

Abstract
In this work, the hydrophobic small molecule NF-κB inhibitor celastrol was loaded into poly(ethylene glycol)-b-poly(propylene sulfide) (PEG-b-PPS) micelles. PEG-b-PPS micelles demonstrated high loading efficiency, low polydispersity, and no morphological changes upon loading with celastrol. Encapsulation of celastrol within these nanocarriers significantly reduced cytotoxicity compared to free celastrol, while simultaneously expanding the lower concentration range for effective inhibition of NF-κB signaling by nearly 50,000-fold. Furthermore, celastrol-loaded micelles successfully reduced TNF-α secretion after LPS stimulation of RAW 264.7 cells and reduced the number of neutrophils and inflammatory monocytes within atherosclerotic plaques of ldlr−/− mice. This reduction in inflammatory cells was matched by a reduction in plaque area, suggesting that celastrol-loaded nanocarriers may serve as an anti-inflammatory treatment for atherosclerosis.
Graphical Abstract
Encapsulation within poly(ethylene glycol)-bl-poly(propylene sulfide) micelles reduces the toxicity of celastrol and enhances its anti-inflammatory effect during treatment of atherosclerosis.
1. Introduction
Atherosclerosis is a complex and progressive disorder involving the development, growth, and destabilization of lipid-rich plaques in the arterial intima1. Cholesterol-loaded low-density lipoprotein (LDL) accumulates in the arterial intima where it becomes oxidized and internalized by vascular smooth muscle cells and monocyte-derived macrophages2, 3. Cholesterol efflux from these lipid laden cells is then mediated by extracellular high-density lipoprotein particles (HDL)4. An imbalance in this process results in cholesterol accumulation and crystal formation, transforming these cells into foam cells5. These foam cells ultimately undergo necrotic or apoptotic cell death and contribute to the necrotic core of advanced atherosclerotic plaques6. Vascular smooth muscle cells that are still present in the developing plaque contribute to the formation of a fibrous cap, which can be weakened by proliferating immune cells and fibroblasts that modulate the extracellular matrix. Weakening of the fibrous cap may result in plaque rupture or result in thrombogenic surfaces that can cause clot formation7.
While it has been long recognized that lipids play a role in the etiology of atherosclerosis, it is only more recently that attention has been paid to the immune component of atherosclerosis8. Immune cells are present in very early atherosclerotic lesions and remain for the duration of plaque progression. Immune cells play an active role in cholesterol efflux9, plaque extracellular matrix restructuring10, and plaque stability and size11. Pro-inflammatory signalling can result in the recruitment of more immune cells to vascular lesions and to the exacerbation of atherosclerosis12.
One of the core inflammatory signalling pathways within immune cells is the NF-κB signalling pathway. NF-κB is a transcription factor that is sequestered within the cytoplasm until upstream signalling results in its release and subsequent translocation into the nucleus13. A number of receptors lie upstream of NF-κB, including Toll-like receptors (TLRs) TLR2 and TLR4. These receptors are known to recognize oxidized LDL, a marker of atherosclerosis and a key component of its development and progression14. NF-κB can result in the expression of pro-inflammatory signalling molecules, such as the cytokine TNF-α, which can induce apoptosis in nearby cells and exacerbate oxidative stress1. Mice lacking MyD88, an adaptor protein upstream of NF-κB in many TLR signalling pathways, have reduced atherosclerosis, highlighting the pro-atherogenic result of NF-kB activation15.
While it has been increasingly appreciated that the immune system and inflammation play a significant role in atherosclerosis, most treatments focus on lipid metabolism and flux16. These treatments, mainly involving statin administration, can reduce the risk of acute cardiac events in many patients. Unfortunately, some patients demonstrate statin insensitivity or side effects and may require alternative therapeutic strategies16. As evidenced by the pro-atherogenic NF-kB pathway, anti-inflammatory approaches may present such an alternative or complement to statins and other hyperlipidaemia-focused treatments for cardiovascular disease.
One potential small molecule inhibitor of NF-κB is celastrol, a triterpene extracted from Tryptergium wilfordii17. Celastrol has been used, in its herbal plant form, in Chinese folk medicine for a number of years before it was isolated and recognized as an inhibitor with a number of advantageous targets18. One (or potentially several)17, 19 of those targets is upstream of NF-κB, and inhibition by celastrol prevents the release and translocation of NF-κB20. This makes celastrol particularly attractive, as its mechanism of action is downstream of the numerous receptors capable of activating the NF-κB pathway.
Unfortunately, celastrol possesses numerous properties that hinder its use as a therapeutic agent. Celastrol is very hydrophobic21, with correspondingly poor bioavailability and a relatively short serum half-life (T1/2β) of 8–10 hours22. Celastrol also has many targets unrelated to inflammatory signalling and effects a wide variety of cell types. It can reduce cell survival by inhibiting the HSP90 pathway23, 24, but can also promote cell survival in neuronal cells, potentially through its inhibition of the NF-κB pathway and upregulation of HSP7025. Celastrol can resensitize the body to leptin in obese mice, most likely by affecting cells in the hypothalamus26. In a recent study, celastrol’s ability to reduce lipopolysaccharide (LPS)-induced inflammation in vivo was counterintuitively found to worsen inflammation when administered via intraperitoneal (IP) injection27. Perhaps related to its ability to induce apoptosis, celastrol can be cytotoxic to cells at concentrations relatively close to its EC5028. If celastrol is to be safely utilized as a therapeutic, for atherosclerosis or otherwise, some of these pharmacological properties will need to be adjusted.
Nanobiomaterials can be engineered to address each of the issues listed above. Encapsulation and transport of celastrol within a nanobiomaterial carrier, i.e. nanocarrier, would enhance water solubility, altering its biodistribution and serum half-life21. Nanocarriers can increase the selectivity of encapsulated cargo by targeting specific cells or tissues, which can reduce off-target effects29. Lastly, nanocarriers can encapsulate more than one payload simultaneously, allowing concerted delivery of multiple molecules to specific cells and tissues30. These benefits can significantly decrease the effective dosage of therapeutics to minimize cytotoxicity and improve safety31, 32.
To administer celastrol for the treatment of atherosclerosis, we employed self-assembled micellar nanocarriers composed of the amphiphilic diblock copolymer poly(ethylene glycol)-block-poly(propylene sulfide) (PEG-b-PPS)33. The hydrophilic portion of the block copolymer, PEG, is an FDA-approved and widely utilized polymer that reduces protein adsorption, increases serum half-life, and has low immunogenicity34. PPS, the hydrophobic portion, is oxidation-sensitive, allowing for the release of loaded cargo under oxidative conditions, such as those in the endolysosome of immune cells35. PEG-b-PPS can self-assemble into diverse nanocarrier morphologies, which are preferentially taken up by phagocytic immune cell populations, primarily antigen presenting cells such as macrophages and dendritic cells36. Different morphologies have distinct levels of uptake by specific immune cell subpopulations, with spherical micelles demonstrating the most widespread uptake37, 38. Previous work with PEG-b-PPS nanocarriers have shown them to be non-immunogenic36, 39, non-toxic in mice and non-human primates40, and capable of becoming internalized by cells in developing atherosclerotic plaques37.
In this work, we demonstrate that celastrol can be effectively loaded into PEG-b-PPS micelles (Cel-MC) to significantly improve its efficacy and safety as an anti-inflammatory agent. With these improved properties, safe in vivo therapeutic application of celastrol was possible and found to alter the immune cell populations within atherosclerotic lesions, ultimately resulting in the reduction of plaque cross sectional area.
2. Experimental section
2.1. Materials
Unless specified below, all chemicals for polymer synthesis were purchased from Sigma Aldrich (St. Louis, MO, USA) and all reagents for flow cytometry were purchased from BioLegend (San Diego, CA, USA).
2.2. Animals
Ldlr−/− female mice with a C57Bl/6 background, 4–5 weeks old, were purchased from Jackson Laboratories. All mice were housed and maintained in the Center for Comparative Medicine at Northwestern University. All animal experimental procedures were performed according to protocols approved by the Northwestern University Institutional Animal Care and Use Committee (IACUC), as supported by federal regulations, particularly the Public Health Service Policy on Humane Care and Use of Laboratory Animals. Mice were fed a normal diet until they were 2–3 months old, at which point they were switched to a high-fat diet (Tekklad TD 88137 42% calories from fat). Mice were fed a high-fat diet for 3 months prior to the beginning of treatment.
2.3. Polymer synthesis
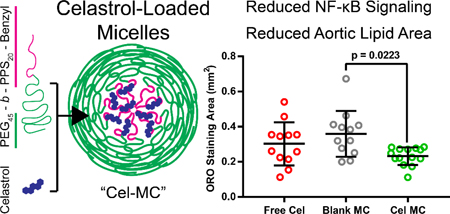
Poly(ethylene glycol)-block-poly(propylene sulphide) (PEG-b-PPS) was synthesized as previously published39, 41. Briefly, commercially available methyl ether PEG (Mn 2000) was functionalized with mesylate and subsequently thioacetate groups. Base-deprotection of the PEG-thioacetate afforded a thiolate anion, which was used to perform living ring-opening polymerization of 20 molar equivalents of propylene sulfide, which was end-capped with benzyl bromide (polymer structure schematic in Fig. 1a).
Fig. 1. Size and morphological characterization of Blank MC and Cel-MC.
(a) Schematic of polymer and celastrol chemical structures and a cartoon figure of an assembled micelle loaded with celastrol. (b) Cryogenic transmission electron micrographs of Blank MC and Cel-MC, scale bars = 50 nm. (c) Small angle x-ray scattering transformed data and polymer micelle model fits. Graphs are vertically offset for ease of visualization.
2.4. Nanocarrier formulation
PEG-b-PPS micelles were formed via thin film hydration, as described previously39. To summarize, 15 mg of PEG45-b-PPS20-Benzyl was weighed into a glass HPLC vial (Thermo Fisher). If the formulation was to contain celastrol, celastrol was added to the vial at this point from a stock solution of 1 mg/mL in tetrahydrofuran (THF). The mixture was dissolved in 1 mL of THF and was left in a vacuum desiccator overnight to remove the THF and coat the walls of the vial in polymer. After desiccation, 1 mL of sterile phosphate buffered saline (1xPBS) was added to each vial. Vials were then shaken for 2 hours at 1000 rpm. Formulations were used immediately or were stored at 4 C.
2.5. Nanocarrier characterization
For cryogenic transmission electron microscopy, 4–5 μL of each formulation was applied to a 400-mesh lacy carbon copper grid. Specimens were then plunge-frozen with a Gatan Cryoplunge freezer. These specimens were imaged using a JEOL 3200FS transmission electron microscope operating at 300 keV at 4000× nominal magnification. All images were collected in vitreous ice using a total dose of ∼10 e− Å−2 and a nominal defocus range of 2.0–5.0 μm.
Small angle X-ray scattering (SAXS) studies were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) beamline at Argonne National Laboratory’s Advanced Photon Source (Argonne, IL, USA) with 10 keV (wavelength λ = 1.24 Å) collimated X-rays. SAXS was performed on undiluted 15 mg/mL polymer formulations, as described previously. Model fitting was performed using SASView and the built-in polymer micelle model.
Dynamic light scattering measurements (DLS) were performed on 15 μg/mL polymer formulations using a Nano 300 ZS zetasizer (Malvern Panalytical, Malvern, UK), using the number average distribution for calculation of the mean diameter and polydispersity of the formulations.
2.6. Quantification of celastrol loading
Celastrol solubility, encapsulation efficiency, and loading capacity were all assessed via high performance liquid chromatography (HPLC) using a Thermo Scientific C18 reverse phase column with a dimethylformamide (DMF) mobile phase at 0.5 mL/min. Area under the curve quantification of celastrol absorbance at 280 nm was performed using Thermo Fisher Chromeleon 7 software. A celastrol standard curve was constructed, demonstrating good linearity between concentrations of 2 mg/mL to 12.5 μg/mL.
To determine the loading capacity of celastrol in micelles, defined here as the highest achievable mass of celastrol that can be stably loaded into 1 mg of micelles in 100 μL of 1xPBS, 1 mg of celastrol was added to 1 mg of PEG45-b-PPS20-Benzyl polymer in 500 uL THF. THF was removed by vacuum desiccation and micelles were formed via thin film hydration with 100 μL of 1xPBS. After micelles were formed, the solution was divided in two, with one half being purified via LH-20 lipophilic column filtration to remove unencapsulated celastrol and the other half being left as is. Both samples (column filtered and unfiltered) were then lyophilized and redissolved in 200 μL DMF and celastrol content was quantified via HPLC.
To determine the encapsulation efficiency of celastrol in micelles, defined as the percentage of the originally added celastrol mass that is stably encapsulated in micelles after filtration to remove unencapsulated celastrol, micelles were formed as described above with variable amounts of celastrol and subsequently filtered using an LH-20 column. Filtered micelles were lyophilized and redissolved in 200 μL DMF and celastrol content was quantified via HPLC.
To determine the solubility of celastrol in aqueous buffer, the standard saturation shake-flask method was employed42. Briefly, 1 mg of celastrol was added to a glass vial along with 10 mL of 1xPBS. This solution was heated to 37° C, stirred for 6 hours, and then left unstirred for 18 hours. The PBS solution was centrifuged at 15,000 RCF to pellet insoluble celastrol aggregates and was subsequently lyophilized. The lyophilized powder was resuspended in 200 μL DMF and celastrol was quantified via HPLC.
Celastrol release from micelles into 1xPBS with or without oxidative trigger was determined as follows. Celastrol micelle formulations (500 μL) placed in Slide-A-Lyzer 10K MWCO MINI dialysis tubes (15 mL tubes, ThermoFisher Scientific) with 13 mL 1xPBS. To each formulation was added either 100 μL of 500 μM H2O2 in water (Sigma Aldrich) or 100 μL of water. Tubes were placed on a shaker (250 rpm) and absorbance readings were obtained at regular intervals. Absorbance at 424 nm, an absorbance peak for celastrol, were taken using a SpectraMax M3 plate reader.
2.7. Confocal microscopy
Cel-MC were formed, as described above, using 10 mg polymer, 10 μg celastrol, and 10 μg DiI with hydration in 1 mL of 1xPBS. DiI is a lipophilic dye that stably loads within PEG-b-PPS nanocarriers43. RAW 264.7 cells (supplied by ATCC, TIB-71) were added to an 8-chamber coverslip-bottom slide at 20,000 cells per chamber. Cells were either left untreated or treated with 1 mg/mL micelles (1 μg/mL celastrol) overnight. Cells were then washed twice with 1xPBS and returned to complete media for an additional 24 hours. Cells were incubated with 100 nM LysoTracker Green DND-26 (ThermoFisher Scientific) and 8 μM Hoechst 33342 (ThermoFisher Scientific) in 1xPBS for 30 minutes prior to being washed twice and returned to complete media. Cells were then imaged using an SP5 Leica confocal microscope at 63x objective magnification. Hoechst nuclear staining was detected using a 405 nm laser with emission detected using a HyD detector set to a 440/470 band. Lysotracker Green was detected using a 488 nm laser and a HyD detector set to a 500/530 band. DiI was detected using a 561 nm laser and a HyD detector set to a 570/630 band.
2.8. Inhibition assays
NF-κB inhibition by celastrol was assayed using RAW Blue cells (supplied by Invivogen, raw-sp), a stably transfected cell line derived from RAW 264.7 macrophage-like cells, which contain the gene for secreted alkaline phosphatase (SEAP) downstream of the NF-κB promoter. Cells were seeded into a 96 well plate at 50,000 cells per well. NF-κB signalling was induced using 100 ng/mL LPS, with Cel-MC and free celastrol (0.1% THF in 1xPBS vehicle) added to the cells concurrent with LPS administration. All micelle formulations contained the same amount of polymer (15 mg/mL) but were loaded with variable amounts of celastrol, and free celastrol formulations were prepared to match the concentration of loaded celastrol within Cel-MC formulations. Free celastrol formulation were made by diluting celastrol stock solutions in THF with 1xPBS to reach the appropriate celastrol concentration and 0.1% THF in 1xPBS. Cells were incubated for 16 hours, as per assay instructions, before supernatant was collected for quantification of SEAP activity, as described by the manufacturer. Colorimetric quantification of SEAP activity was performed on an M3 plate reader (SpectraMax) at an absorbance wavelength of 630 nm.
RAW 264.7 cells were plated in 24 well plates at 500,000 cells per well. TNF-α quantification was performed by treating cells with either 10 ng/mL or 1 μg/mL celastrol in either micelle-loaded or free form (in 0.1% THF/1xPBS) with 100 ng/mL LPS for 6 hours, along with positive control wells, in which LPS was added without celastrol. Supernatant was then collected and stored for ELISA quantification of TNF-α secretion (BioLegend), with TNF-α used to generate a standard curve.
2.9. Cytotoxicity assays
RAW 264.7 cells were plated into 96 well black wall plates at 50,000 cells per well. Cells were then treated with Cel-MC or free celastrol, formulated as described above for the inhibition assays. After 16 h of incubation with free celastrol or Cel-MC formulations, cells were washed and incubated with 4 μM calcein-AM and 2 μM ethidium homodimer (Thermo Fisher), as described by the manufacturer. Readings were performed on an M3 plate reader, at excitation/emission wavelengths of 488/530 nm and 488/635 nm for calcein and ethidium homodimer, respectively. Readings were normalized as described by the manufacturer, accounting for background fluorescence and setting 100% viability for untreated cells and 0% viability for cells incubated with 100% methanol for 15 minutes.
2.10. RNAseq
RAW 264.7 cells were plated at 1×106 cells per well of 6-well plates. Cells were treated with 100 ng/mL LPS to stimulate NF-κB signalling and were then treated in triplicate with one of the following: 1xPBS, 1 μg/mL celastrol in 0.1% THF/1xPBS, 1 ug/mL celastrol in 1 mg/mL micelle formulation in 1xPBS, 0.1% THF/1xPBS, or unloaded ‘blank’ micelles at 1 mg/mL in 1xPBS. Cells were treated for 2 or 6 hours to capture early and later transcriptional events. Cells were washed three times in 1xPBS before having their RNA extracted using a Qiagen RNeasy Mini Kit, as described by the manufacturer.
RNA samples were sent to Admera Health for RNA quality check using an Agilent Bioanalyzer 2100 Eukaryote Total RNA Pico Series II analysis. RNA samples that passed the quality check were used for library preparation (Lexogen QuantSeq 3’ mRNA-Seq) and were sequenced (Illumina Platform 2×150 6–10M PE reads per sample). The RNA-Seq data was aligned and processed using Lexogen QuantSeq data package. Differential gene and pathway analysis utilized DE-Seq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) and GSVA (http://bioconductor.org/packages/release/bioc/html/GSVA.html) using standard default parameters.
2.11. In vivo administration of nanocarriers
Four formulations were made for in vivo use: 15 mg/mL polymer blank micelle formulation, 15 mg/mL polymer 100 ng/mL celastrol micelle formulation, 200 ng/mL celastrol in a 1:1 DMSO:1xPBS formulation, and a vehicle control of 1:1 DMSO:1xPBS formulation. Both micelle formulations were injected intravenously (IV) via tail vein injection (100 μL per injection). The free celastrol and vehicle control formulations were injected intraperitoneally (IP) at 50 μL per injection. Injections were performed on high-fat diet mice (3 months on diet before the beginning of treatment) under isoflurane once a week for 18 weeks. Mice remained on high-fat diet for the duration of treatment. Mice were sacked one week after the end of treatment, and organs were harvested for flow cytometry or were mounted for histology.
2.12. Flow cytometric analysis of immune cell populations
Organs collected from mice were processed for flow cytometry as described previously37. Blood was centrifuged to collect all blood cells. Red blood cells were subsequently lysed using ACK lysis buffer, resulting in a single cell suspension of blood immune cells. Spleens and lymph nodes were mechanically disrupted with a 70 μm nylon filter and a syringe plunger, to form a single cell suspension. Splenocytes were additionally treated with ACK lysis buffer to lyse red blood cells. The aortas were sliced into small pieces (~ 1 mm2) and incubated at 37° C at 300 rpm for 30 minutes in an enzyme cocktail to free cells: 125 U/mL collagenase XI, 60 U/mL hyaluronidase I-S, 60 U/mL DNase I (Roche), and 450 U/mL collagenase I in HBSS buffer. The aorta pieces and buffer were then strained and mechanically disrupted through a 70 μm nylon filter with a syringe plunger.
All single cell suspensions were then incubated for 15 minutes in a blocking buffer containing a fixable viability dye, Zombie Aqua, and an FcR blocking antibody anti-CD16/32. Cells were then stained with one of two antibody panels. Panel 1: FITC anti-CD45, APC/Cy7 anti-CD3, PE anti-CD4, APC anti-CD8, Pacific Blue anti-CD19, PerCP/Cy5.5 anti-NK1.1. Panel 2: FITC anti-CD45, PerCP/Cy5.5 anti-CD11b, Pacific Blue anti-CD11c, PE/Cy5 anti-I-A/I-E, PE/Cy7 anti-F4/80, PE CD86, APC anti-Ly6C, APC/Cy7 anti-Ly6G. Cells were washed, fixed, and analysed using a BD LSR II. Data was analysed using Cytobank online software44. The gating strategy is available in the supporting information (Fig. S4).
2.13. Histological assessment of atherosclerotic plaques
Aortas were carefully dissected from mice to preserve vascular structure and were trimmed and embedded in optimal cutting temperature (OCT) compound for frozen tissue sectioning. Aortas were serially sectioned into 10 μm thick slices, 8–10 sections per slide. Aortic cross sections were stained with Oil Red O, as described previously45, for fluorescence imaging. Images were taken on a Leica DM6B fluorescent microscope at 20x objective magnification with automated image stitching. Quantification of Oil Red O fluorescent staining was performed using a custom Python script.
3. Results and discussion
3.1. Characterization of celastrol-loaded micelles
Micelles formed from PEG-b-PPS typically have a diameter of less than 50 nm and adopt a spherical morphology (Fig. 1a)32, 37, 46, 47. As we and others have previously demonstrated, size and morphology of nanocarriers can drastically alter their organ and cell-level biodistribution after administration in vivo30, 37, 38. Since the loading of cargo may alter the size and morphology of a nanocarrier, we sought to confirm the structure of celastrol-loaded micelles (Cel-MC) as compared to unloaded micelles (Blank MC). We therefore prepared Cel-MC formulations at a fixed concentration of polymer (15 mg/mL), but with increasing amounts of celastrol (1 ng/mL, 100 ng/mL, 10 μg/mL).
Blank MCs and Cel-MCs share the same aggregate morphology, as demonstrated by the small, spherical micellar dots in the cryoTEM micrographs (Fig. 1b) that represent the hydrophobic poly(propylene sulfide) core. This matches previous reports of PEG-b-PPS micelle morphology37, 38. The lack of change in spherical morphology upon loading a hydrophobic cargo is to be expected for PEG-b-PPS micelles, as it has been previously demonstrated for other hydrophobic compounds46, 47. For additional corroboration, SAXS analysis of the formulations was performed and the data was subsequently fitted with a spherical polymer micelle model (Fig. 1c). The modelling parameters indicate a very slight increase in the diameter of micelles upon loading with increasing amounts of celastrol, though it is not statistically significant (Table 1). This is corroborated by DLS data from the same formulations, demonstrating nearly indistinguishable mean diameters, and similarly low polydispersity. Celastrol micelle formulations demonstrated only a slight increase in DLS mean diameter (Table 1), suggesting that all the micelle formulations are of comparable size and that any differences in activity are best explained by the activity of the cargo, celastrol, rather than physical characteristics of the micelles themselves.
Table 1.
Micelle diameter and polydispersity from dynamic light scattering and SAXS modelling.
| Celastrol Loaded (μg) |
DLS Diameter (nm) |
PdI | SAXS Model Diameter (nm) |
|
|---|---|---|---|---|
| Blank MC | 0 | 15.5 | 0.045 | 17.9 |
| Cel-MC | 0.001 | 14.8 | 0.063 | 18.0 |
| 0.1 | 16.4 | 0.053 | 20.2 | |
| 10 | 16.3 | 0.058 | 23.4 | |
| 1000 | 17.9 | 0.032 | Not Performed | |
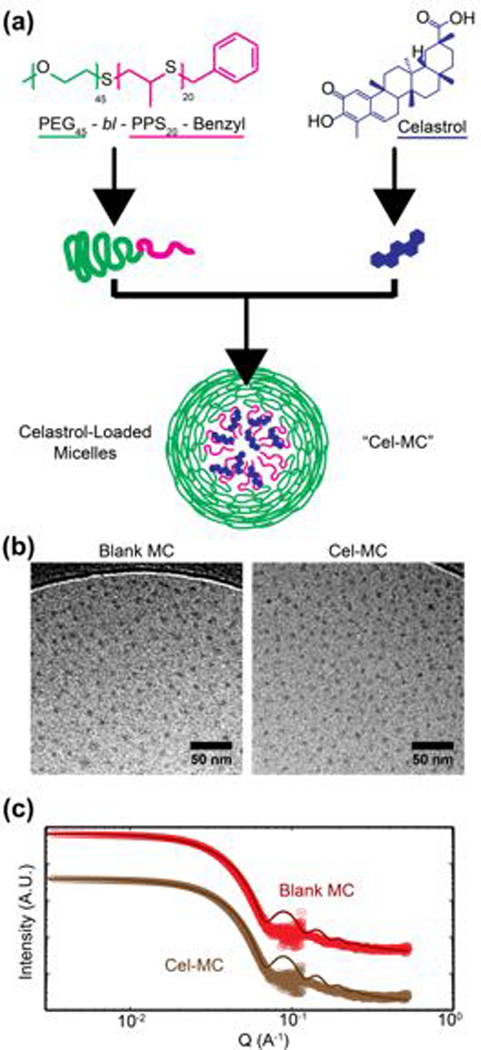
HPLC analysis of formulations before and after removal of unencapsulated celastrol via LH-20 lipophilic column filtration revealed that when celastrol is loaded at 100 μg per 10 mg of polymer the encapsulation efficiency was 96.1 ± 0.8%, confirming the previously demonstrated ability of PEG-b-PPS nanocarriers to efficiently load hydrophobic compounds32, 46, 48. This encapsulation efficiency decreased with higher loading concentrations of celastrol (Fig. 2a). In order to determine the maximum loading capacity, increasing amounts of celastrol were loaded into micelles. When attempting to load 7 mg of celastrol per 10 mg of polymer, the encapsulation efficiency dropped to 31.1 ± 3.4%, which corresponded to a maximum loading capacity of 2.2 mg celastrol per 10 mg polymer (22% loading capacity) (Fig. 2b). Quantification of celastrol dissolved in 1xPBS at 37° C found that celastrol is very sparingly soluble in the aqueous buffer, with only 3.5 μg celastrol detected in 1 mL of 1xPBS. The highest concentration of PEG-b-PPS nanocarriers recorded is 200 mg/mL of polymer, which at a loading capacity of 22% would result in an increase in solubility to 44 mg/mL in 1xPBS for celastrol in Cel-MC, over 10,000 times higher than unencapsulated celastrol. Perhaps due its low water solubility, celastrol remains loaded in micelles for days, exhibiting very slow release (8.0 ± 0.5%) into a 1xPBS sink over 48 hours (Fig. 2c). This is a similar release profile seen with other PEG-b-PPS nanocarriers for hydrophobic cargo48. Celastrol can therefore be stably loaded at high concentrations into PEG-b-PPS micelles, demonstrating nearly 100% encapsulation efficiency when loaded at concentrations less than 500 μg celastrol per 10 mg of PEG-b-PPS polymer.
Fig. 2. Encapsulation efficiency, loading capacity, and release of celastrol from micelles.
(a) Encapsulation efficiency of celastrol in micelles when loaded at different starting amounts of celastrol. ‘Celastrol Added’ represents the amount of celastrol initially available to be loaded into 10 mg of polymer. All data points on graph, n=3. (b) Loading capacity of celastrol in micelles. ‘Celastrol Added’ and ‘Celastrol Loaded’ represent the amount of celastrol initially available to be loaded into micelles and the amount of celastrol actually loaded into micelles, respectively, per 10 mg of polymer. All data points on graph, n=3. (c) Cumulative release of celastrol from celastrol micelles into 1xPBS. Average values plotted on graph, error bars (S.D.) not visible due to low variability compared to y-axis scale, n=3.
3.2. Cel-MC inhibits NF-kB signalling and is less cytotoxic than free celastrol in vitro
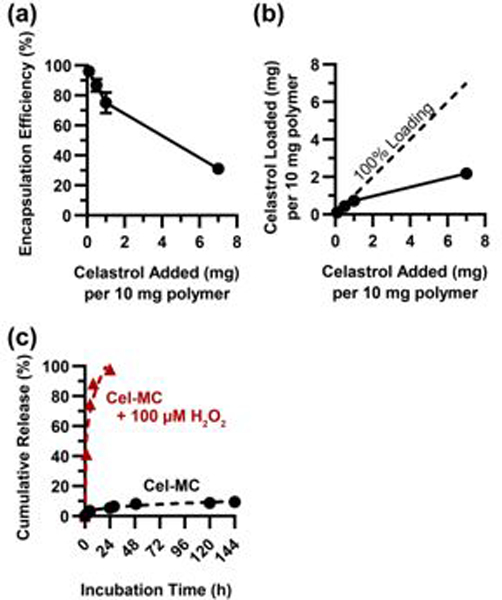
Celastrol is a known inhibitor of NF-κB signalling20, and we sought to confirm that the encapsulation of celastrol within PEG-b-PPS micelles did not negatively impact its ability to function as an inhibitor. Previous work has demonstrated that PEG-b-PPS micelles are internalized by cells through the endolysosomal pathway32, 48. We therefore confirmed that loading of celastrol does not aberrantly affect the uptake and subcellular localization of micelles. Confocal images of Cel-MC were formed and labelled with DiI, a lipophilic dye with spectral properties similar to that of tetramethylrhodamine, which we have previously demonstrated to remain associated with PEG-b-PPS nanocarriers for tracking purposes43. To ensure that the LysoTracker signal is not collected in both the green and red filter sets, leading to an overestimation of colocalization, one well of cells were imaged in the absence of DiI-labeled micelles at the same laser power and detector sensitivity as the micelle-treated cells. These cells showed negligible bleed through into the red channel, ensuring that colocalization observed between the green and red channels accurately reflects the presence of micelles in lysosomes (Fig. S1). Confocal images demonstrate that internalized Cel-MC show strong colocalization with lysosomes, which were stained with LysoTracker Green (Fig. 3a), and this colocalization does not differ between unloaded micelles and Cel-MC.
Fig. 3. Subcellular localization, NF-κB inhibition, and cytotoxicity of Cel-MC in RAW 264.7 cells.
(a) Confocal images of live RAW 264.7 cells incubated with a nuclear stain (blue) and a lysosomal stain (green). Cells were also incubated overnight with blank MC (top row) or Cel-MC (1 μg/mL celastrol, bottom row) labelled with DiI, a lipophilic dye. Composite and brightfield images are included to demonstrate colocalization of micelle and lysosome signal and cell morphology, respectively. (b) RAW Blue colorimetric assay of NF-κB expression at varying concentrations of celastrol. Y-axis is normalized such that 0% represents cells untreated with LPS and 100% represents cells treated with LPS but not treated with any celastrol. X-axis is on a log scale. n=4 (c) ELISA results for TNF-α secretion by RAW 264.7 cells treated with LPS and either free celastrol or Cel-MC. Celastrol treatments were at 10 ng/mL or 1 μg/mL concentrations. All data points shown on graph, n = 5 for treatment conditions, n = 12 for the LPS control. P values shown on graph are from Tukey’s multiple comparison test. (d) RAW 264.7 cell viability with either free celastrol or Cel-MC treatment at varying concentrations of celastrol. Y-axis represents viability normalized by delivery vehicle or formulation, with 100% representing the mean viability of cells treated with vehicle but no celastrol, and 0% representing methanol-treated cells. X-axis is on a log scale. n = 4. (e) Stacked bar graph of RAW 264.7 viability split into three categories: live, dead, or apoptotic. Cells were either LPS treated (+) or not (−). n = 5 for each treatment group. For (b)-(e), error bars represent standard deviation.
Next, we sought to confirm that celastrol is able to maintain its function as an NF-κB inhibitor following micellar encapsulation. To do so, we utilized a reporter cell line, RAW Blue macrophages, in which an NF-κB responsive promoter drives the expression of secreted alkaline phosphatase (SEAP). Upon induction of NF-κB signalling, the cells produce and export SEAP into the supernatant, which can be collected to quantify NF-κB activity using a colorimetric assay of SEAP activity.
Both free (solubilized) celastrol and Cel-MCs were able to inhibit NF-κB signalling in RAW Blue cells treated with LPS (Fig. 3b). However, free celastrol demonstrates a steep decline in its efficacy between 1 μg/mL and 0.1 μg/mL concentrations, with a half maximal effective concentration (EC50) of 0.2 μg/mL. In comparison, Cel-MC has an estimated EC50 of 4.2 pg/mL, a concentration nearly 50,000 times lower. This expansion of the inhibitory concentration range of celastrol is best explained by the increased efficiency of delivery of an inhibitory dose to each cell.
While the RAW Blue cell line functions well as a transcriptional reporter, it was important that we assess the activity of celastrol using an assay with more direct functional relevance. In this case, we chose to perform an enzyme linked immunosorbent assay (ELISA) for TNF-α, a cytokine produced and secreted as a consequence of NF-κB activation. TNF-α plays a key role in both cell survival, apoptosis, stress response, and immune cell recruitment, making its modulation an important part of a potential anti-inflammatory strategy. The RAW Blue assay suggested a drop in inhibitory efficacy for free celastrol between 0.01 – 1 μg/mL celastrol, which was not seen for Cel-MC (Fig. 3b). To confirm this difference, inhibition of RAW 264.7 cells was performed with 10 ng/mL or 1 μg/mL celastrol in either free or Cel-MC form for 6 hours in the presence of LPS. This resulted in a decrease in TNF-α secretion (Fig. 3c). At 1 μg/mL celastrol, both free celastrol and Cel-MC significantly decreased TNF-α levels in the supernatant compared to control cells treated only with LPS. Treatment with free celastrol and Cel-MC at this concentration was not significantly different. At the 10 ng/mL celastrol concentration, however, Cel-MC treatment significantly outperformed free celastrol inhibition, which only partially (but still significantly) inhibited TNF-α production. This corroborates the data from the RAW Blue assay and highlights the finding that Cel-MC remains an effective inhibitor of NF-κB at lower concentrations of celastrol compared to free form celastrol. Notably, unlike the nearly complete inhibition of SEAP activity demonstrated in the RAW Blue assay in Fig. 3b, TNF-α was still at detectable levels in the supernatant of cells after both celastrol treatments. This suggests that either not all cells had their NF-κB signalling completely inhibited, or TNF-α expression was induced by an alternative NF-kB independent mechanism49. The amount of LPS used in these in vitro experiments likely dwarfs the amount of inflammatory stimuli in most in vivo contexts, but serves to illustrate that encapsulated celastrol is able to function efficiently as an inhibitor of NF-κB signalling.
Although beneficial for chemotherapeutic applications50, the high cytotoxicity of celastrol hinders its use as an anti-inflammatory. Since Cel-MC demonstrated high NF-κB inhibition at significantly lower celastrol concentrations than free form celastrol (Fig. 3b), we hypothesized that Cel-MC could serve as a potent anti-inflammatory at nontoxic concentrations of celastrol. We found that at very high doses (0.8 mg/mL celastrol), both free celastrol and Cel-MC are highly cytotoxic to RAW 264.7 cells (Fig. 3d). However, as the dosage of celastrol is decreased, there is a marked increase in cell viability for Cel-MC, but not for free celastrol. When comparing the concentrations that are relevant for successful inhibition of NF-κB, it is apparent that there is only a very narrow range of concentrations for free celastrol that are both effective at inhibiting NF-κB and moderately tolerated by cells. In contrast, Cel-MC has a very broad range of concentrations of loaded celastrol that demonstrate high NF-κB inhibition along with high cell viability.
Celastrol has been investigated as a potential anti-cancer therapeutic due to its ability to induce cell death, and potentially binds to a number of proteins involved in apoptosis28, 51. As the manner of cell death can influence the downstream immune response, we tested for the onset of apoptosis upon treatment with celastrol52. Since NF-κB signalling in RAW 264.7 cells results in the secretion of TNF-α (Fig. 3c), an inducer of apoptosis in some contexts53 as well as cell survival and proliferation in others54, we assessed whether the presence or absence of LPS during celastrol treatment influenced the induction of apoptosis or necrosis. We selected 1 μg/mL of celastrol for testing, which is near the lowest concentration at which both free and micelle-loaded celastrol strongly inhibit NF-κB (Fig. 3b). For cells treated with free celastrol for 4 h, we found 37.5 ± 4.4% of cells to be apoptotic in the presence of LPS, which significantly reduced to 16.5 ± 1.8% (p < 0.0001) in the absence of LPS (Fig. 3e). These results are in line with observations from other groups that found celastrol to induce apoptosis51, 55, and likely reflects a synergy between celastrol-induced and LPS-dependent TNF-induced apoptosis56. Strikingly, there was no significant difference observed when comparing Cel-MC to blank controls with or without LPS, both of which induced less than 4% of cells to be apoptotic and were significantly (p < 0.0001) lower than cells treated with free form celastrol (Fig. 3e).
As celastrol has been shown to target a number of different pathways in different cell types17, we sought to confirm on a transcriptional level that free celastrol and Cel-MC treatments do not have strikingly different transcriptional profiles in inflammatory cells. We again utilized LPS-treated RAW 264.7 cells as our model inflammatory cell type and treated the cells with 1 μg/mL celastrol in free or Cel-MC form, a concentration shown in Fig. 3 to inhibit NF-κB. RNA was extracted from the cells and RNAseq was performed on the mRNA. Heatmap analysis of the 2084 genes significantly altered by free celastrol treatment of LPS-treated RAW 264.7 cells after 2 hours demonstrated that both free celastrol and Cel-MC had similar effects on the inflammatory program (Fig. 4a). Notably, pro-inflammatory genes such as il1b57, tnf58, and nfatc159 were significantly downregulated while anti-inflammatory genes such as lrp160, irf761, and slpi62, were significantly upregulated in both groups (Padj < 0.1; DE-Seq2, Table S1 and S2). Downregulation of il1b and tnf is particularly significant as their products, IL-1β and TNF-α, are highly implicated in the pathogenesis of atherosclerosis. IL-1β, a product of inflammasome signalling, and TNF-α, a product of TLR engagement, are both induced by oxLDL and result in the promotion of foam cell formation and the enrichment of other pro-inflammatory cells in the developing atherosclerotic plaque63. Pathway analysis confirmed that NF-κB target genes were significantly downregulated under both conditions (Padj < 0.1; Fig. 4b,c). This analysis confirms that the encapsulation of celastrol in PEG-b-PPS micelles does not adversely affect its ability to function as a NF-κB inhibitor.
Fig. 4. RNAseq analysis of transcriptional effects of free celastrol and Cel-MC treatment of LPS-stimulated RAW 264.7 cells.
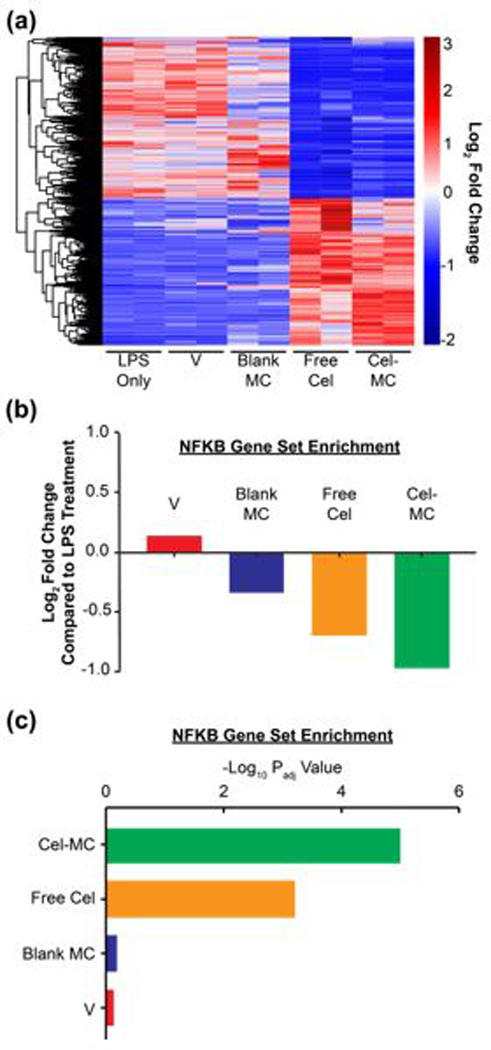
Free celastrol and Cel-MC have similar anti-inflammatory effects on the transcriptomes of LPS-treated RAW 264.7 cells. (a) Heatmap analysis of genes significantly affected by free celastrol. DE-Seq2 analysis identified 2649 genes significantly altered by free celastrol treatment of LPS-treated RAW 264.7 cells after 2 hours. Adjusted P-value (Padj)<0.1. This gene set was used to generate a heatmap with the following conditions: LPS-treated RAW 264.7 cells (LPS), LPS + celastrol vehicle (V), LPS + blank micelles (Blank MC), LPS + free celastrol (Free Cel), and LPS + Cel-MC (Cel-MC). Red and blue colors respectively represent genes that are overexpressed and underexpressed in that sample compared to the other cohorts. (b) Fold Change and (c) Padj of the NF-κB gene set. Gene set variation analysis of the NF-κB pathway (Hinata NF-κB Matrix Gene Set) in LPS-treated RAW 264.7 cells treated for 2 hours with vehicle (V), blank MC, free celastrol, or Cel-MC. Fold change is relative to RAW 264.7 cells treated with only LPS.
3.3. Cel-MC treatment reduces inflammatory immune cell populations in atherosclerotic plaques
Two additional hinderances to the therapeutic use of celastrol are poor solubility/bioavailability and signalling promiscuity in a wide range of cells and tissues. Translation from in vitro to in vivo work highlights these difficulties, as they are difficult to assess in solely mammalian cell culture experiments. Having found non-cytotoxic doses of Cel-MC and tolerable doses of free celastrol (100 ng/mL, Fig. 3), we sought to examine whether controlled delivery of encapsulated celastrol via PEG-b-PPS micelles could ameliorate inflammation within atherosclerotic plaques.
Celastrol is typically administered to humans orally and to mice intraperitoneally (IP). IP injections have limited applicability to humans, so we maintained the free celastrol IP injections as a control and used the more relevant intravenous (IV) route of administration for Cel-MC formulations. As our goal was to alleviate inflammatory signalling in atheromas, we first established early stage atherosclerotic lesions through the feeding of a high fat diet to ldlr−/− mice for 3 months. We subsequently began weekly administrations of the treatments for 3 additional months. Mice were monitored and weighed to discern any changes in appetence or weight due to treatment toxicity, of which neither was detected (Fig. S2). At the end of the experiment, mice had been on high fat diet for 6 months, which typically results in the development of late stage plaques in ldlr−/− mice64. Mice were sacked, and major lymphoid organs (spleen, lymph nodes, blood) and aorta were collected and processed for flow cytometry to characterize the immune cell population profile of the different tissues.
Changes in cell population were compared between celastrol treatments and the blank micelle treatment control, resulting in a log2 fold change heatmap (Fig. 5a). Free celastrol at 33 μg/kg/week had muted effects, with a mixture of cell population increases and decreases compared to blank MC. In contrast, Cel-MC resulted in statistically significant decreases for several key inflammatory cell populations compared to free celastrol and blank MC (Fig. 5b-e, S3). Neutrophils in both the blood (Fig. 5b) and atherosclerotic plaque (Fig. 5c) saw a significant reduction in their share of the immune cell population upon Cel-MC treatment. Neutrophils are highly pro-inflammatory65 and secrete a network of proteins and DNA called neutrophil extracellular traps, a process known as NETosis66. NETosis has been implicated in the progression of atherosclerosis by licensing macrophages to secrete pro-inflammatory cytokines67 and by inducing the cell death of vascular endothelial cells68. As such, this reduction in the neutrophil population could have therapeutic relevance. Similarly, monocytes in the blood are often pro-inflammatory69, and their reduction during the course of Cel-MC treatment (Fig. 5d) could help ameliorate the inflammatory state in the plaque, where monocytes are often recruited and induced to differentiate into macrophages and foam cells70. NK cells in atherosclerotic plaques were also reduced (Fig. 5e), though it is less clear whether this would positively or negatively impact atherosclerosis, if at all71. Intriguingly, NK cells significantly increased in the spleen of Cel-MC treated mice (Fig. S3a), potentially suggesting an alteration in their trafficking.
Fig. 5. Flow cytometric analysis of changes in cell populations in ldlr−/− mice treated with free celastrol or Cel-MC.
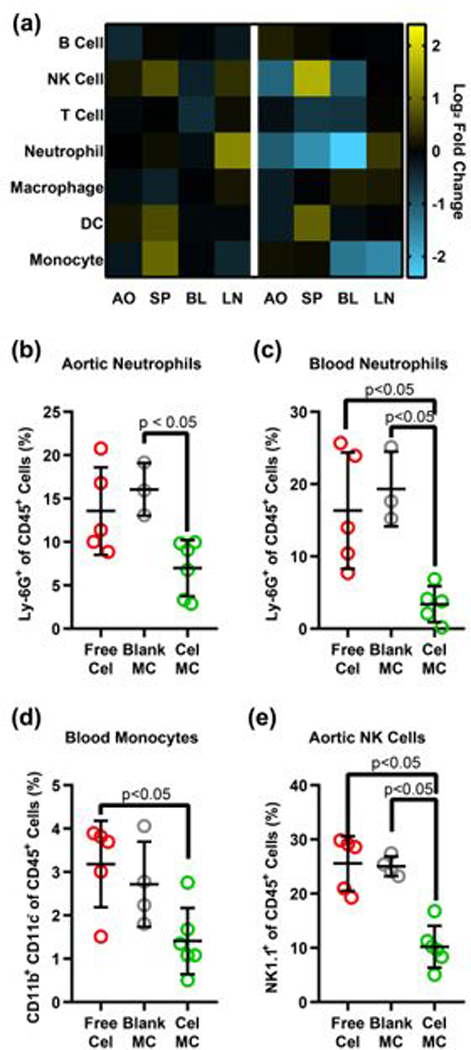
(a) Heatmap of fold change in cell populations. Each row represents an immune cell population, each column represents the organ from which the cells were isolated. Heatmap is on a log2 scale, with yellow representing a fold increase and blue representing a fold decrease in that cell population, compared to the Blank MC control. Cell population as a percent of all immune cells for a given population in a given organ are also provided for: (b) aortic neutrophils, (c) aortic NK cells, (d) blood monocytes, and (e) blood neutrophils. All significant p-values are displayed on their graphs, calculated using Dunn’s multiple comparisons test. Cells were identified as follows: B cells – CD45+ CD19+, NK cells – CD45+ NK1.1+, T cells – CD45+ CD3+, Neutrophils – CD45+ Ly-6G+, Macrophages – CD45+ CD3- NK1.1- CD19- Ly-6G- F4/80+, Dendritic cells – CD45+ CD3- NK1.1- CD19- Ly-6G- F4/80- CD11c+, Monocytes – CD45+ CD3- NK1.1- CD19- Ly-6G- F4/80- CD11c- CD11b+ Ly-6C+.
3.4. Cel-MC treatment reduces plaque area
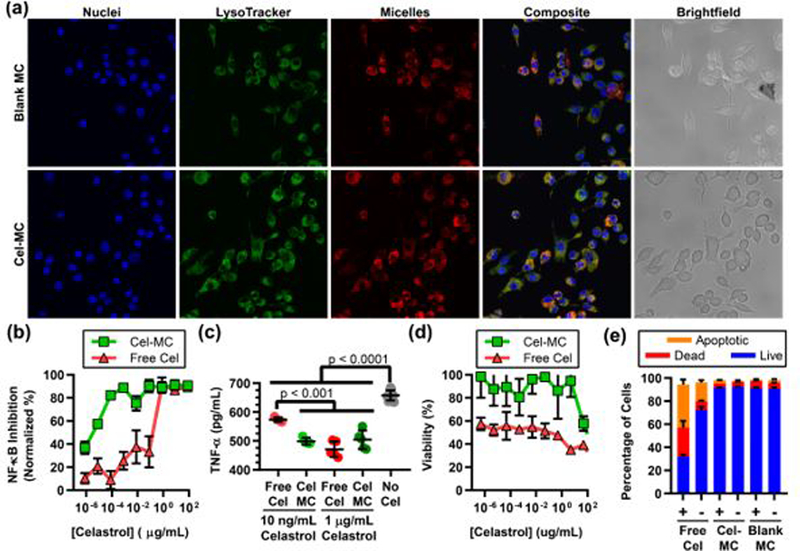
While mouse models of atherosclerosis do not adequately capture the complexity of human atherosclerotic progression, one proxy for plaque progression in mice is plaque area64. To determine whether Cel-MC reduced plaque area, we utilized Oil Red O (ORO) staining on frozen histology cross sections of mouse aorta. ORO is a fluorescent stain for lipid rich regions of atherosclerotic plaques45. Representative sections for Cel-MC and Blank MC treated mouse aortas are shown in Fig. 6a. A quantitative comparison between the ORO staining of different treatment groups was achieved using an automated script written in Python (Fig. 6b), which revealed a significant decrease in plaque area between Cel-MC and Blank-MC control treatments. The administration of free celastrol at this low dosage did not significantly reduce the plaque area compared to the Blank MC control. These results illustrate the benefit of encapsulation within PEG-b-PPS nanocarriers to adjust the therapeutic window of celastrol, achieving targeted therapeutically relevant modulation of key inflammatory cell populations at a sufficiently low dosage to avoid toxicity.
Fig. 6. Oil Red O (ORO) analysis of plaque area in ldlr−/− mice treated with free celastrol or Cel-MC.
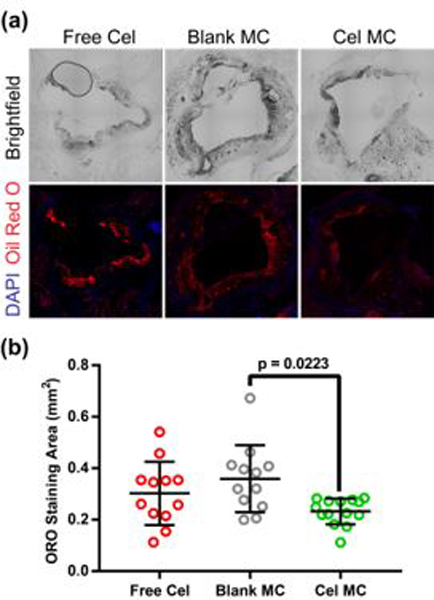
(a) Representative fluorescence microscopy images of ORO stained, frozen aorta sections from free celastrol, blank MC or Cel-MC treated ldlr−/− mice. Top images represent brightfield microscopy, while bottom images were obtained with fluorescence microscopy of DAPI-stained nuclei (blue) and lipid-rich plaques (red). All images were acquired at 20x magnification. (b) Quantification of ORO staining area for free celastrol, Blank MC, and Cel MC treated aorta sections. P-value was calculated using Dunn’s multiple comparisons test. Data points represent imaged sections from discrete portions along the length of the aorta, all data points shown on graph. Bars represent the mean and standard deviation, n = 12 for free celastrol, n = 11 for Blank MC, and n = 14 for Cel-MC.
4. Conclusions
Encapsulation of celastrol into PEG-b-PPS micelles resulted in significant decreases in both effective dose required to inhibit NF-κB as well as cytotoxicity in vitro. In vivo, Cel-MC modulated the proportional makeup of immune cell populations within atherosclerotic plaques and systemically, both of which contribute to the development and progression of atherosclerosis. As a demonstration of therapeutic efficacy, Cel-MC reduced plaque area compared to Blank MC controls in high fat diet fed ldlr−/− mice. Together, these findings provide proof of concept that PEG-b-PPS nanocarriers can drastically enhance the therapeutic utility of celastrol both in vitro and in vivo. With regards to atherosclerosis, we have demonstrated that targeted delivery of an anti-inflammatory small molecule inhibitor to immune cells results in a significant reduction of a marker for plaque progression.
Mouse models of atherosclerosis are useful due to the myriad of tools available for murine research, but they only approximate some elements of human atherosclerosis. More advanced models, such as rabbit or non-human primate models, may be better suited to observe whether inhibition of NF-κB results in plaque changes more relevant to human atherosclerosis, such as an increase in plaque stability. Another topic for further investigation is whether highly selective targeting of immune cell populations within the plaques is preferable to broader systemic delivery to immune cells. This could be investigated with other nanocarrier morphologies, some of which are more likely to target the plaques37, or surface-functionalized nanocarriers which can be directly targeted to particular tissues, organs and cells.
Supplementary Material
Acknowledgements
We would like to acknowledge the aid Dr. Catherine Reardon provided in the form of training for the recovery of aorta from atherosclerotic mice. This work was supported by the Northwestern University – Flow Cytometry Core Facility supported by Cancer Center Support Grant (NCI CA060553). This work made use of the Keck-II facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. Histology services were provided by the Northwestern University Research Histology and Phenotyping Laboratory which is supported by NCI P30-CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. This research was supported by the National Science Foundation grant 1453576 and the National Institutes of Health Director’s New Innovator Award no. 1DP2HL132390–01.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: Fig. S1. Confocal images of RAW 264.7 cells not treated with DiI-labeled micelles. Fig. S2 Mouse body weight and food consumption analysis, Fig. S3. additional cell population treatment comparisons, Fig. S4. flow cytometry gating strategy contour plots. Table S1. List of differentially expressed genes between LPS and free celastrol treatment by DE-Seq2, Padj < 0.1. Table S2. List of differentially expressed genes between LPS and Cel-MC treatment by DE-Seq2, Padj < 0.1. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A and Nasri H, Int J Prev Med, 2014, 5, 927–946. [PMC free article] [PubMed] [Google Scholar]
- 2.Chaabane C, Coen M and Bochaton-Piallat ML, Curr Opin Lipidol, 2014, 25, 374–379. [DOI] [PubMed] [Google Scholar]
- 3.Allahverdian S, Pannu PS and Francis GA, Cardiovasc Res, 2012, 95, 165–172. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR and Rader DJ, J Clin Invest, 2007, 117, 2216–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MS and Goldstein JL, Annu Rev Biochem, 1983, 52, 223–261. [DOI] [PubMed] [Google Scholar]
- 6.Burke-Gaffney A, Brooks AV and Bogle RG, Vascul Pharmacol, 2002, 38, 283–292. [DOI] [PubMed] [Google Scholar]
- 7.Dzau VJ, Braun-Dullaeus RC and Sedding DG, Nat Med, 2002, 8, 1249–1256. [DOI] [PubMed] [Google Scholar]
- 8.Libby P, Arterioscler Thromb Vasc Biol, 2012, 32, 2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH and Rader DJ, N Engl J Med, 2011, 364, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galis ZS and Khatri JJ, Circ Res, 2002, 90, 251–262. [PubMed] [Google Scholar]
- 11.van der Wal AC and Becker AE, Cardiovasc Res, 1999, 41, 334–344. [DOI] [PubMed] [Google Scholar]
- 12.Libby P, Ridker PM, Hansson GK and Leducq A Transatlantic Network on, J Am Coll Cardiol, 2009, 54, 2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilmore TD, Oncogene, 1999, 18, 6842–6844. [DOI] [PubMed] [Google Scholar]
- 14.Chavez-Sanchez L, Garza-Reyes MG, Espinosa-Luna JE, Chavez-Rueda K, Legorreta-Haquet MV and Blanco-Favela F, Hum Immunol, 2014, 75, 322–329. [DOI] [PubMed] [Google Scholar]
- 15.Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, Means T, Halmen K, Luster AD, Golenbock DT and Freeman MW, Nat Med, 2004, 10, 416–421. [DOI] [PubMed] [Google Scholar]
- 16.Shapiro MD and Fazio S, Circ Res, 2016, 118, 732–749. [DOI] [PubMed] [Google Scholar]
- 17.Salminen A, Lehtonen M, Paimela T and Kaarniranta K, Biochem Biophys Res Commun, 2010, 394, 439–442. [DOI] [PubMed] [Google Scholar]
- 18.Morita T, Am J Hypertens, 2010, 23, 821. [DOI] [PubMed] [Google Scholar]
- 19.Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L, Chen X, Zhang D, Zhou Y, Wang Z, Ye X, Cai L, Zhang F, Chen H, Jiang F, Fang H, Yang S, Liu J, Diaz-Meco MT, Su Y, Zhou H, Moscat J, Lin X and Zhang XK, Mol Cell, 2017, 66, 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, Hong YS and Lee JJ, Biochemical Pharmacology, 2006, 72, 1311–1321. [DOI] [PubMed] [Google Scholar]
- 21.Song J, Shi F, Zhang Z, Zhu F, Xue J, Tan X, Zhang L and Jia X, Molecules, 2011, 16, 7880–7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Li CY, Xu MJ, Wu T, Chu JH, Liu SJ and Ju WZ, J Ethnopharmacol, 2012, 144, 195–200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG and Sun D, Mol Cancer Ther, 2008, 7, 162–170. [DOI] [PubMed] [Google Scholar]
- 24.Sreeramulu S, Gande SL, Gobel M and Schwalbe H, Angew Chem Int Ed Engl, 2009, 48, 5853–5855. [DOI] [PubMed] [Google Scholar]
- 25.Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY and Beal MF, Neurodegener Dis, 2005, 2, 246–254. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Lee J, Salazar Hernandez MA, Mazitschek R and Ozcan U, Cell, 2015, 161, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu M, Chen W, Yu X, Ding D, Zhang W, Hua H, Xu M, Meng X, Zhang X, Zhang Y, Zhang A, Jia Z and Huang S, Am J Transl Res, 2018, 10, 2078–2086. [PMC free article] [PubMed] [Google Scholar]
- 28.Sethi G, Ahn KS, Pandey MK and Aggarwal BB, Blood, 2007, 109, 2727–2735. [DOI] [PubMed] [Google Scholar]
- 29.Allen S, Liu YG and Scott E, Regen Eng Transl Med, 2016, 2, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frey M, Bobbala S, Karabin N and Scott E, Nanomedicine (Lond), 2018, DOI: 10.2217/nnm-2018-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haley B and Frenkel E, Urol Oncol, 2008, 26, 57–64. [DOI] [PubMed] [Google Scholar]
- 32.Allen S, Osorio O, Liu YG and Scott E, J Control Release, 2017, 262, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Napoli A, Tirelli N, Wehrli E and Hubbell JA, Langmuir, 2002, 18, 8324–8329. [Google Scholar]
- 34.Harris JM and Chess RB, Nat Rev Drug Discov, 2003, 2, 214–221. [DOI] [PubMed] [Google Scholar]
- 35.Napoli A, Valentini M, Tirelli N, Muller M and Hubbell JA, Nat Mater, 2004, 3, 183–189. [DOI] [PubMed] [Google Scholar]
- 36.Stano A, Scott EA, Dane KY, Swartz MA and Hubbell JA, Biomaterials, 2013, 34, 4339–4346. [DOI] [PubMed] [Google Scholar]
- 37.Yi S, Allen SD, Liu YG, Ouyang BZ, Li X, Augsornworawat P, Thorp EB and Scott EA, ACS nano, 2016, 10, 11290–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dowling DJ, Scott EA, Scheid A, Bergelson I, Joshi S, Pietrasanta C, Brightman S, Sanchez-Schmitz G, Van Haren SD, Ninkovic J, Kats D, Guiducci C, de Titta A, Bonner DK, Hirosue S, Swartz MA, Hubbell JA and Levy O, J Allergy Clin Immunol, 2017, 140, 1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA and Hubbell JA, Biomaterials, 2012, 33, 6211–6219. [DOI] [PubMed] [Google Scholar]
- 40.Allen SD, Liu Y-G, Bobbala S, Cai L, Hecker PI, Temel R and Scott EA, Nano Research, 2018, 11, 5689–5703. [Google Scholar]
- 41.Vasdekis AE, Scott EA, O’Neil CP, Psaltis D and Hubbell JA, ACS nano, 2012, 6, 7850–7857. [DOI] [PubMed] [Google Scholar]
- 42.Baka E, Comer JE and Takacs-Novak K, J Pharm Biomed Anal, 2008, 46, 335–341. [DOI] [PubMed] [Google Scholar]
- 43.Karabin NB, Allen S, Kwon HK, Bobbala S, Firlar E, Shokuhfar T, Shull KR and Scott EA, Nat Commun, 2018, 9, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen TJ and Kotecha N, Curr Top Microbiol Immunol, 2014, 377, 127–157. [DOI] [PubMed] [Google Scholar]
- 45.Koopman R, Schaart G and Hesselink MK, Histochem Cell Biol, 2001, 116, 63–68. [DOI] [PubMed] [Google Scholar]
- 46.Velluto D, Demurtas D and Hubbell JA, Molecular pharmaceutics, 2008, 5, 632–642. [DOI] [PubMed] [Google Scholar]
- 47.Stack T, Vahabikashi A, Johnson M and Scott E, J Biomed Mater Res A, 2018, 106, 1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bobbala S, Allen SD and Scott EA, Nanoscale, 2018, 10, 5078–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falvo JV, Tsytsykova AV and Goldfeld AE, Curr Dir Autoimmun, 2010, 11, 27–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH and Chun KH, BMB Rep, 2014, 47, 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Chen D, Cui QC, Yuan X and Dou QP, Cancer Res, 2006, 66, 4758–4765. [DOI] [PubMed] [Google Scholar]
- 52.Sauter B, Albert ML, Francisco L, Larsson M, Somersan S and Bhardwaj N, J Exp Med, 2000, 191, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs C and Wendel A, Journal of Immunology, 1994, 153, 1778–1788. [PubMed] [Google Scholar]
- 54.Gentle IE, Wong WW, Evans JM, Bankovacki A, Cook WD, Khan NR, Nachbur U, Rickard J, Anderton H, Moulin M, Lluis JM, Moujalled DM, Silke J and Vaux DL, The Journal of biological chemistry, 2016, 291, 2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mou H, Zheng Y, Zhao P, Bao H, Fang W and Xu N, Toxicol In Vitro, 2011, 25, 1027–1032. [DOI] [PubMed] [Google Scholar]
- 56.Fielhaber JA, Carroll SF, Dydensborg AB, Shourian M, Triantafillopoulos A, Harel S, Hussain SN, Bouchard M, Qureshi ST and Kristof AS, J Immunol, 2012, 188, 4535–4542. [DOI] [PubMed] [Google Scholar]
- 57.C. A. Dinarello, Blood, 2011, 117, 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zelova H and Hosek J, Inflamm Res, 2013, 62, 641–651. [DOI] [PubMed] [Google Scholar]
- 59.Fric J, Zelante T, Wong AY, Mertes A, Yu HB and Ricciardi-Castagnoli P, Blood, 2012, 120, 1380–1389. [DOI] [PubMed] [Google Scholar]
- 60.May P, Curr Opin Lipidol, 2013, 24, 134–137. [DOI] [PubMed] [Google Scholar]
- 61.Litvak V, Ratushny AV, Lampano AE, Schmitz F, Huang AC, Raman A, Rust AG, Bergthaler A, Aitchison JD and Aderem A, Nature, 2012, 490, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doumas S, Kolokotronis A and Stefanopoulos P, Infect Immun, 2005, 73, 1271–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Persson J, Nilsson J and Lindholm MW, BMC Immunol, 2008, 9, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Getz GS and Reardon CA, Arterioscler Thromb Vasc Biol, 2012, 32, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamashiro S, Kamohara H, Wang JM, Yang D, Gong WH and Yoshimura T, J Leukoc Biol, 2001, 69, 698–704. [PubMed] [Google Scholar]
- 66.Papayannopoulos V, Nat Rev Immunol, 2018, 18, 134–147. [DOI] [PubMed] [Google Scholar]
- 67.Warnatsch A, Ioannou M, Wang Q and Papayannopoulos V, Science, 2015, 349, 316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S and Resink TJ, FEBS Lett, 2010, 584, 3193–3197. [DOI] [PubMed] [Google Scholar]
- 69.Shi C and Pamer EG, Nat Rev Immunol, 2011, 11, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Woollard KJ and Geissmann F, Nat Rev Cardiol, 2010, 7, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonaccorsi I, De Pasquale C, Campana S, Barberi C, Cavaliere R, Benedetto F and Ferlazzo G, Immunol Lett, 2015, 168, 51–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.