

Abstract
Hematopoietic stem and progenitor cells (HSPCs) are the progenitor cells that can regenerate the entire blood compartment, including the immune system. Recent studies have unearthed considerable immune‐modulating potential of these cells. They can migrate through chemotactic gradients, differentiate into functional immune cells, and crosstalk with immune cells during infections, autoimmune diseases, and cancers. Although the primary role of HSPCs during solid malignancies is considered immunosuppressive, recent studies have discovered immune‐activating HSPCs and progeny. In this review, we will discuss the recent evidence that HSPCs act as immunomodulators during solid cancers and highlight the future directions of discovery. Stem Cells 2019;37:166–175
Keywords: Hematopoietic stem and progenitor cell, Hematopoietic stem cell, Hematopoietic progenitor cell, Myeloid progenitor, Immunotherapy, Solid cancer
Significance Statement.
Hematopoietic stem and progenitor cells have been used during solid cancers to rescue myeloablated bone marrow but not as an independent immune cell. Additionally, most immunotherapy strategies have focused on lineage‐committed or terminal effector immune cells. This article highlights recent work demonstrating that hematopoietic stem and progenitor cells are key modulators of cancer immunity that should be incorporated in cellular immunotherapy strategies.
Introduction
Hematopoietic stem and progenitor cells (HSPCs) at various stages of multipotency and lineage‐commitment have been implicated as key modulators of immunity 1. Namely, it has been suggested that HSPCs can regulate cancer progression 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, drive autoimmunity 18, 19, 20, and regulate infections and inflammation 1, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30. In each of these disease settings, multiple reports have described HSPC‐mediated immune‐suppressive and stimulatory functions. In addition, there is a well‐documented dependence on HSPCs for emergency hematopoiesis, the reconstitution and employment of the blood system during inflammatory conditions 1, 21, 22, 23, 24, 25, 26.
In the context of cancers, hematopoietic stem cell (HSC) transplantation is largely thought of as doing two things: (a) providing the HSPCs to rescue the bone marrow (BM) after toxic therapies or (b) providing graft‐versus‐leukemia effect, an immune response driven by the immune response of contaminating T or natural killer (NK) cells present in the graft that target minor human leukocyte antigen (HLA) mismatching between graft and tumor 31, 32, 33. Although the clinical use of HSPC transfer for a number of malignant and nonmalignant conditions is life‐saving 31, 32, 33, 34, 35, 36, 37, there is an underappreciated role for autologous, nonmismatched HSPCs as a cellular immunotherapy for solid cancers. In this review, we attempt to shift the narrative on HSPCs from a simple reconstituting cell or the mediator of graft‐versus‐leukemia to a multifunctional cell capable of immense immunomodulation. Additionally, with the advent of immunotherapy, there is an important need to evaluate all hematologic cells that are integral to the cancer immune response, including the HSPC that gives rise to all innate and adaptive immune cells.
There are multiple reports of the protumor functions of HSPCs in the context of peripheral cancers. On the contrary, there are multiple reports of the antitumor function of HSPCs in brain tumors, a site traditionally considered to be immunoprivileged. Therefore, we have divided this review into sections dealing with HSPC functions in peripheral tumors or brain tumors. Although there is some overlap in the two settings, we anticipate that through this division cross‐comparison of mechanisms between the two settings can inform future investigation into the untapped immunotherapeutic potential of HSPCs.
HSPC Differentiation and Lineage Commitment
Generation of the blood compartment requires an intricately coordinated system of migration, self‐renewal, lineage commitment, differentiation, and proliferation known as hematopoiesis. Natively, this process generates all blood cells in the immature child and swiftly repopulates any blood cell deficits in the mature adult 38, 39, 40, 41, 42. Although the majority of this process occurs in the BM, extramedullary hematopoiesis can occur in distant sites such as the spleen. In patients that require a rescue of the blood‐forming hematopoietic cells, stem cell transplantation is performed. Within this stem cell transplant is HSPCs, a heterogeneous population that encompasses HSCs and hematopoietic progenitor cells (HPCs). Dogma dictates that HSPCs follow an orderly and linear path of differentiation into terminal effector cells. Recent work has demonstrated considerable discrepancies with this notion and instead promotes the global theory that most effector cells come from a milieu of genetically heterogeneous HPCs 39.
One of the theories of HSPC differentiation is that HSCs become multipotent progenitors (MPP). These MPPs serve as the primary blood‐forming component of the body that generates all lineages and plays the largest role in reconstituting myeloablated hosts during HSPC rescue 38, 39. Recent studies have suggested that there may be considerable early lineage‐restriction in MPPs; specifically, the MPP2 and MPP3 may be myeloid‐restricted, whereas the MPP4 is lymphoid‐restricted 39. Downstream of the MPP is a traditionally binary split between the common myeloid progenitor (CMP) or the common lymphoid progenitor (CLP) 40. The CLP then narrows into small lymphocyte differentiation and progresses into the T and B cell differentiation and maturation pathways. This review will ignore the role of HSPC‐derived lymphoid cells given the predominant focus of HSPC immunology research being focused on the myeloid progenitor populations. Downstream of the CMP are the megakaryocyte erythrocyte progenitor (MEP) and the granulocytic–monocytic progenitor (GMP) 40. Given the limited knowledge about the role of platelets or red blood cells in immunity, the MEP was ignored. However, considerable attention has been paid to the GMP and its progeny: the progenitor that generates macrophages, neutrophils, dendritic cells, eosinophils, and basophils 38, 39, 40, 41, 42.
One of the key cell populations that has a strong link to HSPCs is the myeloid‐derived suppressor cell (MDSC). MDSCs are a heterogenous population of myeloid cells that are halted in immature states of development. Although MDSCs themselves lack mature myeloid cell markers, they can eventually differentiate into nearly all myeloid cell types 4, 27. One of the key forms of cancer‐mediated influence on HSPCs is to differentiate HSPCs into MDSCs 4. In the mouse system, MDSCs can be characterized as CD11b+Ly‐6G+Ly‐6Cint (polymorphonuclear, PMN‐MDSC) or CD11b+Ly‐6G−Ly‐6Chi (monocytic, Mo‐MDSCs) 2, 4, 27. Although in the human system MDSCs are often characterized as CD11b+CD14−CD33+ or Lin−HLA‐DR−CD33+. Additionally, PMN‐MDSCs have upregulated signal transducer and activator of transcription 3 (STAT3) and nicotinamide adenine dinucleotide phosphate that can promote reactive oxygen species (ROS) but low nitric oxide (NO), whereas Mo‐MDSCs are known to have upregulated STAT1 and inducible nitric oxide synthase that promotes high NO and low ROS. In relation to cancer, MDSCs can promote metastases, angiogenesis, and a more stem‐like phenotype of tumor cells 11, 27, 43. The hallmark protumor functions of MDSCs that are most relevant to immunity revolve around impairing T cell function to allow cancer progression and metastasis. MDSCs can impair T cell function through: (a) arginase 1‐mediated metabolism of l‐arginine, a key substrate for T cell proliferation, (b) NO‐mediated inhibition of T cell Janus kinase‐STAT signaling, (c) inhibition of MHC II expression, (d) induction of T cell apoptosis, and (e) expression of coinhibitory molecules including programmed death‐ligand 1 (PD‐L1; Fig. 1) 4, 27, 44.
Figure 1.
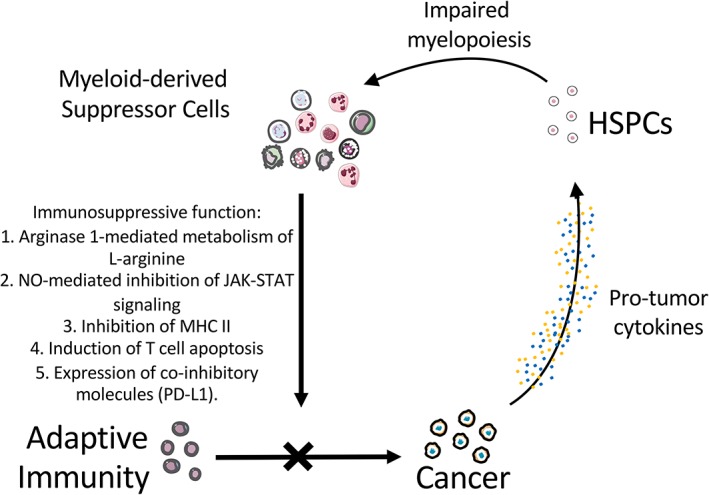
Immunosuppressive function of myeloid‐derived suppressor cells. Abbreviations: HSPCs, hematopoietic stem and progenitor cells; PD‐L1, programmed death ligand‐1; MHC II, major histocompatibility complex II.
Clinical Context
The historical context of BM transplants as an oncologic therapy begins with the graft‐versus‐tumor effect concept. First popularized in the 1960s by Nobel Prize winner E. Donnall Thomas, this soon emerged as a leading therapy for leukemias that remains effective to this day 31, 33, 45. Soon after, George Canellos and Emil Frei III popularized the use of high‐dose “megadose” chemotherapy with autologous HSPC transfers for breast cancer patients from 1985 to the late 90s 45, 46. Throughout this period, a multitude of participants were enrolled in breast cancer trials that were committed to prove the efficacy of this therapy. Despite this massive effort, the lack of survival benefit with high‐dose therapy coupled with future risk of leukemia induced by the megadoses of chemotherapy left the breast cancer field with an understandable resistance to continue megadose chemotherapy with autologous HSPC rescue 47, 48, 49, 50, 51.
There are still many clinicians that promote high‐dose chemotherapy with autologous HSPC rescue for some incredibly fatal solid malignancies based on multiple reports of marked success. For instance, high‐dose chemotherapy with autologous HSC transplant has seemed effective for triple negative breast cancer patients 52 and is still being tested (NCT 02670109). Autologous HSC transplant is also especially effective in a subset of heavily pretreated pediatric brain tumor patients 32, 53, 54, 55, 56, 57, 58. Finlay et al. advocate for chemotherapeutics that specifically have BM toxicity, including Thiotepa, Etoposide, Melphalan, and Busulfan and follow with allogeneic or autologous HSPC rescue 56, 57, 58.
Regardless of the type of cancer, the discussion surrounding chemotherapy with salvage HSPC transplant typically focuses on two mechanisms of action: (a) ability of high‐dose chemotherapy to mediate greater tumor regression as compared with normal chemotherapy and (b) the graft‐versus‐tumor effect if allogeneic stem cell transplantation is performed because of contaminating T/NK cell immune responses against minor HLA mismatching between graft and tumor 32, 33, 47, 48, 49, 50. Outside of these two mechanisms, there has been little to no clinical evaluation of the ability of the HSPC transplant cells to independently mediate an immunologic effect on cancers. Despite this, preclinical investigation on the role of HSPCs in cancers, infections, and autoimmunity has continued to evolve.
Experimental Use of HSPCs
There are at least two distinct settings in which HSPCs are studied: native (or homeostatic) and transplant. The native human system in which all HSPCs are derived from self and have not been transferred or transplanted from donor to recipient. In that setting, studies determine the role of HSPCs in contexts of disease that occur to that single host. The converse setting in which HSPCs are studied is the transplant setting. In that setting, HSPCs are either harvested from BM or peripheral blood of a donor, then transplanted to a recipient. There are two variations on this model: transplant‐treatment and transplant‐delay. In transplant‐treatment, HSPCs are used as a therapeutic intervention and their role is studied subsequent to the treatment. In transplant‐delay, HSPC transplants are used to specifically label the BM cells with a trackable tag to distinguish the BM‐derived cells from peripheral cells in murine systems. Once these chimeras of two types of BM cells are stably engrafted, the transplant‐delay experiment can begin.
Immunomodulatory Capacity of HSPCs
HSPCs respond to inflammation with emergency hematopoiesis, a process that replaces immune effectors that are destroyed, exhausted, or otherwise occupied with clearance of inflammatory stimuli 22, 23, 24, 26. In a more nuanced role, HSPCs can enhance or diminish the highly educated adaptive immune system through MDSC‐like immune suppression or myeloid antigen‐presenting cell‐like immune activation 1, 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 27, 28, 30, 59, 60, 61. In each of these functions, HSPCs can enable or prevent infections, autoimmunity, cancers, and even modulate surgical site healing responses 29.
HSPCs are also proficient at migrating to sites of inflammation through chemokine axes. In the case of malignant gliomas, brain tumors can capitalize on the stromal cell‐derived factor (SDF)‐1‐CXCR4 axis to attract HSPCs to the brain tumor microenvironment through glioma‐secreted SDF‐1 62, 63, 64, 65, 66, 67. This HSPC tropism for certain inflammatory sites has promoted the concept that HSPCs can be a carrier or vehicle cell that will package desired particles, molecules or nucleotides 68, 69. Once packaged, HSPCs could theoretically traffic or migrate through a known axis and deliver its contents to a desired site. Additionally, HSPCs are highly sensitive to exogenous cues and express receptors for key inflammatory cytokines 11. Therefore, HSPCs can become activated and differentiate in response to cytokines including interferon (IFN)‐γ, IFN‐α, and granulocyte–monocyte colony‐stimulating factor (GM‐CSF) 1, 2, 21, 23, 60, 70, 71, 72, 73, 74. Once activated or differentiated by such cytokines, HSPCs and their progeny can then act as immunomodulatory cells. Although HSPCs have distinct roles in a multitude of inflammatory settings, herein we will detail the role of these cells in the context of solid cancers.
HSPCs in Solid Peripheral Cancers
Clinical Correlations in Peripheral Cancers
Immunosuppression can promote cancer formation and progression 3, 4. Specifically, immature myeloid cells (IMCs) including suppressive macrophages or MDSCs play a major role in preventing homeostatic immune surveillance 3, 4, 27. The subset of HSPCs that are myeloid‐restricted can oftentimes overlap in function with immature suppressive myeloid cells and similarly promote cancer formation and progression (Fig. 2). In an important clinical PNAS paper by Wu et al., they noted significant dysregulation of myelopoiesis in 133 untreated patients with 7 different types of cancers including hepatocellular, breast, cervical, esophageal, gastrointestinal, lung, and ovarian 11. In this dysregulation, they observed four to sevenfold increased numbers of GMPs and a preference for granulocyte differentiation. In addition, the degree of dysregulation in the circulation was correlated with severity of disease. Key drivers to the dysregulation were GM‐CSF and IL‐6, components that promoted the HSPC or GMP differentiation into MDSCs in vitro and in vivo. Additionally, in the tumor microenvironment of the colon cancer patients, there was significant infiltration of CD133+CD14+ and CD133+CD15+ MDSCs. In the same setting, the CD34+ subsets appeared to migrate to the tumor site via the CXCR4 chemokine receptor. Altogether this study indicates that human HSPCs and the early myeloid progenitors found in the circulation and tumor stroma are intimately involved in promoting progression of at least seven major peripheral cancers.
Figure 2.
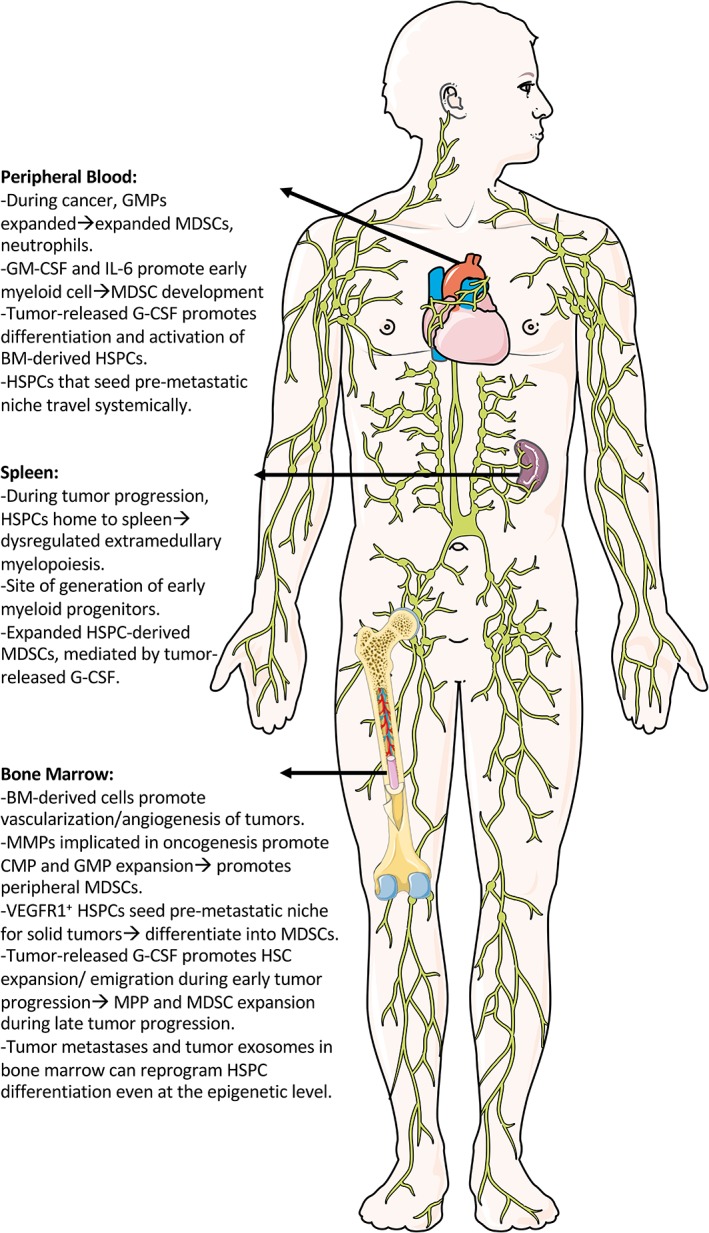
Immunosuppressive pressure on HSPCs during solid cancers. Abbreviations: HSPCs, hematopoietic stem and progenitor cells; GMPs, granulocyte–monocyte precursors; MDSCs, myeloid‐derived suppressor cells; GM‐CSF, granulocyte–monocyte colony‐stimulating factor; G‐CSF, granulocyte colony‐stimulating factor; BM, bone marrow; MPP, multipotent progenitor; CMP, common myeloid progenitor, MMPs, matrix metalloproteinases.
Premetastatic Niche
In early studies in a transplant‐delay model, Lyden et al. demonstrated a significant role of BM‐derived cells in driving vascularization and angiogenesis in B6RV2 lymphoma or Lewis lung carcinoma (LLC) 7. In the follow‐up of those studies, Kaplan et al. demonstrate that the VEGFR1(Flt1)+ HSPCs from the BM are key drivers of premetastatic niches, sites that have a protumor microenvironment that promotes seeding of metastases 6. In murine hosts with intradermally injected LLC and B16 melanoma, this HPC subset promotes metastases to the lung. In follow‐up studies in the same transplant‐delay model, Giles et al. demonstrate the mobilization of HSPCs out of the BM and into the bloodstream that engraft in premetastatic niches 5. Once in these premetastatic niches, the HSPCs proliferate and differentiate into MDSCs that suppress immunity and promote metastasis. They demonstrate that in newly diagnosed rhabdomyosarcoma patients, the number of progenitor cells in the blood capable of generating the myeloid lineage was increased (granulocytic/monocytic colony‐forming unit [CFU‐GM]). In addition, in patients with invasive subtypes of breast cancer there were higher levels of VEGFR1+CD34+ HSPCs as compared with noninvasive nonmetastatic breast cancer, demonstrating a correlation between cancer presence or metastases and increased numbers of HSPCs. In total, BM progenitors are one of the many drivers of the premetastatic niche and one primary mechanism by which this occurs is through HPC‐derived MDSCs.
Dysregulated Myelopoiesis
Casbon et al. demonstrated in a native system that when mice are given breast cancer, they have a significant expansion of immunosuppressive MDSCs in the tumor, blood, spleen, and premetastatic lung 2. They found that the key driver of this expansion was the release of G‐CSF by the tumor itself that coordinated the differentiation and activation of BM HSPCs. Importantly, the differentiation program of the HSPCs (LSK, Lin−sca‐1+c‐kit+) began with an expansion of the HSC compartment (LSK Flk2−CD150+CD48−) during early tumor progression, whereas the MPPs (LSK Flk2+; LSKFlk2−CD48+) and CD11b+Ly‐6G+Ly‐6Cint PMN‐MDSC or CD11b+Ly‐6G−Ly‐6Chi Mo‐MDSCs expanded during later tumor progression. An additional component of this breast cancer study that was corroborated by Cortez‐Retamozo et al. in lung adenocarcinoma is that HSPCs can home to the spleen during tumor progression and undergo a dysregulated myelopoiesis including expansion of HSPC‐derived tumor‐associated macrophages (TAMs) and neutrophils 2, 3. During this extramedullary myelopoiesis, early myeloid progenitors, including the GMP, can accumulate and develop into tumor‐promoting MDSCs. In a similar study by Sio et al., breast cancer induced similarly impaired hematopoiesis and drove early myeloid progenitors out of BM and into the spleen for extramedullary myelopoiesis 9. They noted that tumor progression in these mice was correlated with histone methylation changes in lin−c‐kit+ HSPCs in the BM. Specifically, BM cells had histone methylation at sites that drove genetic dysregulation of lineage commitment and hematopoiesis, including altered regulation of the Hox family genes and Polycomb repressive complex 2 (PCR2). Furthermore, they demonstrated that this process was also dependent on G‐CSF and synergy with GM‐CSF and FLT3L.
Qu et al. demonstrated that innate immune system inflammatory regulators like matrix metalloproteinases (MMP) can drive dysregulation of BM myelopoiesis 75. In their transgenic model in which MMP12 was overexpressed, there was an expansion of CMPs and GMPs in the BM with a concomitant decrease in MEPs. Meanwhile, in multiple peripheral organs there was an expansion of functionally suppressive CD11b+Gr‐1+ MDSCs. In addition, they discovered that in MMP12‐overexpressing animals, IL‐6 in the lung drove a Stat3‐mediated adenocarcinoma formation. Altogether, these findings demonstrated that multiple peripheral tumor models can cause significant migration of HSPCs from the BM toward distant sites where they undergo extramedullary myelopoiesis into tumor‐promoting myeloid cells.
Metastasis and Exosome Effects on BM
In a separate line of questioning, it has been demonstrated by Shiozawa et al. that prostate cancer can establish metastases in the BM that outcompete HSPCs 76. In related studies, Peinado et al. demonstrated that melanoma exosomes are significant contributors to metastasis. In an elegant system of treating mice with melanoma exosomes, they “educate” the BM of melanoma exosome‐treated mice 77. In turn, when the BM of exosome‐educated mice was transplanted into a transplant‐delay model, the tumor volume was markedly higher, HSPCs were increased in tumor, vasculature was more pronounced, and metastases were enhanced. The exosomes educate through horizontally transferring the oncogenic MET tyrosine kinase from B16 melanoma exosomes into HSPCs. Given the oncogenic role of MET in promoting migration, invasion, and angiogenesis in tumors, exosome‐treated BM HSPCs promoted these phenomena. In addition, marked BM mobilization was induced by MET, including an increase in MET‐transferred vasculogenic (c‐kit+Tie2+) and hematopoietic (c‐kit+sca‐1+) HSPCs in the circulation.
HSPCs in the BM can therefore be significantly impacted by peripheral solid malignancies through metastases to the BM, epigenetic regulation of HSPCs, induction of HSPC migration to premetastatic sites or the spleen, and polarizing HSPC differentiation toward MDSCs. HSPCs can then in turn significantly modulate the success of immunologic rejection of peripheral cancers.
Immunostimulatory Role of HSPCs in Peripheral Cancers
There is an overwhelming amount of evidence supporting the idea that HSPCs in peripheral solid malignancies are immunosuppressive. There is, however, one groundbreaking report by Wrzesinski et al. in a melanoma model that counters this paradigm. In 2007, they reported that intravenously administered lin−c‐kit+ HSPCs can drive the proliferation of adoptively transferred CD8+ T cells through an IL‐7 and Il‐15 mediated mechanism 15. Importantly, this was independent of the effects of 9Gy myeloablation. Interestingly, the activation state of the adoptively transferred T cells also did not influence the ability of HSPCs to modulate proliferation. In describing the effect of HSPCs on host immune compartments, they do indicate an increase in host CD4 T cells, Gr‐1 granulocytes, NK1.1 NK cells, and B220 B cells, a series of cell types they claim can impair the function of HSPC‐expanded CD8+ T cells. Most importantly, the HSPC‐mediated expansion of CD8+ T cells promoted antitumor immunity as measured by tumor volume over time.
Although the higher‐dose host conditioning is perceived as the ostensible antitumor component of host conditioning‐HSPC therapies, Wrzesinski et al. changed that focus and highlighted the immunologic function of HSPCs. This falls in stark contrast to the historical backdrop in which this paper was presented.
HSPCs in Brain Tumors
Background
Standard therapy including surgical resection, chemotherapy, and radiation yields a median survival of 18 months for malignant gliomas 78. The addition of tumor‐treating fields has recently yielded a median survival of 20.5 months 79. There have been some reports of success in using autologous HSPCs for some heavily pretreated incredibly fatal pediatric solid malignancies 32, 53, 54, 55, 56, 57, 58. Although promising, these therapeutic strategies are focused on the value of high‐dose chemotherapies that can successfully treat brain tumors. In this setting, HSPCs are used because the primary toxicity of Thiotepa, Etoposide, Melphalan, and Busulfan is in the BM which can be rescued by transplant 56, 57, 58. Although promising, future therapies will require novel combinatorial approaches that employ modification of the immune system for antitumor activity with minimal toxicity to patients.
Stem cell therapies present a novel therapeutic approach because gliomas are proficient at capitalizing on the SDF‐1‐C‐X‐C chemokine receptor 4 (CXCR4) axis to recruit stem cells to the brain tumor microenvironment 63, 64, 65, 66, 67. Of significance to many in the brain tumor field is the capability of mesenchymal stem cells (MSCs) and neural stem cells (NSCs) to both carry antitumor compounds or molecules and traffic successfully to the brain tumor microenvironment 68, 80, 81. Of the more underappreciated stem cell types that also traffic to the brain tumor microenvironment, HSPCs follow the same SDF‐1‐CXCR4 migratory axis 63, 67, 82. Although this is also a rather unique vehicle to the brain tumor site, only a couple of groups have tested the ability of HSPC‐carriers to enact antitumor functions.
The role HSPCs may play in this system is reliant on two key characteristics. First, HSPCs are at their core the precursor cell for the immune system. This means that given the right microenvironment, HSPCs will differentiate into progeny that will have their own immunologic function. Second, HSPCs have a unique and strong tropism for gliomas that is not readily found or well‐characterized in all peripheral organs or cancers. Therefore, if HSPCs were used either as a carrier cell with an immune‐modulating compound or molecule, HSPC‐derived cells could deposit antitumor molecules and would likely have an additional intrinsic role as a differentiated immune cell in the brain tumor microenvironment. There is therefore untapped potential for altering brain tumor immunity with HSPCs.
HSPC Trafficking to Brain Tumor
In a series of seminal studies, Tabatabai et al. demonstrated the mechanisms behind HSPC tropism to gliomas 63, 64, 65, 66, 67. They first acknowledged that HSPCs in the BM depend on SDF‐1 (CXCL12) and CXCR4 expressed on HSPCs to maintain tethering in the marrow. And in 2005, they noted that gliomas express both molecules as well but most importantly noted glioma‐mediated release of SDF‐1. In prior studies, the expression of these molecules has been linked to glioma migration and invasiveness. However, in their study, determined that IV‐administered HSPCs capitalized upon TGF‐β‐dependent release of SDF‐1 (CXCL12) in the brain tumor microenvironment and the SDF‐1 receptor, CXCR4, on HSPCs for successful migration of HSPCs toward human (T189 and T204 primary GBM lines) and murine gliomas (LNT‐229 and SMA‐560) 63. They next delineated in 2006 that there is a potential for synergy of the HSPC homing with irradiation and tumor hypoxia. They determined that 24 hours after 8 Gy irradiation and 12 hours after 1% oxygen exposure, LNT‐229 glioma induces hypoxia inducible factor‐1 (HIF‐1α)‐dependent enhanced HSPC migration. Importantly, they determined that the migratory capacity remained dependent on TGF‐β signaling. At this point, their conclusions maintained that HSPCs were a strong candidate as a cellular vector for the treatment of gliomas and that there was potential for strong synergy with standard of care including temozolomide and irradiation as well as hypoxia‐inducing antiangiogenic drugs.
After determining the interactions of HSPCs with irradiation and hypoxia, they turned their attention to the method of extravasation from the vessels into the brain tumor microenvironment. They determined that gliomas induce an upregulation of CD62E (E‐selectin) on the endothelium near the glioma microenvironment that is reliant on VEGF and TGF‐β signaling. The combined system of SDF‐1‐CXCR4 chemotaxis and glioma‐induced E‐selectin expression allow for HSPCs to circulate nearby and extravasate from inside the blood vessel into the glioma microenvironment.
Of vital importance to the success of any antitumor therapy is to have a therapy that engages the target site and leaves normal stroma unharmed. In the most recent iteration of HSPC‐glioma migration research, the same group demonstrated by 2‐photon and PET imaging that HSPCs are proficient at migrating to gliomas, but refrain from trafficking to normal tissue including spleen, lung, liver, and muscle 67. This targeted feature of HSPCs indicates the potential to use an HSPC‐carrier that through the aforementioned mechanisms can specifically target the desired glioma microenvironment, while minimizing errant migration in the periphery. This is of vital importance for therapeutic modalities that may be carried by HSPCs that have well‐described toxicity profiles in other organs and need to be selectively targeted to glioma tissue.
HSPCs as Vehicles
Tabatabai et al. next focused on proof of principle that HSPCs are efficient carriers that can serve as a transport vehicle for antitumor molecules or compounds. This entailed using a GFP‐lentivirus to transduce lin−HSPCs ex vivo prior to HSPC transfer. In a transplant‐delay setting, they implanted SMA‐560 gliomas and observed noninferior migration of HSPCs to the brain tumor microenvironment when compared with control HSPCs 65. In addition, they demonstrated that GFP‐transduced HSPCs were not tumorigenic and did not impact the survival of glioma‐bearing animals. Through this proof‐of‐principle study and the preceding mechanistic studies, Tabatabai et al. opened up the capability of utilizing HSPCs as cell‐based, efficient carrier of antitumor therapies to the glioma microenvironment.
In a more recent study by Noyan et al., they used an HSPC‐carrier to induce a dominant negative mutant of TGF‐β receptor II (TβRIIDN) in the tumor microenvironment. This entailed a transplant‐delay model in which transplant with the TβRIIDN HSPCs replaced normal marrow with cells deficient in TGF‐β signaling. Afterward, GL261 glioma was subcutaneously injected and analyzed for antitumor immune response. After successfully diminishing TGF‐β signaling in the tumor microenvironment, the HSPC therapy promoted antitumor efficacy compared with a vector control 83, 84. Although these results indicate the capabilities of HSPC carriers, the authors used subcutaneous GL261, not intracranially implanted GL261, thereby limiting the translation to the true brain tumor microenvironment.
Although these studies are both promising, they focus on the influence of the object being carried by the HSPCs. Therefore, much of the speculation about HSPC‐carrier therapies has persisted without attention to the intrinsic immunomodulatory functions of HSPCs.
HSPCs as Components of Tumor Vascularization
Although the investigation of fate of HSPCs in brain tumors is minimal, there has been some investigation of HSPC‐derived endothelial cells in brain tumors. Udani et al. used a transplant‐delay model with rat glioma (RT‐2/RAG) in a murine system to investigate incorporation of HSPCs in the glioma. They determined that the HSPC‐derived cells gave rise to CD45+ endothelial‐like cells that incorporated into the blood vessels in the glioma 85. Although there was reliable engraftment of these HSPC‐derived cells, there was no engraftment in normal brain. This study identifies an obvious next step in the investigation for HSPC‐glioma research: investigating the differentiation, fate, and functions of HSPC‐derived immunologic cells in gliomas.
HSPC‐Mediated Immune‐Activation in Brain Tumors
Although reports in peripheral solid malignancies demonstrate that HSPCs are largely immunosuppressive, there are three reports of HSPCs used in transplant‐treatment models that indicate HSPCs act as immune‐effectors by stimulating T cell responses (Fig. 3). The functions described in these reports are akin to the findings in Wresinksi et al. that described HSPC‐mediated expansion of CD8+ T cells in a melanoma model.
Figure 3.
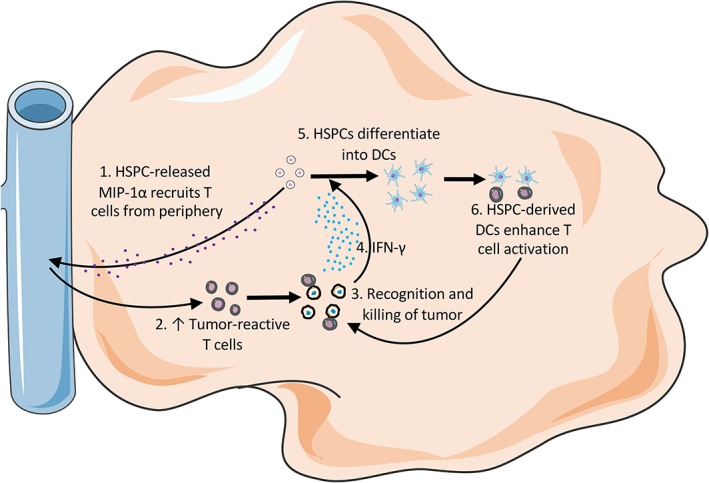
Brain tumor microenvironment after HSPC transfer during adoptive cellular therapy. HSPCs migrate to brain tumor as previously described. HSPCs chemoattract T cells from periphery via MIP‐1α. T cells release IFN‐γ that drives HSPC differentiation intratumorally. HSPC‐derived dendritic cells then promote T cell activation in situ. Abbreviations: HSPCs, hematopoietic stem and progenitor cells; DCs, dendritic cells.
In Flores et al., HSPCs synergize with adoptive T cell immunotherapy, dendritic cell vaccine, and host conditioning to promote glioma rejection 13. The glioma model used, KR158B, was intracranially implanted and had considerable resemblance to the histological features of glioblastoma. A few key roles for HSPCs in this system are identified. Of primary interest, HSPCs promoted the recruitment of tumor‐reactive T cells to the brain tumor microenvironment. This was demonstrated through imaging and flow cytometry of brain tumors of mice that were treated intravenously with DsRed+ HSPCs and GFP+ tumor‐reactive T cells. In the experiments that included HSPCs, there was a marked increase in T cell engraftment in the brain tumor. This was further demonstrated to occur through migration assays to be dependent on CCL3 (MIP‐1α) that was released by HSPCs to attract T cells to gliomas. Altogether, the synergistic combination of adoptive T cell immunotherapy, dendritic cell vaccine, and host conditioning lead to prevention of early glioma growth and mediated long‐term cures.
In a recent report by Wildes et al., HSPC differentiation into immune cells in brain tumors was investigated in the same system. It was demonstrated that HSPCs that migrate to malignant gliomas can differentiate into immune‐stimulating dendritic cells (DCs) that are required for antitumor efficacy 14. Interestingly, 3 hours after intravenous injection, HSPCs that had migrated to the brain tumor microenvironment retained multipotency, whereas after 24 hours post‐transplant, HSPCs did not retain multipotency. Additionally, HSPC‐derived cells were largely CD11c+CD11b+MHCII+ DCs with high CD86 and low CD80, CD8α, and CD103 expression. This tumor‐infiltrating DC that is CD8loCD103lo is very different from the CD103+ DCs implicated in adoptive cellular therapy for peripheral malignancies 86. The HSPC differentiation occurred through T cell‐released IFN‐γ in the brain tumor microenvironment that then in‐turn further activated tumor‐reactive T cells in a feedback loop. In this setting, HSPC‐derived cells expressed very low levels of F4/80, Ly‐6G/6C, PD‐L1, CD3, and c‐kit. Of additional interest is the impact of these HSPCs on host immunity during brain tumor progression. Although Wresinski et al. noted an increase in host CD4 T cells, Gr‐1 granulocytes, NK1.1 NK cells, and B220 B cells, Wildes et al. noted a decrease in host CD11b+Ly‐6G/6C+PD‐L1+ MDSCs among other innate immune cells in the brain tumor microenvironment. Although this investigation did not analyze peripheral organs, future studies should be developed to study peripheral and intracranial malignancies in parallel. It will be interesting to continue to develop our understanding of how HSPCs behave differently depending on their microenvironmental niche.
The findings described above by have been further expanded to include analyses in the combination therapy of HSPC transplant with PD‐1 checkpoint blockade 87. Interestingly, in brain tumors that are refractory to PD‐1 blockade, CCR2+ HSPC transfer mediates immunologic rejection of tumors that generates long‐term cures. Interestingly, the CCR2+ population is the population that migrates to brain tumors, differentiates into immune‐activating DCs, and promotes tumor rejection. Although the therapeutic PD‐1 blockade models in this article did not use total body irradiation, the CCR2+ HSPC population was less capable of rescuing myeloablated hosts than the CCR2− population. This may indicate that the subset of the HSPC population that drives antitumor immunity may be more of a hematopoietic progenitor cell than a pure HSC. Regardless, these studies indicate that HSPCs have an independent role in inducing antitumor immunity.
Although the complete mechanisms of HSPC function in brain tumors is unclear, future studies will determine how these findings translate to human HSPCs and their use in treating human brain tumors. Additionally, new articles continue to identify novel pathways through which the brain can communicate with the immune system. Although brain lymph vessels were identified in 2015 88, 89, recent studies have identified channels that connect brain to the skull BM 90. Considering the immunomodulatory role of HSPCs in brain tumors, future discovery needs to incorporate analysis of these novel circulation and migration pathways.
Conclusion
HSPCs can migrate to tumors, differentiate into immune cells, and crosstalk with the immune system. HSPCs are therefore a multifunctional cell that can be transplanted for specific settings with immense combinatorial capabilities. Given the sometimes contradictory functions of HSPCs during solid cancers, therapeutic takeaways are different depending on the disease, the anatomic location of disease, and the microenvironmental niche that makes up the HSPC environment. For peripheral cancers, tumor‐bearing host HSPCs appear immunosuppressive and should be targeted to impair protumor functions. On the contrary, the fact that HSPC transfers can synergize with T cells in peripheral and CNS tumors indicates that we should optimize this HSPC function. Perhaps future therapeutic development for both peripheral and CNS malignancies should focus on creating environmental niches either in lymphoid organs or in tumor sites that facilitate immune‐activating, antitumor HSPC functions. Regardless of the disease context, we anticipate considerable therapeutic development focused on HSPCs to drive antitumor immunity.
Author Contributions
T.J.W.: conception and design, financial support, administrative support, provision of study material or patients, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; C.T.F.: conception and design, financial support, administrative support, data analysis and interpretation, manuscript writing, final approval of manuscript; D.A.M.: conception and design, financial support, administrative support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
C.T.F. and D.A.M. hold interests in and have patents related to material disclosed in this publication that have been licensed to iOncologi, Inc. The other author indicated no potential conflicts of interest.
Acknowledgments
This research was supported by the University of Florida Health Cancer Center Predoctoral Award (T.J.W.); National Cancer Institute R01 CA195563 (D.A.M.); American Brain Tumor Association Research Collaboration Grant (C.T.F.); Alex's Lemonade Stand Young Investigator Grant (C.T.F.); Florida Center for Brain Tumor Research Grant (C.T.F.).
References
- 1. King KY, Goodell MA. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat Rev Immunol 2011;11:685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casbon AJ, Reynaud D, Park C et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci USA 2015;112:E566–E575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cortez‐Retamozo V, Etzrodt M, Newton A et al. Origins of tumor‐associated macrophages and neutrophils. Proc Natl Acad Sci USA 2012;109:2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gabrilovich DI, Ostrand‐Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giles AJ, Reid CM, Evans JD et al. Activation of hematopoietic stem/progenitor cells promotes immunosuppression within the pre‐metastatic niche. Cancer Res 2016;76:1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaplan RN, Riba RD, Zacharoulis S et al. VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature 2005;438:820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lyden D, Hattori K, Dias S et al. Impaired recruitment of bone‐marrow‐derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med 2001;7:1194–1201. [DOI] [PubMed] [Google Scholar]
- 8. Pang WW, Price EA, Sahoo D et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid‐biased with age. Proc Natl Acad Sci USA 2011;108:20012–20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sio A, Chehal MK, Tsai K et al. Dysregulated hematopoiesis caused by mammary cancer is associated with epigenetic changes and hox gene expression in hematopoietic cells. Cancer Res 2013;73:5892–5904. [DOI] [PubMed] [Google Scholar]
- 10. Starzynska T, Dabkowski K, Blogowski W et al. An intensified systemic trafficking of bone marrow‐derived stem/progenitor cells in patients with pancreatic cancer. J Cell Mol Med 2013;17:792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu WC, Sun HW, Chen HT et al. Circulating hematopoietic stem and progenitor cells are myeloid‐biased in cancer patients. Proc Natl Acad Sci USA 2014;111:4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Luca K, Frances‐Duvert V, Asensio MJ et al. The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate. Leukemia 2009;23:2063–2074. [DOI] [PubMed] [Google Scholar]
- 13. Flores C, Pham C, Snyder D et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 2015;4:e994374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wildes TJ, Grippin A, Dyson KA et al. Cross‐talk between T cells and hematopoietic stem cells during adoptive cellular therapy for malignant glioma. Clin Cancer Res 2018;24:3955–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wrzesinski C, Paulos CM, Gattinoni L et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest 2007;117:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Young MR, Wright MA. Myelopoiesis‐associated immune suppressor cells in mice bearing metastatic Lewis lung carcinoma tumors: Gamma interferon plus tumor necrosis factor alpha synergistically reduces immune suppressor and tumor growth‐promoting activities of bone marrow cells and diminishes tumor recurrence and metastasis. Cancer Res 1992;52:6335–6340. [PubMed] [Google Scholar]
- 17. Talmadge JE, Gabrilovich DI. History of myeloid‐derived suppressor cells. Nat Rev Cancer 2013;13:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Allakhverdi Z, Delespesse G. Hematopoietic progenitor cells are innate Th2 cytokine‐producing cells. Allergy 2012;67:4–9. [DOI] [PubMed] [Google Scholar]
- 19. Griseri T, McKenzie BS, Schiering C et al. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin‐23‐driven chronic intestinal inflammation. Immunity 2012;37:1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer KD, Agrawal DK. Hematopoietic stem and progenitor cells in inflammation and allergy. Front Immunol 2013;4:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baldridge MT, King KY, Boles NC et al. Quiescent haematopoietic stem cells are activated by IFN‐gamma in response to chronic infection. Nature 2010;465:793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boettcher S, Manz MG. Regulation of inflammation‐ and infection‐driven hematopoiesis. Trends Immunol 2017;38:345–357. [DOI] [PubMed] [Google Scholar]
- 23. MacNamara KC, Oduro K, Martin O et al. Infection‐induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN‐gamma signaling. J Immunol 2011;186:1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takizawa H, Boettcher S, Manz MG. Demand‐adapted regulation of early hematopoiesis in infection and inflammation. Blood 2012;119:2991–3002. [DOI] [PubMed] [Google Scholar]
- 25. Zhao JL, Ma C, O'Connell RM et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress‐induced hematopoiesis. Cell Stem Cell 2014;14:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao JL, Baltimore D. Regulation of stress‐induced hematopoiesis. Curr Opin Hematol 2015;22:286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang X, Gao M, Schouteden S et al. Hematopoietic stem/progenitor cells directly contribute to arteriosclerotic progression via integrin beta2. Stem Cells 2015;33:1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu JS, Bury MI, Fuller NJ et al. Bone marrow stem/progenitor cells attenuate the inflammatory milieu following substitution urethroplasty. Sci Rep 2016;6:35638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pu S, Qin B, He H et al. Identification of early myeloid progenitors as immunosuppressive cells. Sci Rep 2016;6:23115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thomas ED, Lochte HL Jr, Lu WC et al. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med 1957;257:491–496. [DOI] [PubMed] [Google Scholar]
- 32. Chabannon C, Kuball J, Bondanza A et al. Hematopoietic stem cell transplantation in its 60s: A platform for cellular therapies. Sci Transl Med 2018;10:eaap9630. [DOI] [PubMed] [Google Scholar]
- 33. Appelbaum FR. Hematopoietic‐cell transplantation at 50. N Engl J Med 2007;357:1472–1475. [DOI] [PubMed] [Google Scholar]
- 34. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–1152. [DOI] [PubMed] [Google Scholar]
- 35. Goldman JM, Apperley JF, Jones L et al. Bone marrow transplantation for patients with chronic myeloid leukemia. N Engl J Med 1986;314:202–207. [DOI] [PubMed] [Google Scholar]
- 36. Kersey JH, Weisdorf D, Nesbit ME et al. Comparison of autologous and allogeneic bone marrow transplantation for treatment of high‐risk refractory acute lymphoblastic leukemia. N Engl J Med 1987;317:461–467. [DOI] [PubMed] [Google Scholar]
- 37. Walters MC, Patience M, Leisenring W et al. Bone marrow transplantation for sickle cell disease. N Engl J Med 1996;335:369–376. [DOI] [PubMed] [Google Scholar]
- 38. Sun J, Ramos A, Chapman B et al. Clonal dynamics of native haematopoiesis. Nature 2014;514:322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herault A, Binnewies M, Leong S et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 2017;544:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ji H, Ehrlich LI, Seita J et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010;467:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell 2007;1:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 2013;13:102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cui TX, Kryczek I, Zhao L et al. Myeloid‐derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013;39:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu C, Savage N, Singh N et al. The expression profiles and regulation of PD‐L1 in tumor‐induced myeloid‐derived suppressor cells. J Immunol 2017;198:124.129–124.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mukherjee S. The Emperor of All Maladies: A Biography of Cancer. New York: Scribner, 2011:573. [Google Scholar]
- 46. Bishop MR. Allogeneic hematopoietic stem cell transplantation for metastatic breast cancer. Haematologica 2004;89:599–605. [PubMed] [Google Scholar]
- 47. Armitage JO. Bone marrow transplantation. N Engl J Med 1994;330:827–838. [DOI] [PubMed] [Google Scholar]
- 48. Antman KH, Rowlings PA, Vaughan WP et al. High‐dose chemotherapy with autologous hematopoietic stem‐cell support for breast cancer in North America. J Clin Oncol 1997;15:1870–1879. [DOI] [PubMed] [Google Scholar]
- 49. Hryniuk W, Levine MN. Analysis of dose intensity for adjuvant chemotherapy trials in stage II breast cancer. J Clin Oncol 1986;4:1162–1170. [DOI] [PubMed] [Google Scholar]
- 50. Stadtmauer EA, O'Neill A, Goldstein LJ et al. Conventional‐dose chemotherapy compared with high‐dose chemotherapy plus autologous hematopoietic stem‐cell transplantation for metastatic breast cancer. Philadelphia Bone Marrow Transplant Group. N Engl J Med 2000;342:1069–1076. [DOI] [PubMed] [Google Scholar]
- 51. Howard DH, Kenline C, Lazarus HM et al. Abandonment of high‐dose chemotherapy/hematopoietic cell transplants for breast cancer following negative trial results. Health Serv Res 2011;46:1762–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nitz UA, Mohrmann S, Fischer J et al. Comparison of rapidly cycled tandem high‐dose chemotherapy plus peripheral‐blood stem‐cell support versus dose‐dense conventional chemotherapy for adjuvant treatment of high‐risk breast cancer: Results of a multicentre phase III trial. Lancet 2005;366:1935–1944. [DOI] [PubMed] [Google Scholar]
- 53. Abdel‐Azim H, Kapoor N, Mahadeo KM et al. Graft versus tumor effect in the brain of a child with recurrent metastatic medulloblastoma. Pediatr Blood Cancer 2015;62:1667–1669. [DOI] [PubMed] [Google Scholar]
- 54. Massimino M, Cohen KJ, Finlay JL. Is there a role for myeloablative chemotherapy with autologous hematopoietic cell rescue in the management of childhood high‐grade astrocytomas? Pediatr Blood Cancer 2010;54:641–643. [DOI] [PubMed] [Google Scholar]
- 55. Gururangan S, Dunkel IJ, Goldman S et al. Myeloablative chemotherapy with autologous bone marrow rescue in young children with recurrent malignant brain tumors. J Clin Oncol 1998;16:2486–2493. [DOI] [PubMed] [Google Scholar]
- 56. Finlay JL, Dhall G, Boyett JM et al. Myeloablative chemotherapy with autologous bone marrow rescue in children and adolescents with recurrent malignant astrocytoma: Outcome compared with conventional chemotherapy: A report from the Children's Oncology Group. Pediatr Blood Cancer 2008;51:806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Finlay JL, Goldman S, Wong MC et al. Pilot study of high‐dose thiotepa and etoposide with autologous bone marrow rescue in children and young adults with recurrent CNS tumors. The Children's Cancer Group. J Clin Oncol 1996;14:2495–2503. [DOI] [PubMed] [Google Scholar]
- 58. Finlay JL, Zacharoulis S. The treatment of high grade gliomas and diffuse intrinsic pontine tumors of childhood and adolescence: A historical—and futuristic—perspective. J Neurooncol 2005;75:253–266. [DOI] [PubMed] [Google Scholar]
- 59. Davoust N, Vuaillat C, Cavillon G et al. Bone marrow CD34+/B220+ progenitors target the inflamed brain and display in vitro differentiation potential toward microglia. FASEB J 2006;20:2081–2092. [DOI] [PubMed] [Google Scholar]
- 60. MacNamara KC, Jones M, Martin O et al. Transient activation of hematopoietic stem and progenitor cells by IFN‐gamma during acute bacterial infection. PLoS ONE 2011;6:e28669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 2014;14:460–472. [DOI] [PubMed] [Google Scholar]
- 62. Levesque JP, Hendy J, Takamatsu Y et al. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest 2003;111:187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tabatabai G, Bahr O, Mohle R et al. Lessons from the bone marrow: How malignant glioma cells attract adult haematopoietic progenitor cells. Brain 2005;128:2200–2211. [DOI] [PubMed] [Google Scholar]
- 64. Tabatabai G, Frank B, Mohle R et al. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF‐beta‐dependent HIF‐1alpha‐mediated induction of CXCL12. Brain 2006;129:2426–2435. [DOI] [PubMed] [Google Scholar]
- 65. Tabatabai G, Hasenbach K, Herrmann C et al. Glioma tropism of lentivirally transduced hematopoietic progenitor cells. Int J Oncol 2010;36:1409–1417. [DOI] [PubMed] [Google Scholar]
- 66. Tabatabai G, Herrmann C, von Kurthy G et al. VEGF‐dependent induction of CD62E on endothelial cells mediates glioma tropism of adult haematopoietic progenitor cells. Brain 2008;131:2579–2595. [DOI] [PubMed] [Google Scholar]
- 67. Hasenbach K, Wiehr S, Herrmann C et al. Monitoring the glioma tropism of bone marrow‐derived progenitor cells by 2‐photon laser scanning microscopy and positron emission tomography. Neuro Oncol 2012;14:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bexell D, Svensson A, Bengzon J. Stem cell‐based therapy for malignant glioma. Cancer Treat Rev 2013;39:358–365. [DOI] [PubMed] [Google Scholar]
- 69. Gardner SL. Application of stem cell transplant for brain tumors. Pediatr Transplant 2004;8:28–32. [DOI] [PubMed] [Google Scholar]
- 70. Belyaev NN, Brown DE, Diaz A‐IG et al. Induction of an IL7‐R+c‐Kithi myelolymphoid progenitor critically dependent on IFN‐[gamma] signaling during acute malaria. Nat Immunol 2010;11:477–485. [DOI] [PubMed] [Google Scholar]
- 71. de Bruin AM, Demirel O, Hooibrink B et al. Interferon‐gamma impairs proliferation of hematopoietic stem cells in mice. Blood 2013;121:3578–3585. [DOI] [PubMed] [Google Scholar]
- 72. de Bruin AM, Libregts SF, Valkhof M et al. IFNgamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood 2012;119:1543–1554. [DOI] [PubMed] [Google Scholar]
- 73. de Bruin AM, Voermans C, Nolte MA. Impact of interferon‐gamma on hematopoiesis. Blood 2014;124:2479–2486. [DOI] [PubMed] [Google Scholar]
- 74. Essers MA, Offner S, Blanco‐Bose WE et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009;458:904–908. [DOI] [PubMed] [Google Scholar]
- 75. Qu P, Yan C, Du H. Matrix metalloproteinase 12 overexpression in myeloid lineage cells plays a key role in modulating myelopoiesis, immune suppression, and lung tumorigenesis. Blood 2011;117:4476–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shiozawa Y, Pedersen EA, Havens AM et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 2011;121:1298–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Peinado H, Aleckovic M, Lavotshkin S et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro‐metastatic phenotype through MET. Nat Med 2012;18:883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013;310:1842–1850. [DOI] [PubMed] [Google Scholar]
- 79. Stupp R, Taillibert S, Kanner AA et al. Maintenance therapy with tumor‐treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. JAMA 2015;314:2535–2543. [DOI] [PubMed] [Google Scholar]
- 80. Bovenberg MS, Degeling MH, Tannous BA. Advances in stem cell therapy against gliomas. Trends Mol Med 2013;19:281–291. [DOI] [PubMed] [Google Scholar]
- 81. Grauer OM, Wesseling P, Adema GJ. Immunotherapy of diffuse gliomas: Biological background, current status and future developments. Brain Pathol 2009;19:674–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Guo KT, Juerchott K, Fu P et al. Isolation and characterization of bone marrow‐derived progenitor cells from malignant gliomas. Anticancer Res 2012;32:4971–4982. [PubMed] [Google Scholar]
- 83. Noyan F, Diez IA, Hapke M et al. Induced transgene expression for the treatment of solid tumors by hematopoietic stem cell‐based gene therapy. Cancer Gene Ther 2012;19:352–357. [DOI] [PubMed] [Google Scholar]
- 84. Shah AH, Tabayoyong WB, Kundu SD et al. Suppression of tumor metastasis by blockade of transforming growth factor beta signaling in bone marrow cells through a retroviral‐mediated gene therapy in mice. Cancer Res 2002;62:7135–7138. [PubMed] [Google Scholar]
- 85. Udani VM, Santarelli JG, Yung YC et al. Hematopoietic stem cells give rise to perivascular endothelial‐like cells during brain tumor angiogenesis. Stem Cells Dev 2005;14:478–486. [DOI] [PubMed] [Google Scholar]
- 86. Spranger S, Dai D, Horton B et al. Tumor‐residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017;31:711.e714–723.e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Flores CT, Wildes TJ, Drake JA et al. Lin(−)CCR2(+) hematopoietic stem and progenitor cells overcome resistance to PD‐1 blockade. Nat Commun 2018;9:4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Aspelund A, Antila S, Proulx ST et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015;212:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Louveau A, Smirnov I, Keyes TJ et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Herisson F, Frodermann V, Courties G et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci 2018;21:1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]