

Abstract
Clinical trials repurposing lysosomotropic chloroquine (CQ) derivatives as autophagy inhibitors in cancer demonstrate encouraging results, but the underlying mechanism of action remains unknown. Here we report a novel dimeric CQ (DC661) capable of deacidifying the lysosome and inhibiting autophagy significantly better than HCQ. Using an in situ photoaffinity pulldown strategy, we identified palmitoyl-protein thioesterase 1 (PPT1) as a molecular target shared across monomeric and dimeric CQ derivatives. HCQ and Lys05 also bound to and inhibited PPT1 activity, but only DC661 maintained activity in acidic media. Knockout of PPT1 in cancer cells using CRISPR-Cas9 editing abrogates autophagy modulation and cytotoxicity of CQ derivatives, and results in significant impairment of tumor growth similar to that observed with DC661. Elevated expression of PPT1 in tumors correlates with poor survival in patients in a variety of cancers. Thus, PPT1 represents a new target in cancer that can be inhibited with CQ derivatives.
Keywords: CQ, depalmitoylation, autophagy, lysosome, cancer
Introduction
Lysosomal activity is elevated in advanced cancers and preserves anabolic programs by supporting mTORC1-dependent protein translation and catabolic programs such as autophagy (1, 2). Translational efforts to target the lysosome have predominantly focused on CQ derivatives such as hydroxychloroquine (HCQ) (3–8). However, pharmacokinetic-pharmacodynamic studies have demonstrated that HCQ produces only modest lysosomal inhibition in patients treated with the highest FDA-allowed dose. In addition, the mechanism by which HCQ inhibits the lysosome remains poorly understood. We recently demonstrated that palmitoyl-protein thioesterase 1 (PPT1) is the molecular target of the potent lysosomal inhibitor DQ661, a dimeric quinacrine (9). This raised the question of whether or not PPT1 is the molecular target of chloroquine derivatives as well. Here we utilize a dimeric chloroquine compound possessing improved potency relative to monomeric HCQ, and CRISPR cas9 genome editing to identify and validate the molecular target of chloroquine derivatives.
Results
Elongation of the linker length of Lys05 significantly enhances anti-autophagy and anti-cancer efficacy.
We designed a novel dimeric CQ (DC661) based upon a previously reported dimeric CQ (Lys05) (10). Lys05 exhibited in vivo activity in a number of xenograft mouse models including melanoma, colon cancer, and breast cancer (9–11). Our previous work consisting of systematic modification of the linker length and central nitrogen substitution in a series of dimeric quinacrines identified DQ661 as the most potent anti-lysosomal agent. DQ661 has 6 carbon atoms between each pair of nitrogens in the triamine linker that connects the two acridine heterocycles. The methylation of the central nitrogen was critical for lysosomal localization of DQ661 (9). Based on these findings we synthesized DC661 (Fig. 1A). Treatment of melanoma cells with DC661 resulted in a more striking accumulation of the autophagic vesicle marker LC3BII at lower concentrations compared to either Lys05 or HCQ, reflecting more pronounced accumulation of autophagic vesicles at concentrations between 0.1 and 10 μM (12). All cells died at concentrations above 10 μM for DC661 in contrast to Lys05 and HCQ (Fig. 1B). Compared to HCQ or Lys05, DC661 treatment induced a significantly more potent inhibition of autophagic flux in melanoma cells expressing the mCherry-eGFP-LC3B reporter (Fig. 1C), and significantly higher levels of free GFP in melanoma cells expressing GFP-LC3B (Supplementary Fig. S1A). These results demonstrate that DC661 inhibits autophagic flux more potently than either Lys05 or HCQ. DC661 treatment resulted in significantly greater lysosomal deacidification compared to either HCQ or Lys05 (Fig. 1D). A significantly higher percentage of cancer cells treated with DC661 demonstrated evidence of lysosomal membrane permeabilization relative to Lys05 or HCQ (Fig 1E). The IC50 of DC661 in 72 hr MTT assays was 100-fold lower than that of HCQ across multiple cancer cell lines including colon and pancreas cancer cell lines (Supplementary Fig. S1B). DC661 suppressed long-term clonogenic growth of melanoma cells more effectively (Fig. 1F), and induced significantly more apoptosis than Lys05, HCQ, or combined BRAF- and MEK-inhibition in BRAF-mutant melanoma cells (Supplementary Fig. S1C). To characterize the in vivo antitumor activity of DC661, HT29 colorectal xenografts were established in the flanks of NOD.Cg-Prkdcscid Il2rgtm1Wjl/Szl (NSG) mice. After one to two weeks, tumors were palpable and mice were treated intraperitoneally (i.p.) with two daily doses of vehicle control (water), Lys05 (10 mg/kg) or DC661 (10 mg/kg) (Supplementary Fig. S1D). Unlike continued dosing of Lys05 as was previously demonstrated (10), two doses of Lys05 had only a transient effect on tumor growth compared with vehicle control. In contrast, two doses of DC661 treatment resulted in a significant and sustained growth impairment of tumors. However, all mice treated with DC661 10 mg/kg dose were euthanized due to lethargy. Therefore, another HT29 xenograft experiment was performed in which mice were treated with vehicle control (water) or DC661 3 mg/kg i.p. daily. Treatment with the reduced dose of DC661 resulted in a significant reduction in tumor volume and almost complete suppression of daily tumor growth rate (Fig. 1G-H) compared with control mice without significantly affecting mouse weight (Supplementary Fig. S1E). Evidence of in vivo autophagy inhibition and apoptosis induction was observed through immunoblotting of lysate from treated tumors (Supplementary Fig. S1F). Altogether, elongation of the linker length of Lys05, which contains two carbon linkers between each of the two nitrogens of the linker, to DC661, which contains six carbon atoms between the nitrogen atoms linkers, significantly increased the anti-autophagy and anti-cancer properties of DC661.
Figure 1:
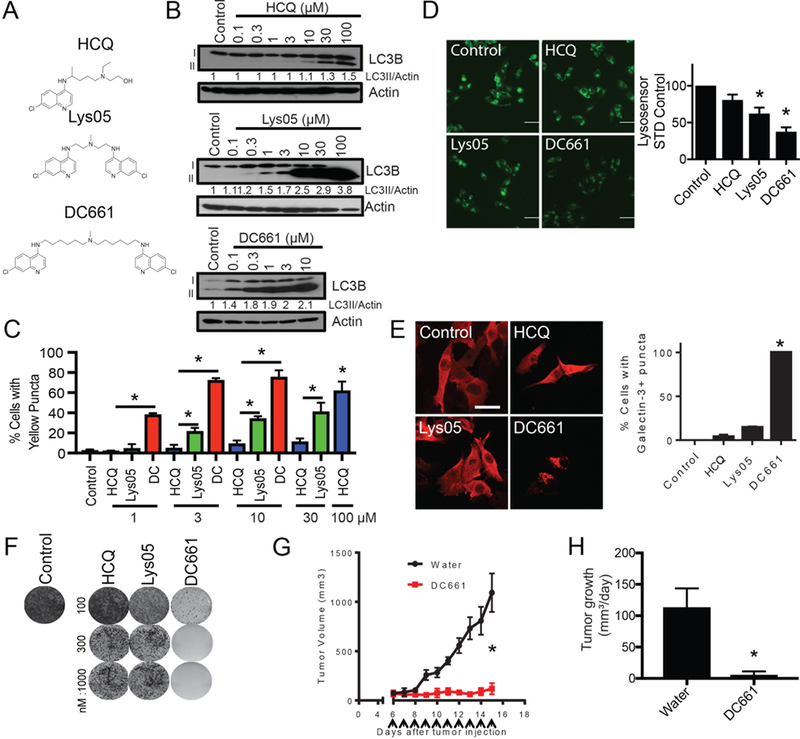
DC661 possesses exquisite anti-lysosome activity. (A) Chemical structures of HCQ, Lys05 and DC661. (B) A375P cells were treated with doses shown of HCQ, Lys05, or DC661 for 6 hours before lysates were immunoblotted. (C) A375P cells expressing the mCherry-eGFP-LC3B reporter were treated with HCQ, Lys05, or DC661 (0 – 100 μM, 1 hr) and analyzed by microscopy. (D) A375P cells were treated with HCQ, Lys05, or DC661 (3 μM, 6 hr) and stained with Lysosensor and imaged with fluorescent microscopy. Shown to the right is quantitation. (E) A375P cells were treated with HCQ, Lys05, or DC661 (3 μM, 6 hr) and subsequently stained with galectin-3 and imaged via fluorescent microscopy. Shown to the right is quantitation of Galectin-3+ puncta expressing cells. (F) A375P cells were treated chronically with HCQ, Lys05 or DC661 (0 – 1000 nM, 2 weeks) in a colony formation assay. Cells were subsequently stained with crystal violet and imaged. (G) HT-29 (colorectal cancer) cells were injected into the flanks of NSG mice. Once palpable, mice were treated daily with 3 mg/kg of DC661 or vehicle (water). Tumor volumes are shown. (H) Mean +/− SEM tumor growth rate. All data is representative from at least 2 experiments. * indicates p<0.05. Students T test was used.
PPT1 identified as the common target of HCQ, Lys05 and DC661.
A previous study demonstrated that Lys01, the free base of Lys05 used for in vitro studies, penetrates into cells and elicits cytotoxicity more effectively than HCQ in cells that are plated in acidic medium, mimicking the acidic tumor microenvironment (13). To determine whether the increased activity of DC661 in vitro and in vivo compared to Lys05 or HCQ could be attributed to improved cell penetration by DC661 in the acidic tumor microenvironment, cancer cells were cultured under acidic conditions. Under these conditions, DC661 was the only compound capable of inhibiting autophagy (Fig. 2A) and decreasing cell viability (Fig. 2B) (13). We next functionalized DC661 to prepare a pulldown probe in an effort to identify the molecular target. A photoprobe, consisting of a DC661 warhead attached to benzophenone (as a photoaffinity label), linked to desthiobiotin (as an affinity handle) was synthesized (Fig. 2C; see Methods for chemical strategy). Treatment of A375P cells with the DC661-photoprobe produced accumulation of LC3II, demonstrating that the photoprobe was cell permeable, localized to the lysosome, and retained activity (Supplementary Fig. S2A). A375P cells were treated with this photoprobe in the absence and presence of free DC661 compound (competition), followed by UV irradiation to conjugate the probe to the bound proteins. Treatment of the resulting mixture with neutravidin resin facilitated collection of proteins for analysis by mass spectrometry (Supplementary Fig. S2B see methods). Candidate protein targets were identified using the criteria of proteins that had >10-fold intensity ratios of +UV/-UV and –competition/+competition samples (Fig. 2D). Cathepsin D exhibited the highest signal intensity, but binding did not change with competition, indicating that the observed Cathepsin D-photoprobe binding was not specific to the DC661-warhead. This had also previously been observed with the analogous DQ661 photoprobe(9). In DC661-photoprobe experiments, palmitoyl-protein thioesterase 1 (PPT1) was pulled down and partially competed off the photoprobe by free drug. Immunoblotting against PPT1 further demonstrated that the DC661 photoprobe specifically bound to and could be UV conjugated to PPT1 in cells (Fig. 2E). Similar experiments utilizing either a Lys05-based or CQ-based photoprobe revealed that PPT1 was a shared protein target across each of the monomeric and dimeric CQ derivatives (Fig. 2F-2I, Supplementary Fig. S2C-D). Differential scanning calorimetry (DSC) of recombinant PPT1 in the absence or presence of a 4-fold molar excess of HCQ, Lys05, or DC661 revealed a statistically significant decrease in PPT1 melting temperature for HCQ, Lys05, and DC661, respectively. These results are consistent with direct binding of each of the CQ derivatives tested to PPT1 (Fig. 2J). Additionally, inhibition of PPT1 enzymatic activity in cells was observed with HCQ, Lys05, and DC661, respectively, with increasing potency (Fig. 2K). Finally, treatment of melanoma cells with either HCQ or DC661 resulted in the accumulation of the palmitoylated form of CD44, providing further evidence that CQ derivatives inhibit PPT1-dependent depalmitoylation (Fig. 2L).
Figure 2:
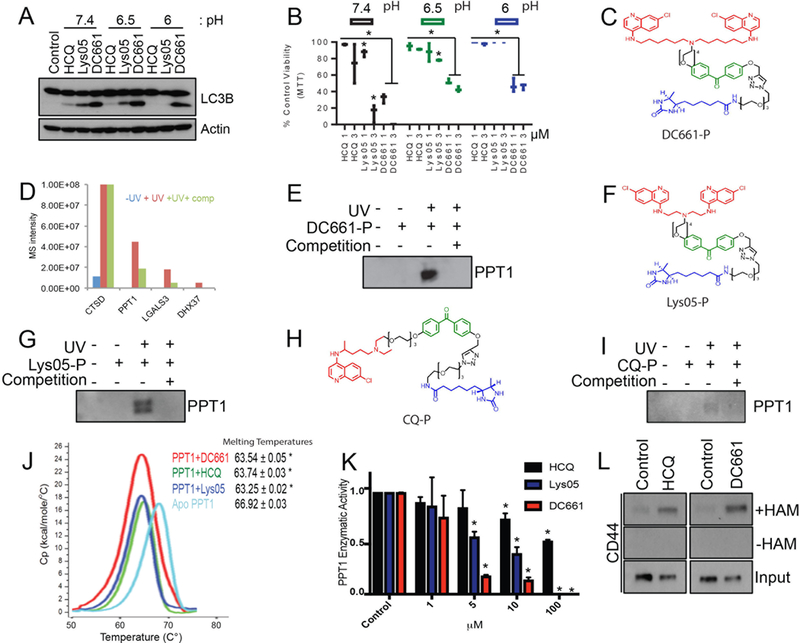
PPT1 is the unified target of CQ-derivatives. (A) A375P cells were treated with HCQ, Lys05 or DC661 (3 μM, 6 hrs) in media with a pH of 7.4, 6.5, or 6, and lysate was subsequently immunoblotted. (B) A375P cells were treated in an MTT assay with HCQ, Lys05 or DC661 (0 – 3 μM, 72 hrs) in media with a pH of 7.4, 6.5, or 6. (C) Chemical structure of the DC661-photoprobe (DC661-P). Red signifies the chemical warhead, green signifies the photoaffinity label, and blue signifies the desthiobiotin probe. (D) Proteomic results of eluents generated from A375P cells treated with either DC661-P (400 nM) minus UV, DC661-P plus UV, or DC661-P plus UV and plus DC661 competition (4 μM, 1 hr). (E) Lysate from A375P cells treated as described from figure 1D was immunoblotted for PPT1. (F) Chemical structure of the Lys05-photoprobe (Lys05-P). (G) Lysate generated from A375P cells treated with either Lys05-P (400 nM) minus UV, Lys05-P plus UV, or Lys05-P plus UV and plus Lys05 competition (40 μM, 1 hr) was immunoblotted for PPT1. (H) Chemical structure of the CQ-photoprobe (CQ-P). (I) Lysate generated from A375P cells treated with CQ-P (400 nM) minus UV, CQ-P plus UV, or CQ-P plus UV and plus HCQ competition (40 μM, 1 hr) was immunoblotted for PPT1. (J) Differential scanning calorimetry was performed using HCQ+PPT1, Lys05+PPT1, and DC661+PPT1 against apo-PPT1 ligand. (K) PPT1 enzymatic assays were performed in A375P cells treated with HCQ, Lys05, or DC661 (0 – 100 μM, 1 hr). (L) Acyl biotin exchange assays were performed in A375P cells treated with HCQ or DC661 (3 μM, 1 hr). All data is representative of at least 2 experiments. Standard deviation is shown in (B), (J) and (K). * indicates p<0.05. Students T test was used.
PPT1 is critical for lysosomal function.
To determine whether inhibition of PPT1 function was responsible for the observed anti-autophagy effects of both the monomeric and dimeric CQ-derivatives, A375P cells were treated with HCQ, Lys05, or DC661 in the presence of the chemical thioesterase mimetic N-tert-Butylhydroxylamine (NtBuHA) (14). NtBuHA rescued autophagy inhibition associated with HCQ, Lys05 and DC661 as evidenced by a significant decrease in cells with inhibited autophagic flux following treatment (Fig. 3A). Autophagic flux was significantly inhibited in cells treated with the previously reported irreversible PPT1 inhibitor hexadecylsulfonyl fluoride (HDSF) (15), as seen by the rescue of GFP from lysosomal degradation, further demonstrating that the specific inhibition of PPT1 impairs lysosomal degradative capacity (Supplementary Fig. S3A). Next, CRISPR Cas9 genome editing was used to generate A375P Cas9 CRISPR KO PPT1 (KO PPT1) and A375P Cas9 (WT PPT1) cells. A375P KO PPT1 cells demonstrated complete loss of expression of PPT1, decreased phopho-S6 and increased LC3II/LC3I levels compared to WT PPT1 cells, suggesting constitutive defects in mTOR and autophagy (Supplementary Fig. S3B). HDSF treatment of WT PPT1 cells resulted in lysosomal deacidification, but KO PPT1 cells exhibited significantly more pronounced lysosomal deacidification relative to untreated WT PPT1 cells. Treatment with HCQ, Lys05, and DC661 did not produce further deacidification of lysosomes within the KO PPT1 cells (Fig. 3B). Interestingly, LC3II lipidation studies revealed that the ability of HCQ, Lys05 and DC661, respectively, to inhibit the lysosome and cause autophagosomes to accumulate was significantly blunted in KO PPT1 cells compared to WT PPT1 cells (Fig. 3C). This loss of activity of autophagy inhibitors was functional, as KO PPT1 cells were also significantly less sensitive to the anti-proliferative effects of HCQ, Lys05 and DC661, respectively, relative to WT PPT1 cells (Fig. 3D). CRISPR Cas9 genome editing was also used to generate mouse melanoma B16 Cas9 CRISPR KO Ppt1 and B16 Cas9 WT Ppt1 cells (Supplementary Fig. 3C), and demonstrate that the ability of HCQ, Lys05 and DC661 to inhibit the lysosome is blunted with loss of Ppt1 (Fig. 3E).
Figure 3:
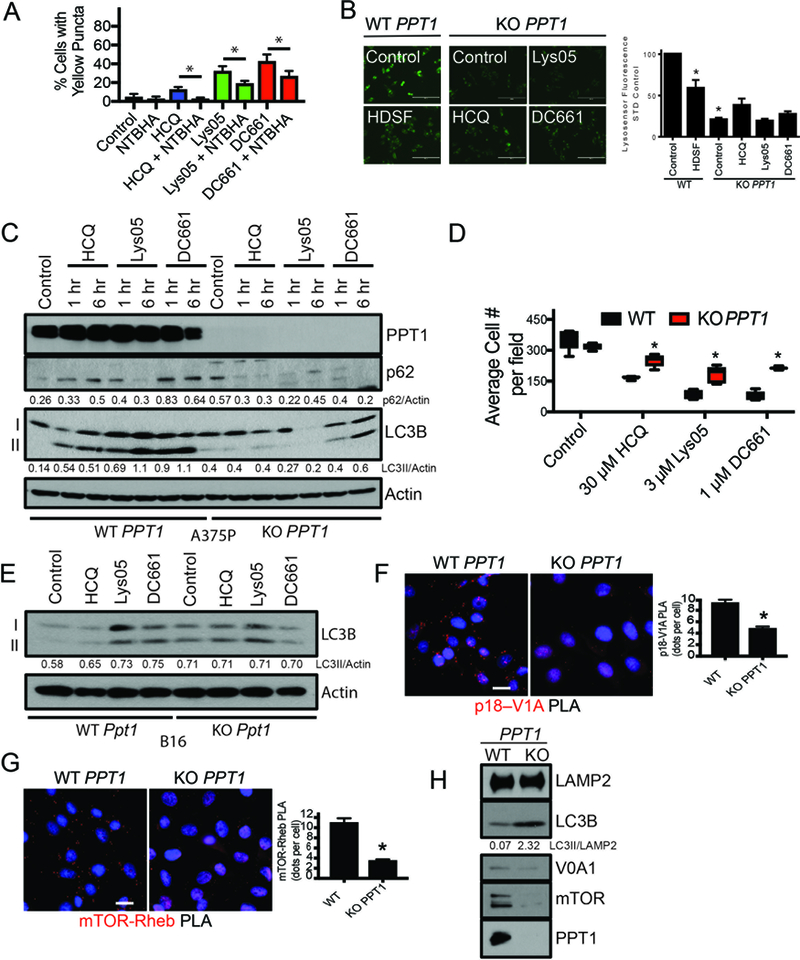
PPT1 inhibition leads to lysosomal inhibition. (A) A375P cells expressing the mCherry-eGFP-LC3B reporter were treated with HCQ (3 μM, 1 hr), Lys05, (3 μM, 1 hr), or DC661 (3 μM, 1 hr) in the presence or absence of N-tert-Butylhydroxylamine (NTBHA, 2 mM), and subsequently analyzed by microscopy. Shown are quantitation. (B) A375P WT PPT1 or KO PPT1 cells were treated with HDSF (60 μM), HCQ (3 μM), Lys05 (3 μM) or DC661 (3 μM) for 6 hours before being stained with Lysosensor. To the right is quantitation of Lysosensor intensity. (C) A375P WT PPT1 or KO PPT1 cells were treated with HCQ, Lys05, or DC661 (3 μM, 0 – 6 hr). Lysate was immunoblotted (D) A375P WT PPT1 or KO PPT1 cells were treated with HCQ (30 μM), Lys05 (3 μM), or DC661 (1 μM, 72 hrs) and cell numbers were quantified as shown. (E) B16 WT Ppt1 or KO Ppt1 cells were treated with HCQ, Lys05, or DC661 (3 μM, 1 hr). Lysate was immunoblotted. (F) Proximity ligation assay for the p18 (Ragulator) – V1A (vATPase subunit) interaction in WT PPT1 and KO PPT1 cells. Shown below is quantitation. (G) Proximity ligation assay for the mTOR – Rheb interaction in WT PPT1 and KO PPT1 cells. Shown below is quantitation. (H) Membrane fractions generated from WT PPT1 and KO PPT1 cells were subsequently immunoblotted. All data is representative of at least 2 experiments. Standard deviation is shown in (D), (F), and (G). * indicates p<0.05. Students T test was used.
To investigate the impact that partial or complete PPT1 inhibition may have on intracellular signaling pathways, reverse-phase protein arrays (RPPA) were performed on A375P cells treated with DC661 or HDSF and compared to signaling changes in untreated or KO PPT1 and WT PPT1 cells. Out of > 300 proteins measured across multiple signaling pathways, only components of the mTORC1 pathway (4E-BP1, S6) emerged as significantly impacted by chemical inhibition or PPT1 KO (Supplementary Fig. S3D-G) (9). Proximity ligation assays (PLAs) in WT PPT1 and KO PPT1 cells established a significant loss of physical interaction between the LAMTOR1/p18 subunit of Ragulator and the ATP6V1A subunit of the vacuolar-type H+-ATPase (v-ATPase), which is critical for and results in the loss of mTOR-Rheb physical interactions (Fig. 3F-G) (16, 17). PPT1 inhibition resulted in the displacement of mTORC1 from the lysosome membrane, inhibiting mTORC1 activity (Fig. 3H, Supplemental Fig. S3H).
Suppression of PPT1 in tumors limits growth, and expression of PPT1 in clinical samples is associated with worse prognosis.
To investigate the role that PPT1 serves in vivo, we leveraged organoid-type culture whereby tumor cells are grown in a 3D collagen matrix with fibroblasts. A375P KO PPT1 cells form significantly smaller spheroids than WT PPT1 cells (Supplementary Fig. S4A). Further, loss of Ppt1 severely limits the viability of B16 spheroids to the point that DC661 could not further enhance death in this system (Supplementary Fig. S4B). We next generated xenograft tumors on the flanks of NSG mice with A375P KO PPT1 and WT PPT1 cells. Relative to WT PPT1 tumors, KO PPT1 tumors displayed significantly blunted tumor growth (Fig. 4A-B). Immunoblotting from lysates generated from in vivo WT PPT1 and KO PPT1 tumors demonstrated increased LC3II/LC3I ratios in all tumors reflective of impaired autophagy (Fig. 4C). To determine if PPT1 expression was differentially expressed in human primary tumor tissue compared to normal tissue, 13 cancers types from The Cancer Genome Atlas (TCGA) were identified for which there were > 10 tumor-normal pairs. Compared to normal tissue, PPT1 expression was significantly higher in breast cancer, clear cell renal cell carcinoma, head and neck squamous cell carcinoma (HNSC), thyroid cancer, colon adenocarcinoma, and esophageal adenocarcinoma (Supplementary Fig.S4C). In a more comprehensive analysis of independent patient groups, PPT1 expression from tumor tissue for patients without matched normal tissue was compared to PPT1 expression from normal tissue for all patients with normal tissue. PPT1 expression was significantly elevated in tumor versus normal in the six cancer types above as well as in gastric adenocarcinoma, hepatocellular carcinoma, and papillary renal cell carcinoma (Fig. 4D). In both analyses, PPT1 expression was found to be significantly lower in tumor versus normal tissue in non-small cell lung cancer. Elevated expression of PPT1 in cancer was further demonstrated by analysis of nearly 1000 cancer cell lines in the Broad Novartis Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle ) where PPT1 expression approached the upper limits of RNA expression across multiple tumor types. In the TCGA database, PPT1 gene amplification is present in 4–10% of ovarian, esophageal, bladder, uterine and stomach cancers (Supplementary Fig. 4D-E). In melanoma, the only TCGA cancer type with both metastases and primary samples available, PPT1 expression was significantly higher in metastases compared to primary melanoma tumors. (Fig. 4E). Survival analyses for TCGA cancer patients demonstrated that tumor expression of PPT1 was associated with shorter overall survival in a variety of cancers including esophageal cancer, head and neck cancer, renal cell carcinoma, and hepatocellular cancer (Fig. 4F-I).
Figure 4:
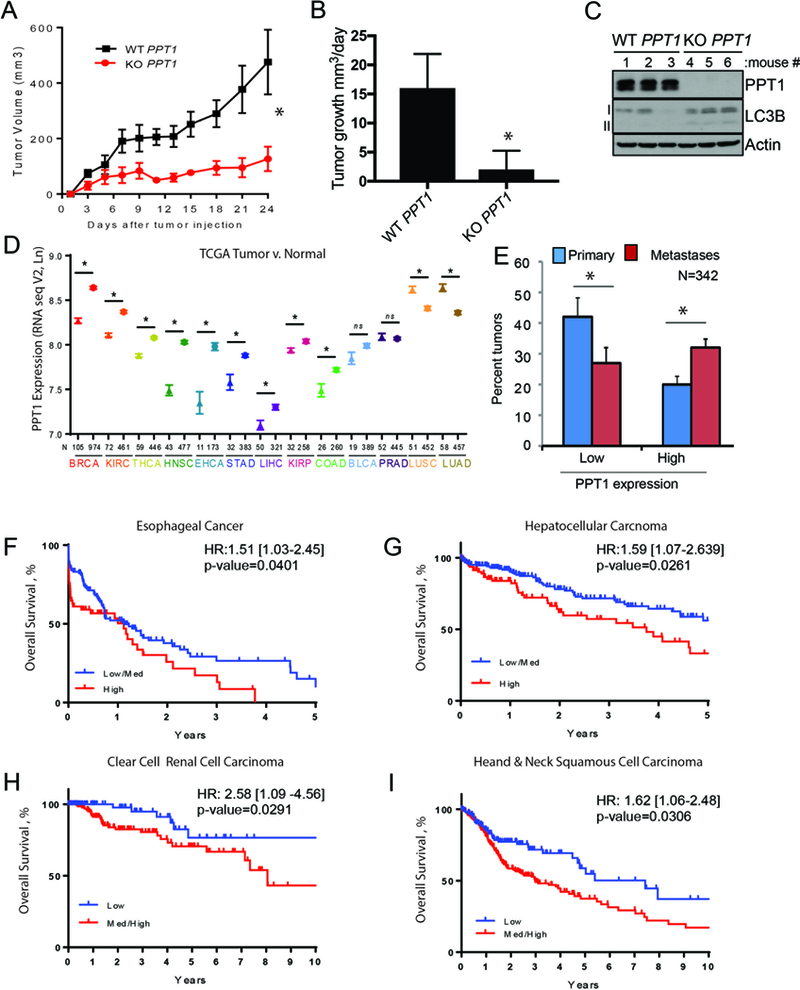
PPT1 expression is correlated with poorer patient survival. (A) WT PPT1 or KO PPT1 cells (1 × 106 cells/mouse were injected into the flanks of NSG mice (n=5, each arm). Tumor volumes were measured every three days as shown. (B) Mean +/− SEM daily tumor growth rate. (C) Lysate generated from WT PPT1 or KO PPT1 cells grown in vivo were immunoblotted. (D) PPT1 expression (TCGA) in unpaired normal (symbol: triangles) and tumor samples (symbol: stars) for the following cancers: BRCA: breast cancer; KIRC: clear cell renal cell carcinoma; THCA: thyroid cancer; HNSC: head and neck squamous cell carcinoma; ESCA: esophageal cancer; STAD: gastric cancer; LIHC: hepatocellular carcinoma; KIRP: Papillary renal cell carcinoma; COAD: colon cancer; BLCA: bladder cancer; PRAD: prostate adenocarcinoma; LUSC: non-small cell lung cancer squamous cell; LUAD: non-small cell lung cancer adenocarcinoma. Mean +/− SEM is presented; *p<0.05 paired t-test. (E) PPT1 expression (TCGA RNAseqV2) in primary versus metastatic melanoma tumor samples. PPT1 values less than the 30th percentile were classified as low and values greater than the 70th percentile were classified as low. (F-I) Kaplan-Meier survival curves were computed for medium/high versus low or high versus medium/low expression of PPT1 in TCGA patients with complete RNASeqV2 expression data as well as overall survival information. Hazard ratio with 95% confidence interval and associated p-value are from a Cox regression analysis. (F) esophageal cancer, (G) hepatocellular carcinoma, (H) clear cell renal cell carcinoma, (I) head and neck cancer. Low, medium and high was defined based on tertiles for PPT1.
Discussion
Chloroquine derivatives have been used as therapeutics for decades without the identification of a protein target (18). Nevertheless, while many weak bases in the pharmacopeia accumulate in the lysosome, few are as effective as CQ derivatives at blocking autophagic flux. These findings suggest that CQ blocks autophagic flux through inhibition of a specific molecular target. We establish herein that PPT1 is the molecular target of both monomeric and dimeric CQ derivatives in cancer cells. These findings reclassify these drugs as targeted therapies, rather than non-specific weak bases.
Our previous work demonstrated that dimerization improved the cytotoxicity of aminoquinoline and acridine-based compounds traditionally viewed as antimalarials (9). More recently we established that extension of length of the linker that connects the two ring systems and central nitrogen methylation significantly enhances lysosomal localization of dimeric quinacrines. Here we disclose that this is also the case for dimeric chloroquines, as DC661 was superior to Lys05 or HCQ in penetrating an acidic medium, blocking autophagy, and eliciting cytoxicity. The increased potency of DC661 was used to perform an in situ pulldown of putative molecular targets in the absence and presence of competition. This pulldown strategy was applied to dimeric Lys05 and monomeric CQ and in each case PPT1 was pulled down and effectively competed off with excess free drug. In cells, PPT1 activity was significantly impaired in a dose-dependent manner, consistent with the anti-autophagy potency of each CQ derivative. KO PPT1 cells demonstrated constitutive lysosomal deacidification, autophagy inhibition and concurrent mTOR inhibition, recapitulating the effects of the chloroquine derivatives in WT PPT1 cells. Treatment of KO PPT1 cells with chloroquine derivatives fails to enhance lysosomal deacidification. Lipidated LC3 reflecting autophagy inhibition is decreased in KO PPT1 cells treated with either HCQ, Lys05 or DC661, respectively, compared to WT PPT1 cells. Ultimately, KO PPT1 cells are resistant to CQ-associated cytotoxicity. Either genetic or pharmacological PPT1 inhibition produces significant antitumor activity demonstrating the considerable promise of PPT1 as a new target for drug development.
The identification of PPT1 as the molecular target of CQ and HCQ is especially interesting when considering that the main toxicity of CQ derivatives that requires monitoring in patients is retinopathy (19). This retinopathy manifests as bull’s eye retinopathy on a clinical exam, and resembles the retinopathy that can be observed in patients who are born with inactivating mutations in PPT1. These patients suffer from infantile neuronal cerebral lipofuscinosis (INCL), a disease that eventually leads to fatal neurodegeneration (20, 21). Importantly, despite its name, INCL patients often live into the second decade of life, and diagnosis can be delayed until age 10. For many years of life, despite PPT1 deficiency present in all cells in the body, the primary manifestation of clinical disease is in the central nervous system (22). These observations suggest that as long as PPT1 inhibitors do not penetrate the blood brain barrier and, in the absence of chronic dosing, a therapeutic window for cancer therapy may be present. Clinical trials in cancer patients with combinations involving high dose HCQ, which has some penetration of the blood brain barrier have thus far reported no cases of neurodegenerative disease, or the accentuation of neuropathy associated when combined with neurotoxic therapies (3–8).
Little is currently understood about the importance of PPT1 expression and activity in cancer. Our analysis of publicly available databases demonstrated that PPT1 expression is high in most cancer cell lines, increased in tumors compared to paired normal tissue, and in metastases versus primary tumors. We and others have recently identified a functional role for PPT1-dependent depalmitoylation in stabilizing the lysosomal localization of v-ATPase subunits. The v-ATPase complex serves to i) maintain lysosomal acidity necessary for cellular catabolism, as well as ii) facilitate machinery critical for mTOR localization and subsequent activation. The results described herein reinforce the role that dynamic palmitoylation serves in stabilizing autophagy-lysosome function and mTOR signaling, via regulating appropriate vesicular trafficking of v-ATPase subunits. Finally, in the TCGA database, stage IV patients with multiple tumor types had significantly shorter overall survival if they exhibited high expression of PPT1. Taken together, these studies establish the potential of PPT1 inhibition strategies in cancer therapy. Future studies are warranted to determine whether PPT1 plays a critical role in tumorigenesis, driving and maintaining metastases, tumor immunity and therapeutic resistance.
Methods
Chemical Methods
Please see Supplemental Methods
Cell Culture and Reagents
Melanoma cell lines A375P, WM3918 and WM983B were obtained from Meenhard Herlyn (Wistar Institute) between 2008 and 2015. HT-29 (ATCC HTB-38) cells were acquired from the ATCC in 2015 and authenticated by the ATCC by way of short-tandem repeat profiling. PANC1 cells were obtained from Tetralogics Pharmaceuticals in 2012. Cell lines are tested for Mycoplasma biannually and authenticated using short-tandem repeat fingerprinting. All cell lines are cultured in RPMI-164 (Invitrogen, 11875) supplemented with 10% fetal bovine serum (FBS; Sigma Aldrich, F6178) in the presence of 5% CO2 at 37°C. Commercially purchased compounds included HCQ, CQ (Spectrum Chemicals), NTBHA (Sigma-Aldrich, and HDSF (Santa Cruz Biotechnology), LysoSensor (ThermoFisher #L7535).
Immunoblotting, Lysosome Fractionation, and Fluorescence Microscopy
Whole-cell lysates and lysosomal extracts were immunoblotted as previously described (Ref 11 Can Disc). Cell Signaling Technology antibodies included RHEB (#13879), β-actin (#3700), phospho-S6 S240_244 (#5364), S6 (#13443), p18 (#8975), mTOR (#2983), LC3B (#3868). Galectin-3 was purchased from BD Biosciences (#556904). PPT1 was purchased from Sigma (HPA021546). Lysosomal fractions were isolated according to the manufacturer’s instructions (Sigma #LYSIS01). All fluorescent microscopy was performed as described previously(9).
PLA
PLA (Sigma #DUO92008) was performed as previously described (23).
MTT Assay and Clonogenic Assay
For MTT assays, cells were plated (2,000 cells/well) in 96-well plates and the clonogenic assay were performed as described previously described (9). Medium was changed with fresh drug every 3 to 4 days for weeks before staining of colonies with crystal violet.
RPPA
RPPA was conducted as previously described (9).
Proteomics
DC661-P pulldowns from cell lysate were performed as previously described (9)
Purification of PPT1
Full length PPT1 plasmid was obtained from Addgene (Plasmid #25205). Sf9 cells were grown at a density of 3×106 cells per milliliter of media were infected with 10 mL/L of P2 viral stock. Cell culture medium was collected following 96 hours of incubation at 27o C. Final cell viability was 48%. The cultured medium was centrifuged at 4000 x g for 30 minutes. PPT1 protein was collected using ammonium sulfate precipitation at 60% saturation. Media was stirred with ammonium sulfate for 1 hour at 4o C and precipitated protein was collected by filtration. The precipitate was resuspended in Resuspension Buffer (20 mM HEPES pH 7, 150 mM NaCl), centrifuged at 4000 x g for 10 minutes to remove remaining cell pellet, and then purified on Ni-NTA resin. Following washing (Resuspension Buffer plus 20 mM imidazole) and elution (Resuspension Buffer plus 300 mM imidazole), the protein was further purified on an s200 gel filtration column (20 mM Bis-Tris pH 6, 150 mM NaCl).
DSC
Purified recombinant protein was concentrated to 1 mg/mL (29.4 μmol/L) and dialyzed for 18 hours into 20 mM NaOAc pH 5, 50 mM NaCl. DSC experiments used a MicroCal VP-Capillary DSC with and without the addition of 100 μM DC661, HCQ, and Lys05. Scans were run from 10°C to 90°C using a scan rate of 60°C/hour and a filter period of 10 seconds.
ABE Assay
The acyl biotin exchange (ABE) assay was performed as previously described (24).
3D Culture
The 3D spheroid assay was performed as previously described (23).
In Vivo Mouse Studies
Xenograft studies were performed with NSG mice (The Jackson Laboratory). Tumor generation, measurement, and harvesting were performed as previously described (Ref 48). Briefly, HT-29 (colorectal cancer) cells were subcutaneously injected (1 × 106) over the right flank of each mouse with an equal volume of Matrigel (BD). Treatment commenced once tumors became palpable. Tumors were measured using electric calipers. Tumor volume was calculated as L x W2 × 0.5. All animal experiments were performed in accordance to the protocols approved by the University of Pennsylvania Institute of Animal Care and Use Committee.
Statistical Analysis
Statistical significance was determined by performed Student two-tailed, unpaired t test unless otherwise noted. Differences of in vivo longitudinal growth data amongst treatment groups was analyzed using linear mixed-effect models in figures 1 and 4. A P value of less than 0.05 was considered as significantly different form the null hypothesis across all experiments and was indicated by an * in all figures.
Supplementary Material
Statement of Significance.
This study identifies PPT1 as the previously unknown lysosomal molecular target of monomeric and dimeric CQ derivatives. Genetic suppression of PPT1 impairs tumor growth, and PPT1 levels are elevated in cancer and associated with poor survival. These findings provide a strong rationale for targeting PPT1 in cancer.
Acknowledgement:
This work was entirely supported by NIH grants R01CA169134 (R. Amaravadi; P. Gimotty); P01 CA114046 (R. Amaravadi; J. Winkler, M. Herlyn); P30 CA016520 (R. Amaravadi); SPORE P50 CA174523 (R. Amaravadi, D. Speicher, M. Herlyn); 1R01CA198015 (R. Amaravadi); CA016672 (G. Mills); P30CA010815 (M. Herlyn). RPPA work was performed in collaboration with Gordon Mills, Yiling Lu, and the MD Anderson RPPA Core.
Footnotes
Conflict of Interest Statement: RA and JW are inventors on 3 patent applications related to this work. The patents have been licensed to a biotech company to promote clinical development of dimeric chloroquine derivatives. RA is a consultant for Presage Biosciences, Sprint Biosciences and Immunaccell, LLC.
References
- 1.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO journal. 2012;31:1095–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy. 2014;10:1415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10:1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. The oncologist. 2014;19:637–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A, et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 2017; 11: 1266–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O’Dwyer PJ. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res. 2013;19:2995–3007. [DOI] [PubMed] [Google Scholar]
- 12.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellegrini P, Strambi A, Zipoli C, Hagg-Olofsson M, Buoncervello M, Linder S, et al. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy. 2014;10:562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkar C, Chandra G, Peng S, Zhang Z, Liu A, Mukherjee AB. Neuroprotection and lifespan extension in Ppt1(−/−) mice by NtBuHA: therapeutic implications for INCL. Nat Neurosci. 2013;16:1608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potts MB, McMillan EA, Rosales TI, Kim HS, Ou YH, Toombs JE, et al. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nature chemical biology. 2015;11:401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bray PG, Ward SA, O’Neill PM. Quinolines and artemisinin: chemistry, biology and history. Curr Top Microbiol Immunol. 2005;295:3–38. [DOI] [PubMed] [Google Scholar]
- 19.Marmor MF, Kellner U, Lai TY, Lyons JS, Mieler WF. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118:415–22. [DOI] [PubMed] [Google Scholar]
- 20.Bellizzi JJ 3rd, Widom J, Kemp C, Lu JY, Das AK, Hofmann SL, et al. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc Natl Acad Sci U S A. 2000;97:4573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, et al. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci U S A. 2001;98:13566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez Poyato MS, Mila Recansens M, Ferrer Abizanda I, Domingo Jimenez R, Lopez Lafuente A, Cusi Sanchez V, et al. Infantile neuronal ceroid lipofuscinosis: follow-up on a Spanish series. Gene. 2012;499:297–302. [DOI] [PubMed] [Google Scholar]
- 23.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124:1406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protoc. 2007;2:1573–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.