

Abstract
Background
The epidemic Clostridioides difficile ribotype 027 strain resulted from the dissemination of 2 separate fluoroquinolone-resistant lineages: FQR1 and FQR2. Both lineages were reported to originate in North America; however, confirmatory large-scale investigations of C difficile ribotype 027 epidemiology using whole genome sequencing has not been undertaken in the United States.
Methods
Whole genome sequencing and single-nucleotide polymorphism (SNP) analysis was performed on 76 clinical ribotype 027 isolates obtained from hospitalized patients in Texas with C difficile infection and compared with 32 previously sequenced worldwide strains. Maximum-likelihood phylogeny based on a set of core genome SNPs was used to construct phylogenetic trees investigating strain macro- and microevolution. Bayesian phylogenetic and phylogeographic analyses were used to incorporate temporal and geographic variables with the SNP strain analysis.
Results
Whole genome sequence analysis identified 2841 SNPs including 900 nonsynonymous mutations, 1404 synonymous substitutions, and 537 intergenic changes. Phylogenetic analysis separated the strains into 2 prominent groups, which grossly differed by 28 SNPs: the FQR1 and FQR2 lineages. Five isolates were identified as pre-epidemic strains. Phylogeny demonstrated unique clustering and resistance genes in Texas strains indicating that spatiotemporal bias has defined the microevolution of ribotype 027 genetics.
Conclusions
Clostridioides difficile ribotype 027 lineages emerged earlier than previously reported, coinciding with increased use of fluoroquinolones. Both FQR1 and FQR2 ribotype 027 epidemic lineages are present in Texas, but they have evolved geographically to represent region-specific public health threats.
Keywords: anaerobe infections, antimicrobial stewardship, molecular epidemiology, whole genome sequencing
Broad spectrum of activity, favorable safety profiles, and multiple indications favored fluoroquinolone use as a widely prescribed class of antimicrobials in the 1990s and early 2000s [1]. However, continued use of fluoroquinolones is associated with development of resistance [2]. Introduction of the first quinolone, nalidixic acid, rapidly led to the development of resistance, especially in Escherichia coli [2, 3]. Resistance mutations were genetically mapped to subunits of deoxyribonucleic acid (DNA) gyrase, which were found to be the target of fluoroquinolone antibiotics [4].
Clostridioides difficile, a Gram-positive, anaerobic, spore-forming bacterium, is also associated with fluoroquinolone resistance. The epidemic caused by the ribotype 027 strain was primarily characterized by the acquisition of a transposable element and a mutation in the gyrA gene that ultimately led to high-level fluoroquinolone resistance within ribotype 027 C difficile strains [5]. Distinct mutation acquisition events led to the generation of 2 unique fluoroquinolone-resistant lineages designated FQR1 and FQR2 [6]. Both lineages emerged with high probability in the United States in the early 1990s. However, the use of whole genome sequencing to assess national and global dissemination have been limited by a low number of ribotype 027 isolates available for investigation [5]. In addition, previous reports are limited by the lack of United States-derived ribotype 027 C difficile strains, especially of the FQR1 lineage.
Considering this, we sought to conduct a large, epidemiologic study to better understand the emergence and dissemination of ribotype 027 in the United States. Since 2011, we have conducted several Texas state-wide surveillance studies to type C difficile isolates obtained from hospitalized patients with a focus on healthcare centers in Houston, the fourth largest city in the United States [7, 8]. Clostridioides difficile ribotype 027 comprised approximately 20% of isolates obtained from these studies. Given the international healthcare nature of Houston, which includes the Texas Medical Center—the largest medical center in the world—we investigated whether both ribotype 027 lineages are present, providing a unique opportunity to study the evolution of this epidemic strain in the United States.
METHODS
Sample Collection
Institutional Review Boards at the University of Houston and participating hospitals approved this study. Fecal samples were collected from hospitalized patients diagnosed with C difficile infection (CDI) between 2011 and 2018. All patient samples were deidentified. Isolates were purified and ribotyped as previously described [7–9]. Previously sequenced C difficile strains from He et al [5] and Steglich et al [10] were used for comparative bioinformatics analyses.
Sequencing and Analyses
Deoxyribonucleic acid was extracted using either the QIAamp DNA mini kit (QIAGEN) or AnaPrep automated DNA extractor (BioChain). Deoxyribonucleic acid was quantified by NanoDrop (ThermoFisher Scientific) and Qubit (ThermoFisher Scientific), and DNA quality was assessed using a BioAnalyzer (Agilent). The DNA libraries were prepared according to Illumina’s protocols, multiplexed on a flow cell, and run on a NextSeq (Illumina) using paired-end sequencing. The generated fastq files were trimmed using Trimmomatic [11] and sequencing quality was examined by software FastQC. The presence of known antimicrobial resistance genes was determined from cleaned reads using the ARG-ANNOT database [12] and SRST2 pipeline [13].
For whole genome SNP analysis, cleaned sequence reads were mapped to the R20291 reference genome (GenBank accession number FN545816) using the RedDog pipeline according to the developer’s guidelines (https://github. com/katholt/RedDog). In brief, Bowtie2 version 2.2.3 was used for mapping [14] and SAMtools version 0.1.19 was used for calling SNPs [15]. Only high-quality SNPs were used for phylogenetic analyses [5, 10, 16–19]. Phylogenetic trees were created in FigTree and heat maps were generated using R. STRING (version 9.05) was used for pathway analyses. BEAST 2.4.7 was used to incorporate spatial and temporal variables into the phylogeny [20]. Analyses were done after the tutorial “Ancestral reconstruction/discrete phylogeography” with BEAST 2.0 (available at http://www.beast2.org/ wiki/index.php/Tutorials) and by using previously published manuscripts as guides [10]. Phylogeographic reconstruction was done using SpreaD3 [21]. Quark (https://zhanglab.ccmb.med.umich.edu/QUARK/) was used for modeling SNP effects on protein structure [22].
RESULTS
Whole genome sequencing data from a total of 108 ribotype 027 (NAP1/BI) C difficile isolates were analyzed from 23 different geographic regions worldwide. Among the 108 samples, 83 (77%) were isolated from the United States, 76 of which were collected from 15 different regional hospitals in Texas (Table 1), primarily from the greater Houston metropolitan area. After mapping to the R20291 reference genome, SNP calling identified 2841 high-quality discriminatory SNPs, which defined the maximum-likelihood phylogeny (Figure 1A). The SNPs consisted of 900 nonsynonymous mutations, 1404 synonymous substitutions, and 537 intergenic changes. Two disparate groupings of isolates affected the resolution of the phylogeny; however, the FQR lineages were prominent after removing these outlier groups (Figure 1B). One group of outliers was identified as of pre-epidemic origin (Figure 1A, green), whereas the other grouping (Figure 1A, blue) clustered amongst other FQR1 strains. Upon further examination of these distant FQR1 strains, we identified 760 SNP sites, >500 indels, and significant homologous recombination that was shared amongst these 2 distant FQR1 isolates compared with other FQR1 strains (Supplemental Figure 1A). These changes were highly related and corresponded to shared changes in metabolic pathways (Supplemental Figure 1B). In total, the phylogenetic tree (Figure 1B) consists of 49 FQR1 strains (n = 35 Texas-specific), 54 FQR2 strains (n = 36 Texas-specific), and 5 pre-epidemic strains. To our knowledge, this is the largest collection of FQR1 strains that has been analyzed thus far and, overall, the largest collection of ribotype 027 strains that has been sequenced from the United States. It is interesting to note that the maximum-likelihood phylogeny tended to closely group the Texas strains within each lineage resulting in a sublineage, which suggests that there is a local genetic uniqueness to these strains.
Table 1.
Strain Information
| Isolate | Isolation Country | Isolation Region | FQR Lineage | Reference |
|---|---|---|---|---|
| 07-00080 | Germany | Stuttgart | FQR2 | Steglich et al [10] |
| 08-00070 | Germany | Landau/Pfalz | FQR2 | Steglich et al [10] |
| 09-00022 | Germany | Bad Langensalza | FQR2 | Steglich et al [10] |
| 09-00077 | Germany | Sindelfingen | FQR2 | Steglich et al [10] |
| 10-00484 | Germany | Leipzig | FQR1 | Steglich et al [10] |
| 12-00001 | Germany | Homburg | FQR2 | Steglich et al [10] |
| 12-00004 | Germany | Bielefeld | FQR2 | Steglich et al [10] |
| 12-00014 | Germany | Leipzig | FQR1 | Steglich et al [10] |
| 12-00017 | Germany | Bad Berka | FQR1 | Steglich et al [10] |
| 12-00018 | Germany | Radebeul | FQR1 | Steglich et al [10] |
| 12-00019 | Germany | Zittau | FQR1 | Steglich et al [10] |
| Aus001 | Australia | Melbourne | FQR2 | He et al [5] |
| Aus005 | Australia | Melbourne | FQR2 | He et al [5] |
| Bir002 | United Kingdom | Birmingham | FQR2 | He et al [5] |
| BMC-18 | United States | Texas | FQR1 | From this study |
| Can010 | Canada | Montreal | FQR2 | He et al [5] |
| Exe014 | United Kingdom | Exeter | FQR2 | He et al [5] |
| Exe015 | United Kingdom | Exeter | FQR2 | He et al [5] |
| FCH-1 | United States | Texas | FQR2 | Endres et al [9] |
| FCH-2 | United States | Texas | FQR2 | Endres et al [9] |
| FCH-4 | United States | Texas | FQR2 | Endres et al [9] |
| Ham005 | United Kingdom | London | FQR2 | He et al [5] |
| Kor002 | Korea | Korea | FQR1 | He et al [5] |
| Kor003 | Korea | Korea | FQR1 | He et al [5] |
| Kor004 | Korea | Korea | FQR1 | He et al [5] |
| Kor005 | Korea | Korea | FQR1 | He et al [5] |
| LSTM002 | United States | Pennsylvania | FQR1 | He et al [5] |
| LSTM005 | United States | Arizona | FQR1 | He et al [5] |
| LSTM006 | United States | Arizona | FQR1 | He et al [5] |
| LSTM022 | United States | Oregon | FQR1 | He et al [5] |
| LSTM025 | United Kingdom | Dundee | FQR2 | He et al [5] |
| LTC10 | United States | Texas | FQR2 | Endres et al [9] |
| LTC-14 | United States | Texas | FQR2 | Endres et al [9] |
| LTC15 | United States | Texas | FQR2 | Endres et al [9] |
| LTC15A | United States | Texas | FQR2 | Endres et al [9] |
| LTC15B | United States | Texas | FQR2 | Endres et al [9] |
| LTC19 | United States | Texas | FQR2 | Endres et al [9] |
| LTC-36 | United States | Texas | FQR2 | Endres et al [9] |
| LTC39 | United States | Texas | FQR2 | Endres et al [9] |
| LTC39A | United States | Texas | FQR2 | Endres et al [9] |
| LTC44 | United States | Texas | FQR2 | Endres et al [9] |
| LTC5A | United States | Texas | FQR2 | Endres et al [9] |
| LTC7B | United States | Texas | FQR2 | Endres et al [9] |
| MCD-116 | United States | Texas | FQR1 | From this study |
| MCD-173 | United States | Texas | FQR1 | From this study |
| MCD-60 | United States | Texas | FQR1 | From this study |
| MDA133 | United States | Texas | FQR2 | From this study |
| MHS-301 | United States | Texas | FQR2 | From this study |
| MHS-322 | United States | Texas | Pre-epidemic | From this study |
| MHS-479 | United States | Texas | FQR2 | From this study |
| MHS-480 | United States | Texas | FQR2 | From this study |
| MHS490 | United States | Texas | Pre-epidemic | From this study |
| MT1039 | United States | Texas | FQR1 | From this study |
| MT1300 | United States | Texas | FQR2 | From this study |
| MT1344 | United States | Texas | FQR2 | From this study |
| MT1349 | United States | Texas | FQR1 | From this study |
| MT1410 | United States | Texas | FQR2 | From this study |
| MT1433 | United States | Texas | FQR2 | From this study |
| MT1443 | United States | Texas | FQR2 | From this study |
| MT1453 | United States | Texas | FQR2 | From this study |
| MT1470 | United States | Texas | FQR2 | From this study |
| MT1641 | United States | Texas | FQR2 | From this study |
| MT1753 | United States | Texas | FQR1 | From this study |
| MT1821 | United States | Texas | FQR1 | From this study |
| MT188 | United States | Texas | FQR1 | From this study |
| MT189 | United States | Texas | FQR2 | From this study |
| MT201 | United States | Texas | FQR1 | From this study |
| MT2040 | United States | Texas | FQR1 | From this study |
| MT207 | United States | Texas | FQR1 | From this study |
| MT2252 | United States | Texas | FQR1 | From this study |
| MT2626 | United States | Texas | FQR1 | From this study |
| MT2710 | United States | Texas | FQR1 | From this study |
| MT2715 | United States | Texas | FQR2 | From this study |
| MT2780 | United States | Texas | FQR1 | From this study |
| MT2795 | United States | Texas | FQR2 | From this study |
| MT3133 | United States | Texas | FQR1 | From this study |
| MT3227 | United States | Texas | FQR2 | From this study |
| MT3678 | United States | Texas | FQR1 | From this study |
| MT443 | United States | Texas | FQR1 | From this study |
| MT-4447 | United States | Texas | FQR1 | From this study |
| MT-5025 | United States | Texas | FQR1 | From this study |
| MT-5051 | United States | Texas | FQR2 | From this study |
| MT-5055 | United States | Texas | FQR1 | From this study |
| MT-5064 | United States | Texas | FQR1 | From this study |
| MT5314 | United States | Texas | FQR1 | From this study |
| MT5337 | United States | Texas | FQR1 | From this study |
| MT5342 | United States | Texas | FQR2 | From this study |
| MT5370 | United States | Texas | FQR1 | From this study |
| MT785 | United States | Texas | FQR1 | From this study |
| P23 | United States | Pennsylvania | FQR2 | He et al [5] |
| P31 | United States | Pennsylvania | FQR2 | He et al [5] |
| P45 | United States | Pennsylvania | FQR2 | He et al [5] |
| R20291 | United Kingdom | Stoke Mandeville | FQR2 | He et al [5] |
| SH1171 | United States | Texas | FQR1 | From this study |
| SH515 | United States | Texas | FQR1 | From this study |
| SH541 | United States | Texas | FQR2 | From this study |
| SH-769 | United States | Texas | FQR1 | From this study |
| SH-797 | United States | Texas | FQR2 | From this study |
| SH-804 | United States | Texas | FQR1 | From this study |
| VM-2-S41 | United States | Texas | FQR1 | From this study |
| VM-2-S60 | United States | Texas | FQR1 | From this study |
| VM-5-S43 | United States | Texas | FQR1 | From this study |
| VM-5-S59 | United States | Texas | FQR1 | From this study |
| VM-8-S42 | United States | Texas | FQR1 | From this study |
| VM-8-S58 | United States | Texas | FQR1 | From this study |
| WH-28 | United States | Texas | FQR1 | From this study |
| WH-29 | United States | Texas | FQR2 | From this study |
| ZKS-32 | United States | Kansas | FQR1 | From this study |
Figure 1.

Maximum-likelihood phylogeny based on 2841 core genome single-nucleotide polymorphisms amongst ribotype 027 Clostridioides difficile samples. (A) The phylogenetic tree includes 108 samples, which depicts some isolates as outliers. (B) A zoomed in phylogenetic tree clearly separates the FQR1 and FQR2 lineages.
The major SNP difference that delineates the pre-epidemic strain from the epidemic strains is the gyrA mutation (c.245C>T) at position 6310 (R20291 genome location), which confers high-level fluoroquinolone resistance in the FQR1 and FQR2 lineages. The primary SNP differences between the FQR1 and FQR2 could be titrated to 28 SNP differences (Table 2), 21 of which are nonsynonymous and unique compared with the SNPs identified by He et al [5]. Of the 21 nonsynonymous SNP differences, 8 SNPs were located to genes linked to drug resistance in orthologous species, including the DNA-directed ribonucleic acid polymerase beta chain (encodes rifampicin resistance), putative penicillin-binding proteins, a putative 5-nitroimidazole reductase, and a putative drug/sodium antiporter [23–25]. Using predictive protein modeling, the SNP in putative 5-nitroimidazole reductase within FQR1 strains affects protein folding and may impact nitroimidazole (metronidazole) action (Supplemental Figure 2). Other SNPs identified occurred more frequently in FQR1 strains and were found in genes involved in cell metabolism, which suggests that this lineage’s worldwide dissemination may have been hampered by cell fitness.
Table 2.
Prominent SNP Differences Between FQR1 and FQR2 Strains
| Position (bp) | Gene | Gene Product | Change | R20291 | FQR1 | FQR2 |
|---|---|---|---|---|---|---|
| 95422 | CDR20291_0060 | DNA-directed RNA polymerase beta chain | Nonsynonymous | G | A | G |
| 95552 | CDR20291_0060 | DNA-directed RNA polymerase beta chain | Nonsynonymous | A | G | A |
| 118571 | CDR20291_0090 | Putative ribosomal protein | Nonsynonymous | G | A | G |
| 563962 | CDR20291_0466 | Chemotaxis protein methyltransferase | Nonsynonymous | A | C | A |
| 879963 | CDR20291_0712 | Penicillin-binding protein | Nonsynonymous | C | T | C |
| 1026853 | CDR20291_0842 | 2-isopropylmalate synthase | Synonymous | G | A | G |
| 1202866 | CDR20291_0985 | Putative penicillin-binding protein | Nonsynonymous | C | G | C |
| 1203554 | CDR20291_0985 | Putative penicillin-binding protein | Nonsynonymous | C | T | C |
| 1232712 | CDR20291_1013 | Conserved hypothetical protein | Nonsynonymous | C | A | C |
| 1460490 | CDR20291_1231 | Probable transporter | Nonsynonymous | A | C | A |
| 1547479 | CDR20291_1308 | Putative 5-nitroimidazole reductase | Nonsynonymous | T | A | T |
| 1600434 | CDR20291_1354 | Putative drug/sodium antiporters | Nonsynonymous | G | C | G |
| 1600436 | CDR20291_1354 | Putative drug/sodium antiporters | Nonsynonymous | T | C | T |
| 1794733 | CDR20291_1522 | 2-component response regulator | Nonsynonymous | A | G | A |
| 2448413 | CDR20291_2088 | ATP-dependent RNA helicase | Nonsynonymous | A | G | A |
| 2459187 | CDR20291_2096 | Cyclomaltodextrinase (maltogenic alpha-amylase) | Synonymous | A | G | A |
| 2568546 | Intergenic | G | A | G | ||
| 2649551 | CDR20291_2259 | Quinolinate synthetase A | Nonsynonymous | G | T | G |
| 2665592 | CDR20291_2272 | Putative signaling protein | Nonsynonymous | T | G | T |
| 2803020 | CDR20291_2394 | Putative histidinol-phosphate aminotransferase | Nonsynonymous | G | A | G |
| 2931889 | CDR20291_2499 | Hypothetical protein | Nonsynonymous | A | G | A |
| 2942446 | Intergenic | G | A | G | ||
| 3111866 | CDR20291_2643 | Phosphoenolpyruvate-protein phosphotransferase | Synonymous | C | A | C |
| 3128507 | CDR20291_2657 | Capsular polysaccharide biosynthesis protein | Synonymous | A | G | A |
| 3163982 | CDR20291_2682 | Cell surface protein (S-layer precursor protein) | Nonsynonymous | G | A | G |
| 3248655 | CDR20291_2744 | d-alanine–poly(phosphoribitol) ligase subunit 1 (d-alanine-activating enzyme) | Nonsynonymous | A | C | A |
| 3500659 | Intergenic | G | A | G | ||
| 3600925 | CDR20291_3018 | Ribonuclease R | Nonsynonymous | G | T | G |
Abbreviations: bp, base pair; DNA, deoxyribonucleic acid; RNA, ribonucleic acid; SNP, single-nucleotide polymorphism.
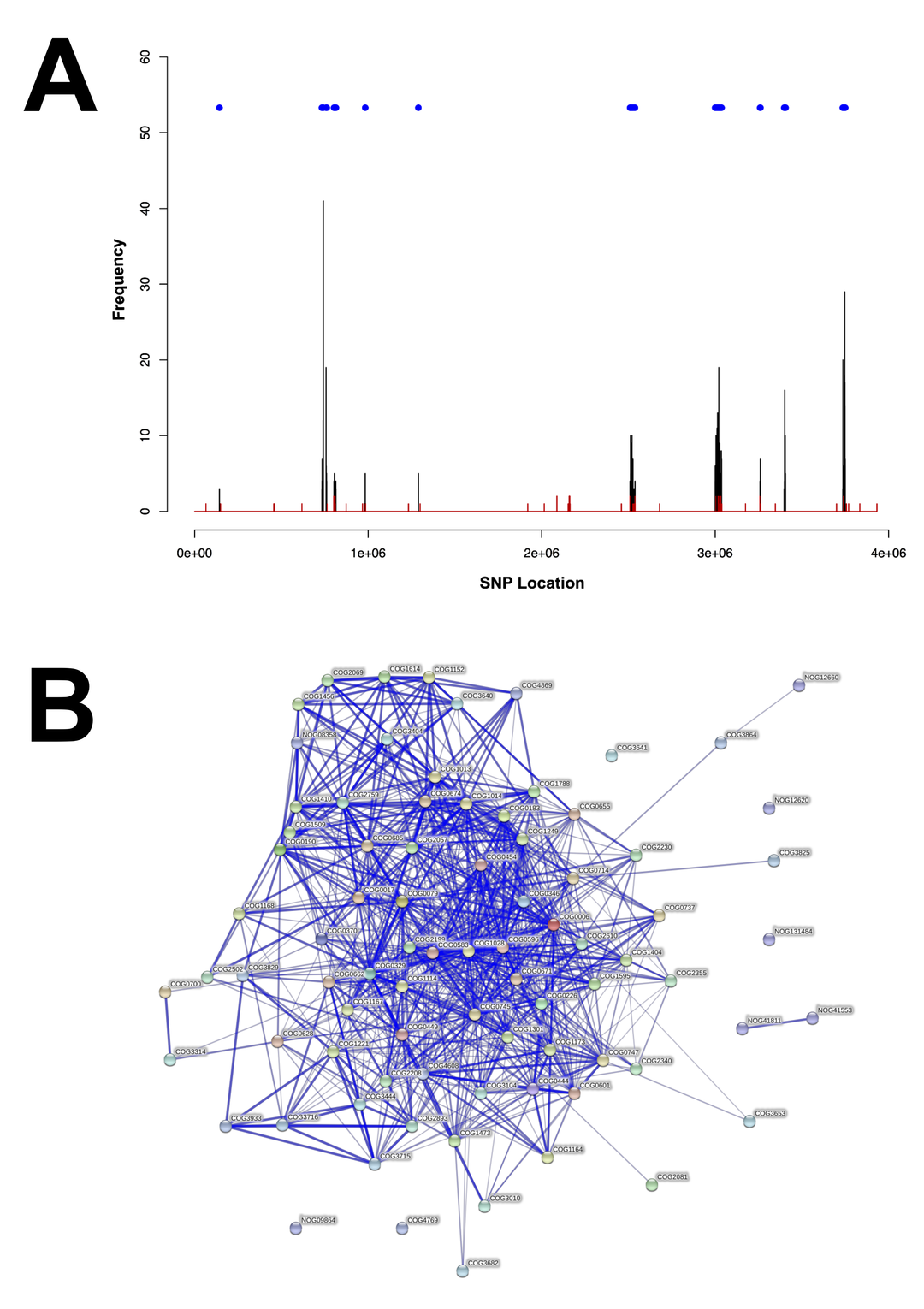
Gene presence or absence was also used to discriminate ribotype 027 strains [5]. This analysis separated strains into distinct groups, possibly due to differences in sequence coverage in some strains (Figure 2A). However, targeted analysis of high variability regions separated strains into 2 prominent groups, corresponding to FQR1 and FQR2 lineages (Figure 2A). The region shared among epidemic strains (FQR1 and FQR2) that was absent in the pre-epidemic strain was the transposable element, Tn6192. In contrast, Tn6105 was present in FQR2 strains and was absent in FQR1 strains. The presence or absence of a genomic region corresponding to ABC transporter proteins was not able to discriminate between the FQR1 and FQR2 strains because this was present in approximately half of the strains. This region was more commonly found in strains outside of the United States. Another noticeable missing feature in 6 US strains was CDR20291_1079, which encodes for a putative teicoplanin resistance protein. Finally, 3 strains were missing a gene cluster corresponding to ethanolamine metabolism. Two of the 3 strains were of the FQR1 lineage and were found to be outliers by maximum-likelihood phylogeny using whole genome SNP analysis. Clinical data were available for these 2 patients, 1 of which had fatal refractory CDI. Coincidentally, these strains were also found to harbor SNPs in several other metabolic pathways that may impact virulence properties (Supplemental Figure 1B).
Figure 2.

Gene presence or absence analysis amongst 108 clinical strains. (A) Genes that were covered by sequencing in the R20291 genome for each strain were plotted as green, whereas gene absence is plotted red. Gene absence may have been due to low coverage in some of the strains. A closer view of regions of high variability on the right allows for complete discrimination between FQR1 and FQR2 strains. (B) Antimicrobial resistance genes were found in the genomes of the samples, and (C) overall gene presence is shown for FQR1, FQR2, and the pre-epidemic strains.
Because the FQR strains are genetically resistant to fluoroquinolones, we also determined whether other known antimicrobial resistance genes were coinherited. In total, 69 of 108 samples (64%) had at least 1 other antimicrobial resistance gene with the most common being ermB (67%) encoding the MLSb (macrolide, lincosamide, and streptogramin B) phenotype (Figure 2B). Other genes encoding aminoglycoside (AAC(6’)-Ie-APH(2”)-Ia, AAC(6’)-Im, APH2-Ib, and Sttr), β-lactam (TEM-1d and BlaZ), trimethoprim (DfrC), MLSb (MsrA and VgaA), and tetracycline resistance (tetM and tetW) were also present in some strains. Tetracycline resistance was more common in US strains compared with other worldwide strains (n = 17 of 18 total). In general, antimicrobial resistance genes could be used as a means of separating the 2 lineages (Figure 2B). In total, FQR1 strains contained more antimicrobial resistance genes compared with the FQR2 strains (Figure 2C). It is interesting to note that 31.5% of FQR2 strains from Houston were positive for ermB, whereas no geographically removed strain was positive for this.
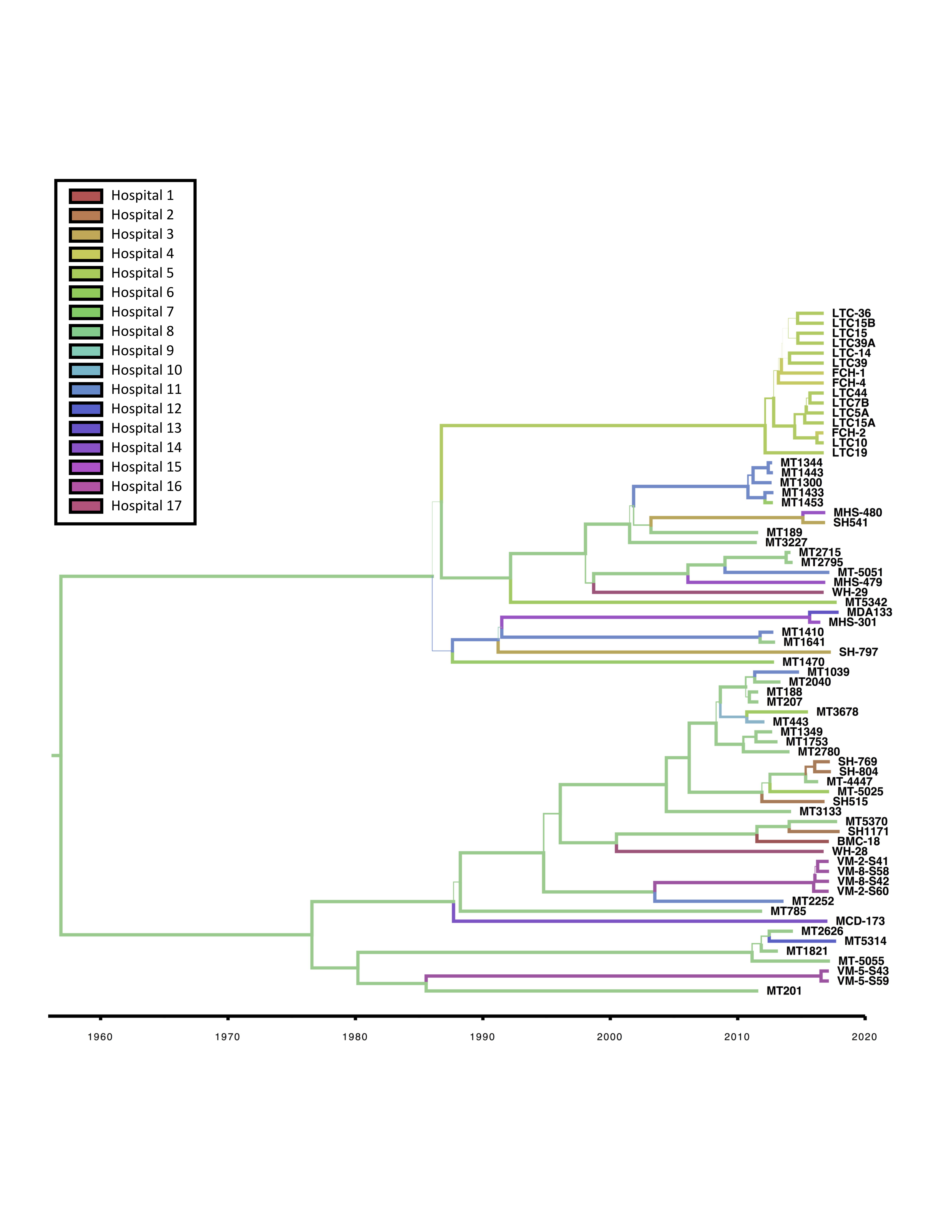
Bayesian phylogenetic and phylogeographic analyses were carried out on the FQR1 and FQR2 strains as previously reported [5, 10]. Consistent with these prior findings, our analysis indicated that mutations arose in the C difficile ribotype 027 core genome at a rate of 1.4 × 10−7 SNPs per site per year corresponding to approximately 1 mutation per genome per year. Bayesian analyses confirmed the distribution of isolates within the fluoroquinolone resistance lineages and also demonstrated sublineages of Houston isolates within FQR1 and FQR2. Emergence of the FQR1 strain was calculated to be as early as 1985, whereas emergence of the FQR2 strain was calculated to be in 1990 (Figure 3A). Ciprofloxacin and norfloxacin were approved shortly before 1985 by the US Food and Drug Administration, suggesting that use of these antibiotics may have contributed to the ribotype 027 epidemic. Although these are predictions for the emergence of the first fluoroquinolone-resistant strains, the strains began proliferating and spreading in the mid-1990s [5, 10]. Both lineages are predicted to have emerged from the United States with high probability (Figure 3B). Considering the number of hospital systems in Houston and Texas-wide, we also investigated the transmission events that have occurred within the city and to neighboring cities. Most of the isolates were derived from Texas Medical Center hospitals and were transmitted to other hospitals/sites (Figure 3C). There was significant mixing amongst hospital systems within Houston likely resulting in the unique Houston strains that are described (Supplemental Figure 3).
Figure 3.

Bayesian phylogenetic and phylogeographic analyses of FQR1 and FQR2 strains. The computational tool BEAST2 was used to incorporate time and geography variables with the single-nucleotide polymorphism analysis. (A) Phylogenetic analyses are plotted with US Food and Drug Administration approval dates for certain fluoroquinolones. (B) Predictive dissemination of FQR1 (red lines) and FQR2 (blue lines) strains worldwide and (C) within Texas.
DISCUSSION
Prior investigations into the worldwide dissemination of the ribotype 027 epidemic strain are limited by a low number of globally available strains for typing, specifically FQR1 lineage isolates from the United States. Considering this, we sought to conduct a large, epidemiologic study to better understand the emergence of ribotype 027 in the United States. Using whole genome sequencing, we demonstrated that both fluoroquinolone resistance lineages (FQR1 and FQR2) were equally present in hospitalized patients with CDI in Texas. Core genome SNP analysis was able to discriminate between the 2 lineages, as well as determine the presence or absence of specific genes (Tn6192, Tn6105, putative teicoplanin resistance gene, and the ethanolamine gene cluster). Worldwide resistance differences were noted including teicoplanin resistance that was relatively absent in US strains. The ethanolamine gene cluster was absent in only 3 strains in our collection, all isolated from the United States. Mechanistic gene mutation studies have shown that absence of this gene cluster increases virulence, suggesting that this may have also contributed to more aggressive infections in humans [26]. Several antimicrobial resistance traits unique to Houston strains were also identified in our collection, including increased presence of tetracycline resistance genes. In general, FQR1 strains had more antimicrobial resistance genes compared with FQR2 strains, which may be partially explained by this strain emerging earlier. All of these findings will need to be expanded and confirmed in future studies.
Although previous reports provided a global overview of the 2 epidemic ribotype 027 lineages, they do not show how isolates differ genetically at the geographic level, especially in the United States. In this study, we demonstrated that sublineages of FQR1 and FQR2 have become unique to the greater Houston vicinity compared with worldwide comparators. This study also provides data to support a hypothesis that extends the potential emergence of the ribotype 027 lineage back to the 1980s and early 1990s. These dates coincide with the clinical use of fluoroquinolone antibiotics in the United States, suggesting that resistance developed quickly after introduction of these drugs into clinical practice [2]. Fluoroquinolone resistance was first documented in E coli as early as 1969 after the introduction of nalidixic acid into clinical practice in 1967, and thus resistance in C difficile approximately 15 years later may be expected. Compared to the relatively slow mutation rate observed in C difficile, E coli mutate at a rate almost 10 000 times faster, which likely contributed to the earlier accelerated resistance [27]. However, as fluoroquinolones became more widely used in the 1990s and early 2000s, increased antimicrobial selection pressure on C difficile strains increased the likelihood that a fluoroquinolone-resistant mutant could emerge as an epidemic ribotype. This hypothesis will require further testing.
An additional notable finding was that both FQR1 and FQR2 strains were present in equal abundance in the United States, compared with other global sites. FQR1 strains harbored more antimicrobial resistance genes, and 2 FQR1 isolates were identified that predicted more serious infection course. Further studies will be needed to establish whether FQR1 strains remained in the United States because of changes in cellular metabolism (increased SNPs found in metabolic genes) and natural selection due to antibiotic use. This is indicated from the Bayesian genetic analysis (Figure 3A) as a stepwise evolution of the FQR1 strains compared with the FQR2 strains in which sublineages emerged at the same time. Regardless of this, our study shows that these strains are still present in the United States and represent a continued urgent threat.
This study has limitations. We sampled a limited number of regional isolates and compared them to the worldwide collection of ribotype 027 isolates that have already been reported. Increasing sample size would strengthen the Bayesian and phylogeographic analyses and allow more precise mapping of isolates within the United States. In addition, because of the highly international nature of healthcare in the Texas Medical Center Houston, many patients travel to Houston from other countries and may have impacted the phylogeographic analyses. The geographic clustering that we observed in this study will need to be demonstrated in other large regional medical centers as well.
CONCLUSIONS
In conclusion, we found that both FQR1 and FQR2 epidemic C difficile ribotype 027 lineages were present in our sample cohort and emerged earlier than previous reports coinciding with increased use of fluoroquinolones. Our findings also indicate that local prescribing patterns may have led to antibiotic resistance that is present in these genomes. Looking forward, more epidemiological tracking studies are needed to understand the evolving dynamics of C difficile.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the University of Houston Seq-N-Edit core for help with sequencing. We also thank the University of Houston Center for Advanced Computing and Data Science, specifically Peggy Lindner, for their assistance and support with the bioinformatics analyses.
Author contributions.All authors contributed equally and made substantial contributions to the following: the conception or design of the work; the acquisition, analysis, or interpretation of data for the work; and drafting the work or revising it critically for important intellectual content and final approval of the version to be published. They agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Financial support. This work was funded by the National Institutes of Health National Institute of Allergy and Infectious Diseases (U01AI124290-01 and 1R56AI126881-01A1) and the Centers for Disease Control and Prevention (CDC-RFA-CK17-1701).
Potential conflict of interests. K. W. G. has received consulting fees and research grant support from Merck & Co., Synthetic Biologics, and Summit, PLC. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Neuhauser MM, Weinstein RA, Rydman R, et al. . Antibiotic resistance among Gram-negative bacilli in US intensive care units: implications for fluoroquinolone use. JAMA 2003; 289:885–8. [DOI] [PubMed] [Google Scholar]
- 2. Emmerson AM, Jones AM. The quinolones: decades of development and use. J Antimicrob Chemother 2003; 51(Suppl 1):13–20. [DOI] [PubMed] [Google Scholar]
- 3. Hooper DC. Clinical applications of quinolones. Biochim Biophys Acta 1998; 1400:45–61. [DOI] [PubMed] [Google Scholar]
- 4. Fàbrega A, Madurga S, Giralt E, Vila J. Mechanism of action of and resistance to quinolones. Microb Biotechnol 2009; 2:40–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He M, Miyajima F, Roberts P, et al. . Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 2013; 45:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McDonald LC, Killgore GE, Thompson A, et al. . An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 2005; 353:2433–41. [DOI] [PubMed] [Google Scholar]
- 7. Aitken SL, Alam MJ, Khaleduzzaman M, et al. . In the endemic setting, Clostridium difficile ribotype 027 is virulent but not hypervirulent. Infect Control Hosp Epidemiol 2015; 36:1318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alam MJ, Walk ST, Endres BT, et al. . Community environmental contamination of toxigenic Clostridium difficile. Open Forum Infect Dis 2017; 4:ofx018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Endres BT, Dotson KM, Poblete K, et al. . Environmental transmission of Clostridioides difficile ribotype 027 at a long-term care facility; an outbreak investigation guided by whole genome sequencing. Infect Control Hosp Epidemiol 2018; 39:1322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steglich M, Nitsche A, von Müller L, et al. . Tracing the spread of Clostridium difficile ribotype 027 in Germany based on bacterial genome sequences. PLoS One 2015; 10:e0139811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta SK, Padmanabhan BR, Diene SM, et al. . ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother 2014; 58:212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Inouye M, Dashnow H, Raven LA, et al. . SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med 2014; 6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H, Handsaker B, Wysoker A, et al. . The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knetsch CW, Kumar N, Forster SC, et al. . Zoonotic transfer of Clostridium difficile harboring antimicrobial resistance between farm animals and humans. J Clin Microbiol 2018; 56: pii: e01384-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cairns MD, Preston MD, Hall CL, et al. . Comparative genome analysis and global phylogeny of the toxin variant Clostridium difficile PCR ribotype 017 reveals the evolution of two independent sublineages. J Clin Microbiol 2017; 55:865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar N, Miyajima F, He M, et al. . Genome-based infection tracking reveals dynamics of Clostridium difficile transmission and disease recurrence. Clin Infect Dis 2016; 62:746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knetsch CW, Connor TR, Mutreja A, et al. . Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011. Euro Surveill 2014; 19:20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bouckaert R, Heled J, Kühnert D, et al. . BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 2014; 10:e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bielejec F, Baele G, Vrancken B, et al. . SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol Biol Evol 2016; 33:2167–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012; 80:1715–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria: an update. Drugs 2009; 69:1555–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–85. [DOI] [PubMed] [Google Scholar]
- 25. Leiros HK, Kozielski-Stuhrmann S, Kapp U, et al. . Structural basis of 5-nitroimidazole antibiotic resistance: the crystal structure of NimA from Deinococcus radiodurans. J Biol Chem 2004; 279:55840–9. [DOI] [PubMed] [Google Scholar]
- 26. Nawrocki KL, Wetzel D, Jones JB, et al. . Ethanolamine is a valuable nutrient source that impacts Clostridium difficile pathogenesis. Environ Microbiol 2018; 20:1419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee H, Popodi E, Tang H, Foster PL. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A 2012; 109:E2774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.