

Abstract
CRISPR/Cas9-mediated gene editing of human hematopoietic stem cells (hHSCs) is a promising strategy for the treatment of genetic blood diseases through site-specific correction of identified causal mutations. However, clinical translation is hindered by low ratio of precise gene modification using the corrective donor template (homology-directed repair, HDR) to gene disruption (non-homologous end joining, NHEJ) in hHSCs. By using a modified version of Cas9 with reduced nuclease activity in G1 phase of cell cycle when HDR cannot occur, and transiently increasing the proportion of cells in HDR-preferred phases (S/G2), we achieved a 4-fold improvement in HDR/NHEJ ratio over the control condition in vitro, and a significant improvement after xenotransplantation of edited hHSCs into immune-deficient mice. This strategy for improving gene editing outcomes in hHSCs has important implications for the field of gene therapy, and can be applied to diseases where increased HDR/NHEJ ratio is critical for therapeutic success.
Keywords: Adult haematopoietic stem cells, CD34+, Cell cycle, Clinical translation, CRISPR, Gene therapy, Hematopoietic stem cells (HSCs), Stem/progenitor cell
GRAPHICAL ABSTRACT
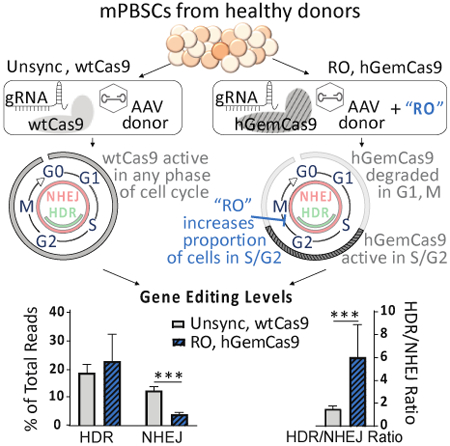
A modified version of Cas9 (hGemCas9) with reduced nuclease activity in G1 phase of cell cycle, when HDR cannot occur, and transient synchronization of mobilized peripheral blood stem cells (mPBSCs) in HDR-preferred phases (S/G2) resulted in a 4-fold improvement in HDR/NHEJ ratio over the control condition in vitro, and a significant improvement after xenotransplantation of edited hHSCs into immune-deficient mice.
INTRODUCTION
Autologous hematopoietic stem cell (HSC) transplantation, combined with gene editing, could provide an ideal therapeutic option for the treatment of congenital blood diseases, such as hemoglobinopathies, primary immune deficiencies, and storage disorders. Site specific gene editing is achieved by inducing a double-stranded break (DSB) in the DNA near the mutation using targeted nucleases, such as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated (Cas9) system, and subsequently taking advantage of the innate cellular repair pathways to fix the break. The two major DNA repair pathways are: error-prone non-homologous end joining (NHEJ), which is available throughout the cell cycle [1], and precise homology-directed repair (HDR), which uses a repair template to accurately correct the DSB, and is restricted to S/G2 phases [2, 3]. Once modified, HSCs can be re-infused into the patient and provide a long-term self-renewing population to generate a life-long supply of healthy cells.
DSB repair via NHEJ pathway is efficient in human HSCs due to their quiescent status [2, 4, 5], and approaches to disrupt genes for the treatment of certain diseases have already moved to clinical trials [6, 7]. However, for diseases where a precise correction of the disease-causing mutation is required, the HDR pathway must be employed to incorporate the provided repair template. To date, HDR-mediated gene editing has been constrained by relatively low efficiency in primitive long-term reconstituting HSCs [8–10].
In this study, we modeled the correction of the sickle cell disease (SCD)-causing mutation in human hematopoietic stem and progenitor cells (HSPCs). A single nucleotide substitution in the beta-globin gene results in the formation of sickle hemoglobin, HbS, giving rise to rigid and misshaped red blood cells under low oxygen conditions. This devastating disease has lifelong physical and psycho-social consequences: repeated painful crises and frequent costly hospitalization, anemia, kidney failure, stroke and neuro-cognitive deficiencies. There are approximately 300,000 new cases of SCD worldwide every year [11, 12], and despite some available therapies, the remaining morbidity and mortality present a significant need for developments of improved treatment options [12]. Based on results from allogeneic HSC transplants, it has been predicted that gene correction levels of approximately 10–30% in long-term primitive HSCs are required to cure SCD patients [13–15]. Although HDR levels of almost 50% are achievable in vitro currently [9, 16–18], only 5–7% are retained in long-term cells after transplant into NOD/SCID/IL2rgnull (NSG) mice [9, 17]. Therefore, it is critical to understand the reason for lower HDR levels in HSCs, and to develop methods for improving gene editing outcomes in human HSCs.
Increasing the absolute amount of HDR is an obvious goal for improving gene editing outcomes. Additionally, it is equally important to consider simultaneously limiting the levels of NHEJ, as NHEJ-mediated repair may result in large deletions or genomic rearrangements [19]. Moreover, insertions and deletions (indels) in the beta-globin gene could convert a sickle allele to a beta-thalassemia allele, potentially resulting in a worse phenotype than the initial disease. Although an acceptable background value for NHEJ in patient cells has not been established in the field yet, it is clear that the relative proportion of precise edits to indels needs to be increased in order to ensure positive therapeutic outcomes. We used a metric, designated “HDR/NHEJ ratio” to evaluate approaches of improving gene editing outcomes.
Here, we studied the temporal regulation of DNA repair pathway utilization in human HSPCs. We hypothesized that cell cycle phase-dependent manipulation of DNA repair will improve HDR/NHEJ ratio in primary human HSPCs. We showed that by using a Cas9 variant with decreased nuclease activity in G1 and M phases, and synchronizing the cells in S/G2, we can obtain a 4-fold increase in HDR/NHEJ ratio in primary human HSPCs in vitro, compared to standard conditions. More importantly, these manipulations led to a significant improvement in HDR/NHEJ ratio in vivo.
MATERIALS AND METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Donald B. Kohn (DKohn@mednet.ucla.edu)
Experimental Model and Subject Details
Primary Human CD34+ Cells
Leukopaks from healthy donors were purchased from HemaCare (HemaCare BioResearch Products; Van Nuys, CA). Mobilized peripheral blood (mPB) was collected from normal, healthy donors on days 5 and 6 after 5 days of stimulation with granulocyte-colony stimulating factor (G-CSF). mPB was processed to enrich for CD34+ cells using magnetic beads and cryopreserved as described previously [9].
Method Details
Cell Culture
Healthy human CD34+ cells from mPB (PBSCs) were thawed in pre-warmed X-VIVO 15 medium (Lonza; Basel, Switzerland) with PSQ (penicillin, streptomycin, glutamine, (Gemini Bio-Products; Sacramento, CA)), pelleted at 500g for 5 mins, and resuspended at 5×105cells/mL in pre-warmed X-VIVO 15 medium with PSQ and SFT cytokines (50 ng/mL stem cell factor (SCF), 50 ng/mL fms-related tyrosine kinase 3 ligand (Flt3-L), and 50 ng/mL thrombopoietin (TPO)) (Peprotech; Rocky Hill, NJ). Cells were prestimulated at 37°C and 5% CO2 incubator for 48 hours. Where indicated, 30uM RO (RO-3306, Sigma Aldrich; St. Louis, MO) was added to cell culture medium 28 hours after thawing. Cells were washed twice with PBS after 20 hours of RO treatment.
Electroporation
For electroporation, 2×105 (or 1×106 for mouse experiments) cells per condition were pelleted at 90g for 15 mins at RT, resuspended in 100ul of BTXpress Electroporation buffer (Harvard Bioscience, Inc; Holliston, MA), combined with pre-aliquoted mRNA (kept on ice), and pulsed once at 250 V for 5 milliseconds in the BTX ECM 830 Square Wave Electroporator (Harvard Apparatus; Holliston, MA). Unless otherwise specified, 4ug of Cas9 mRNA and 4ug of sgRNA targeting the human beta-globin gene were used (See Fig 2A). GFP mRNA was electroporated at 0.3ug. After electroporation, cells were rested in cuvettes for 10 mins at RT, and then recovered with 400ul (or 2.4mL, for 1e6 cells) of X-VIVO 15 medium (with PSQ and SFT cytokines), containing AAV6 (multiplicity of infection, MOI=2e4) to introduce 4 SNPs (see Fig 2B for details) (UNC vector core; Chapel Hill, NC). The cells were cultured in a 24-well (or 6-well, for 1×106 cells) plate at 37°C, 5% CO2 incubator. 24 hours post electroporation, the cells were counted by hemocytometer using 1:2 dilution with trypan blue to determine viability (# of live cells/# of total cells × 100) and fold expansion (# of cells 24 hours after electroporation/# of cells before electroporation). Cells were replated into 1mL (or 5mL, for 1×106 cells) of myeloid expansion medium (Iscove’s Modified Dulbecco’s Medium (IMDM, Thermo Fisher Scientific; Waltham, MA) + 20% FBS (HI FBS, Gibco/ThermoFisher; Waltham, MA) + 5ng/mL Interleukin 3 (IL3), 10ng/mL Interleukin 6 (IL6), 25ng/mL SCF (Peprotech; Rocky Hill, NJ)), and cultured for 4 days (unless otherwise specified) prior to harvesting for gDNA. Samples that were FACS-sorted into HSPC sub-populations were cultured 2–4 weeks post sort (as indicated in figure legends) to obtain enough cells for gDNA and subsequent high throughput sequencing (HTS) (electroporation of nuclease and donor template was more toxic that nuclease alone, therefore longer expansion time was required). gDNA was extracted using PureLink Genomic DNA Mini Kit (Invitrogen/ThermoFisher Scientific; Carlsbad, CA).
Figure 2: Overview of Gene Editing Reagents.

A. A map of the beta globin locus on human Chr 11 (not to scale). The inset illustrates the beginning of Exon 1. The blue cloud represents Cas9 protein and the gRNA binding site is indicated. The PAM sequence is underlined in blue. The scissors and the dashed line represent the Cas9 cut site. The red “A” represents the location of the sickle mutation.
B. A schematic of the AAV donor template (not to scale). The inset shows the four mutations that are introduced by the donor template. These include: a common SNP (29bp upstream from cut site), a sickle mutation (18bp upstream from cut site), a PAM mutation to prevent re-cleavage by Cas9 after successful gene modification (5bp upstream from cut site), and a HhaI RFLP (5bp downstream of cut site) to be used as a surrogate marker for gene modification.
mRNA/sgRNA production
To make mRNA template, maxi-prepped expression plasmids were linearized with SpeI (NEB; Ipswitch, MA), and purified using PCR purification kit according to manufacturer’s protocol. In vitro transcription was carried out using mMessage T7 Ultra Kit (ThermoFisher Scientific; Waltham, MA). mRNA product was purified using the RNeasy MinElute Cleanup Kit (Qiagen; Valencia, CA) following the manufacturer’s protocol.
sgRNA template was prepared as previously described (dx.doi.org/10.17504/protocols.io.hdrb256). RNA was purified using the RNeasy MinElute Cleanup Kit (Qiagen; Valencia, CA) following manufacturer’s protocol.
hGemCas9
hGemCas9 was created by cloning a 110-amino acid sequence of human geminin domain to C-terminus of Cas9, based on the strategy used by Gutschner et al., 2016. Gene blocks were ordered from IDT to include homology arms for NEBuilder cloning, and cloned into SpCas9 plasmid [16, 20].
High-Throughput Sequencing (HTS)
Library prep
DNA library for HTS was prepared as described previously [9]. Briefly, an outer PCR was performed on genomic DNA to amplify a 1.1kb region of interest (using Outer PCR Fwd and Rev primers). A second PCR was performed to add a unique index to the PCR product of each sample to be sequenced (read1/read 2 and P5/P7 primers). The PCR products with the indexes were mixed at equal concentrations, which was determined by densitometry of the PCR products and analyzed by gel electrophoresis, to create a pooled library. The pooled library was purified twice using AMPure XP beads (Beckman Coulter Inc.; Brea, CA) and then quantified using ddPCR (QX 200; Bio-Rad Laboratories Inc.; Hercules, CA). High-thoughput sequencing was performed at UCLA Technology Center for Genomics & Bioinformatics (TCGB) using MiSeq 2×150 paired-end reads (Illumina Inc; San Diego, CA).
Sequencing Analysis and Calculations
Analysis of sequencing data was performed as in Hoban et al, 2015; 2016 [9, 16]. Percentage of HDR was calculated as the (number of sequence reads containing a sickle change)/(total reads for that sample)*100. Percentage of NHEJ was calculated as the frequency of sequence reads containing an insertion or deletion −50/+36 bases around the nuclease cut site.
Flow Cytometry/FACS
All flow cytometry analysis and FACS were performed on the following instruments: BD LSRII, BD LSRFortessa, BD FACS Aria II, all with the similar 5-laser configurations: UV 355 nm, Violet 405 nm, Blue 488 nm, Yel-Grn 561 nm, Red 633 nm.
Analysis of HSC/MPP/Progenitor Populations
The cells were collected and washed with PBS+2%FBS, and stained for 30 minutes at 4°C with the following antibodies: CD34, CD38, CD45RA, CD90 (detailed antibody information in supplemental methods). Cells were washed twice with 2ml PBS+2%FBS, and resuspended in 200ul FBS+2%FBS for flow cytometry analysis, or 500ul FBS+2%FBS for FACS. Representative gating strategy is illustrated in Figure S1A. Cells were sorted into myeloid expansion medium + PSQ. After sort, the cells were spun at 300g for 15 mins, and replated into myeloid expansion medium + PSQ + cytokines (IL-3, IL-6, SCF).
Cell Cycle
For cell cycle flow cytometry analysis, 2×104 cells were collected after RO or DMSO treatment, fixed in 1mL ice-cold 70% EtOH and stored at −20°C for at least 2 hours (and up to 2 weeks). Fixed cells were washed twice with PBS+2%FBS and stained with 5ug/mL Hoechst 33342 (BD Biosciences; San Jose, CA) in PBS+2%FBS at a final volume of 250ul for at least 45 minutes.
For FACS-sorting of live cells, cells were cultured at 5×105-1×106 cells/mL and stained with 5ug/mL Hoechst 33342 for 45–60 mins at 37°C. Cells were washed with PBS + 2%FBS and resuspended at 5×106 cells/mL in PBS + 2%FBS + 5ug/mL Hoechst 33342.
Mouse Transplants
All research designs and procedures were reviewed by the UCLA institutional animal care and use committee (IACUC). Mouse work was performed according to the ARC protocol number 2008–167
Transplant
To evaluate the engraftment of gene-edited cells, and the levels of gene editing in long-term HSC population, the cells were transplanted into NSG mice (The Jackson Laboratory; Bar Harbor, ME). The cells were treated and electroporated as described earlier using 1×106-cell electroporation protocol. For RO-treated cells, 3ug CtIP* was co-electroporated with gene editing reagents. 24 hours after electroporation, the cells were washed with PBS and counted. 5×104 cells were kept in myeloid culture for evaluating gene editing levels prior to transplant. The remaining cells were resuspended in PBS + 1% OKT3 (Biolegend; SanDiego, CA) at 1.25×106 cells per 125ul. Cells were transplanted retro-orbitally into mice 3–5 hours after 250-cGy total body irradiation. The number of mice to transplant per arm was determined to give sufficient power to indicate statistically meaningful differences.
Harvest and Analysis
Retro-orbital bleeding was performed at 8 weeks post-transplant to determine the levels of human engraftment. Red blood cells were lysed using the lysing buffer (BD Biosciences, San Jose, CA). The levels of human engraftment were determined by flow cytometry (detailed flow panels in supplementary methods), and calculated as (hCD45+/(hCD45++mCD45+))×100. Mice were euthanized 16 weeks post-transplant. Two femurs were collected and flushed with MACS buffer. Spleens were squeezed though a 70um filter. Red blood cells were lysed using the lysing buffer (BD Biosciences, San Jose, CA). The levels of human engraftment were determined by flow cytometry. The human cells from mouse BM were isolated using hCD45+ microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), gDNA was extracted, and the levels of gene editing in human cells were determined by HTS, as described earlier. Mouse harvest, flow cytometry and sequencing analyses were performed in a blinded manner.
Quantification and Statistical Analysis
For quantitative measurements related to HSPC sub-populations, gene editing levels, evaluation of hGemCas9 nuclease, engraftment, gene editing and lineage analysis in NSG mice, descriptive statistics such as number of donors/experiments/animals, mean, standard deviation were reported and presented in figures. A two-step approach was adopted to test difference among multiple experimental groups/conditions. In the first step, a non-parametric Kruskal-Wallis test was used to evaluate if there was an overall difference. If result from the first step was significant, then unpaired Wilcoxon rank sum test was performed to identify which two groups were different. If normality assumption was met and the sample size was too small, unpaired t test was used instead. Chi-square test was used to assess if the distributions of cells are the same between experimental groups. For all statistical investigations, tests for significance were two-tailed. A p-value less than the 0.05 significance level was considered to be statistically significant. All statistical analyses were carried out using SAS version 9.4 (SAS Institute Inc. 2013).
RESULTS
HDR Levels are Significantly Lower in Human HSCs, Compared to Progenitors
To understand the reason for the drop in gene correction levels after transplantation of edited cells into NSG mice [9, 17], we studied whether gene editing efficiency in long-term HSCs differs from the progenitors.
We began by comparing different phases of gene editing in the following human immunophenotypically-defined HSPC sub-populations: HSCs (CD34+/CD38−/CD90+/CD45RA−); multipotent progenitors (MPPs) (CD34+/CD38−/CD90−/CD45RA−); and mature stem/progenitor populations (CD34+/CD38+) [21] (Figure S1A). We measured cell cycle profile of immunophenotypic HSPC sub-populations after two days of prestimulation with cytokines (Figure S1B). HSC and MPP sub-populations mainly comprised of G1 cells (75% and 87%, respectively). In contrast, in the progenitor sub-population, 55% of cells were in G1 and ~40% in S/G2 phases. To assess the efficiency of delivery and translation of Cas9 mRNA among the HSPC sub-populations, we compared the delivery and translation of electroporated green fluorescent protein (GFP) mRNA. Bulk CD34+ cells from healthy human granulocyte colony stimulating factor (G-CSF)-mobilized peripheral blood cells (“PBSCs”) were electroporated with in vitro-transcribed (IVT) mRNA encoding GFP. No statistically significant differences were observed among the HSPC sub-populations in regards to percentage of GFP-positive (GFP+) cells or the mean fluorescence intensity (MFI) on days 1–2 post electroporation, indicating that delivery and translation of electroporated mRNA is equivalent in HSCs, MPPs and progenitors (Figure 1A-B).
Figure 1. HDR Levels are Significantly Lower in Human HSCs, Compared to Progenitors.

A. GFP expression in sub-populations (HSCs, MPPs, progenitors) of human PBSCs, measured by flow cytometry on days 1 and 2 after electroporation of GFP mRNA. N=3. Data are represented as mean ± standard deviation (SD). Differences between sub-populations are not significant, based on Kruskal-Wallis test. See Fig S1A for flow cytometry gating strategy.
B. Mean fluorescence intensity (MFI) of GFP expression in Fig 1A. N=3. Data are represented as mean ± SD. Differences between sub-populations are not significant, based on Kruskal-Wallis test.
C. GFP expression in sub-populations of human PBSCs, measured by flow cytometry on days 1 and 2 after transducing cells with GFP-AAV6. N=4. Data are represented as mean ± SD. Differences between sub-populations are not significant if not specified, based on Kruskal-Wallis test, * P<0.05, based on Wilcoxon Rank-Sum test.
D. MFI of GFP expression in Fig 1C. N=4. Data are represented as mean ± SD. Differences between sub-populations are not significant, based on Kruskal-Wallis test.
E. Percentage of insertions and deletions (indels) in FACS-sorted human PBSC sub-populations (HSCs, MPPs, progenitors), measured by high throughput sequencing (HTS) two weeks after electroporating bulk PBSCs with Cas9 mRNA and sgRNA (see also Fig 2A). % indels are calculated as described in Methods. N=2. Data are represented as individual points, bars indicate mean.
F. Percentage of gene editing (HDR and NHEJ) in FACS-sorted human PBSC sub-populations (HSCs, MPPs, progenitors), measured by HTS 4 weeks after electroporating bulk PBSCs with Cas9 mRNA and sgRNA, and transducing with AAV6 donor template (see Fig 2B for a map of AAV6). % HDR and NHEJ are calculated as described in Methods. N=4. Data are represented as mean ± SD. Differences are not significant if not specified, based on Kruskal-Wallis test. * P<0.05, based on Wilcoxon Rank-Sum test.
Prior to utilizing adeno-associated virus serotype 6 (AAV6) as a delivery vehicle for the donor template, we tested the transduction efficiency of GFP-AAV6 in the three sub-populations as a proxy for donor template availability in the cells. No differences in the percentage of GFP+ cells or MFI were observed among HSCs, MPPs and progenitors one day post transduction, suggesting that the delivery of the donor template would be equivalent in the three sub-populations (Figure 1C-D). On day 2, the percentage of GFP+ cells in the MPPs and progenitors was higher than in HSCs, however the MFI was the same.
To determine whether the three sub-populations are equally amenable to Cas9 nuclease activity, IVT mRNA encoding Cas9 and a single guide RNA (sgRNA) targeting exon 1 of the human beta-globin locus were delivered to bulk PBSCs by electroporation (refer to Figure 2A) [16, 17]. The following day, HSCs, MPPs and progenitors were separated by fluorescence-activated cell sorting (FACS), and each sub-population was cultured in myeloid expansion medium for two weeks prior to performing high-throughput sequencing (HTS) of the human beta-globin locus. The percentages of beta-globin alleles containing insertions/deletions (indels) in HSPC sub-populations are shown in Figure 1E, demonstrating that similar endonuclease activity was achieved in the HSCs, MPPs and progenitors.
Next, to measure the levels of HDR and NHEJ in the presence of donor template, PBSCs were electroporated with Cas9 mRNA and sgRNA, and subsequently transduced with AAV6 donor template carrying a 1.1kb insert, homologous to the exon 1 of beta-globin gene, with a single-nucleotide polymorphism (SNP) to convert wild-type (wt) beta globin to sickle (along with three other SNPs described in Figure 2B). HSCs, MPPs and progenitors were separated by FACS the next day, and cultured in myeloid expansion medium for four weeks, prior to HTS. Unlike NHEJ levels, which were variable, but generally similar among sub-populations, gene editing via HDR pathway was significantly lower in the HSCs and MPPs, compared to the progenitors (Figure 1F). Of note, the levels of HDR and NHEJ in the progenitor sub-population were repeatedly measured during the four-weeks to ensure that prolonged culture time does not affect gene editing levels (data not shown). Taken together, these data demonstrate an inherent difference between HSCs and progenitors in their ability to utilize HDR pathway for DSB repair.
HDR/NHEJ Ratio is Highest in S/G2 Phases of Cell Cycle
HDR is described to be restricted to S/G2 phases of cell cycle in cell lines [1], however it has not been previously shown whether the same temporal regulation exists in primary human CD34+ cells. To assess this, PBSCs were FACS-sorted into G1 and S/G2/M fractions (Figure 3A), and electroporated with gene editing reagents immediately after sort. It should be noted that the cells were not cell cycle-arrested, and continued normal cell cycle progression after FACS, therefore the populations were merely enriched for the indicated cell cycle phases at the time of electroporation. As expected based on cell line data, the levels of HDR were significantly higher in S/G2/M-sorted cells, compared to G1-sorted and bulk unsorted PBSCs (Figure 3B). Additionally, NHEJ was slightly lower in S/G2/M, resulting in a ~5-fold higher HDR/NHEJ ratio (Figure 3B, C).
Figure 3: HDR/NHEJ Ratio is Highest in S/G2 Phases of Cell Cycle.

A. Cell cycle analysis of human PBSCs, immediately after FACS-sorting into G1 and S/G2/M populations. See also Fig S2A for analysis at a later time point.
B. Percentage of gene editing (HDR and NHEJ) in unsorted, or FACS-sorted G1 or S/G2/M PBSCs, electroporated with Cas9 mRNA and sgRNA immediately after sort, and subsequently transduced with AAV6 donor template. N=5. Data are represented as mean ± SD. Differences are not significant if not specified, based on Kruskal-Wallis test. * P<0.05, ** P≤0.01, based on Wilcoxon Rank-Sum test.
C. HDR/NHEJ ratio, calculated from Fig 3B. Data are represented as mean ± SD. Differences are not significant if not specified, based on Kruskal-Wallis test. ** P≤0.01, based on Wilcoxon Rank-Sum test.
hGemCas9 Allows for Lower Nuclease Activity in G1, while Retaining Specificity
Since Cas9 can generate DSBs in any phase of cell cycle, but HDR is confined to S/G2, we sought to decrease nuclease activity in G1 phase to limit unwanted NHEJ. Similar to Gutschner et al., a fragment of the human geminin domain was fused to the C-terminus of Cas9 (termed hGemCas9) (Figure 4A) to allow the protein to be ubiquitinated and degraded by APC/Cdh1 complex in G1 and late M phases [22]. To confirm the reduced nuclease activity of hGemCas9 in G1, human PBSCs were FACS-sorted into G1 or S/G2/M phases and electroporated immediately after sort with either wtCas9 or hGemCas9 IVT mRNA and sgRNA. In G1-sorted cells, nuclease activity of hGemCas9 was significantly lower than of wtCas9 (Figure 4B). There was also a slight decrease in activity during S/G2/M phases, possibly due to the cells progressing through cell cycle prior to analysis.
Figure 4: hGemCas9 Allows for Lower Nuclease Activity in G1, while Retaining Specificity.

A. Schematic of a portion of wtCas9 and hGemCas9 plasmids used for IVT (not to scale). hGem insert at C-terminus of Cas9 consisted of the first 110 amino acids of human geminin.Percentage of nuclease activity (% indels), measured by HTS, in
B. unsorted and FACS-sorted G1 or S/G2/M PBSCs, electroporated immediately after sort with wtCas9 or hGemCas9 mRNA. N=3. Data are represented as mean ± SD. ns – not significant, * P<0.05, based on unpaired t-test. (See also Figure S2B, D)
C. Nuclease specificity of wtCas9 and hGemCas9 in different phases of cell cycle, calculated from data in Fig 4B. (See also Figure S2C,E)
Additionally, to compare the specificity of wtCas9 vs hGemCas9, indels were quantified by HTS at the previously-identified off-target sites for this guide, termed OT1 and OT2 [17], in the absence and presence of donor template (Figure S2B and S2D, respectively). Nuclease specificity, calculated as the ratio of on-target to off-target indels, was similar between wtCas9 and hGemCas9 in bulk (unsorted) cells (Figure 4C, Figure S2C, E). However, hGemCas9 appeared to be more specific than wtCas9 in G1-sorted cells (Figure 4C), demonstrating a previously unreported role of hGemCas9 in preferentially decreasing off-target nuclease activity in G1 phase of cell cycle, potentially due to lower affinity for OT1.
Human HSPCs Can Be Transiently Synchronized in S/G2 Phases of Cell Cycle
HSCs are principally quiescent while occupying their natural niches in the body [23–26]; after cytokine stimulation in vitro, the majority of HSPCs are in G1 phase of cell cycle, with only about 35% present in S/G2 after two days of culture (Figure 5A). To increase the proportion of cells available for HDR, we used a selective inhibitor of CDK1, RO-3306 (“RO”), which transiently arrests cells at the G2-M transition, increasing the number of cells in S and G2 phases [27, 28]. RO concentration (30uM) and treatment time (20 hours) were optimized to result in a 10–12% increase in the number of cells in S/G2 phases, compared to vehicle control (DMSO), and a concomitant decrease in the number of cells in G1 phase (Figure 5A-B). RO treatment resulted in initial toxicity (Figure S3A). However, colony forming unit (CFU) assay and a competitive transplant of RO-treated and control cells indicated that the surviving cells retained their hematopoietic potential (Figure S3B-G).
Figure 5: Human HSPCs Can Be Transiently Synchronized in S/G2 Phases of Cell Cycle.

(See also Figure S3). A. Representative flow cytometry plot of cell cycle analysis of PBSCs treated with 30uM RO or DMSO (volume equivalent) for 20 hours. SubG1 population represents apoptotic cells.
B. Quantification of flow cytometry analysis in Fig 5A. N=6. Data are represented as mean ± SD. Numbers indicate means. Differences are not significant, based on chi-square test.
Reducing Nuclease Activity in G1 and Synchronizing Human HSPCs in S/G2 Improves HDR/NHEJ Ratio in Vitro
After confirming that hGemCas9 decreased nuclease activity in G1 and that RO treatment increased the percentage of cells in S/G2 phases, we went on to test the effects of these manipulations on gene editing levels in human PBSCs, as compared to unsynchronized (unsync) cells with wtCas9 control. As expected, hGemCas9 did not affect HDR, but significantly reduced NHEJ levels from 12.1±1.4% to 6.9±0.7% (Figure 6A). Cell synchronization with RO increased HDR slightly from 18.1±3.5% to 23.5±8.3%, and decreased NHEJ from 12.1±1.4% to 7.5±1.7%. Additionally, there was an additive effect between hGemCas9 and cell synchronization at reducing NHEJ to below 4%, thus improving HDR/NHEJ ratio about 4-fold over the control condition (Figure 6B). The use of hGemCas9 did not have any negative effects on cell viability and fold expansion (Figures 6C-D). Similar to earlier observations, RO treatment resulted in significant initial decrease in viability and fold expansion, measured 24 hours post electroporation.
Figure 6: Reducing Nuclease Activity in G1 and Synchronizing Human HSPCs in S/G2 Improves HDR/NHEJ Ratio in Vitro.

A. Levels of gene editing, measured by HTS, in PBSCs, pre-treated with DMSO or RO for 20 hours, electroporated with wtCas9 or hGemCas9, and subsequently transduced with AAV6 donor template. For wtCas9 – N=10, for hGemCas9 – N=8. Data are represented as mean ± SD. Differences are not significant if not specified, based on Kruskal-Wallis test, *** P≤0.001, based on Wilcoxon Rank-Sum test. See also Fig S2D-E.
B. Ratio of HDR to NHEJ, calculated from data in Fig 6A. For wtCas9 – N=10, for hGemCas9 – N=8. Data are represented as mean ± SD. Differences are not significant if not specified, ** P≤0.01, *** P≤0.001, based on Wilcoxon Rank-Sum test.
C. Percent viability (from cells in Fig 6A) measured by trypan blue 24 hours post electroporation. For wtCas9 – N=9, for hGemCas9 – N=7. Data are represented as mean ± SD. ns – not significant, ** P≤0.01, *** P≤0.001, based on Wilcoxon Rank-Sum test.
D. Fold expansion (from cells in Fig 6A), measured 24 hours post electroporation. For wtCas9 – N=9, for hGemCas9 – N=7. Data are represented as mean ± SD. ns – not significant, ** P≤0.01, *** P≤0.001, based on Wilcoxon Rank-Sum test.
E. Indel spectrum, normalized to %NHEJ (in Fig 6A) for each sample. Only the top indels (making up ~75% of all indels) are plotted. See also Fig S4A. Numbers in parentheses indicate position of an indel relative to cut site, numbers following parentheses indicate the length of an indel; indel – insertion or deletion (individual values for insertions or deletions events of the same length occurring at the same position were summed), del – deletion only. For wtCas9 – N=10, for hGemCas9 – N=8. Data are represented as mean ± SD.
In addition to successfully decreasing nuclease activity, hGemCas9 also did not alter nuclease specificity (Figure S2C,E), or the types or proportion of indels formed in bulk, unsorted PBSCs (Figure 6E, S4A). There is a 5-base pair (bp) microhomology around the cut site, identified by an online prediction algorithm [29], which results in a 9-bp deletion via microhomology-mediated end joining (MMEJ) pathway (Figure S4B). Interestingly, RO pre-treatment resulted in proportionally less indels produced via MMEJ, compared to unsynchronized cells, possibly as a consequence of CDK1 inhibition [30, 31] (Figure 6E, S4A). To overcome the potential decrease of CtIP phosphorylation (which is essential for DNA end resection in homologous recombination [32–35]) by CDK1 inhibition upon RO treatment, we sought to express phosphomimic CtIP (T847E) in the cells. This constitutively active mutation has been shown to be sufficient for DSB end processing [32], which is required for HDR [3]. CtIP T847E (CtIP*) IVT mRNA was co-electroporated with gene editing reagents. However, no differences in HDR or NHEJ levels, indel distribution or viability were observed with the addition of CtIP* mRNA (Figure S5A-E).
Reducing Nuclease Activity in G1 and Synchronizing Human HSPCs in S/G2 Improves HDR/NHEJ Ratio in Vivo
Finally, to determine the effects of reducing nuclease activity in G1 (hGemCas9) and/or synchronizing cells in S/G2 (RO) on gene editing levels in long-term HSC population, bulk PBSCs were treated and electroporated as described earlier, and transplanted into 6–9-week-old sub-lethally irradiated mice 24 hours later. A small fraction of cells was kept in myeloid culture for 2 weeks to determine the levels of gene modification prior to transplantation (Figure 7A-B).
Figure 7: Reducing Nuclease Activity in G1 and Synchronizing Human HSPCs in S/G2 Improves HDR/NHEJ Ratio in Vivo.

A. Levels of gene editing (HDR and NHEJ), measured by HTS in vitro before transplant, in human PBSCs electroporated with wtCas9 mRNA and sgRNA, and subsequently transduced with AAV6. N=1.
B. Ratio of HDR/NHEJ from data in Fig 7A. N=1.
C. Levels of human engraftment in NSG mice at 8 and 16 weeks post-transplant in the indicated organs, measured by flow cytometry, and calculated as (%hCD45+/(%hCD45++mCD45+))x100. PB – peripheral blood, BM – bone marrow. See also Fig S6A-B. N=6–7 (3–4 female mice + 3–4 male mice). Individual points are plotted. Line – geometric (geo) mean ± geo SD. ND – zero value is not plotted on a log scale, but was used in calculating mean and SD. Differences between experimental arms are not significant, based on Kruskal-Wallis test.
D. Levels of gene editing (HDR and NHEJ) in the engrafted human cells in mouse BM at 16 weeks post-transplant. N=6–7 (3–4 female mice + 3–4 male mice). Individual points are plotted. Line – geo mean ± geo SD. Data point from one of the mice in “Unsync, hGemCas9” cohort was excluded due to very low human engraftment (0.02%) resulting in unreliable gene editing values. Differences are not significant if not specified, * P<0.05, based on Wilcoxon Rank-Sum test.
E. Ratio of HDR/NHEJ from data in Fig 7D. N=6–7 (3–4 female mice + 3–4 male mice). Data are represented as box-and-whisker plot, horizontal line indicates mean, boundaries of the box – the upper and lower quartiles, whiskers – the lowest and highest observations. Differences are not significant if not specified, * P<0.05, based on Wilcoxon Rank-Sum test.
At 8 weeks post-transplant, the percentage of human engraftment in peripheral blood (PB) was ~3% (geometric mean) in the “RO, hGemCas9” group, and ~1% (geometric mean) in the other experimental arms (Figure 7C, S6A). The mice were euthanized at 16 weeks post-transplant, and the levels of human engraftment were quantified in the mouse PB, bone marrow (BM) and spleen (Figure 7C). The human engraftment varied from 0.1% to 20%, typically with lower values in the male mice (Figure S6B), as previously observed by other groups [36]. No statistically significant differences in human engraftment or lineage distribution of engrafted cells were observed among treatment groups (Figure 7C, S6A, C-D).
Gene editing levels in the human engrafted cells in the mouse BM were generally lower than before transplant (Figure 7D). The HDR levels in the control arm (Unsync, wtCas9) decreased from over 20% in vitro to 0.03–0.4% after transplant. Similar to the in vitro data, RO pre-treatment allowed for slightly higher HDR levels, reaching 2.5%. Comparable to previously-published data [9, 17], the levels of NHEJ remained high after transplant, with a geometric mean of ~5% in the “Unsync, wtCas9” group. Analogous to the in vitro data, “Unsync, hGemCas9” and “RO, hGemCas9” groups had lower NHEJ levels with a geometric mean of 0.5% and 0.7%, respectively (Figure 7D). Furthermore, there was an additive effect between Cas9 manipulation (hGemCas9) and cell cycle synchronization (RO), thus improving the HDR/NHEJ ratio on an average of 20-fold over the control condition (Figure 7E). Interestingly, the HDR levels in RO-treated cells were slightly higher than in control, despite the lower HDR pre-transplant. Because of an unexpected decrease in HDR levels in RO-treated samples before transplant, the levels of gene editing after transplant were normalized to the input values to calculate the percentage of gene editing maintained after transplant (Figure S7). Our data suggest that RO-treated cells have an increased ability to maintain HDR levels after transplant, and hGemCas9 is able to preserve lower NHEJ frequencies.
DISCUSSION
In this study, we took advantage of the temporal regulation of DNA repair pathways and utilized them to improve the HDR/NHEJ ratio from gene editing in primary human HSPCs. Several methods to increase HDR and decrease NHEJ have been proposed previously. One attempt is through inhibition of the NHEJ pathway by inhibiting DNA-PK with NU7441 or KU-0060648 [37], or by blocking Ligase IV with SCR7 [38–40]. However, we and other groups have not been able to achieve an improvement in HDR with SCR7 in our experimental systems [22, 37, 41, 42]. Moreover, DNA-PK and Ligase IV act relatively late in the NHEJ, after pathway commitment. Another potential method to improve editing outcomes is to initiate HDR in the G1 phase of cell cycle by manipulating DNA repair factors to control repair pathway choice at the decision-making stage [43–45]. Additional alternative strategies including controlling the timing of delivery and activity of nuclease reagents based on cell cycle progression, synchronizing cells in the desirable cell cycle phase, and modifying Cas9 expression based on cell cycle progression, have led to an improvement in HDR in cell lines or iPSCs [22, 42, 46]. While all these manipulations were shown to be successful at increasing HDR in cell lines, their effects in primary human HSPCs have not yet been reported.
Since the goal is to provide a life-long treatment for monogenic diseases of the blood, the mutation must be corrected in the primitive stem cell sub-population of HSPCs, termed HSCs. The rare HSCs are outnumbered by progenitors, so the readout of the bulk HSPCs in vitro is only representative of the progenitor sub-populations, masking any differences in the HSCs. To determine gene editing levels in long-term HSCs, gene-modified HSPCs must be transplanted into NSG mice and assessed at 14–16 weeks post-transplant. While sufficiently high levels of editing are currently achievable in vitro [9, 17, 18], we and others have observed that transplantation of gene-edited cells into NSG mice results in a decrease in gene correction values [9, 17], unless a selectable marker was included to enrich the edited population prior to transplant [18]. These observations suggest that the gene editing occurs most efficiently in the progenitor population, and less in the rare HSCs.
To determine why the HSCs are less amenable to gene editing than the progenitors, we studied the efficiency of delivery and expression of gene editing reagents in the immunophenotypically-defined HSPC sub-populations (HSCs, MPPs, progenitors). Our results suggested that the delivery and translation of nuclease mRNA are not the likely limiting factors. Also, the donor template cellular availability is unlikely restrictive, based on delivery of a GFP reporter virus of the same serotype (AAV6) to HSPCs. Additionally, the percentage of indels were similar among the sub-populations following delivery of CRISPR/Cas9 alone, indicating that the nuclease can cut DNA in HSCs as well as the progenitors. However, when combining nuclease and a donor template, the HDR levels were much lower in the HSCs and MPPs, compared to progenitors. These results indicated that the factor limiting HDR in HSCs is likely an intrinsic property of DNA repair pathway choice.
One of the characteristics that makes HSCs different from progenitors is their lack of cell cycle progression. Unlike progenitors, which are actively cycling, HSCs are normally quiescent in vivo [5, 47]. In culture, cytokine-stimulated HSCs enter cell cycle, but at a lower frequency than progenitors [48]. We observed that a two-day pre-stimulation with cytokines is required to increase the number of cells in S/G2 phases and to achieve detectable HDR levels (unpublished data). Interestingly, NHEJ levels were similar in HSC and progenitor sub-populations, and across different phases of cell cycle, supporting the observation that percentages of NHEJ events do not decrease in the long-term HSCs after transplant [9, 49]. This observation can be explained by the fact that NHEJ can occur in any phase of cell cycle and is therefore not restricted in quiescent HSC sub-population.
The DNA repair pathways are temporally regulated, with NHEJ being active throughout the cell cycle, and HDR being confined to S/G2 phases. To take advantage of this temporal regulation, we took two approaches.
First, to decrease nuclease activity in G1 phase of cell cycle when HDR cannot occur, we produced a modified version of Streptococcus pyogenes (Sp) Cas9, termed hGemCas9. As expected, there were no differences in the levels of HDR, but we observed a significant decrease in NHEJ, thus resulting in a significant improvement in HDR/NHEJ ratio, both in vitro and in vivo. Additionally, the specificity of hGemCas9 was similar to that of wtCas9 in bulk unsorted cells. Interestingly, in G1 phase of cell cycle, hGemCas9 was more specific than wtCas9, suggesting that decreasing nuclease activity might result in more specificity. However, it will be necessary to compare the specificity of hGemCas9 vs wtCas9 at other loci to determine whether this observation is generally true.
In the second approach, we sought to increase the proportion of cells that are available for HDR. We used a selective inhibitor of CDK1, RO-3306, to temporarily synchronize cells in the S/G2 phases of cell cycle. Other groups have shown that synchronization of cells in the S/G2 phases increases HDR [46]; however, this has not been previously tested in primary human HSPCs. We observed a >2-fold increase in the HDR/NHEJ ratio in the cells pre-treated with RO. Moreover, we observed an additive effect between hGemCas9 and cell synchronization (RO), resulting in ~4-fold improvement in the HDR/NHEJ ratio in vitro, and a significant improvement in vivo.
Although a CFU assay and a competitive mouse transplant experiment suggested that cell synchronization with RO does not affect the long-term viability and hematopoietic potential of HSCs, this drug resulted in significant initial toxicity and was only effective at increasing the S/G2 population by ~15%. Additionally, inhibition of CDK1 may be disadvantageous for improving HDR/NHEJ ratio due to its roles in DNA repair, such as phosphorylation of key proteins involved in HDR, including CtIP and MRN complex [32–35]. Based on our data, replenishing phosphomimic CtIP by co-electroporation of CtIP* mRNA was not sufficient to significantly increase HDR levels. Therefore, to expand on encouraging results of improved HDR/NHEJ ratio in S/G2-synchronized cells, and in order for this strategy to be translatable to patients, it will be necessary to search for and test other synchronization reagents that do not counteract critical HDR factors and do not result in as much initial toxicity.
While the absolute levels of gene editing in NSG mice were still sub-optimal, the improvement in the HDR/NHEJ ratio relative to the control (unsync, wtCas9) provides an important proof-of-principle finding. It is essential to note that the amounts of gene editing reagents used here were chosen to provide non-plateau gene editing levels for comparative purposes, and the HDR levels may be further increased by optimizing the amounts, formulation, and delivery methods of Cas9, gRNA and donor template [50]. Several combinations for delivery of gene editing reagents are currently available, with the most popular being IVT mRNA Cas9 and guide RNA, or ribonucleoprotein (RNP) complex; and AAV6 or oligonucleotide for donor template. There is no clear ideal choice – each has its own advantages and drawbacks for the future use in patients. While in these studies we used mRNA+sgRNA+AAV6 delivery system for Cas9, guide and donor template, respectively, it may be of interest to test the effectiveness of the approaches described here utilizing other, potentially more effective, gene editing reagent delivery systems to achieve an increase in absolute HDR levels, or an even further improvement in the HDR/NHEJ ratio.
In summary, this work provides a proof-of-principle finding that HDR/NHEJ ratio can be improved in primary human HSPCs by controlling the Cas9 activity in cell cycle phase-specific manner and regulating cell cycle progression. In the future, the approaches described here can be tested with other gene editing reagent delivery systems and can be scaled up for clinical gene therapy applications.
CONCLUSION
The goal of this paper was to study the mechanistic reason for a decrease in targeted gene correction (HDR levels) in HSCs revealed after transplantation. We sought to influence the choice of DNA repair pathways to increase HDR and decrease gene disruption via NHEJ pathway, thus improving HDR/NHEJ ratio. Specifically, we focused on cell cycle phase-specific regulation of DNA repair pathways. We showed that temporal regulation of Cas9 nuclease activity and transient synchronization of primary human CD34+ cells in HDR-preferred phases resulted in a significant improvement in HDR/NHEJ ratio in vitro. Moreover, these manipulations resulted in a significant increase in the HDR/NHEJ ratio after transplant of gene-edited cells into immune-deficient mice.
Supplementary Material
SIGNIFICANCE STATEMENT.
This study reports an improvement in CRISPR/Cas9-mediated gene editing outcomes in human hematopoietic stem cells. Here, we show that cell cycle-dependent control of nuclease activity and DNA repair pathways can influence gene editing outcomes to favor the precise DNA modification over faulty repair events in human hematopoietic stem cells.
This work provides important proof-of-principle findings, and can improve gene editing outcomes for the treatment of congenital diseases of the blood system.
ACKNOWLEDGEMENTS
The authors thank Felicia Codrea, Jessica Scholes and Jeffrey Calimlim of the UCLA Eli & Edythe Broad Center for Regenerative Medicine & Stem Cell Research Flow Cytometry Core for flow cytometry and FACS assistance, and the UCLA Technology Center for Genomics & Bioinformatics Core for DNA sequencing. We also thank Alejanda Davila, Amalia Icreverzi and Allen Yu for processing mobilized peripheral blood units. S.F.G. acknowledges support from a QCB Collaboratory Postdoctoral Fellowship, and the QCB Collaboratory community directed by Matteo Pellegrini.
This work was supported by the grants from Doris Duke Charitable Foundation (Sickle Cell Disease/Advancing Cures Award, to D.B.K., for in vitro studies only), the California Institute for Regenerative Medicine (CIRM) TR4–06823 and TR4–09292 (to D.B.K.), Dr. Ursula Mandel Scholarship (to A.L.), and the National Heart, Lung, And Blood Institute (NHLBI) of the National Institutes of Health (NIH) under Award Number F31HL134208 (to A.L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding:
Doris Duke Charitable Foundation (Sickle Cell Disease/Advancing Cures Award, to D.B.K. for in vitro studies only), the California Institute for Regenerative Medicine (CIRM) TR4–06823 and TR4–09292 (to D.B.K.), Dr. Ursula Mandel Scholarship (to A.L.), and the National Heart, Lung, And Blood Institute (NHLBI) of the National Institutes of Health (NIH) under Award Number F31HL134208 (to A.L.).
Footnotes
DECLARATION OF INTERESTS
J.E.C declared leadership position with Spotlight Therapeutics, Consultant/Advisory role with Inari Agriculture SAB, honoraria from Bioverativ honoraria, Sangamo honoraria and research funding from AstraZeneca. R.P.H. declare consultant and intellectual property rights holder. E.Y.M. is an employee of UCLA. All other authors declared no competing financial interests.
REFERENCES
- 1.Lieber MR, Ma Y, Pannicke U, et al. Mechanism and regulation of human non-homologous DNA end-joining. NAT. REV. MOL. CELL BIOL 2003;4(9):712–720. [DOI] [PubMed] [Google Scholar]
- 2.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. NAT. REV. MOL. CELL BIOL 2008;9(4):297–308. [DOI] [PubMed] [Google Scholar]
- 3.Heyer W-D, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. ANNU. REV. GENET 2010;44:113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. COLD SPRING HARB PERSPECT BIOL 2013;5(5):a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. J. CELL BIOL 2011;195(5):709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang K-H, Smith SE, Sullivan T, et al. Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34+ Hematopoietic Stem and Progenitor Cells. MOL THER METHODS CLIN DEV 2017;4(C):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiGiusto DL, Cannon PM, Holmes MC, et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. MOL THER METHODS CLIN DEV 2016;3:16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genovese P, Schiroli G, Escobar G, et al. Targeted genome editing in human repopulating haematopoietic stem cells. NATURE 2014;510(7504):235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoban MD, Cost GJ, Mendel MC, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. BLOOD 2015;125(17):2597–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Exline CM, DeClercq JJ, et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. NATURE BIOTECHNOLOGY 2015;33(12):1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ribeil J-A, Hacein-Bey-Abina S, Payen E, et al. Gene Therapy in a Patient with Sickle Cell Disease. N ENGL J MED 2017;376(9):848–855. [DOI] [PubMed] [Google Scholar]
- 12.Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N ENGL J MED 2017;376(16):1561–1573. [DOI] [PubMed] [Google Scholar]
- 13.Andreani M, Nesci S, Lucarelli G, et al. Long-term survival of ex-thalassemic patients with persistent mixed chimerism after bone marrow transplantation. BONE MARROW TRANSPLANT 2000;25(4):401–404. [DOI] [PubMed] [Google Scholar]
- 14.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation for Severe Sickle Cell Phenotype. JAMA 2014;312(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fitzhugh CD, Cordes S, Taylor T, et al. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. BLOOD 2017;130(17):1946–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoban MD, Lumaquin D, Kuo CY, et al. CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells. MOL. THER 2016;24(9):1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeWitt MA, Magis W, Bray NL, et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. SCI TRANSL MED 2016;8(360):360ra134–360ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. NATURE 2016;539(7629):384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. NATURE BIOTECHNOLOGY 2018;36(8):765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bjurstrom CF, Mojadidi M, Phillips J, et al. Reactivating Fetal Hemoglobin Expression in Human Adult Erythroblasts Through BCL11A Knockdown Using Targeted Endonucleases. MOL THER NUCLEIC ACIDS 2016;5:e351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. CELL STEM CELL 2007;1(6):635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutschner T, Haemmerle M, Genovese G, et al. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. CELLREPORTS 2016;14(6):1555–1566. [DOI] [PubMed] [Google Scholar]
- 23.Cheshier SH, Morrison SJ, Liao X, et al. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. PROC NATL ACAD SCI USA 1999;96(6):3120–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. CELL 2008;135(6):1118–1129. [DOI] [PubMed] [Google Scholar]
- 25.Walter D, Lier A, Geiselhart A, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. NATURE 2015;520(7548):549–552. [DOI] [PubMed] [Google Scholar]
- 26.Takizawa H, Manz MG. In vivo divisional tracking of hematopoietic stem cells. ANN. N. Y. ACAD. SCI 2012;1266:40–46. [DOI] [PubMed] [Google Scholar]
- 27.Vassilev LT, Tovar C, Chen S, et al. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. PROC NATL ACAD SCI USA 2006;103(28):10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vassilev LT. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. CELL CYCLE 2006;5(22):2555–2556. [DOI] [PubMed] [Google Scholar]
- 29.Bae S, Kweon J, Kim HS, et al. Microhomology-based choice of Cas9 nuclease target sites. NAT. METHODS 2014;11(7):705–706. [DOI] [PubMed] [Google Scholar]
- 30.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. TRENDS GENET. 2008;24(11):529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava M, Raghavan SC. DNA Double-Strand Break Repair Inhibitors as Cancer Therapeutics. CHEM. BIOL 2015;22(1):17–29. [DOI] [PubMed] [Google Scholar]
- 32.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. BIOL. CHEM 2009;284(14):9558–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson SE, Li Y, Chait BT, et al. Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J. CELL BIOL 2011;194(5):705–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson SE, Li Y, Wu-Baer F, et al. Activation of DSB processing requires phosphorylation of CtIP by ATR. MOLECULAR CELL 2013;49(4):657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hustedt N, Durocher D. The control of DNA repair by the cell cycle. NAT. CELL BIOL 2016;19(1):1–9. [DOI] [PubMed] [Google Scholar]
- 36.Notta F, Doulatov S, Dick JE. Engraftment of human hematopoietic stem cells is more efficient in female NOD/SCID/IL-2Rgc-null recipients. BLOOD 2010;115(18):3704–3707. [DOI] [PubMed] [Google Scholar]
- 37.Robert F, Barbeau M, Éthier S, et al. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. GENOME MED 2015;7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srivastava M, Nambiar M, Sharma S, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. CELL 2012;151(7):1474–1487. [DOI] [PubMed] [Google Scholar]
- 39.Chu VT, Weber T, Wefers B, et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. NATURE BIOTECHNOLOGY 2015;33(5):543–548. [DOI] [PubMed] [Google Scholar]
- 40.Maruyama T, Dougan SK, Truttmann MC, et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. NATURE BIOTECHNOLOGY 2015;33(5):538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pinder J, Salsman J, Dellaire G. Nuclear domain “knock-in” screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. NUCLEIC ACIDS RESEARCH 2015;43(19):9379–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang D, Scavuzzo MA, Chmielowiec J, et al. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. SCI REP 2016;6:21264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. MOLECULAR CELL 2013;49(5):872–883. [DOI] [PubMed] [Google Scholar]
- 44.Orthwein A, Noordermeer SM, Wilson MD, et al. A mechanism for the suppression of homologous recombination in G1 cells. NATURE 2015;528(7582):422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Canny MD, Moatti N, Wan LCK, et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. NATURE BIOTECHNOLOGY 2018;36(1):95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin S, Staahl BT, Alla RK, et al. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. ELIFE 2014;3:e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uchida N, He D, Friera AM, et al. The unexpected G0/G1 cell cycle status of mobilized hematopoietic stem cells from peripheral blood. BLOOD 1997;89(2):465–472. [PubMed] [Google Scholar]
- 48.Rossi DJ, Seita J, Czechowicz A, et al. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. CELL CYCLE 2007;6(19):2371–2376. [DOI] [PubMed] [Google Scholar]
- 49.Holt N, Wang J, Kim K, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. NATURE BIOTECHNOLOGY 2010;28(8):839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richardson CD, Ray GJ, DeWitt MA, et al. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. NATURE BIOTECHNOLOGY 2016;34(3):339–344. [DOI] [PubMed] [Google Scholar]
- 51.Cooper AR, Patel S, Senadheera S, et al. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. JOURNAL OF VIROLOGICAL METHODS 2011;177(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.