferred by the Sts-knockout, an alternative mechanism for the observed antifungal effect of tetracycline derivatives could be explained by the inhibition of the Sts-1 phosphatase activity. Further studies are needed to confirm if the inhibition of the Sts enzymes is indeed a factor in the observed antifungal activity of tetracyclines.
Multiple sulfonated azo dyes (Fig. 4) also appeared with high frequency ((6 of the 51 active compounds) in our screening results. These compounds had IC50 values from the low micromolar to sub-micromolar (Table 3). Likely owing to their inherent fluorescence, some of these compounds also show up in a large number of other screens (Tables 3 and S2). Two of the compounds, NF-279 and PPNDS, had a reasonably low promiscuity index (≤ 5). These two compounds are both potent and selective inhibitors of P2X receptors (41, 42). To rule out that these compounds were non-specifically interfering with the assay, we determined the inhibition constants for two representative sulfonated azo dyes using the PNPP assay. Both Congo red and Evans blue were potent inhibitors with nanomolar Ki values (Table 3). As observed in the primary screen, Congo red was several-fold more potent than Evans blue. Unlike the tetracyclines, the sulfonated azo dyes show high levels of inhibition regardless of the substrate used in these assays. This indicates that these compounds more fully occupy the active site, precluding even the smaller PNPP substrate. Analysis of the conserved structural features of these azo dyes suggests that the naphthalenesulfonate group may act as a phosphotyrosine surrogate. The catalytic histidine residues are likely to bind the sulfonate group, as observed in the sulfate-bound crystal structure of Sts-1HP (20). The conserved arginine residues would then be oriented to make cation-π interactions with the naphthyl group. Additional protein-inhibitor interactions would likely occur between the peripheral aromatic residues in the active site and the phenyl, benzyl, pyridoxal or methoxybenzyl group (see further discussion below).
Mechanism of action and selectivity:
To examine MOA we determined the kinetics of the Sts-1HP phosphatase reaction at several inhibitor concentrations for doxycycline and Congo Red. Analysis of the double reciprocal plots of the data, in both cases, show a reduction in Km and no effect on the maximal rate, indicating competitive inhibition (Fig. 5). The initial rates of progress curves were linear, suggesting rapid equilibrium inhibition, and confirmatory dilution experiments did not suggest any time dependence of the inhibition (43, 44).
Figure 5.

Mechanism of action. Double reciprocal plots for inhibition of Sts-1HP by the tetracycline derivative doxycycline (A) and the azo dye Congo red (B). For each, three concentrations of the inhibitor were used (shown as inset on each graph) and a linear fit was used. The lines fit to the data intersect the X-axis at a value close to zero, in both cases, indicating a competitive mode of inhibition.
Drug discovery efforts targeting PTPs are often hampered by off-target effects from non-selective binding of inhibitors. While Sts-1 is a protein tyrosine phosphatase, it is structurally distinct from canonical PTPs (20). To determine the selectivity of the inhibitors for Sts-1HP, we quantified the effect of a tetracycline derivative and an azo dye on the activity of the non-receptor type PTPs PTP1B and SHP1. The data, in Table 4, shows that Congo red is approximately 20- and 50-fold more active against Sts-1 than either PTP1B or SHP1, respectively. Similarly, doxycycline is more than 200-fold more potent against Sts-1 than the other PTPs. These data demonstrates that these compounds are relatively potent and selective competitive inhibitors of Sts phosphatase activity. In addition, this illustrates the overall effectiveness of the screening approach as a means of hit generation for lead development.
Table 4.
Selectivity of inhibitors for Sts-1HP over other PTPs
| Protein | Km (mM) | Ki Doxycycline (mM) | Ki Congo red (μM) |
|---|---|---|---|
| Sts-1HP | 2.64 ± 0.48 | 0.099 ± 0.019 | 0.079 ± 0.02 |
| PTP-1B | 5.39 ± 1.07 | No Inhibition | 1.47 ± 0.34 |
| SHP1 | 3.28 ± 0.26 | No Inhibition | 3.88 ± 0.59 |
Key structural features of Sts-1 that contribute to inhibitor binding:
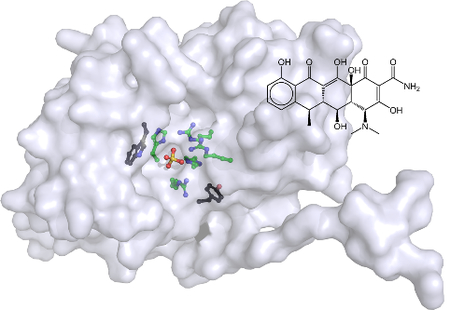
Structural comparison of the hits from the screen, and the two classes highlighted above in particular, yielded several initial structure-activity relationship observations. Both the tetracycline derivatives and sulfonated azo dyes all possess a high number of hydrogen bond donors and acceptors. The azo dyes, specifically, also have multiple sulfonates, which can potentially act as phosphate surrogates in the Sts active site. The stereochemistry of the compounds also played a key role, something that is most evident in the comparison of the tetracycline analogs (Fig. 3 and Table 2). Subtle stereochemical changes yield several-fold differences in potency. Perhaps the most striking similarity amongst the inhibitor classes discovered in the screen was the almost universal presence of relatively planar, polyaromatic groups. This propensity for Sts-1 to bind flat, polyaromatic compounds is also seen in the substrate specificity. The larger coumarin derivative DiFMUP and fluorescein derivative OMFP are significantly better substrates (kcat/Km = 5 × 107 and 4 × 106, respectively) than the phosphotyrosine mimic p-nitrophenyl phosphate (kcat/Km = 5 × 103) (20). This trend likely is indicative of structural features of the protein that confer selectivity for the native substrate(s). Examination of the structure of Sts-1HP reveals the presence of a tryptophan residue (W494) and a tyrosine residue (Y596) at the periphery of the active site (Fig. 6).
Figure 6.

Active site of the histidine phosphatase domain of Sts-1. In addition to the catalytic histidine residues (H379 and H565; shown in ball-and-stick representation with green carbon atoms and blue nitrogen atoms) and conserved arginine residues (R379, R383 and R462; shown in ball-and-stick representation with green carbon atoms and blue nitrogen atoms), two aromatic residues (W494 and Y596; shown in ball-and-stick representation with black carbon atoms, blue nitrogen atoms and red oxygen atoms) occupy the phosphatase active site of Sts-1 (A). A sulfate molecule (shown in ball-and-stick representation with the sulfate atom shown in orange and oxygen atoms shown in red) in the structure acts as a phosphate surrogate and marks the site where catalysis occurs. An overall view of the Sts-1HP dimer (B); the left protomer shows a surface representation where the conserved His and Arg residues are colored blue, while W494 and Y596 are colored black) illustrates how Trp494 and Tyr596 are at the periphery of the active site and could serve a role in positioning planar aromatic inhibitors to block access to the active site.
To determine if specific residues at or near the active site make significant contributions to the binding of polycyclic aromatic inhibitors, we reexamined the inhibition kinetics using several Sts-1 mutants (Table 5). Mutation of W494 to either phenylalanine, histidine or leucine reduces the potency (measured by the ratio of Ki of mutant to Ki of native Sts-1HP) of doxycycline by 7 – 15 fold and of Congo red by 13 – 18 fold. The contribution of Y596 to inhibitor binding was much smaller. Mutation of Y596 to phenylalanine or leucine led to only small decreases (< 5-fold) in inhibitor potency. The single exception was the Y596L mutant, which was inhibited by Congo red with a 12-fold lower potency. These results indicate that both of these residues contribute to ligand binding, and that W494 plays a more significant role in this regard. Comparison of the sulfate-bound to the unliganded Sts-1HP structures show that, while the conserved histidine and arginine sidechains remain fixed, both W494 and Y596 move upon substrate binding (20). W494, in particular, undergoes a slight (~10°) rotation leaving the nitrogen atom more than 2Å from its position in the unliganded structure. In addition, in the X-ray crystal structure, W494 had one of the highest B-factor values (which measure the extent to which the electron density is spread out in the model). This indicates that this tryptophan residue has a high degree of local mobility. This flexibility may contribute to how the protein recognizes and binds to substrates.
Table 5.
Inhibition of Sts-1HP mutants
| Construct | Km (mM) | Ki Doxycycline (mM) | Doxycycline Ki Ratioa | Ki Congo Red (μM) | Congo Red Ki Ratioa |
|---|---|---|---|---|---|
| Sts-1HP | 2.64 ± 0.48 | 0.099 ± 0.019 | 1 | 0.079 ± 0.02 | 1 |
| W494F | 5.70 ± 1.05 | 1.42 ± 0.11 | 14.3 | 1.42 ± 0.32 | 18.0 |
| W494L | 6.90 ± 0.95 | 0.73 ± 0.04 | 7.4 | 1.46 ± 0.13 | 18.5 |
| W494H | 6.32 ± 0.83 | 1.48 ± 0.09 | 14.9 | 1.07 ± 0.03 | 13.5 |
| Y596F | 3.65 ± 0.49 | 0.39 ± 0.04 | 3.9 | 0.23 ± 0.02 | 2.9 |
| Y596L | 3.88 ± 0.74 | 0.49 ± 0.05 | 4.9 | 0.94 ± 0.05 | 11.9 |
Defined as the ratio of the Ki of the mutant Sts-1HP to that of the Ki of the wild-type Sts-1HP enzyme
In Sts-1, the SH3 and UBA domains are believed to be protein-protein interacting domains that may help to recruit specific protein targets for dephosphorylation. Despite the presence of these domains, a selectivity mechanism would almost certainly be required to distinguish between the many phosphotyrosine residues on the target protein. Zap-70, a known substrate of Sts-1, has over 25 tyrosine residues, many of which are clustered together. Both Sts-1 and Sts-2 show varying levels of phosphatase activity for different tyrosine residues on Zap-70, indicating that a mechanism for selectivity is in place (45, 46). Our data suggests that the aromatic residues at the active site, particularly W494, may serve as a selectivity filter for native substrates. Further analysis of protein-ligand interactions and the structural features that determine these interactions is needed in order to fully define this selectivity mechanism.
Immunotherapies have become of increasing interest of late, particularly in their promise to treat infection and different types of cancers (47–51). For the treatment of infectious disease, there are definite advantages in the use of immunomodulatory agents, either alone or in combination with standard of care antibiotics. Central among these is the decreased potential to generate antibiotic resistance (48). While there are a number of immunostimulatory therapies currently in use, they are primarily in the form of peptides and other biomolecules, and new targets are actively being sought. The Sts proteins are a highly promising new target for the development of immunomodulatory therapies. Genetic inactivation of these proteins leads to a profound resistance phenotype and accelerated clearance of several types of dangerous human pathogens (8, 9). As a first step towards developing Sts-targeted drugs, we have identified two new classes of selective inhibitors of Sts-1 phosphatase activity using a high-throughput approach. These compounds, a series of tetracycline derivatives and several sulfonated azo dyes, are the first reported inhibitors that are both potent and selective for Sts-1. The results of the screen and accompanying characterization of the hits provides important insights into substrate binding and is strong evidence supporting the druggability of this class of protein.
MATERIALS AND METHODS
Cloning, expression and purification of Sts-1HP and mutants
Sts-1HP (residues 373–636) was cloned with a 6xhis tag in the pTHT vector (a modified form of pET-28 with a Tobacco Etch Virus protease site in place of the thrombin site). To generate mutants, wild-type Sts-1HP construct was used as DNA templates for PCR-based site-directed mutagenesis. All constructs were sequence-verified to ensure accuracy. Wild-type Sts-1HP and mutants were expressed using BL21(DE3) cells and purified as previously described (20).
Fluorescence assay protocol in 1,536-well plate format
The optimized conditions for the HTS assay in 1,536-well plate format are summarized in Table 6. Assay buffer was prepared fresh daily and contained 30 mM Tris HCl, 75 mM NaCl, 1 mM EDTA and 1m M DTT (all reagents from Sigma Aldrich). STS-1HP was prepared in assay buffer to a final concentration of 750 ng/ml. 4 μL of STS-1HP, or buffer only, were dispensed to the appropriate wells of a 1,536-well black solid bottom microtiter plates (Greiner BioOne) using an FRD (Aurora Biosciences). Test compounds in DMSO or DMSO alone were added to the appropriate wells using the automated GNF/Kalypsis robotic platform. A 10 mM stock of OMFP, freshly prepared in neat DMSO, was diluted in water and the assay was started by dispensing 2 μL of OMFP fluorescent substrate to all wells to yield a 50 μM final concentration. Plates were centrifuged and incubated for 20 minutes at room temperature. To stop the reaction, a 0.7 N NaOH solution was added at 1 μL to a final concentration of 100 mM. After centrifugation, the plates were read at 485 nm excitation and 525 nm emission using a Viewlux plate reader (PerkinElmer Life Sciences).
Table 6.
Summary of Sts-1 HTS assay protocol for 1,536-well plate format
| Step | Description | Quantity/Condition | Comments |
|---|---|---|---|
| 1 | Sts-1HP addition in buffer | 4 μL/well | 750 ng/mL STS-1 in Assay buffer (30 mM Tris HCl, 75 mM NaCl, 1 mM EDTA, 1 mM DTT) |
| 2 | Pin compound or DMSO | 35 nL/well | 5.8 μM compound in 0.4% DMSO final |
| 3 | OMFP addition in water | 2 μL/well | 50uM final |
| 4 | Incubation | 20 minutes | Room temperature in the dark |
| 5 | NaOH addition | 1 μL/well | 100 mM final, stops reaction |
| 6 | Read fluorescence | Viewlux | Ex: 480 nm; Em: 520 nm |
Compound library
For the pilot screen, a 1,280 compound library known as the Library of Pharmacologically Active Compounds (LOPAC®1280, Sigma Aldrich) was used. The compounds were tested in triplicate at a single nominal concentration of 5.8uM. In the 20,580-compound screen, a subset of the Scripps Drug Discovery Library (SDDL) was profiled in the HTS campaign. The SDDL currently consists of >650K unique compounds, representing a diversity of drug-like scaffolds targeted to traditional and non-traditional drug-discovery biology. The SDDL has been curated from over 20 commercial and academic sources and contains more than 20,000 compounds unique to Scripps. The SDDL compounds have been selected based on scaffold novelty, physical properties and spatial connectivity (52–57). We selected compounds for the 20K pilot effort that included a heavy concentration of approved drugs (~3,200), or drug like molecules such as the Cayman bioactive lipids (>1,000), TOCRIS, Prestwick, and for diversity, portions of the Chembridge, Enamine, Life Chemicals and Microscources collections.
Data Analysis
In both the LOPAC pilot screen and the 20K HTS, compounds were tested at a single concentration at a final nominal concentration of 5.8 μM using the protocol summarized in Table 6. The activity of each well was normalized on a per-plate basis using the following equation:
Where Test well is defined as wells containing enzyme/substrate in the presence of test compound, Median Low Control is defined as the median of the wells containing test compounds and Median High Control is defined as the wells containing DMSO and OMFP in the absence of the Sts-1HP enzyme.
The reference compound, PHPS1, was used to monitor the stability of the assay, but was not used in the calculation of the controls or the normalization. Data was normalized on a per plate basis, and each assay plate underwent a quality control check. A value greater than 0.5 for Z´ was required before further analysis (58). Similar to other HTS campaigns, nominally inhibiting compounds (hit cutoff) were selected as those that had response values above three times the standard deviation of the average percent response from all compounds tested (59). Data was visualized with a randomized scatterplot of all compound activity (Figs. 1A and 2B), generated using Spotfire Pro (TIBCO Software Inc.).
Counterscreen and concentration response assays
The concentration response curves (CRCs) were generated using assays that employed the same reagents, protocols, and detection systems as the primary assay, but also utilized a counterscreen assay where the compounds were added after the NaOH step to help identify fluorescent quenchers and other non-specific inhibitors. The assays tested compounds as 10-point dose-response titration (3-fold dilutions) in triplicate. For each test compound, percent activity was plotted against compound concentration. A four parameter equation describing a sigmoidal dose-response curve was then fitted with adjustable baseline using Assay Explorer software (Symyx Technologies Inc.). The reported IC50 values were generated from fitted curves by solving for the X-intercept value at the 50% activity level of the Y-intercept value. Compounds with an IC50 greater than 10 μM were considered inactive while compounds with an IC50 equal to or less than 10 μM were considered active. Only compounds that were active (IC50 < 10 uM) in the primary screen and inactive (IC50 > 10 uM) in the counterscreen were considered hits and given further consideration.
QA/QC
All samples in the SDDL have been tested for purity and structural confirmation via LC-MS and/or NMR to provide adequate QA/QC. In addition, after completion of the 20K screen, all compounds that proceeded through the titration phase were subjected to LC-MS analysis using samples obtained directly from the same source plates used in the assay. This allowed for reconfirmation of hit sample purity (single peak) and mass (correct molecular weight). Only those samples that passed LC-MS analysis for both purity and mass were advanced for further analysis.
Steady-State Kinetics
The steady-state kinetics were conducted using a spectrophotometric phosphatase assay making use of p-nitrophenyl phosphate (pNPP). The assay was run as described previously (20). Briefly, Sts-1HP was assayed in a buffer containing a final concentration of 20 mM Tris (pH 7.5), 150 mM sodium chloride, 5 mM magnesium chloride and 1 mM βME and varying concentrations of pNPP as a substrate. Activity was measured by following the increase in absorbance at 405 nm due to the production of pNP. The initial reaction rate (<10 % reaction completion) was plotted against pNPP concentration. The data were fit with a rectangular hyperbola using the Michaelis-Menten equation and values for Km and kcat were determined from the graph (Kaleidagraph, Synergy Software).
Inhibition Analysis
The pNPP assay was used for the follow-up inhibition analysis after completion of the 20K pilot screen and for MOA studies. The initial velocity, as a function of substrate concentration, was measured at several fixed concentrations of the inhibitor of interest (20). In all cases, a series of control reactions were run to ensure that the optical properties of the compound being tested were not interfering with the assay or changing in the presence of the enzyme. An example of our raw data and analysis are given in the Supplemental Information (Figs. S1 – S3). Lineweaver-Burk (double reciprocal) plots were constructed in each case and a linear fit was used. All the kinetic experiments were conducted in triplicate, and the error bars shown are the standard error from triplicate measurements. Error values given in tables report the error in the fit to the equation. For the double reciprocal plots, the data was fit using equation (1) for competitive inhibition:
| (1) |
Ki was calculated from the Km value determined in the presence of inhibitor using equation (2):
| (2) |
The error values provided in all tables are standard errors from triplicate measurements.
Supplementary Material
Table S1 - Promiscuity of tetracycline compounds in PubChem BioAssays
Table S2 - Promiscuity of azo dye compounds in PubChem BioAssays
Figure S1 - Raw data for the measurement of the inhibition of Sts-1HP by Congo Red using the PNPP-based absorbance assay
Figure S2 - Analysis of inhibition of Sts-1HP phosphatase activity by Congo Red measured by the PNPP absorbance-based assay
Figure S3 - Analysis of inhibition of Sts-1HP phosphatase activity by Congo Red measured by the OMFP fluorescence-based assay
ACKNOWLEDGEMENTS
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH) through Award Number U01HL127522 (JBF, NC), the National Institute of Allergy and Infectious Disease of the NIH through Award Number R21AI130859 (JBF, NC), and the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH17-1-0147 (JBF, NC). In addition, parts of this work were supported by the National Institutes of General Medical Sciences of the NIH under grant R35GM124898 (JBF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Defense. Additional support was provided by the Center for Biotechnology, a New York State Center for Advanced Technology, Stony Brook University, Cold Spring Harbor Laboratory, Brookhaven National Laboratory and Feinstein Institute for Medical Research.
ABBREVIATIONS
- Sts
Suppressor of TCR Signaling
- TCR
T cell receptor
- PTP
protein tyrosine phosphatase
- HP
histidine phosphatase
- UBA
ubiquitin association domain
- SH3
Src-homology 3 domain
- PGM
phosphoglycerate mutase
- PHPS1
4-[2-[1,5-dihydro-3-(4-nitrophenyl)-5-oxo-1-phenyl-4H-pyrazol-4-ylidene]hydrazinyl]-benzenesulfonic acid
- OMFP
3-O-methylfluorescein phosphate
- LOPAC
Library of pharmacologically active compounds
- HTS
high throughput screening
- PNPP
p-nitrophenylphosphate
- MOA
mechanism of action
- SAR
structure activity relationship
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interest.
REFERENCES
- 1.Mustelin T, Tasken K. 2003. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem J 371:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith-Garvin JE, Koretzky GA, Jordan MS. 2009. T cell activation. Annu Rev Immunol 27:591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singer AL, Koretzky GA. 2002. Control of T cell function by positive and negative regulators. Science 296:1639–40. [DOI] [PubMed] [Google Scholar]
- 4.Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, Doherty P, Ihle JN. 2004. Regulation of ZAP-70 Activation and TCR Signaling by Two Related Proteins, Sts-1 and Sts-2. Immunity 20:37–46. [DOI] [PubMed] [Google Scholar]
- 5.Mikhailik A, Ford B, Keller J, Chen Y, Nassar N, Carpino N. 2007. A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol Cell 27:486–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpino N, Naseem S, Frank DM, Konopka JB. 2017. Modulating Host Signaling Pathways to Promote Resistance to Infection by Candida albicans. Front Cell Infect Microbiol 7:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frank D, Naseem S, Russo GL, Li C, Parashar K, Konopka JB, Carpino N. 2018. Phagocytes from Mice Lacking the Sts Phosphatases Have an Enhanced Antifungal Response to Candida albicans. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naseem S, Frank D, Konopka JB, Carpino N. 2015. Protection from systemic Candida albicans infection by inactivation of the Sts phosphatases. Infect Immun 83:637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parashar K, Kopping E, Frank D, Sampath V, Thanassi DG, Carpino N. 2017. Increased Resistance to Intradermal Francisella tularensis LVS Infection by Inactivation of the Sts Phosphatases. Infect Immun 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. [DOI] [PubMed] [Google Scholar]
- 11.Brown GD, Denning DW, Levitz SM. 2012. Tackling human fungal infections. Science 336:647. [DOI] [PubMed] [Google Scholar]
- 12.Mayer FL, Wilson D, Hube B. 2013. Candida albicans pathogenicity mechanisms. Virulence 4:119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asare R, Kwaik YA. 2010. Exploitation of host cell biology and evasion of immunity by francisella tularensis. Front Microbiol 1:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLendon MK, Apicella MA, Allen LA. 2006. Francisella tularensis: taxonomy, genetics, and Immunopathogenesis of a potential agent of biowarfare. Annu Rev Microbiol 60:167–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oyston PC. 2008. Francisella tularensis: unravelling the secrets of an intracellular pathogen. J Med Microbiol 57:921–30. [DOI] [PubMed] [Google Scholar]
- 16.Hicke L, Schubert HL, Hill CP. 2005. Ubiquitin-binding domains. Nat Rev Mol Cell Biol 6:610–21. [DOI] [PubMed] [Google Scholar]
- 17.Musacchio A 2002. How SH3 domains recognize proline. Adv Protein Chem 61:211–68. [DOI] [PubMed] [Google Scholar]
- 18.Tsygankov AY. 2013. TULA-family proteins: a new class of cellular regulators. J Cell Physiol 228:43–9. [DOI] [PubMed] [Google Scholar]
- 19.Rigden DJ. 2008. The histidine phosphatase superfamily: structure and function. Biochem J 409:333–48. [DOI] [PubMed] [Google Scholar]
- 20.Zhou W, Yin Y, Weinheimer AS, Kaur N, Carpino N, French JB. 2017. Structural and Functional Characterization of the Histidine Phosphatase Domains of Human Sts-1 and Sts-2. Biochemistry 56:4637–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bond CS, White MF, Hunter WN. 2001. High resolution structure of the phosphohistidine-activated form of Escherichia coli cofactor-dependent phosphoglycerate mutase. J Biol Chem 276:3247–53. [DOI] [PubMed] [Google Scholar]
- 22.Bond CS, White MF, Hunter WN. 2002. Mechanistic implications for Escherichia coli cofactor-dependent phosphoglycerate mutase based on the high-resolution crystal structure of a vanadate complex. J Mol Biol 316:1071–81. [DOI] [PubMed] [Google Scholar]
- 23.San Luis B, Nassar N, Carpino N. 2013. New insights into the catalytic mechanism of histidine phosphatases revealed by a functionally essential arginine residue within the active site of the Sts phosphatases. Biochem J 453:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Q, Jiang D, Zhang W, Zhang Q, Zhao Q, Jin J, Li X, Yang H, Bartlam M, Shaw N, Zhou W, Rao Z. 2014. Mechanism of dephosphorylation of glucosyl-3-phosphoglycerate by a histidine phosphatase. J Biol Chem 289:21242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luis BS, Carpino N. 2014. Insights into the suppressor of T-cell receptor (TCR) signaling-1 (Sts-1)-mediated regulation of TCR signaling through the use of novel substrate-trapping Sts-1 phosphatase variants. FEBS J 281:696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Jakoncic J, Carpino N, Nassar N. 2009. Structural and functional characterization of the 2H-phosphatase domain of Sts-2 reveals an acid-dependent phosphatase activity. Biochemistry 48:1681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Jakoncic J, Parker KA, Carpino N, Nassar N. 2009. Structures of the phosphorylated and VO(3)-bound 2H-phosphatase domain of Sts-2. Biochemistry 48:8129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivanova E, Carpino N. 2016. Negative regulation of TCR signaling by ubiquitination of Zap-70 Lys-217. Mol Immunol 73:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman TN, Liverani E, Ivanova E, Russo GL, Carpino N, Ganea D, Safadi F, Kunapuli SP, Tsygankov AY. 2014. Members of the novel UBASH3/STS/TULA family of cellular regulators suppress T-cell-driven inflammatory responses in vivo. Immunol Cell Biol 92:837–50. [DOI] [PubMed] [Google Scholar]
- 30.Imoto M, Kakeya H, Sawa T, Hayashi C, Hamada M, Takeuchi T, Umezawa K. 1993. Dephostatin, a novel protein tyrosine phosphatase inhibitor produced by Streptomyces. I. Taxonomy, isolation, and characterization. J Antibiot (Tokyo) 46:1342–6. [DOI] [PubMed] [Google Scholar]
- 31.Umezawa K, Kawakami M, Watanabe T. 2003. Molecular design and biological activities of protein-tyrosine phosphatase inhibitors. Pharmacol Ther 99:15–24. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe T, Takeuchi T, Otsuka M, Tanaka S, Umezawa K. 1995. Synthesis and protein tyrosine phosphatase inhibitory activity of dephostatin analogs. J Antibiot (Tokyo) 48:1460–6. [DOI] [PubMed] [Google Scholar]
- 33.Gupta SC, Phromnoi K, Aggarwal BB. 2013. Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells through activation of protein tyrosine phosphatase SHP1. Biochem Pharmacol 85:898–912. [DOI] [PubMed] [Google Scholar]
- 34.Liang F, Huang Z, Lee SY, Liang J, Ivanov MI, Alonso A, Bliska JB, Lawrence DS, Mustelin T, Zhang ZY. 2003. Aurintricarboxylic acid blocks in vitro and in vivo activity of YopH, an essential virulent factor of Yersinia pestis, the agent of plague. J Biol Chem 278:41734–41. [DOI] [PubMed] [Google Scholar]
- 35.Fiori A, Van Dijck P. 2012. Potent synergistic effect of doxycycline with fluconazole against Candida albicans is mediated by interference with iron homeostasis. Antimicrob Agents Chemother 56:3785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Y, Li H, Liu S, Zhang X, Sun S. 2014. Synergistic effect of fluconazole and doxycycline against Candida albicans biofilms resulting from calcium fluctuation and downregulation of fluconazole-inducible efflux pump gene overexpression. J Med Microbiol 63:956–61. [DOI] [PubMed] [Google Scholar]
- 37.Gu W, Yu Q, Yu C, Sun S. 2018. In vivo activity of fluconazole/tetracycline combinations in Galleria mellonella with resistant Candida albicans infection. J Glob Antimicrob Resist 13:74–80. [DOI] [PubMed] [Google Scholar]
- 38.Miceli MH, Bernardo SM, Lee SA. 2009. In vitro analyses of the combination of high-dose doxycycline and antifungal agents against Candida albicans biofilms. Int J Antimicrob Agents 34:326–32. [DOI] [PubMed] [Google Scholar]
- 39.Gao Y, Zhang C, Lu C, Liu P, Li Y, Li H, Sun S. 2013. Synergistic effect of doxycycline and fluconazole against Candida albicans biofilms and the impact of calcium channel blockers. FEMS Yeast Res 13:453–62. [DOI] [PubMed] [Google Scholar]
- 40.Shi W, Chen Z, Chen X, Cao L, Liu P, Sun S. 2010. The combination of minocycline and fluconazole causes synergistic growth inhibition against Candida albicans: an in vitro interaction of antifungal and antibacterial agents. FEMS Yeast Res 10:885–93. [DOI] [PubMed] [Google Scholar]
- 41.Damer S, Niebel B, Czeche S, Nickel P, Ardanuy U, Schmalzing G, Rettinger J, Mutschler E, Lambrecht G. 1998. NF279: a novel potent and selective antagonist of P2X receptor-mediated responses. Eur J Pharmacol 350:R5–6. [DOI] [PubMed] [Google Scholar]
- 42.Lambrecht G, Rettinger J, Baumert HG, Czeche S, Damer S, Ganso M, Hildebrandt C, Niebel B, Spatz-Kumbel G, Schmalzing G, Mutschler E. 2000. The novel pyridoxal-5’-phosphate derivative PPNDS potently antagonizes activation of P2X(1) receptors. Eur J Pharmacol 387:R19–21. [DOI] [PubMed] [Google Scholar]
- 43.Copeland RA. 2003. Mechanistic considerations in high-throughput screening. Anal Biochem 320:1–12. [DOI] [PubMed] [Google Scholar]
- 44.Copeland RA. 2005. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem Anal 46:1–265. [PubMed] [Google Scholar]
- 45.Chen X, Ren L, Kim S, Carpino N, Daniel JL, Kunapuli SP, Tsygankov AY, Pei D. 2010. Determination of the substrate specificity of protein-tyrosine phosphatase TULA-2 and identification of Syk as a TULA-2 substrate. J Biol Chem 285:31268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.San Luis B, Sondgeroth B, Nassar N, Carpino N. 2011. Sts-2 is a phosphatase that negatively regulates zeta-associated protein (ZAP)-70 and T cell receptor signaling pathways. J Biol Chem 286:15943–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuge O, Vasdev N, Allchorne P, Green JS. 2015. Immunotherapy for bladder cancer. Res Rep Urol 7:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masihi KN. 2001. Fighting infection using immunomodulatory agents. Expert Opin Biol Ther 1:641–53. [DOI] [PubMed] [Google Scholar]
- 49.Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, Levy R. 2018. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Syn NL, Teng MWL, Mok TSK, Soo RA. 2017. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 18:e731–e741. [DOI] [PubMed] [Google Scholar]
- 51.Yang Y 2015. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest 125:3335–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lovering F, Bikker J, Humblet C. 2009. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem 52:6752–6. [DOI] [PubMed] [Google Scholar]
- 53.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. [DOI] [PubMed] [Google Scholar]
- 54.Kolb HC, Sharpless KB. 2003. The growing impact of click chemistry on drug discovery. Drug Discov Today 8:1128–37. [DOI] [PubMed] [Google Scholar]
- 55.Ding S, Gray NS, Wu X, Ding Q, Schultz PG. 2002. A combinatorial scaffold approach toward kinase-directed heterocycle libraries. J Am Chem Soc 124:1594–6. [DOI] [PubMed] [Google Scholar]
- 56.Congreve M, Carr R, Murray C, Jhoti H. 2003. A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today 8:876–7. [DOI] [PubMed] [Google Scholar]
- 57.Baell JB, Holloway GA. 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53:2719–40. [DOI] [PubMed] [Google Scholar]
- 58.Zhang JH, Chung TD, Oldenburg KR. 1999. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4:67–73. [DOI] [PubMed] [Google Scholar]
- 59.Madoux F, Tanner A, Vessels M, Willetts L, Hou S, Scampavia L, Spicer TP. 2017. A 1536-Well 3D Viability Assay to Assess the Cytotoxic Effect of Drugs on Spheroids. SLAS Discov 22:516–524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 - Promiscuity of tetracycline compounds in PubChem BioAssays
Table S2 - Promiscuity of azo dye compounds in PubChem BioAssays
Figure S1 - Raw data for the measurement of the inhibition of Sts-1HP by Congo Red using the PNPP-based absorbance assay
Figure S2 - Analysis of inhibition of Sts-1HP phosphatase activity by Congo Red measured by the PNPP absorbance-based assay
Figure S3 - Analysis of inhibition of Sts-1HP phosphatase activity by Congo Red measured by the OMFP fluorescence-based assay