

Abstract
The WWOX gene has a WW domain containing oxidoreductase, which is located at the common fragile site FRA16D at chromosome 16q23. WWOX is a tumor suppressor gene that has been associated with several types of cancer such as hepatic, breast, lung, prostate, gastric, and ovarian. Recently WWOX has been implicated in epilepsy, where studies show homozygous loss-of-function mutation lead to early-infantile epileptic encephalopathy, spinocerebellar ataxia, intractable seizures and developmental delay, and early lethal microcephaly syndrome with epilepsy. Here we investigate two consanguineous Saudi families and we identified three probands with epileptic encephalopathy. Whole exome sequencing revealed a novel homozygous mutation in the WWOX gene in one proband. In addition, we identified a previously reported WWOX mutation in two probands. Later on these findings were confirmed with Sanger sequencing. The underlying mechanism on how WWOX mutations lead to seizure remains elusive. To date very few WWOX mutations have been associated with neurological disorder and our newly identified mutations support the notion that WWOX play an important role in neurons and will aid in better diagnosis and genetic counseling.
Keywords: WWOX, Cancer, WW domain, epilepsy, Tumor suppressor gene, Encephalopathy, Oxidoreductase
Introduction
The WWOX gene (WW domain-containing oxidoreductase[MIM 605131]) is a tumor suppressor gene that have been associated with several types of cancer such as breast, hepatic, lung, prostate, ovarian, colon, gastric, and esophageal carcinomas [1, 2, 3, 4, 5, 6, 7, 8, 9]. The WWOX gene is located on chromosome 16q23 and encodes 414 amino acids protein (46 kDa), which contains two N-terminal WW domains that has highly conserved proline and tryptophan residues responsible of mediating protein-protein interactions, a short chain of dehydrogenase reductase domain at the C-terminal, and a nuclear localization sequence [2, 10, 11]. When cells are exposed to apoptotic stresses, WWOX is up-regulated and activated by pY33 phosphorylation via Tyrosine Kinase Src [12, 13]. The pY33-WWOX act as cancer suppressor by inhibiting tumor growth and supporting normal cell physiology [14]. On the other hand, phosphorylation of Ser14 in the WWOX protein is necessary for T cell maturation [15]. However, WWOX lose the tumor suppression function when Ser14 is phosphorylated and involved in cancer progression, but when mouse model of melanoma is treated with Zfra (a WWOX inhibitor) the cancer cell growth and metastasis is blocked [16, 17].
Recently, several studies indicated an important role of WWOX in neuronal development, function, and diseases [18, 19, 20, 21]. Gribaa and colleagues, identified two WWOX homozygous mutations in highly conserved residues p.Pro47Thr and p.Gly372Arg in two consanguineous Saudi family resulting in autosomal recessive cerebellar ataxia with epilepsy and mental retardation [22, 23]. They demonstrated that the proline residue at position 47 is part of the hydrophobic core that stabilizes the WW fold and required to maintain the WWOX protein fully functional [23]. An additional two nonsense mutations have been identified a W44* and Arg54*, which result in non-functional protein and lead to intractable seizures and early lethal microcephaly syndrome with epilepsy respectively [24, 25].
In this study we investigated two consanguineous Saudi families and we identified three affected children with epileptic encephalopathy. One patient had a novel homozygous mutation that has not been previously described in the WWOX gene.
Method
Human Subjects
We investigated three probands from two Saudi families with WWOX mutation. All patients underwent a comprehensive clinical evaluation by a clinical geneticist and a neurologist. Samples were taken and sent to Centogene, Germany for WES, and validation by Sanger sequencing.
Whole Exome sequencing
First, approximately 37 Mb (214,405 exons) of the Consensus Coding Sequences (CCS) were enriched from fragmented genomic DNA by >340,000 probes designed against the human genome. Then, the sample has been processed on the NextSeq Platform (Illumina). Consequently, an end to end bioinformatics pipelines was applied. This included base calling, primary filtering of low quality reads and probable artifacts, and annotation of variants. For the medical evaluation, all disease causing variants reported in HGMD®, in ClinVar or in CentoMD® (classes 1 and 0) as well as all variants with minor allele frequency (MAF) of less than 1% in ExAc database are considered. Variants that possibly impair the protein sequence, i.e. disruption of conserved splice sites, missense, nonsense, readthroughs, or small insertions/deletions, are prioritized. All relevant inheritance patterns are considered based on the information given in the request is probed. For the inherited autosomal dominant disorders only variants/mutations of the classes 0, 1, and 2 are considered. Only variants related to the phenotype are reported.
Sanger sequencing
The findings were confirmed by PCR and sequencing of both DNA strands of the entire coding region and the highly conserved exonintron splice junctions.
Ethical statement
This study was approved by Institutional review board office at King Abdullah International Medical Research Centre (KIMARC) (Study number: RC16/113/R). Written consent was obtained from the participating patient or their parents.
Result
Clinical Report Patient 1
The proband is a 1-year and 9 months old male, is a second child of 1st cousin parents with no family history of the disorder born by normal spontaneous vaginal delivery with birth weight 2.7 kg (<3rd centile), Length: 49cm (<3rd centile) and head circumference (HC): 35cm (25th centile). Immediately after birth, he was admitted to NICU due to transient tachypnoea of newborn (TNN), cephalhematoma and indirect hyperbilirubinemia and discharge after few weeks. At two months he started to have focal seizure with secondary generalization followed by infantile spasms. The seizure was refractory to antiepileptic medications. Subsequently the patient continued to have global developmental delay, not fixing or following and just lying in the bed not acquiring milestones. On examination at 1 year 9 months of age, his height and weight 77cm and 10 kg were below the 5th percentile. He was microcephalic HC: 45cm and showed subtle dysmorphic features, in a form of low set ears, long eyelashes, long philtrum, high arch palate, depressed nasal bridge, micrognathia, gingival hypertrophy and low hairline. The skeletal survey revealed kyphosis with short clavicles, scoliosis, fibrochondrogenesis type 2, stickler syndrome type III, and platyspondyly. The neurological examination revealed that he is not fixing, not following, not rolling over, shows no social smile, he is not sitting and not reacting. Also he shows occasional horizontal nystagmus with fisting and spasticity in both upper and lower limbs with decreased deep tendon reflexes, plantar down going, and exhibits axial hypotonia. Brain Magnetic resonance image (MRI) showed brain atrophy and increased white matter signal in cerebellar area. All of the following biochemical and genetics investigations were unremarkable including: ammonia, lactic acid, creatine Kinase (CK) level, chromosomal analysis, acylcarnitine profile, arylsulfatase A and B, hexosaminidase A and B, and urine for mucopolysaccharidosis. Comparative genomic hybridization (CGH) array did not detect any copy number variants (CNV) which could be relevant for the phenotype of the patient. Whole exome sequencing (WES) revealed novel homozygous variant in intron 4 of the WWOX gene, NM_016373.3: c.409+1G > T (Ch16:78149052 G>T). Parents and healthy sister tested and they are carrier for the same mutation.
Patient 2
A 3 months old male, product of preterm (35 weeks) and normal vaginal delivery. He was admitted to neonatal intensive care unit (NICU) for respiratory distress secondary to congenital pneumonia. At age of 6 weeks, he started to have vomiting after each feeding, non-bilious, sometimes projectile, moderate to large amount, mixed with streaks of blood, for which he was admitted to the hospital for 5 days and conducted laboratory and imaging studies. The study concluded that the vomiting was due to an allergy to dairy protein. The patient was discharged on special formula, and since then he partially improved. At the age of 2 months, he started to have decreased activity, decreased interaction that progressed until he was not having any spontaneous movement, moving only when he is hungry, and having feeble cry. Then he started to have infantile spasms for few seconds, occur in clusters, 3 times/cluster each lasting for about 3 minutes, occurring once per week. Upon physical examination he was not fixing or following, not dysmorphic, has axial hypotonia, symmetrical face, spastic, peripheral hypertonia, +2 reflexes with no clonus, and no neurocutaneous stigmata. The MRI brain revealed brain atrophy, moderate corpus callosum thinning, unremarkable spectroscopy, electroencephalogram (EEG) low voltage interval with burst suppression. Whole exome sequencing revealed previously reported homozygous mutation at genomic location c.160G > T (p.Arg54*)[24], which resulted in a premature termination.
Patient 3
18 months old female (sister of patient 2). She is a product of full-term pregnancy and normal vaginal delivery. She was admitted to NICU for 5 days secondary to suspected sepsis. At age of 2 months she became hypoactive and the symptom progressed gradually. She started to have abnormal movements in form of tonic flexion of the upper and lower limbs lasting for few seconds, occurring in clusters 3-4 time/cluster/day and each cluster last for about 3 minutes. Frequency of the attacks increased gradually, which resulted in an attack every 30 minutes –1hour. At age of 4 months, she was having no spontaneous movements, non-reacting, and her head was fixed to the right side. She was having recurrent episodes of vomiting, non-projectile and nonbilious (8-10 times/day). Upper GI study showed swallowing incoordination, which was treated by gastrostomy tube with fundoplication. There is no history of recurrent infections and no special odor was noticed. She has an older sister which died at the age of 7 days with unknown cause, and one younger brother with a similar condition. Upon physical examination she was vitally stable, dysmorphic in form of hypertelorism, large ears, high arched palate, and hypoactive. She was conscious, non-attentive not following objects, not responding to voices, pupils 3 mm in diameter and reactive bilaterally, normal extra ocular muscle movement, normal gag reflex, symmetrical face, spastic, hypertonic, contractures of achilles tendon, knee, hand and wrist, no reflexes could be elicited with no clonus and no neurocutanious stigmata. MRI brain revealed generalized asymmetrical brain tissue volume loss and asymmetrical ventricular dilatation, thinning of corpus callosum and periventricular white matter more left side (Figure 1). Patient’s EEG recordings showed hypsarrhythmia. WES revealed previously reported homozygous non sense mutation c.160G > T (p.Arg54*), which resulted in a premature termination.
Figure 1.
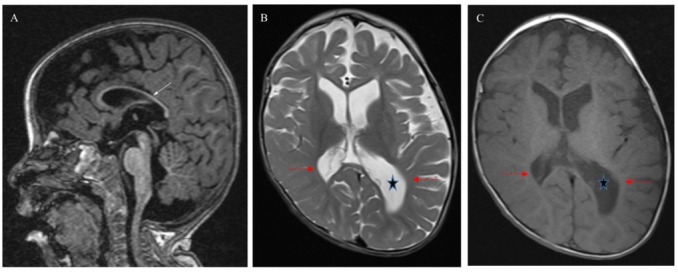
Brain MRI patient 3 at 18 months of age: (A) Sagittal T1 MPRAGE –weighted image shows thinning of corpus callousm (white arrow). (B) and (C) axial T2 and T1-weighted images show brain volume loss, paucity white matter (red arrows) and asymmetry enlargement of lateral ventricles (star) left more than right. Skull deformity.
Discussion
Mutations in the WWOX gene are associated with early infantile epileptic encephalopathy, which is inherited in an autosomal recessive manner [22, 24, 25,26]. Some of the clinical features associated with WWOX mutation are early death, intellectual disability, seizures, pharmacoresistant epilepsy, poor or absent eye contact, spasticity, hypotonia, and hypomyelination, ataxia, psychomotor delay [22, 24, 25, 26, 27, 28]. In addition, MRI imaging revealed that most patients suffer from Microcephaly and brain atrophy (Table 1). So far 10 mutations in the WWOX gene have been identified to be associated with epilepsy (Table 1) [22, 23, 24, 25, 26, 29, 30, 31]. All of these mutations are homozygous and most of them are located in or near the first WW domain, suggesting that one functional copy of the WWOX gene is sufficient for conducting normal neuronal activity.
Table 1.
Comparison of clinical phenotypes in patients with WWOX mutations.
| current study | Johannsen et al. | Tarta-Arsene et | Elsaadany et al. | Tabarki et al. | Ben-Salem et | Mignot et al. 201526 | Mylene valduga | Mallaret et al. | Abdel-Salam et | % | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2018" | al. 2017 28 | 201625 | 2015 30 | al. 2015 29 | et al. 2015 31 | 201423 | al. 201424 | ||||
| # of patients | 3 | 2 | 1 | 2 | 5 | 1 | 5 | 2 | 6 | 1 | 28 |
| Gender | 2M, 1F | 2F | M | 2F | 4F, 1M, | M | 5F, 1M | F | 4F, 2M | F | 20:8 |
| Nucleotide change | (a)c.409+1G<T (b) c.160G<T | c.689A<C | (a)c.173-1G<T (b)Single bp DEL c.918del | C.131 G<A | c.606-1G<A | MicroDEL removing exon 5 | (a)131-KB DEL (b)82-KB DEL (c)157-KB DEL (d)c.1005G<A (e) 4-BP DEL | 594-KB DEL | (a)c.139C< A (b)c.1114G<C | c.160G<T | |
| Amino acid change | (a) Might cause intron insertion or exon skipping (b) p.Arg54* | p.Gly230Pro | (a) Might cause intron insertion or exon skipping (b) Creates a frameshift starting at codon Glu306 | Trp44* | Abolishes splice acceptor, which result in the loss of 62 amino acids | Frame-shit resulting in a stop codon at amino acid position 212 | (a) Deletion encompassing exon 1-5 (b) Deletion encompassing exon 6-8 (c) Deletion encompassing exon 6 (d) Trp335* (e) Frameshift and premature termination | Deletion encompassing exon 1-6 | (a) p.Pro47Thr (b) p.Gly372Arg | p.Arg54* | |
| Nature of the mutation | Homozygous | Homozygous | Compound heterozygous for both mutations in trans configuration | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Recessive mutation |
| Ethnicity | Saudi | Afghani | Romanian | Qatari | Saudi | Emirati | NA | Turkish | Saudi and Palestinian | Egyptian | Predominantly Saudi |
| Microcephaly | + | + | - | - | + | + | Only 2 cases | + | NA | + | 57.1 |
| Ataxia | + | + | 25 | ||||||||
| Psychomotor delay | + | + | + | + | + | + | + | + | - | + | 78.6 |
| axial hypotonia | + | + | + | + | + | + | + | + | + | + | 100 |
| Spasticity | + | + | + | + | + | + | Only 2 cases | + | + | + | 92.9 |
| Ophthalmological involvement | + | + | + | + | NA | + | Only 3 cases | + | + | + | 75 |
| Age of onset | 2M | 2M | 1M | 7W | 2-3M | 2W | 2-5M | 3M | NA | 2M | Usually at 2M |
| Seizure type | Focal/secondary generalization/ infantile spasms | tonic/myoclonic/infantile spasms | Focal/secondary generalization/ infantile spasms | Myoclonic seizure | Multifocal/Infantile Spasms/LGS/ Focal/ secondary generalization | Early seizure | Focal and generalized tonic clonic and myoclonic | Infantile spasm | Generalized tonic-clonic | Focal and generalized tonic clonic and myoclonic | Predominantly Infantile Spasms |
| Response to AED | Refractory | Refractory | Refractory | Partial | Partial | NA | Partial | NA | Yes (2/4), partial (2/4) | Partial | Mostly partial |
| Brain MRI | Brain atrophy | Brain atrophy, hypoplasia of the corpus callosum, and myelination delay | Brain atrophy, hypoplasia of the corpus callosum, and myelination delay | Brain atrophy | Brain atrophy | Brain atrophy | Brain atrophy (3 cases) | Brain atrophy | Posterior white matter hyperintensities | Posterior white matter hyperintensities | Predominantly brain atrophy |
| Premature death | NA | + | Only 2 cases | + | + | 28.6 |
Abbreviations: LGS: Lennox-Gastaut syndrome, M: month, W:week, NA: not available.
In the first family using WES we identified a novel homozygous variant in intron 4 of the WWOX gene, c.409+1G>T in patient 1, both parents were heterozygous for the variant. This mutation might cause intron sequence to be included in the mRNA or the elimination of exon. Both scenarios can alter protein function/activity or rendering it nonfunctional. The patient presented similar clinical features such as seizure, brain atrophy, and developmental delay, which have been observed in other patients with WWOX mutation. However, other skeletal abnormalities, that have not been reported in WWOX patients, have been observed such as fibrochondrogenesis type 2, stickler syndrome type III, and platyspondyly. There are two possibilities that might explain the skeletal phenotypes. One, it is possible that some phenotypes are unique to certain WWOX mutations. In fact, it has been observed that some WWOX mutations cause stronger phenotypes than other WWOX mutations, such as death. The second possibility is thatthe severity of the phenotypes depends on the patient’s genetics make up. Unfortunately, the number of cases with WWOX mutations is low, thus at this point we are not certain if some WWOX mutations have their own unique phenotypes.
In the second family, we identified a previously reported mutation R54* in two siblings, which created a stop codon and resulted in premature termination. Abdel-Salam and collages were the first group to report this mutation. The main clinical phenotypes were severe syndrome of growth retardation, microcephaly, epileptic seizures, retinopathy and early death [24].
WWOX is a cytoplasmic protein that is involved in several cellular processes, such as tumor suppression, cell growth and differentiation, protein–protein interactions, and induction of apoptosis [23, 30]. However, the underlying mechanism remains unknown but there is several factors support that WWOX play a role in epilepsy pathway. First WWOX is expressed in brain areas that are susceptible to seizures, such as hippocampus and cortical neurons. Second, WWOX binds to tau and regulates tau phosphorylation, which is important for cell viability and differentiation. Thus, WWOX loss-of-function may compromise some neuronal processes, which may lead to shrinkage of some areas in the brain and cause in cell death. As we mentioned earlier WWOX is a tumor suppressor gene; however no tumors have been observed in epileptic patients with WWOX mutation. This might be explained by the fact that most of these patients die early, which might not be sufficient time for the patient to develop cancer. Further, functional studies need to be conducted to elucidate the underlying mechanism of WWOX associated epilepsy.
Conclusion
In conclusion, we have identified two consanguineous Saudi families with one novel intronic mutation and one previously reported mutation at the WW domain. The intronic mutation is predicted to result in either exon skipping or including the intron in the protein sequence. Thus, this may lead to frame shift and alter the protein sequence or result in premature protein termination. Further functional studies are needed to understand the underlying mechanism of WWOX associated epilepsy.
Acknowledgments
We are grateful to the patients and his families reported in this article for their genuine support.
Footnotes
Funding sources
No funding for this article from any institution or agency.
Author Contributions
SA: write the first draft and edit the subsequent version. MJA: edit the manuscript and contributed to the clinical diagnosis and management of the patients. JJA: edit the manuscript and read the MRI findings of the patients. KMA: edit the manuscript and contributed to the clinical diagnosis and management of the patients. MAF: edit the manuscript and contributed to the clinical diagnosis and management of the patients. All authors have read and approved the final draft of the manuscript.
Competing interests:
None declared.
References
- [1].Aqeilan RI, Kuroki T, Pekarsky Y, Albagha O, Trapasso F, Baffa R. Loss of WWOX expression in gastric carcinoma. Clin Cancer Res. 2004;10:3053. doi: 10.1158/1078-0432.ccr-03-0594. et al. –. [DOI] [PubMed] [Google Scholar]
- [2].Bednarek AK, Laflin KJ, Daniel RL, Liao Q. Hawkins KA Aldaz CM WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 2000;60:2140. –. [PubMed] [Google Scholar]
- [3].Carter BS, Ewing CM, Ward WS, Treiger BF, Aalders TW, Schalken JA. Allelic loss of chromosomes 16q and 10q in human prostate cancer. Proc Natl Acad Sci U S A. 1990;87:8751. doi: 10.1073/pnas.87.22.8751. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kuroki T, Trapasso F, Shiraishi T, Alder H, Mimori K, Mori M. Genetic alterations of the tumor suppressor gene WWOX in esophageal squamous cell carcinoma. Cancer Res. 2002;62:2258. et al. –. [PubMed] [Google Scholar]
- [5].Kuroki T, Yendamuri S, Trapasso F, Matsuyama A, Aqeilan RI, Alder H. The tumor suppressor gene WWOX at FRA16D is involved in pancreatic carcinogenesis. Clin Cancer Res. 2004;10:2459. doi: 10.1158/1078-0432.ccr-03-0096. et al. –. [DOI] [PubMed] [Google Scholar]
- [6].Nishida N, Fukuda Y, Kokuryu H, Sadamoto T, Isowa G, Honda K. Accumulation of allelic loss on arms of chromosomes 13q, 16q and 17p in the advanced stages of human hepatocellular carcinoma. Int J Cancer. 1992;51:862. doi: 10.1002/ijc.2910510605. et al. –. [DOI] [PubMed] [Google Scholar]
- [7].Paige AJ, Taylor KJ, Taylor C, Hillier SG, Farrington S, Scott D. WWOX: a candidate tumor suppressor gene involved in multiple tumor types. Proc Natl Acad Sci U S A. 2001;98:11417. doi: 10.1073/pnas.191175898. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum Mol Genet. 2000;9:1651. doi: 10.1093/hmg/9.11.1651. et al. –. [DOI] [PubMed] [Google Scholar]
- [9].Yendamuri S, Kuroki T, Trapasso F, Henry AC, Dumon KR, Huebner K. WW domain containing oxidoreductase gene expression is altered in non-small cell lung cancer. Cancer Res. 2003;63:878. et al. –. [PubMed] [Google Scholar]
- [10].Ilsley JL. Sudol M Winder SJ The WW domain: linking cell signalling to the membrane cytoskeleton. Cell Signal. 2002;14:183. doi: 10.1016/s0898-6568(01)00236-4. –. [DOI] [PubMed] [Google Scholar]
- [11].Macias MJ. Wiesner S Sudol M WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002;513:30. doi: 10.1016/s0014-5793(01)03290-2. –. [DOI] [PubMed] [Google Scholar]
- [12].Chang NS, Doherty J, Ensign A, Lewis J, Heath J, Schultz L. Molecular mechanisms underlying WOX1 activation during apoptotic and stress responses. Biochem Pharmacol. 2003;66:1347. doi: 10.1016/s0006-2952(03)00484-2. et al. –. [DOI] [PubMed] [Google Scholar]
- [13].Aqeilan RI, Pekarsky Y, Herrero JJ, Palamarchuk A, Letofsky J, Druck T. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci U S A. 2004;101:4401. doi: 10.1073/pnas.0400805101. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chang NS, Hsu LJ, Lin YS. Lai FJ Sheu HM WW domain-containing oxidoreductase: a candidate tumor suppressor. Trends Mol Med. 2007;13:12. doi: 10.1016/j.molmed.2006.11.006. –. [DOI] [PubMed] [Google Scholar]
- [15].Huang SS, Su WP, Lin HP, Kuo HL. Wei HL Chang NS Role of WW Domain-containing Oxidoreductase WWOX in Driving T Cell Acute Lymphoblastic Leukemia Maturation. J Biol Chem. 2016;291:17319. doi: 10.1074/jbc.M116.716167. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huang SS. Chang NS Phosphorylation/de-phosphorylation in specific sites of tumor suppressor WWOX and control of distinct biological events. Exp Biol Med (Maywood) 2018;243:137. doi: 10.1177/1535370217752350. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee MH, Shih YH, Lin SR, Chang JY, Lin YH, Sze CI. Zfra restores memory deficits in Alzheimer’s disease triple-transgenic mice by blocking aggregation of TRAPPC6ADelta, SH3GLB2, tau, and amyloid beta, and inflammatory NF-kappaB activation. Alzheimers Dement (N Y) 2017;3:189. doi: 10.1016/j.trci.2017.02.001. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chang HT, Liu CC, Chen ST, Yap YV. Chang NS Sze CI WW domain-containing oxidoreductase in neuronal injury and neurological diseases. Oncotarget. 2014;5:11792. doi: 10.18632/oncotarget.2961. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen ST, Chuang JI, Cheng CL. Hsu LJ Chang NS Light-induced retinal damage involves tyrosine 33 phosphorylation, mitochondrial and nuclear translocation of WW domain-containing oxidoreductase in vivo. Neuroscience. 2005;130:397. doi: 10.1016/j.neuroscience.2004.07.054. –. [DOI] [PubMed] [Google Scholar]
- [20].Chen ST, Chuang JI, Wang JP, Tsai MS. Li H Chang NS Expression of WW domain-containing oxidoreductase WOX1 in the developing murine nervous system. Neuroscience. 2004;124:831. doi: 10.1016/j.neuroscience.2003.12.036. –. [DOI] [PubMed] [Google Scholar]
- [21].Lo CP, Hsu LJ, Li MY, Hsu SY, Chuang JI, Tsai MS. MPP+-induced neuronal death in rats involves tyrosine 33 phosphorylation of WW domain-containing oxidoreductase WOX1. Eur J Neurosci. 2008;27:1634. doi: 10.1111/j.1460-9568.2008.06139.x. et al. –. [DOI] [PubMed] [Google Scholar]
- [22].Gribaa M, Salih M, Anheim M, Lagier-Tourenne C, H’Mida D, Drouot N. A new form of childhood onset, autosomal recessive spinocerebellar ataxia and epilepsy is localized at 16q21-q23. Brain. 2007;130:1921. doi: 10.1093/brain/awm078. et al. –. [DOI] [PubMed] [Google Scholar]
- [23].Mallaret M, Synofzik M, Lee J, Sagum CA, Mahajnah M, Sharkia R. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain. 2014;137:411. doi: 10.1093/brain/awt338. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abdel-Salam G, Thoenes M, Afifi HH, Korber F. Swan D Bolz HJ The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J Rare Dis. 2014;9:12. doi: 10.1186/1750-1172-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Elsaadany L, El-Said M, Ali R. Kamel H Ben-Omran T W44X mutation in the WWOX gene causes intractable seizures and developmental delay: a case report. BMC Med Genet. 2016;17:53. doi: 10.1186/s12881-016-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mignot C, Lambert L, Pasquier L, Bienvenu T, Delahaye-Duriez A, Keren B. WWOX-related encephalopathies: delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J Med Genet. 2015;52:61. doi: 10.1136/jmedgenet-2014-102748. et al. –. [DOI] [PubMed] [Google Scholar]
- [27].Johannsen J, Kortum F, Rosenberger G, Bokelmann K, Schirmer MA, Denecke J. A novel missense variant in the SDR domain of the WWOX gene leads to complete loss of WWOX protein with early-onset epileptic encephalopathy and severe developmental delay. Neurogenetics. 2018 doi: 10.1007/s10048-018-0549-5. et al. [DOI] [PubMed] [Google Scholar]
- [28].Tarta-Arsene O, Barca D. Craiu D Iliescu C Practical clues for diagnosing WWOX encephalopathy. Epileptic Disord. 2017;19:357. doi: 10.1684/epd.2017.0924. –. [DOI] [PubMed] [Google Scholar]
- [29].Ben-Salem S, Al-Shamsi AM, John A. Ali BR Al-Gazali L A novel whole exon deletion in WWOX gene causes early epilepsy, intellectual disability and optic atrophy. J Mol Neurosci. 2015;56:17. doi: 10.1007/s12031-014-0463-8. –. [DOI] [PubMed] [Google Scholar]
- [30].Tabarki B, AlHashem A, AlShahwan S, Alkuraya FS. Gedela S Zuccoli G Severe CNS involvement in WWOX mutations: Description of five new cases. Am J Med Genet A 167A: 3209-3213. 2015 doi: 10.1002/ajmg.a.37363. [DOI] [PubMed] [Google Scholar]
- [31].Valduga M, Philippe C, Lambert L, Bach-Segura P, Schmitt E, Masutti JP. WWOX and severe autosomal recessive epileptic encephalopathy: first case in the prenatal period. J Hum Genet. 2015;60:267. doi: 10.1038/jhg.2015.17. et al. –. [DOI] [PubMed] [Google Scholar]