

Abstract
The term ferroptosis was coined in 2012 to describe an iron-dependent regulated form of cell death caused by the accumulation of lipid-based reactive oxygen species; this type of cell death was found to have molecular characteristics distinct from other forms of regulated cell death. Features of ferroptosis have been observed periodically over the last several decades, but these molecular features were not recognized as evidence of a distinct form of cell death until recently. Here, we describe the history of observations consistent with the current definition of ferroptosis, as well as the advances that contributed to the emergence of the concept of ferroptosis. We also discuss recent implications and applications of manipulations of the ferroptotic death pathway.
Keywords: ferroptosis, iron, cysteine, lipid peroxidation, cell death, metabolism, chemical biology
Graphical Abstract
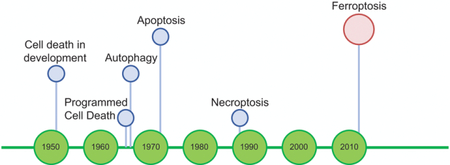
Introduction
Death is a common fate of all life, from organisms to cells. The understanding that cell death can be regulated by molecular mechanisms and can yield physiological benefits and pathological consequences for multicellular organisms emerged early in the 1960s, with the concept of ‘programmed cell death’ [1–3]. It is now established that such programmed cell death is essential for normal development and homeostasis and, when dysregulated, contributes to a variety of pathological conditions.
Regulated cell death is defined as a death process that relies on dedicated molecular machinery, and that thus can be modulated (delayed or accelerated) by specific pharmacological and genetic interventions. Programmed cell death refers to physiological instances of regulated cell death that occur in the context of development and tissue homeostasis, in the absence of any exogenous perturbations. Programmed cell death is therefore a subset of regulated cell death. Regulated cell death is used to describe cell death that originates from perturbations of the intracellular or extracellular microenvironment, executed by molecular mechanisms when other adaptive responses are incapable of restoring cellular homeostasis [4]. Regulated cell death is thus mechanistically distinct from classic necrosis, or unregulated cell death caused by overwhelming stresses, such as dramatic heat shock, use of detergents, pore forming reagents, or highly reactive alkylating agents.
One may ask whether induced loss, through chemical or genetic perturbation for example, of an essential cellular factor represents regulated cell death or simple loss of the homeostasis needed for life. For example, deletion of the Mdm2 gene, or inhibition of the MDM2 protein, results in cell death in many cell lines, and in embryonic lethality in mice due to the lack of this essential protein [5–9]. However, this death and embryonic lethality can be entirely reversed by the simultaneous deletion of the TP53 tumor suppressor gene, which is the major target of MDM2 [6,8,10]. Thus, in this case, the idea that normal life requires MDM2, and that loss of MDM2 represents a sabotaging of life’s homeostatic machinery is not accurate. Indeed, cells and mice can exist, albeit not as well, without MDM2, as long as they also lack the p53 protein. Similarly, one might wonder if the cell death caused by deletion of an essential metabolic gene represents the loss of an essential factor needed for life (e.g., hexokinases [11]). However, in this case, again, such cells may survive if they are provided with the product of the metabolic enzyme, or an alternative means to satisfy this metabolic need. The important point is that the “normal” requirements for life depend on the network of other cellular and environmental factors present at a given point in time.
One might nonetheless suppose that although the cellular factors needed for normal homeostasis are context-dependent, the mechanism of death resulting from the loss of a context-dependent essential factor may not be of interest per se. However, if the cell death mechanism in such a situation reveals the relationship between the missing factor and cellular regulators of its essential function, then this mechanism may indeed be of interest. Moreover, it is possible in some cases to trigger regulated cell death for therapeutic benefit, such as in some cancers. Some pathological processes also act through depletion of essential factors and the resulting cell death process, and inhibition of such death mechanisms may be beneficial.
Finally, the surprising finding over the last two decades is that there appears to be a limited number of execution mechanisms for cell death, despite the wealth of possible triggers of cell death. Although there are thousands of essential genes in most organisms, cell death after loss of such genes occurs by unregulated necrosis, or converges on one of a handful of execution mechanisms involving activation of proteases, permeabilization of the plasma membrane, or overwhelming lipid peroxidation. In summary, the concept of regulated cell death encompasses the notions that normal homeostasis is context-dependent, that the mechanisms driving cell death are intrinsically of interest, and that such mechanisms converge on a limited number of execution mechanisms.
Historically, cell death was divided into three main categories, based on distinctive morphological features: type I cell death, referred to as apoptosis, type II cell death, referred to as cell death involving autophagy, and type III cell death, referred to as necrosis. Regulated cell death was considered synonymous with apoptosis [12], and only more recently was it demonstrated that there are multiple forms of regulated cell death, which are distinct in their molecular mechanisms and morphological characteristics.
In contrast to type I and type II cell death, which were considered to be ‘programmed’ and ‘regulated’, necrosis was categorized as an ‘accidental cell death’, and believed to be passive, unregulated, and commonly associated with pathological death caused by exposure to severe insults of a physical, chemical, or mechanical nature that could not be reversed by molecular perturbations [12].
The Nomenclature Committee of Cell Death (NCCD) recommended in 2012 that researchers replace the morphologic classification of cell death with a new classification based on molecular events associated with cell death [13]. This change contributed to a classification of regulated cell death that uses biochemical mechanisms. For instance, even though the notion that death with a necrotic morphology can be molecularly regulated began to emerge in the late 1980s [14], it took two decades until it was widely accepted that ‘necroptosis’ should be classified as a form of regulated cell death [15,16]. Contributing to this notion were the discoveries of the involvement of molecular mechanisms such as death receptors (e.g., Fas, TNFR1 [17,18]) and specific kinases (e.g., RIP1, RIP3 [19]) that can be genetically or chemically inhibited to delay or inhibit necroptosis [20]. Necroptosis also has normal physiological functions, such as the modulation of inflammation and the maintenance of adult T-cell homeostasis [15,21,22], and may thus be categorized as programmed cell death [23].
We focus here on the historical discoveries that led to the development of the concept of ferroptosis. Ferroptosis is defined as an iron-dependent form of regulated cell death, which occurs through the lethal accumulation of lipid-based reactive oxygen species (ROS) when glutathione (GSH)-dependent lipid peroxide repair systems are compromised [24]. Ferroptosis involves genetic, metabolic, and protein regulators, triggers, and execution mechanisms that for the most part do not overlap with other forms of regulated cell death [25]. Ferroptotic cell death can be inhibited by lipophilic antioxidants, iron chelators, inhibitors of lipid peroxidation, and depletion of polyunsaturated fatty acyl phospholipids (PUFA-PLs), which are prime substrates driving lethal lipid peroxidation [24,26].
Early observations consistent with ferroptosis
Ferroptosis has been observed a number of times over the years prior to the detailed molecular understanding of this cell death process, and the concept that it exists. Yet, until it was termed as such in 2012, reports describing what we now know as cell death with ferroptotic characteristics were attributed to other cell death mechanisms, or not recognized as being biologically significant. For example, metabolic dependencies leading to cell death were noted in the decades before ferroptosis was discovered: early in the 1950’s, studies were performed by Harry Eagle and colleagues to test the requirements for specific metabolites, such as amino acids, vitamins and other nutrients, to support growth and proliferation of mammalian cells in culture [27–29]. These reports showed that starvation of just a single amino acid out of 13 different amino acids tested inhibited the growth of human and mouse cells in culture: cells deprived of cystine exhibited a unique microscopic morphology that was different from the morphologies apparent upon deprivation of other amino acids, which the authors speculated to be similar to death caused by viral infection [28].
In 1959, these investigators found that in cystine-free medium, incorporation of cysteine in de novo protein biosynthesis did not suffice to restore cell growth, thus concluding that cystine entails an additional metabolic function besides incorporation into protein. These studies linked cystine deprivation with the disappearance of glutathione and showed that glutathione supplementation could promote growth in cystine-free media [29,30]. Presumably, cysteine did not suffice because it is taken up by cells through different mechanisms than cystine. In the early 1970’s, there were reports of hepatic necrosis (that today would be referred to as ferroptosis) in mice, which was accompanied by glutathione depletion and could be rescued by pretreatment of glutathione or cysteine [31].
In the 1970’s, Shiro Bannai and colleagues showed that cystine starvation led to reduction in cellular glutathione and cell death [32]. Supporting the contribution of reactive oxygen species (ROS) accumulation in the induction of cell death was the investigators’ observation that this cystine-deprivation-induced death could be rescued by the addition of the lipophilic antioxidant α-tocopherol (a component of vitamin E), without restoring glutathione levels [32]. These results implied that cysteine, derived from reduction of cystine, was needed to sustain glutathione levels and to prevent lipid-ROS-based toxicity, which could also be prevented by lipophilic antioxidants.
In the following years, several studies confirmed the crucial role of cellular cysteine deprivation and glutathione depletion in inducing cell death, and demonstrated that both iron chelators (or serum-deprivation due to lack of iron) and lipophilic antioxidants could block such death from occurring [33–41], which are now recognized as the cardinal features of ferroptosis. These common dependencies and features were demonstrated in many types of mammalian cells, including human embryonic fibroblasts [42], hybridomas and myelomas [33], cortical neurons [34–36], oligodendroglia [37], oligodendrocytes [38,39] and hair cells [43]. Remarkably, these numerous studies recognized reactive oxygen species as drivers of the death process, as well as several distinct triggers and rescuers of cell death, but were nonetheless not interpreted as evidence for a distinct cell death process that does not overlap with apoptosis or necrosis, although it was speculated upon in some cases (e.g., [44]). The situation is similar to the history of protein degradation, which was assumed to be largely unregulated prior to the pioneering studies of Avram Hershko, Aaron Ciechanover and Irwin Rose [45]. Indeed, numerous pieces of the ubiquitin-dependent pathway of protein degradation were known, but not recognized as such [46–49].
At the heart of ferroptosis is a process of lethal lipid peroxidation, which is the oxidative addition of molecular oxygen (O2) to lipids, such as polyunsaturated fatty acyl tails in phospholipids. The first descriptions of such enzymatic reactions were in 1955 by E. Peterson and colleagues [50] and S. Rothberg and colleagues [51] independently; since then, lipid peroxidation by lipoxygeneses and other mechanisms for the peroxidation of lipids have received a great deal of attention in diverse biological contexts [52–54]. The first suggestion of lipid peroxidation as a prime cause of cellular damage was in 1965, when two separate groups studying drug toxicology showed increases in lipid peroxidation in the liver of CCl4-treated rats [55,56]. By the 1980s, it was well established that lipid peroxidation is one of the major forms of oxidative damage through destruction of unsaturated lipid moieties of cell membranes, lipoproteins and other structures [57,58], and was correlated with a number of pathological conditions [59]. However, these events were associated with other cellular damage mechanisms and not recognized as a cell death mechanism per se.
Due to the dual contribution of cellular ROS to signaling mechanisms and cell lethality, enzymatic control of redox regulation is an essential regulatory mechanism for normal cell homeostasis [60]. Redox-sensitive cysteine residues in enzymes exploit the unique ability of sulfur to cycle between oxidation states. Selenoproteins that carry a catalytically active selenocysteine can also contribute to redox control [61].
In a seminal discovery, the membrane-associated ‘phospholipid hydroperoxide glutathione peroxidase 4’ (PHGPX or GPX4) was isolated as the second known selenoperoxidase, after the identification of the cytosolic glutathione peroxidase (GPX1) [62,63]. GPX4 was first isolated and purified from pig liver in 1982 by Urisini and colleagues [64] and was identified on the basis of its ability to inhibit iron-catalyzed lipid peroxidation in microsomes. The enzyme was first given the functional name ‘peroxidation-inhibiting protein’, that was later changed to its current name [65]. This enzyme was described as a glutathione peroxidase that protects phosphatidylcholine-containing liposomes and biological membranes from peroxidative degradation, and is now known to be the key enzymatic inhibitor of ferroptosis. Early work also showed a similar protective role for the lipophilic antioxidant α-tocopherol (vitamin E) in rat liver mitoplasts and microsomes, which was supported by previous similar observations [66]. In the late 1980s, GPX4 was shown to have lipid-peroxidation-protective effects in mammalian spermatozoa, which were known to be sensitive to the deleterious effects of oxygen free radicals, due to their rich polyunsaturated fatty acid content [67,68]. This finding was in line with the notion that selenium is essential for male fertility [69,70], and was further demonstrated by the identification of GPX4 as a major structural component of the mitochondrial capsule, which embeds sperm mitochondria and is thus required for structural sperm stability [71].
The cloning of GPX4 in 1991 revealed its similarity to the classical cytosolic glutathione peroxidase (GPX1), but also its unique properties in inhibiting peroxidation of lipids due to its hydrophobic nature and monomeric form [72]. By the beginning of 1990s, there were several lines of evidence supporting the notion that GPX4 plays a unique role in protecting cells against the damaging and lethal effects of lipid peroxidation [73–76].
Shortly after the isolation of human GPX4 from human liver in 1994 [77], several groups demonstrated that overexpression of this enzyme in human cells results in resistance to lipid peroxide cytotoxicity compared to parental cells [78–82], an effect that was consistently attributed to being protective of oxidative-stress-induced apoptotic cell death [83]. Transgenic and knockout mouse models of glutathione-dependent peroxidases stressed the importance of GPX4 in protecting against cell lethality, as GPX4 knockout was the only one (out of GPX1-, GPX2-, GPX3- [84,85] and GPX4-knockout mouse models) to induce embryonic lethality [86–88]. Heterozygous GPX4 mouse models (GPX4+/−) have contributed to the understanding of its protective role against a unique form of cell death, as these mice were shown to be more sensitive to death induced by γ-irradiation and tert-butyl hydroperoxide [88,89]. It was surprising that these heterozygous GPX4 knockout mice live longer than wild-type mice due to delayed pathologies such as fatal lymphoma [90], that can now be explained as increased sensitivity of such nascent cancer cells to ferroptotic death (see below), implying that GPX4 may be oncogenic. In line with GPX4 overexpression in cell culture, overexpression of GPX4 in mouse models was also shown to be protective from oxidative-stress-induced death [91].
A breakthrough in the understanding of the unique properties of GPX4-downregulation-induced cell death was accomplished when Seiler et al. in 2008 demonstrated the role of 12/15-lipoxygenase (12/15-LOX), a polyunsaturated fatty acid metabolizing enzyme [92], in the execution of GPX4-knockout-mediated cell death [87]. This finding was supported by previous evidence of LOX involvement in deleterious pathologic situations [93–96], and provided a mechanistic explanation for several observations of what was then considered to be ‘oxidative-stress-induced apoptosis’, which was not dependent on activation of the Bcl-2 family proteins [97–99]. This led to the notion of a unique cell death mechanism linking membrane lipid peroxidation, arachidonic acid metabolism, glutathione peroxidase activity (or lipophilic antioxidants; vitamin E) and oxidative stress.
The emergence of the concept of ferroptosis
Throughout the years, there have been several puzzling reports of non-apoptotic caspase-independent cell death with necrotic-like morphology, that seemed to be an active form of cell death that could be regulated [100–103]. One example is ceramide-induced cell death that was shown to involve the accumulation of ROS and did not exhibit apoptotic characteristics, but could be rescued by radical scavengers in human glioma cells [104]. This observation was supported by earlier descriptions of ceramide-induced cell death [105,106]; here too, investigators recognized the distinct nature of this cell death, but tried unsuccessfully to fit it into the existing framework of cell death modes. Ceramide may be involved in the induction of ferroptosis, although this remains unclear [107].
In 2001–2003, the Stockwell Lab performed a screen for small molecules that could selectively kill human BJ fibroblasts that had been engineered to be tumorigenic, but not their otherwise isogenic parental precursors. This screen was designed to identify lethal compounds with selectivity for cells expressing oncogenic mutant HRAS, as well as the large and small T oncoproteins [108–110]. The most selectively lethal compound to emerge from the screen was a novel compound from a newly generated small molecule combinatorial library with no known activity. Stockwell and colleagues named this compound “erastin” because of its apparent ability to Eradicate RAS-and Small T transformed cells. The lab members became interested in determining the mechanism of action for this new compound erastin, as it might illuminate a way of selectively killing RAS-transformed cancer cells. The other compounds that emerged from this screen induced apoptosis; the default assumption was that erastin would as well. However, when Dolma and Stockwell performed typical apoptosis assays with erastin in these engineered tumor cells, they consistently found no evidence of caspase activation, no cleavage of caspase substrates, no Annexin V staining, no nuclear morphological changes, or any other hallmarks of apoptosis. Nonetheless, erastin’s lethality could be potently suppressed by iron chelators and lipophilic antioxidants. Thus, the lab members began to consider the possibility that erastin induced a regulated, but non-apoptotic, form of cell death [108,111], which was a somewhat heretical notion at the time.
Yang and Stockwell screened additional small molecule libraries in the same assay and identified another compound, which was named RAS synthetic lethal 3 (RSL3), that induced a similar form of non-apoptotic, iron-dependent cell death [109]. This confirmed that erastin was not unique in its ability to activate this type of cell death, and that perhaps this form of cell death was a more generally important phenomenon. A series of experiments by Dixon and Stockwell led to the idea that erastin acted by inhibiting the cystine/glutamate antiporter, system Xc−, which reduces cysteine-dependent synthesis of reduced glutathione (GSH) [25,112]. Reinforcing that conclusion was the finding that the unique cell death pathway induced by erastin was similar tocell death induced by sullfasalazine (SAS) [25], a known system Xc− inhibitor [113]. This was the first mechanistic insight into a trigger for what became known as ferroptosis, showing that erastin inhibits cystine import, leading to deregulated cellular redox homeostasis. Distinct from apoptotic cell death, this cell death mechanism did not require caspase activation or the involvement of other apoptotic effectors, such as BAX or BAK, and was not accompanied by apoptotic morphological features or biochemical processes. Moreover, there was no inhibitory effect on erastin- and RSL3-induced cell death by either small molecule inhibitors of necroptosis (necrostatin-1) or autophagy (chloroquine or 3-methyladenine) [25,114,115].
Following these discoveries, the term ferroptosis was coined in 2012 [25], to describe this iron-dependent, non-apoptotic form cell death induced by erastin and RSL3. This discovery was accompanied by the development of the first small molecule ferroptosis inhibitor, termed ferrostatin-1, and the demonstration of glutamate-induced ferroptosis in organotypic rat brain slices, suggesting the potential function of ferroptosis in neurodegeneration.
Elucidation of the RSL3 mechanism of action provided the next major insight in the regulation of the emerging mechanism of ferroptosis. Chemoproteomic studies using (1S,3R)-RSL3 (the active diastereomer out of the four possible RSL3 stereoisomers) as an affinity reagent identified the crucial role for the glutathione-dependent selenoprotein glutathione peroxidase 4 (GPX4) in the regulation of ferroptosis [114].
Subsequently, numerous other studies began identifying a similar ferroptotic process underlying diverse phenomena. In multiple cell types, amino acid starvation was reported to induce cell death that is non-apoptotic and non-necroptotic, but only in the presence of serum [116,117]. It was shown that deprivation of cystine was sufficient to induce the same serum-dependent cell death pathway, and that the serum component crucial for cell killing was transferrin, an iron carrier. Deprivation of cystine was thus equivalent to system Xc− inhibition. These findings led to the confirmation that the cell death mechanism induced by amino acid starvation in the presence of an exogenous iron source is ferroptosis.
It was later found that not all ROS function equally in ferroptosis, and that lipid peroxidation is the main driver for ferroptotic death [118], supported by the previous identification of lipophilic antioxidants as suppressors of ferroptotic death induced by erastin and other compounds [111].
Identification of positive regulators of ferroptosis
Iron has an essential role in life on Earth dating back billions of years. However, emergence of iron-dependent oxygen utilization and polyunsaturated fatty acid metabolism to drive processes in living cells created a deadly paradox: while these processes of energy production, lipid metabolism, and signaling are valuable for the existence of complex life forms, they are also associated with generation of harmful and ultimately lethal species. The oxidation of organic substrates by iron (II) with hydrogen peroxide (H2O2) is referred to as Fenton chemistry (or the Fenton reaction) and was first described by H. J. H. Fenton in 1894 [119] (reviewed in [120]). This reaction partially explains the dependency of ferroptosis on iron, as redox-active iron pools are able to directly catalyze propagation of lipid peroxidation to form damaging species that lead to death.
Early in the 1960s, iron was shown to contribute to lipid peroxidation-associated pathological changes in rats, that could be prevented by vitamin E [121]. As implied by the name ‘ferroptosis’, the existence of high levels of intracellular iron is a requirement for the execution of this type of cell death. An indication of this necessity is that ferroptotic death, whether induced by system Xc− inhibition, direct GPX4 inhibition, cystine deprivation, or extracellular glutamate, can be suppressed by iron chelators, knockdown of the iron transporter transferrin or its receptor, or the lack of iron in serum [25,109,114,122,123], as well as inhibition of iron availability to lipoxygenases [124], which drive ferroptosis through peroxidation of PUFA-PLs (see below). Moreover, addition of iron to the growth medium [25], as well as of iron-bound transferrin [117], were shown to accelerate erastin-induced ferroptosis, and administration of a bioavailable iron form enhances ferroptotic death in mouse models defective in system Xc− [125].
Mechanistically, intracellular redox-active iron promotes ferroptosis by catalyzing the formation of soluble lipid radicals that can initiate or propagate oxidative PUFA fragmentation, enzymatically and non-enzymatically [126]. Intracellular iron homeostasis is strictly regulated by the iron-binding and mRNA-regulatory proteins named iron-regulatory proteins 1 and 2 (IRP1 and IRP2), that can sense the cellular concentration of free iron and respond by altering the expression of proteins governing iron export, import, storage and release [127]. Ferroptotic death is often linked to the disruption of delicate iron homeostasis, which causes an undesired increase of free cellular iron concentration (Fe2+; also known as the ‘labile iron portion’ or ‘LIP’). This fine-tuning of iron levels is mostly attributed to an impaired activity of IRP2, coupled with increased expression of the iron carrier transferrin, and transferrin receptor [117].
One of the main mechanisms enabling recycling of intracellular redox-active iron is lysosomal degradation of ferritin in the process of autophagy, often referred to as ‘ferritinophagy’ [128]. Autophagy was suggested to contribute to ferroptotic cell death by promoting the degradation of iron storage proteins such as ferritin, which results in release of free iron [129,130]. Reinforcing this hypothesis, recent evidence indicates that ferroptotic induction requires the presence of active lysosomes [123]. The importance of autophagy (specifically ‘ferritinophagy’) for inducing ferroptosis is demonstrated by the complete blockage of ferroptotic death by inhibition of NCOA4, a specific autophagy cargo receptor that mediated the delivery of ferritin to lysosomes Confirming the central role of lysosomal iron in ferroptosis, is the ability of the membrane impermeable iron chelator deferoxamine (DFO) to inhibit erastin-induced and RSL3-induced death [25,109]. DFO is taken up by the cell through endocytosis and accumulates in lysosomes [131], suggesting that it prevents ferroptosis by chelation of the ‘redox active’ lysosomal iron pool or by inhibiting specific iron-dependent lipid-ROS promoting enzyme.
One of the central organelles where significant amounts of ROS can be generated is mitochondria, where ROS are formed as a result of normal metabolism and energy production through the electron transport chain. There is some evidence of possible mitochondrial involvement in processes supporting ferroptotic death, starting from the first description of a distinctive morphological feature of erastin-treated cells that showed smaller mitochondria with increased membrane density [25]. Moreover, several mitochondrial genes were found to be associated with ferroptotic cell death, and there is a suggestion of peroxidation of cardiolipin, a mitochondria-specific phospholipid, linking mitochondrial lipid peroxidation to ferroptosis [132–134]. Nevertheless, there are other cellular sources for ROS, and cells with mitochondrial DNA depleted can still undergo potent ferroptosis [25]. Recently, it was found that removing mitochondria from cells does not prevent ferroptosis, suggesting that mitochondria are not needed for ferroptosis [135].
Both glutamate and glutamine play important roles in ferroptosis induction. High concentrations of extracellular glutamate can block cystine uptake through system Xc− and induce oxidative-stress-mediated cell death which can be rescued by α-tocopherol supplementation [40,136]. This cell death mechanism was described in cells of the central nervous system and shown to be distinct from apoptosis, thus it was termed ‘oxidative glutamate toxicity’ or ‘oxytosis’ [137]. Glutamate-induced cell death shares several characteristics with ferroptosis, mainly in the mechanism of initiation by cystine deprivation and glutathione depletion, leading to accumulation of lipid-based ROS [25]. However, the terminal phases of death by glutamate toxicity may involve apoptotic, and not ferroptotic features, in some neuronal cells, and are dependent on calcium rather than iron. This death mechanism may overlap with ferroptosis in other cells [138]. A more general analysis of the degree of overlap between these two death mechanisms requires further investigation.
The role of glutamine in ferroptosis is complex. Although glutamine can be converted to glutamate by glutaminases (GLS1 and GLS2), high concentrations of extracellular glutamine alone cannot induce ferroptosis. Instead, glutamine was shown to drive ferroptosis through glutaminolysis, in combination with cystine deprivation in MEFs [116,117]. Moreover, inhibition of glutamine uptake and of glutaminolysis was recently suggested to contribute to ferroptosis resistance in cancer cells [139]. In the absence of glutamine, or when glutaminolysis is inhibited, cystine starvation and blockage of cystine import cannot induce ferroptosis or the associated rapid accumulation of ROS and lipid peroxidation. The necessity for glutaminolysis in ferroptosis is further reinforced by the observation that α-ketoglutarate (aKG), a product of glutaminolysis, can replace the requirement of glutamine for ferroptosis. Interestingly, although both GLS1 and GLS2 can convert glutamine to glutamate, only GLS2 was shown to be required for ferroptosis [117]. These glutaminases differ in their intracellular localization: GLS1 is a cytosolic protein, whereas GLS2 localizes in the mitochondria [140], suggesting that, at least in this cellular context, the induction of ferroptosis seem to be dependent on mitochondrial GSL2. Bearing in mind that mitochondria were shown to not be essential for ferroptosis induction in other cellular models [25,135], determination of the centrality of mitochondria-mediated ROS production in ferroptosis requires further study. Of note, glutaminolysis is considered to be a mitochondrial process that promotes (mainly cancer) cell survival by maintaining ATP production and inhibiting ROS production [141]. Thus, data suggesting an involvement of glutaminolysis as a driver of ferroptosis through the activity of mitochondrial GLS2 [117], was surprising and counterintuitive. However, glutaminolysis was also shown to stimulate autophagy, which can drive ferroptotic death as discussed above.
A common feature of ferroptosis is the iron-dependent accumulation of lipid-ROS and the subsequent depletion of polyunsaturated fatty acid phospholipids (PUFA-PLs) [118]. The PUFA chains of membrane lipids are more susceptible to both enzymatic and non-enzymatic oxidation, which results in PUFA fragmentation into a variety of products [126]. Indeed, several cell types that contain relatively high levels of PUFAs, such as cells of the retina and spermatozoa, were known for a few decades to be more sensitive to lethal oxidative stress that can be reduced by vitamin E or GPX4 [68,142]. This is in line with earlier observations that inhibition of arachidonate 12-lipoxygenase (Alox12), an iron-containing lipid dioxygenase, inhibited oxidative glutamate toxicity and cell death in neurons, while treatment of cells with arachidonic acid (AA), a substrate of Alox12, further potentiated such death [143]. Arachidonic acid (AA) is now known to be the most frequently depleted PUFA in cells undergoing ferroptosis [144,145]. One study showed that deletion of crucial enzymes involved in the insertion of AA into membrane phospholipids can prevent ferroptosis induction [146]. Acyl-CoA synthetase long-chain family member 4 (ACSL4), thus drives ferroptosis by contributing to the accumulation of oxidizable cellular membrane phospholipids [26,147]. The currently suggested mechanism for the involvement of lipid metabolic pathways in ferroptosis induction is the following: ACSL4, which prefers AA as its main substrate, promotes ferroptosis by producing oxidized phosphatidylethanolamines (PE) in endoplasmic-reticulum-associated oxygenation centers. ACSL4 catalyzes the ligation of an arachidonyl (AA) or adrenoyl (AdA) to produce AA or AdA acyl Co-A derivatives. These derivatives are then esterified into PE to form AA-PE and AdA-PE by lysophosphatidylcholine acyltransferase 3 (LPCAT3), and subsequently oxidized by 15-lipoxygenase (15-LOX) to generate lipid hydroperoxides, which execute ferroptosis [26,124,147]. Although 15-LOX is thought to play a central role in catalyzing lipid peroxidation that leads to ferroptotic death, this role may be attributed to multiple LOXs, since deletion in 15-LOX fails to rescue the renal phenotype of GPX4 null mice [144]. The idea that lipid metabolism and membrane lipid composition affect susceptibility of cells to ferroptosis is further supported by molecular dynamics modeling of membrane lipid peroxidation [148].
Although the essential role for lipid peroxides in induction of ferroptotic death has been established, there is still no definitive evidence that this class of ROS is the most downstream factors that execute ferroptosis. Therefore, the question of what the ultimate molecular executioner of ferroptotic cell death is (e.g., as caspases for apoptosis), remains to be resolved.
Identification of negative regulators of ferroptosis
The first description of ferroptosis induction, by Dixon et al. in 2012, was in the context of cystine deprivation by inhibiting cystine (Cys2) import via the system Xc− antiporter with the small molecule erastin [25]. System Xc− is a plasma membrane cystine/glutamate antiporter composed of a twelve-pass transmembrane transporter protein, SLC7A11 (xCT), linked to the transmembrane regulatory protein, SLC3A2, by a disulfide bridge [149]. The lethal effect of erastin (and also of SAS, see above) can be reversed by β-mercaptoethanol (β-ME) [25,112], which bypasses the need for system Xc− by forming mixed disulfides with Cys2 that can be imported into the cell by a different transporter [150]. Additionally, some cells can use the transsulfuration pathway to biosynthesize cysteine from methionine when system Xc− is inactivated. This pathway was recently shown to also be upregulated upon knockdown of cystenyl-tRNA synthetase (CARS). Cells that use the transsulfuration biosynthetic pathway were thus found to be resistant to ferroptosis induced by system Xc− inhibitors, but could still undergo ferroptosis through GPX4 inhibition [151]. Consistent with the need of cellular cystine to protect from ferroptotic death, according to a recent series of experiments, the redox-sensitive transcription factor Nrf2 [152,153] can protect cells from ferroptotic death by upregulating system Xc− [132,154], and was found to be commonly overactivated in various cancers [155,156]. In contrast, induction of ferroptosis through inhibition of system Xc− was suggested to be a mechanism of tumor suppression by p53 [157].
The precise mechanism by which erastin inhibits SLC7A11-mediated cystine import it still unknown. The initially proposed mechanism, by which erastin binds a related transport protein, SLC7A5, and inhibits SLC7A11 in trans [25], was revised soon after, and it was suggested that erastin inhibits SLC7A11 perhaps directly [112]. An additional inhibitor of system Xc−, the FDA-approved multi-kinase inhibitor sorafenib (Nexavar), was shown to induce a GSH-depletion-mediated ferroptosis in cancer cell lines [112,158–160] and enhance ROS accumulation in cancer patients [161]. Although the clinical benefit of sorafenib was in part caspase-independent [160,161], this drug was shown to trigger classic apoptosis in some cells [162]. The specific contribution of ferroptotic cell death to the therapeutic effects of sorafenib in cancer patients is unknown [163]. A more potent inhibitor for system Xc−, imidazole ketone erastin (IKE), was designed by introducing a stable ketone to erastin [164]. In addition to the improved potency of IKE compared to erastin, IKE was shown to have improved metabolic stability placing this compound as a good candidate for in vivo studies of ferroptosis induction.
Downstream of system Xc− is a central cellular guard against lipid ROS-induced ferroptotic death, GPX4. GPX4 acts as a guardian of cell membranes and converts potentially toxic lipid hydroperoxides (L-OOH) to non-toxic lipid alcohols (L-OH) [64]. Small molecule inhibition of GPX4 was first observed with the small molecule (1S, 3R)-RSL3, and resulted in uncontrolled polyunsaturated fatty acid phospholipid (PUFA-PL) oxidation and fatty acid radical generation, leading to ferroptotic cell death [114,124]. Genetic or pharmacological inhibition of GPX4 leads to ferroptotic death, and hence is independent of cystine supply [114,144]; deletion of GPX4 in mice is embryonically lethal [87]. Concomitantly, overexpression of GPX4 blocks RSL3-induced ferroptosis [114], and the lethality of GPX4-depleted mice could be rescued with ferroptosis inhibitors (e.g., ferrostatin-1) [87,144]; further confirming that GPX4 activity is essential to prevent ferroptosis. The fact that elimination of this important cellular guard against lipid ROS is sufficient to induce ferroptotic death suggests that cells are continually exposed to the threat of radical-mediated lipid destruction. In addition to RSL3, other less potent small molecule inhibitors of GPX4 were identified (e.g., altretamine [165]), yet the mechanism of action of these inhibitors is not clear [138].
Although both cys-mediated GSH synthesis and GPX4 activity were shown to be essential for protecting cells against ferroptotic death, alternative antioxidant pathways maintain cell survival in the absence of GSH-GPX4. Indeed, the majority of commonly used ferroptosis inhibitors, including the original ferrostatin-1 and the more recently discovered liproxstatin-1 [144], are believed to function by trapping lipid radicals [122,166]. Radical-trapping antioxidants are molecules that react with chain-carrying radicals and break the oxidation chain reaction [167]. Vitamin E, a lipophilic antioxidant, was discovered almost a century ago [168] and its beneficial antioxidant effect in human health was recognized since. The most biologically active form of vitamin E, α-tocopherol [169], was shown to inhibit ferroptotic death both in vitro and in vivo [170–173], and vitamin E deficiency was linked to ferroptosis-mediated premature onset of neurodegeneration [174,175]. In some cells, both in vitro and in vivo, high levels of SLC7A11-mediated cystine import, in conjugation with the GSH-independent thioredoxin (Txn) system, maintains endogenous α-tocopherol (vitamin E) in a reduced state and prevent lethal lipid-ROS accumulation [176–179]. Additionally, it was recently suggested that in addition to the direct antioxidant activity of vitamin E in limiting lipid-ROS, it can also act as inhibitor of lipogeneses by competing for their substrate binding site, further inhibiting ferroptosis [26].
The recent identification of new mechanisms for triggering ferroptosis, by compounds termed FIN56 and FINO2, provided new insights into regulation of ferroptosis. FINO2 acts through a distinct mechanism: it promotes lipid peroxidation by oxidizing iron and indirectly inactivating GPX4 [180]. FIN56 was found to trigger ferroptosis by inducing a combined effect of mediating the depletion of both GPX4 protein and the mevalonate-derived antioxidant coenzyme Q10 (CoQ10) [172]. CoQ10 is an endogenously-produced lipid-soluble antioxidant, which was shown to prevent the harmful oxidation of proteins, lipids and DNA [181,182]. Importantly, in addition to its direct antioxidant activity, CoQ10 also contributes to regeneration of other antioxidants such as ascorbate and α-tocopherol [183]. The role for cellular CoQ10 pool in regulating ferroptotic death remains to be explored.
Possible biological functions of ferroptosis
Although a low and controlled level of lipid ROS is perhaps acceptable for normal cellular and organismal function, the aberrant accumulation of lipid ROS is associated with a number of chronic degenerative conditions and acute organ injuries. Such conditions are caused by the imbalance between radical-generating and radical-scavenging cellular systems and pathways described above, leading to oxidative stress that results in cell death. Thus, mechanisms of controlling ferroptotic cell death are being investigated in recent years as therapeutic means for multiple pathologies.
Ferroptosis in neurodegeneration
Although there has been evidence for the involvement of lipid peroxidation and oxidative stress in numerous neurological conditions for a few decades before ferroptosis was described [184– 186], brain cell death in neurological and neuropsychiatric conditions was attributed to apoptosis, and acute central nervous system (CNS) cell death events such as traumatic brain injury and infection, were considered to be due to necrosis [187]. Following the discovery of ferroptosis, this paradigm is under challenge, with ferroptosis now being suggested as the main driver of neurological cell death in diseased such as Parkinson’s disease (PD) and Alzheimer disease (AD) [188–191]. It is now being appreciated that dysregulation in iron homeostasis might be a central driver for such neurodegenerative diseases [192,193]. Even before the discovery of ferroptosis, there was already evidence of the central role of oxidative stress in neuropsychiatric conditions such as bipolar disorder [194], schizophrenia [195] and depression [196], disorders for which ferroptosis is now considered to be a possible driver [197]. Ferroptosis was also suggested to contribute to the toxic effect of mutant Huntingtin (HTT) that cause Huntington’s disease [198], which was shown to correlate with dysregulation of iron, glutamate and glutathione [199,200]. Further supporting the notion that ferroptosis plays a detrimental role for cell death in Huntington’s disease is the ability of the ferroptosis inhibitor ferrostatin-1 to abrogate cell death induced by mutant HTT [145].
Accumulating evidence of the correlation between elevated iron levels in the brains of Alzheimer’s disease patients and cognitive decline [201–204], along with evidence of increased lipid peroxidation [205–207], have raised the hypothesis that ferroptosis is the main form of cell death in this pathology (reviewed in [191]). The development of novel ferroptosis inhibitors, with improved ADME properties (relatively to the original ferrostatin-1) [208], are currently considered promising treatments for neurodegenerative and neuropsychiatric diseases.
Ferroptosis in cancer
Cancers often exhibit high proliferation and imbalanced redox consumption and signaling, since various oncogenic pathways (such as proliferation and evading cell death) converge on redox-dependent signaling [209]. Additionally, recent epidemiological and animal studies have shown that the Iron-rich microenvironment that often characterizes malignancies supports rapid proliferation and contributes to carcinogenesis [210,211]. This creates an ‘addiction’ of cancer cells to high iron levels and places tumors under persistent oxidative stress [212], which often drives addiction to genes and pathways that protect from ferroptotic death providing growth advantage and contributing to cancer chemoresistance. For example, various cancer types harbor somatic mutations in Nrf2 or Keap1 which lead to enhanced transcription of antioxidant enzymes [213], and glutathione and thioredoxin antioxidant systems are commonly activated in cancers [214,215]. Thus, a fine control of body iron levels was recently suggested as a strategy for reduction of cancer-risk in healthy populations [210,211].
A link between ferroptosis sensitivity and overexpressed oncogenic HRAS activity served as the basis for the original discovery of ferroptosis. As stated above, ferroptosis was discovered through the study of erastin and RSL3, which were found in a small molecule screen to be selectively more lethal to cancer cells harboring oncogenic RAS mutants. In addition, silencing of oncogenic mutant KRAS or BRAF was found to reduce sensitivity to erastin-induced death in two different cancer cell models [111], suggesting that the RAS-RAF-MEK pathway plays a role in determining ferroptosis sensitivity, at least in some cancer cell lines. A suggested mechanistic explanation for this relationship is that constitutive activity of the RAS signaling pathway mediates an increase in cellular iron content through elevated expression of transferrin receptor and down regulation of the iron storage protein ferritin [109]. Of note, this was only demonstrated in a model of engineered tumor cell line, while following studies showed no correlation between ferroptosis sensitivity and oncogenic RAS genes across many cancer types [114], and even increased resistance to erastin and RSL3 in some contexts [216].
Recent discoveries of ferroptosis-inducing agents and further identification of regulation mechanisms and genes involved in ferroptosis induction [217] serve as a foundation for developing strategies for cancer therapy through induction of ferroptosis [218,219]. The iron-enriched (ferroptosis-promoting) tumor environment [220], along with accumulating evidence of overexpression of ferroptosis-inhibiting mechanisms in cancer cells, is suggestive for increased sensitivity of cancer cells to ferroptosis induction [114], which is counteracted by cancer cell addiction to tumor suppressor pathways. For example, the pronounced addiction of triple-negative breast carcinoma to glutamine relates (at least in part) to its ability to drive cysteine uptake via system xc−, implying that system xc− may constitute a therapeutic target in this setting [221,222]. In addition, B cell-derived lymphomas and a subset of triple-negative breast cancer cell lines were shown to acquire a strong dependency on GPX4 and system Xc− [223,224] (reviewed in [217]).
An additional example for the role of ferroptosis in tumorigenesis is the ability of the tumor suppressor p53 to induce ferroptosis. The p53 protein is a crucial tumor suppressor that mediates cell cycle arrest, cell death and/or senescence in response to various stress conditions [225]. Recent studies link ferroptotic cell death to a non-conventional p53-mediated activity of tumor suppression [226,227]. Years before the acknowledgment that cancer cells may be more prone to ROS-induced death, and that tumor suppressor mechanisms may involve evading ferroptosis, it was suggested that p53 can regulate ROS accumulation [228,229]. The mechanism for that regulation was recently suggested by Jiang at al., which showed that p53 represses the expression of the system Xc− component, SLC7A11, causing a reduction in cystine uptake, which results in increased sensitivity to ferroptosis [230]. The importance of this tumor suppressive role of p53 is further reinforced by the ability of SLC7A11 overexpression, observed in several forms of human cancer [223,224,231], to protect from ROS-induced death. However, p53 was also shown to prevent ROS accumulation by upregulating antioxidant genes, a function that was attributed to prevention of ROS-mediated DNA damage and tumor suppression, but also contributes to ferroptosis evasion in malignancies.
Ferroptosis in development
The occurrence of several types of morphogenetic death processes during development was demonstrated decades ago [232]. However, apoptotic death is still considered to be the main form of cell death that contributes to proper tissue and organ formation. The notion that ROS act as major intracellular signal mediators during development emerged in the 1990s [233,234]. Since then, the important functions of ROS in embryonic development, as well as the crucial role for ROS scavengers (specifically lipid ROS) has been extensively studied [235]. Observations that embryonic tissues undergoing cell death show increased levels of ROS, and that such death can be controlled by GPX4 expression and inhibited by lipophilic antioxidants (i.e., in the developing limb [236,237]), supported the involvement of lipid ROS-mediated cell death in development. To date, the role of ferroptotic death in embryonic development is considered to be more relevant for maintaining tissue integrity and homeostasis, rather than organ formation per se.
Recent studies also point to homeostatic and ROS-induced cell death in plants, that encompass ferroptotic characteristics [238,239]. This further demonstrates the relevance of ferroptosis to maintain organismal homeostasis and supports the evolutionary conservation of this cell death mechanism. Although it is becoming clear that ferroptotic death is an essential process for the multicellular organism, the specific roles for ferroptosis in development are currently not understood.
In summary, the concept of ferroptosis encapsulates the idea that iron-dependent lethal lipid peroxidation is a distinct form of cell death, with unique triggers, regulators, and effectors. This concept provides a framework for elucidating additional molecular controls of this process, its involvement in disease, therapeutic strategies, and its physiological and evolutionary functions.
Figure 1. Ferroptosis-related scientific discoveries before it was termed.
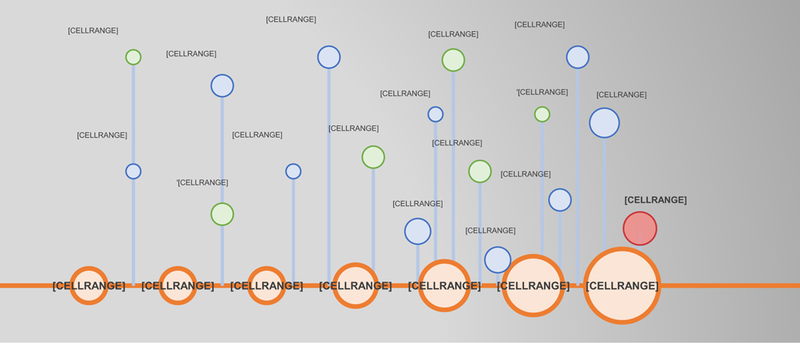
Schematic timeline highlights key discoveries that contributed to the emergence of the concept of ferroptosis (green) and important observations that are consistent with the current definition of ferroptosis (blue), before it ferroptosis termed.
Table 1.
Main findings describing cell death of ferroptotic nature, before ferroptosis was coined.
| Year | Main Finding | Ref | |
|---|---|---|---|
| Lipid peroxidation |
1962 | Iron contributes to lipid peroxidation-associated pathology in rats | [121] |
| 1965 | CCl4-mediated lipid peroxidation is associated with cell death in rat liver |
[55,56] | |
| 1984 | Lipid peroxidation is linked to multiple pathological conditions | [57–59] | |
| 2005 | Ceramide induces non-apoptotic ROS-dependent cell death | [104] | |
| 1955 | First observation that cystine deprivation lead to cell death of unique morphology |
[27,28] | |
| Cystine deprivation |
1973 | Cystine- and glutathione-dependent hepatic necrosis in mice | [31,245] |
| 1977 | Cystine deprivation lead to glutathione depletion-mediated cell death in fibroblasts, which is rescued by α-tocopherol |
[32] | |
| 1979 | Scavenging deleterious oxidative radicals protect from oxidative necrosis induced by glutathione depletion in fibroblasts |
[42] | |
| 1989 | Glutamate-induced cytotoxicity in neuronal cells is due to inhibition of cystine uptake, resulting in lowered glutathione levels leading to oxidative stress and cell death |
[40,41] | |
| 1994 | Cystine deprivation leads to death of embryonal cortical neurons due to reduced formation of glutathione |
[34,35] | |
| 1994 | serum deprivation diminished glutathione-depletion-induced death in embryonal cortical neurons |
[36] | |
| 1996 | Cystine deprivation lead to reduced glutathione-mediated death of oligodendroglia, rescued by antioxidants and iron chelator |
[37] | |
| 2005 | SLC7A11 expression predicts chemosensitivity cells to drug treatment in cancer cell lines |
[223] | |
| 1982 | GPX4 purified from pig liver and characterized as protective of liposomes and biomembranes from peroxidative degradation |
[64] | |
| 1987 | GPX4 protects mammalian spermatozoa from lipid peroxidation- mediated lethality |
[67,68] | |
| 1990 | GPX4, but not GPX1, reduce lipid hydroperoxides in membranes | [73,74] | |
| 1991 | GPX4 protects against lipid hydroperoxides-mediated lethality in murine lymphoid leukemia cells |
[246] | |
| GPX4 | |||
| 1991 | Cloning of GPX4 reveals its distinct nature compared to other GPXs | [72] | |
| 1996– 2004 |
Overexpression of GPX4 in vitro and in vivo results in increased resistance to oxidative stress-induced death |
[79– 82,91,97] |
|
| 2003– 2008 |
GPX4 knockout induce embryonic (E7.5) lethality in mice | [86–88] | |
| 2003 | Heterozygous GPX4 mice are more sensitive to oxidative stress- mediated death |
[88,89] | |
| 2006 | Inhibition of 12/15-lipoxygenase protects from death by vascular injury in the ischemic brain |
[93,94] | |
| PUFAs | |||
| Critical role for 12/15-LOX in GPX4-knockout-mediated distinct form of cell death |
[87] | ||
| 2008 | |||
Table 2.
Main discoveries towards characterization of ferroptotic cell death
| year | Main finding | Ref |
|---|---|---|
| 2003 | Erastin was discovered through a synthetic lethal high-throughput screen to selectively kill engineered tumorigenic cells in a non-apoptotic manner |
[108] |
| 2007 | Erastin treatment causes the appearance of oxidative species | [111] |
| Erastin-induced cell death is inhibited by lipophilic antioxidants | [111] | |
| 2008 | RSL3 was discovered through a synthetic lethal screening of small molecules to have increased lethality in the presence of oncogenic RAS, through a death mechanism similar to erastin |
[109] |
| Iron chelation and lipophilic antioxidants inhibit RSL-3 induced cell death | [109] | |
| 2011 | Modulatory profiling identifies common death induction mechanisms for erastin and RSL3, which are distinct from death mechanisms of other death inducers |
[115,247] |
| 2012 | Erastin was found to inhibit system xc− | [25,112] |
| Ferrostatin-1 was identified as inhibitor of ferroptosis | [25] | |
| Iron accelerates erastin-induced ferroptosis | [25] | |
| Erastin-treated cells show smaller mitochondria with increased membrane density | [25] | |
| Mitochondrial DNA is not required for ferroptosis | [25] | |
| 2013 | Sorafenib induces ferroptotic cell death | [159,160] |
| 2014 | GPX4 was identified as the target for RSL3 | [114] |
| GPX4 knockout triggers ferroptosis-induced acute renal failure in mice | [114] | |
| Arachidonic acid (AA) is the most frequently depleted PUFA in cells undergoing ferroptosis |
[114,145] | |
| Liproxstatin-1 was identified as inhibitor of ferroptosis | [144] | |
| 2015 | The iron carrier transferrin and L-glutamine are the two serum components required for cystine-depletion-mediated ferroptosis |
[117] |
| ACSL4 and LPCAT3, involved in the insertion of PUFAs into membrane phospholipids, contribute to ferroptosis induction |
[146,147] | |
| imidazole ketone erastin (IKE), a potent inhibitor of system Xc−, was designed | [164] | |
| 2016 | Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis | [124] |
| RSL3 inhibit GPX4 by covalently targeting the active site selenocysteine, leading to accumulation of PUFA hydroperoxides |
[124] | |
| ‘Ferritinophagy’ contributes to ferroptotic death trough increase in LIP | [128– 130] |
|
| Upregulation of the transsulfuration pathway (biosynthesize cysteine from methionine) can inhibit ferroptosis when induced by system Xc− inactivation |
[151] | |
| Nrf2 protects against ferroptosis in HCC | [155] | |
| FIN56 was identified as a ferroptosis inducer through depletion of GPX4 and CoQ10 | [172] | |
| Vitamin E protects from ferroptosis in GPX4 null mice | [170] | |
| 2017 | Phosphatidylethanolamines (PEs) are the fatty acids oxidized in the ER through ferroptosis |
[26] |
| 2018 | Mitochondria are not required for ferroptosis | [135] |
Highlights.
Ferroptotic death was observed in multiple contexts prior to coining of the term in 2012
Unique features of ferroptosis were frequently attributed to other death pathways
Ferroptosis appears to be central to multiple pathologies
BOX: Open questions.
What is the exact function of iron in ferroptosis?
Iron is believed to have multiple functions in ferroptosis, and a redox-independent role for iron has not been completely ruled out. The most common proposed model is that iron is involved in the generation of lipid ROS, either through Fenton chemistry, or via the action of iron-dependent oxidases [122]. Nevertheless, the requirement for iron for ferroptosis may reflect the role of various metabolic enzymes in ROS generation, for which iron functions as a cofactor (e.g., the LOX family of enzymes [240] or prolyl 4-hydroxylase isoform 1, PHD1 [241]).
What is the molecular executor of ferroptosis?
The molecular events that occur downstream of lipid oxidation and the specific ‘point of no return’ in ferroptotic cell death are unclear. It may be that oxidative fragmentation of PUFAs and membrane lipid damage are sufficient death inducers, through permeabilization of the plasma membrane and damage to intracellular organelle membranes. Alternatively (or cooperatively), the fragmented products of oxidized PUFAs may promote death by reacting and inactivating essential cellular proteins. For example, the toxic reactive lipid intermediate 4-hydroxynonenal (4-HNE), a byproduct of oxidative PUFA fragmentation, can be detoxified by three aldo-keto reductase family 1, member C (AKR1C) [242] The expression of AKR1C is controlled by the antioxidant master regulatory transcription factor NRF2 [243] and is overexpressed in cell lines selected for resistance to ferroptosis [112], suggesting that the accumulation of 4-HNE may be a key ferroptotic driver. Nevertheless, we cannot exclude the possibility of a death-inducing protein or protein complex activated downstream of lipid-ROS accumulation, thus further study is required to determine the terminal executor(s) for ferroptotic cell death.
Are there molecular markers to identify cells undergoing ferroptosis?
In the study of ferroptosis, there is a great need to identify molecular markers that would classify cells undergoing this process prior to death, such as caspase-activation for apoptosis. To date, the experimental confirmation of ferroptotic process relies mainly on observation of increased cellular ROS and the ability of ferroptosis inhibitors (e.g., ferrostatin-1) and iron chelators (e.g., DFO) to block cell death. The mRNA expression of two genes, prostaglandin E synthase 2 (PTGS2) and ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1), were found to be significantly elevated in cells undergoing ferroptosis [112,114]. Yet, these markers are not suitable for use in live cells or intact tissues. Additionally, an increase in the expression levels of heme oxygenase-1 (HO-1) was observed upon erastin-mediated ferroptosis induction [244], but the generality of this marker for the occurrence of ferroptotic death in various cells and initiation pathways requires further validation. Therefore, researchers are constantly in search for additional ferroptosis markers that could be used for future in vivo studies.
Under what contexts life benefit from ferroptotic cell death?
Since iron-dependent oxidative metabolism became an essential part of life billions of years ago, one can wonder, is ferroptosis the most ancient form of programmed cell death? If so, did any evolutionary force drive organisms to take advantage such ROS/iron-driven cell death and ‘program’ it for their own benefit?
Acknowledgments
The research of B.R.S. was funded by NIH/NCI grants 1R35CA209896 and P01CA087497.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kerr JFR, A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes, J. Pathol. Bacteriol 90 (1965) 419–435. doi: 10.1002/path.1700900210. [DOI] [PubMed] [Google Scholar]
- [2].Lockshin RA, Williams CM, Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths, J. Insect Physiol 10 (1964) 643– 649. doi: 10.1016/0022-1910(64)90034-4. [DOI] [Google Scholar]
- [3].Lockshin RA, Williams CM, Programmed cell death—I. Cytology of degeneration in the intersegmental muscles of the Pernyi silkmoth, J. Insect Physiol 11 (1965) 123–133. doi: 10.1016/0022-1910(65)90099-5. [DOI] [PubMed] [Google Scholar]
- [4].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK-M, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin K-M, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, García-Sáez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jäättelä M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine J-C, Martin SJ, Martinou J-C, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Muñoz-Pinedo C, Nagata S, Nuñez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon H-U, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G, Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ 25 (2018) 486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chavez-Reyes A, Parant JM, Amelse LL, de O RM. Luna SJ Korsmeyer G Lozano, Switching Mechanisms of Cell Death in mdm2- and mdm4-null Mice by Deletion of p53 Downstream Targets, Cancer Res 63 (2003) 8664–8669. [PubMed] [Google Scholar]
- [6].Jones SN, Roe AE, Donehower LA, Bradley A, Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53, Nature 378 (1995) 206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- [7].Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev LT, Andreeff M, MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy, Blood 106 (2005) 3150–3159. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Montes R de Oca Luna, Wagner DS, Lozano G, Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53, Nature 378 (1995) 203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- [9].Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C Klein N Fotouhi EA Liu, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, Science 303 (2004) 844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- [10].Xiong S, Mouse models of Mdm2 and Mdm4 and their clinical implications, Chin. J. Cancer 32 (2013) 371–375. doi: 10.5732/cjc.012.10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey B, Hay N, Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer, Cancer Cell 24 (2013) 213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S, Melino G, Classification of cell death: recommendations of the Nomenclature Committee on Cell Death, Cell Death Differ (2005). doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed]
- [13].Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp RA, Knight S Kumar SA Lipton X. Lu F. Madeo W Malorni P. Mehlen G. Nuñez ME. Peter M. Piacentini DC. Rubinsztein Y. Shi H-U. Simon P. Vandenabeele, White J. Yuan B. Zhivotovsky G. Melino G. Kroemer, Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012, Cell Death Differ 19 (2012) 107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Laster SM, Wood JG, Gooding LR, Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis, J. Immunol. Baltim. Md 1950 141 (1988) 2629–2634. [PubMed] [Google Scholar]
- [15].Christofferson DE, Yuan J, Necroptosis as an alternative form of programmed cell death, Curr Opin Cell Biol 22 (2010) 263–8. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J, Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway, Cell 135 (2008) 1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J, Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule, Nat. Immunol 1 (2000) 489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- [18].Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers P Vandenabeele, Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor, J. Exp. Med 187 (1998) 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J, Identification of RIP1 kinase as a specific cellular target of necrostatins, Nat. Chem. Biol 4 (2008) 313–321 doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J, Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury, Nat. Chem. Biol 1 (2005) 112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- [21].Kaczmarek A, Vandenabeele P, Krysko DV, Necroptosis: the release of damage-associated molecular patterns and its physiological relevance, Immunity 38 (2013) 209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- [22].Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P, Regulated necrosis: the expanding network of non-apoptotic cell death pathways, Nat Rev Mol Cell Biol 15 (2014) 135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- [23].Weinlich R, Oberst A, Beere HM, Green DR, Necroptosis in development, inflammation and disease, Nat. Rev. Mol. Cell Biol 18 (2017) 127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- [24].Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD, Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease, Cell 171 (2017) 273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, Stockwell BR, Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell 149 (2012) 1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayır H, Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis, Nat. Chem. Biol 13 (2017) 81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Eagle H, Nutrition Needs of Mammalian Cells in Tissue Culture, Science 122 (1955) 501–504 doi: 10.1126/science.122.3168.501. [DOI] [PubMed] [Google Scholar]
- [28].Eagle H, The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture, J Exp Med 102 (1955) 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Eagle H, Amino acid metabolism in mammalian cell cultures, Science 130 (1959) 432–437. [DOI] [PubMed] [Google Scholar]
- [30].Eagle H, Piez KA, Oyama VI, The biosynthesis of cystine in human cell cultures, J. Biol. Chem 236 (1961) 1425–1428. [PubMed] [Google Scholar]
- [31].Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB, Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione, J Pharmacol Exp Ther 187 (1973) 211–7. [PubMed] [Google Scholar]
- [32].Bannai S, Tsukeda H, Okumura H, Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium, Biochem Biophys Res Commun 74 (1977) 1582–8. [DOI] [PubMed] [Google Scholar]
- [33].Mercille S, Massie B, Induction of apoptosis in nutrient-deprived cultures of hybridoma and myeloma cells, Biotechnol. Bioeng 44 (1994) 1140–1154. doi: 10.1002/bit.260440916. [DOI] [PubMed] [Google Scholar]
- [34].Ratan RR, Baraban JM, Apoptotic death in an in vitro model of neuronal oxidative stress, Clin. Exp. Pharmacol. Physiol 22 (1995) 309–310. [DOI] [PubMed] [Google Scholar]
- [35].Ratan RR, Murphy TH, Baraban JM, Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione, J. Neurosci. Off. J. Soc. Neurosci 14 (1994) 4385– 4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ratan RR, Lee PJ, Baraban JM, Serum deprivation inhibits glutathione depletion-induced death in embryonic cortical neurons: evidence against oxidative stress as a final common mediator of neuronal apoptosis, Neurochem. Int 29 (1996) 153–157. [DOI] [PubMed] [Google Scholar]
- [37].Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ, Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor, J. Neurochem 67 (1996) 566–573. [DOI] [PubMed] [Google Scholar]
- [38].Rosin C, Bates TE, Skaper SD, Excitatory amino acid induced oligodendrocyte cell death in vitro: receptor-dependent and -independent mechanisms, J. Neurochem 90 (2004) 1173–1185. doi: 10.1111/j.1471-4159.2004.02584.x. [DOI] [PubMed] [Google Scholar]
- [39].Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ, Rosenberg PA, 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes, Eur. J. Neurosci 20 (2004) 2049–2058. doi: 10.1111/j.1460-9568.2004.03650.x. [DOI] [PubMed] [Google Scholar]
- [40].Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT, Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress, Neuron 2 (1989) 1547–58. [DOI] [PubMed] [Google Scholar]
- [41].Murphy TH, Schnaar RL, Coyle JT, Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 4 (1990) 1624–1633. [PubMed] [Google Scholar]
- [42].De Brabander M, Van Belle H, Aerts F, Van De Veire R, Geuens G, Protective effect of levamisole and its sulfhydryl metabolite OMPI against cell death induced by glutathione depletion, Int. J. Immunopharmacol 1 (1979) 93–100. doi: 10.1016/0192-0561(79)90011-0. [DOI] [PubMed] [Google Scholar]
- [43].Sunami K, Yamane H, Konishi K, Iguchi H, Nakagawa T, Shibata S, Takayama M, Nakai Y, Role of amino acids in cochlear degeneration: deprivation of cystine induces death of cochlear hair cells of guinea pigs in vitro, Acta Oto-Laryngol. Suppl 538 (1998) 19–21. [DOI] [PubMed] [Google Scholar]
- [44].Rössler OG, Bauer I, Chung H-Y, Thiel G, Glutamate-induced cell death of immortalized murine hippocampal neurons: neuroprotective activity of heme oxygenase-1, heat shock protein 70, and sodium selenite, Neurosci. Lett 362 (2004) 253–257. doi: 10.1016/j.neulet.2004.03.033. [DOI] [PubMed] [Google Scholar]
- [45].Kresge N, Simoni RD, Hill RL, The Discovery of Ubiquitin-mediated Proteolysis by Aaron Ciechanover, Avram Hershko, and Irwin Rose, J. Biol. Chem 281 (2006) e32–e32. [Google Scholar]
- [46].Chin DT, Kuehl L, Rechsteiner M, Conjugation of ubiquitin to denatured hemoglobin is proportional to the rate of hemoglobin degradation in HeLa cells, Proc. Natl. Acad. Sci. U. S. A 79 (1982) 5857–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Goldberg AL, St. John AC, Intracellular Protein Degradation in Mammalian and Bacterial Cells: Part 2, Annu. Rev. Biochem 45 (1976) 747–804. doi: 10.1146/annurev.bi.45.070176.003531. [DOI] [PubMed] [Google Scholar]
- [48].Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA, Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis, Proc. Natl. Acad. Sci. U. S. A 77 (1980) 1783–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Saus J, Timoneda J, Hernández-Yago J, Grisolía S, Scope of the ATP—ubiquitin system for intracellular protein degradation, FEBS Lett 143 (n.d.) 225–227. doi: 10.1016/0014-5793(82)80104-X. [DOI] [PubMed] [Google Scholar]
- [50].Mason HS, Fowlks WL, Peterson E, OXYGEN TRANSFER AND ELECTRON TRANSPORT BY THE PHENOLASE COMPLEX1, J. Am. Chem. Soc 77 (1955) 2914–2915. doi: 10.1021/ja01615a088. [DOI] [Google Scholar]
- [51].Hayaishi O, Katagiri M, Rothberg S, MECHANISM OF THE PYROCATECHASE REACTION, J. Am. Chem. Soc 77 (1955) 5450–5451. doi: 10.1021/ja01625a095. [DOI] [Google Scholar]
- [52].Barrera G, Pizzimenti S, Dianzani MU, Lipid peroxidation: control of cell proliferation, cell differentiation and cell death, Mol. Aspects Med 29 (2008) 1–8. doi: 10.1016/j.mam.2007.09.012. [DOI] [PubMed] [Google Scholar]
- [53].Niki E, Yoshida Y, Saito Y, Noguchi N, Lipid peroxidation: Mechanisms, inhibition, and biological effects, Biochem. Biophys. Res. Commun 338 (2005) 668–676. doi: 10.1016/j.bbrc.2005.08.072. [DOI] [PubMed] [Google Scholar]
- [54].Romero FJ, Bosch-Morell F, Romero MJ, Jareño EJ, Romero B, Marín N, Romá J, Lipid peroxidation products and antioxidants in human disease, Environ. Health Perspect 106 Suppl 5 (1998) 1229–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Comporti M, Saccocci C, Dianzani MU, Effect of CCl-4 in vitro and in vivo on lipid peroxidation of rat liver homogenates and subcellular fractions., Enzymologia 29 (1965) 185–204. [PubMed] [Google Scholar]
- [56].Ghoshal AK, Recknagel RO, Positive evidence of acceleration of lipoperoxidation in rat liver by carbon tetrachloride: in vitro experiments, Life Sci 4 (1965) 1521–1530. doi: 10.1016/0024-3205(65)90173-6. [DOI] [PubMed] [Google Scholar]
- [57].Girotti AW, Mechanisms of lipid peroxidation, J. Free Radic. Biol. Med 1 (1985) 87–95. doi: 10.1016/0748-5514(85)90011-X. [DOI] [PubMed] [Google Scholar]
- [58].Kappus H, 12 - Lipid Peroxidation: Mechanisms, Analysis, Enzymology and Biological Relevance, in: Sies H (Ed.), Oxidative Stress, Academic Press, London, 1985: pp. 273–310. doi: 10.1016/B978-0-12-642760-8.50016-8. [DOI] [Google Scholar]
- [59].Halliwell B, Gutteridge JMC, Oxygen toxicity, oxygen radicals, transition metals and disease, Biochem. J 219 (1984) 1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].D’Autréaux B, Toledano MB, ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis, Nat. Rev. Mol. Cell Biol 8 (2007) 813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- [61].Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN, Characterization of Mammalian Selenoproteomes, Science 300 (2003) 1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- [62].Flohe L, Günzler WA, Schock HH, Glutathione peroxidase: A selenoenzyme, FEBS Lett 32 (1973) 132–134. doi: 10.1016/0014-5793(73)80755-0. [DOI] [PubMed] [Google Scholar]
- [63].Mills GC, Hemoglobin Catabolism I. Glutathione Peroxidase, an Erythrocyte Enzyme Which Protects Hemoglobin from Oxidative Breakdown, J. Biol. Chem 229 (1957) 189–197. [PubMed] [Google Scholar]
- [64].Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C, Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides, Biochim. Biophys. Acta 710 (1982) 197–211. [DOI] [PubMed] [Google Scholar]
- [65].Ursini F, Maiorino M, Gregolin C, The selenoenzyme phospholipid hydroperoxide glutathione peroxidase, Biochim. Biophys. Acta BBA - Gen. Subj 839 (1985) 62–70. doi: 10.1016/0304-4165(85)90182-5. [DOI] [PubMed] [Google Scholar]
- [66].Combs GF, Noguchi T, Scott ML, Mechanisms of action of selenium and vitamin E in protection of biological membranes, Fed. Proc 34 (1975) 2090–2095. [PubMed] [Google Scholar]
- [67].Aitken RJ, Clarkson JS, Cellular basis of defective sperm function and its association with the genesis of reactive oxygen species by human spermatozoa, J. Reprod. Fertil 81 (1987) 459–469. [DOI] [PubMed] [Google Scholar]
- [68].Alvarez JG, Storey BT, Role of glutathione peroxidase in protecting mammalian spermatozoa from loss of motility caused by spontaneous lipid peroxidation, Gamete Res 23 (1989) 77–90. doi: 10.1002/mrd.1120230108. [DOI] [PubMed] [Google Scholar]
- [69].Gunn SA, Gould TC, Anderson WAD, Incorporation of Selenium into Spermatogenic Pathway in Mice., Proc. Soc. Exp. Biol. Med 124 (1967) 1260–1263. doi: 10.3181/00379727-124-31981. [DOI] [PubMed] [Google Scholar]
- [70].Jacobsson SO, Hansson E, Distribution of selenium in mice studied by whole-body autoradiography after injection ff SE-75-sodium selenite, Acta Vet. Scand 6 (1965) 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, Flohé L, Dual Function of the Selenoprotein PHGPx During Sperm Maturation, Science 285 (1999) 1393–1396. doi: 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- [72].Schuckelt R, Brigelius-Flohé R, Maiorino M, Roveri A, Reumkens J, Strabburger W, Ursini F, Wolf B, Flohé L, Phospholipid Hydroperoxide Glutathione Peroxidase is a Seleno-Enzyme Distinct from the Classical Glutathione Peroxidase as Evident from Cdna and Amino Acid Sequencing, Free Radic. Res. Commun 14 (1991) 343–361. doi: 10.3109/10715769109093424. [DOI] [PubMed] [Google Scholar]
- [73].Thomas JP, Geiger PG, Maiorino M, Ursini F, Girotti AW, Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins, Biochim. Biophys. Acta BBA - Lipids Lipid Metab 1045 (1990) 252–260. doi: 10.1016/0005-2760(90)90128-K. [DOI] [PubMed] [Google Scholar]
- [74].Thomas JP, Maiorino M, Ursini F, Girotti AW, Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides., J. Biol. Chem 265 (1990) 454–461. [PubMed] [Google Scholar]
- [75].Ursini F, Bindoli A, The role of selenium peroxidases in the protection against oxidative damage of membranes, Chem. Phys. Lipids 44 (1987) 255–276. doi: 10.1016/0009-3084(87)90053-3. [DOI] [PubMed] [Google Scholar]
- [76].Zhang L, Maiorino M, Roveri A, Ursini F, Phospholipid hydroperoxide glutathione peroxidase: specific activity in tissues of rats of different age and comparison with other glutathione peroxidases, Biochim. Biophys. Acta BBA - Lipids Lipid Metab 1006 (1989) 140–143. doi: 10.1016/0005-2760(89)90336-6. [DOI] [PubMed] [Google Scholar]
- [77].Chambers SJ, Lambert N, Williamson G, Purification of a cytosolic enzyme from human liver with phospholipid hydroperoxide glutathione peroxidase activity, Int. J. Biochem 26 (1994) 1279–1286. [DOI] [PubMed] [Google Scholar]
- [78].Arai M, Imai H, Koumura T, Yoshida M, Emoto K, Umeda M, Chiba N, Nakagawa Y, Mitochondrial Phospholipid Hydroperoxide Glutathione Peroxidase Plays a Major Role in Preventing Oxidative Injury to Cells, J. Biol. Chem 274 (1999) 4924–4933. doi: 10.1074/jbc.274.8.4924. [DOI] [PubMed] [Google Scholar]
- [79].Brigelius-Flohé R, Friedrichs B, Maurer S, Schultz M, Streicher R, Interleukin-1-induced nuclear factor κB activation is inhibited by overexpression of phospholipid hydroperoxide glutathione peroxidase in a human endothelial cell line, Biochem. J 328 (1997) 199–203. doi: 10.1042/bj3280199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hurst R, Korytowski W, Kriska T, Esworthy RS, Chu F-F, Girotti AW, Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4, Free Radic. Biol. Med 31 (2001) 1051–1065. doi: 10.1016/S0891-5849(01)00685-2. [DOI] [PubMed] [Google Scholar]
- [81].Imai H, Sumi D, Sakamoto H, Hanamoto A, Arai M, Chiba N, Nakagawa Y, Overexpression of Phospholipid Hydroperoxide Glutathione Peroxidase Suppressed Cell Death Due to Oxidative Damage in Rat Basophile Leukemia Cells (RBL-2H3), Biochem. Biophys. Res. Commun 222 (1996) 432–438. doi: 10.1006/bbrc.1996.0762. [DOI] [PubMed] [Google Scholar]
- [82].Yagi K, Komura S, Kojima H, Sun Q, Nagata N, Ohishi N, Nishikimi M, Expression of Human Phospholipid Hydroperoxide Glutathione Peroxidase Gene for Protection of Host Cells from Lipid Hydroperoxide-Mediated Injury, Biochem. Biophys. Res. Commun 219 (1996) 486–491. doi: 10.1006/bbrc.1996.0260. [DOI] [PubMed] [Google Scholar]
- [83].Imai H, Nakagawa Y, Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells, Free Radic. Biol. Med 34 (2003) 145–169. doi: 10.1016/S0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- [84].Cheng WH, Ho YS, Ross DA, Valentine BA, Combs GF, Lei XG, Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidases in various tissues, J. Nutr 127 (1997) 1445–1450. doi: 10.1093/jn/127.8.1445. [DOI] [PubMed] [Google Scholar]
- [85].Esworthy RS, Aranda R, Martín MG, Doroshow JH, Binder SW, Chu F-F, Mice with combined disruption of Gpx1 andGpx2 genes have colitis, Am. J. Physiol.-Gastrointest. Liver Physiol 281 (2001) G848–G855. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- [86].Garry MR, Kavanagh TJ, Faustman EM, Sidhu JS, Liao R, Ware C, Vliet PA, Deeb SS, Sensitivity of mouse lung fibroblasts heterozygous for GPx4 to oxidative stress, Free Radic. Biol. Med 44 (2008) 1075–1087. doi: 10.1016/j.freeradbiomed.2007.12.002. [DOI] [PubMed] [Google Scholar]
- [87].Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M, Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent-and AIF-mediated cell death, Cell Metab 8 (2008) 237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- [88].Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA, The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults, Free Radic. Biol. Med 34 (2003) 496–502. doi: 10.1016/S0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- [89].Ran Q, Van Remmen H, Gu M, Qi W, Roberts LJ, Prolla T, Richardson A, Embryonic fibroblasts from Gpx4+/− mice: a novel model for studying the role of membrane peroxidation in biological processes, Free Radic. Biol. Med 35 (2003) 1101–1109. doi: 10.1016/S0891-5849(03)00466-0. [DOI] [PubMed] [Google Scholar]
- [90].Ran Q, Liang H, Ikeno Y, Qi W, Prolla TA, Roberts LJ, Wolf N, VanRemmen H, Richardson A, Reduction in Glutathione Peroxidase 4 Increases Life Span Through Increased Sensitivity to Apoptosis, J. Gerontol. Ser. A 62 (2007) 932–942. doi: 10.1093/gerona/62.9.932. [DOI] [PubMed] [Google Scholar]
- [91].Ran Q, Liang H, Gu M, Qi W, Walter CA, Roberts LJ, Herman B, Richardson A, Remmen HV, Transgenic Mice Overexpressing Glutathione Peroxidase 4 Are Protected against Oxidative Stress-induced Apoptosis, J. Biol. Chem 279 (2004) 55137–55146. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- [92].Kühn H, Borchert A, Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes1 1This article is part of a series of reviews on “Regulatory and Cytoprotective Aspects of Lipid Hydroperoxide Metabolism.” The full list of papers may be found on the homepage of the journal., Free Radic. Biol. Med 33 (2002) 154–172. doi: 10.1016/S0891-5849(02)00855-9. [DOI] [PubMed] [Google Scholar]
- [93].Jin G, Arai K, Murata Y, Wang S, Stins MF, Lo EH, van Leyen K, Protecting Against Cerebrovascular Injury: Contributions of 12/15-Lipoxygenase to Edema Formation After Transient Focal Ischemia, Stroke 39 (2008) 2538–2543. doi: 10.1161/STROKEAHA.108.514927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].van Leyen K, Kim HY, Lee S-R, Jin G, Arai K, Lo EH, Baicalein and 12/15-Lipoxygenase in the Ischemic Brain, Stroke 37 (2006) 3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- [95].Middleton MK, Zukas AM, Rubinstein T, Jacob M, Zhu P, Zhao L, Blair I, Puré E, Identification of 12/15-lipoxygenase as a suppressor of myeloproliferative disease, J. Exp. Med 203 (2006) 2529–2540. doi: 10.1084/jem.20061444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sun D, Funk CD, Disruption of 12/15-Lipoxygenase Expression in Peritoneal Macrophages ENHANCED UTILIZATION OF THE 5-LIPOXYGENASE PATHWAY AND DIMINISHED OXIDATION OF LOW DENSITY LIPOPROTEIN, J. Biol. Chem 271 (1996) 24055–24062. doi: 10.1074/jbc.271.39.24055. [DOI] [PubMed] [Google Scholar]
- [97].Nomura K, Imai H, Koumura T, Arai M, Nakagawa Y, Mitochondrial Phospholipid Hydroperoxide Glutathione Peroxidase Suppresses Apoptosis Mediated by a Mitochondrial Death Pathway, J. Biol. Chem 274 (1999) 29294–29302. doi: 10.1074/jbc.274.41.29294. [DOI] [PubMed] [Google Scholar]
- [98].Sordillo LM, Weaver JA, Cao Y-Z, Corl C, Sylte MJ, Mullarky IK, Enhanced 15-HPETE production during oxidant stress induces apoptosis of endothelial cells, Prostaglandins Other Lipid Mediat 76 (2005) 19–34. doi: 10.1016/j.prostaglandins.2004.10.007. [DOI] [PubMed] [Google Scholar]
- [99].Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G, Molecular characterization of mitochondrial apoptosis-inducing factor, Nature 397 (1999) 441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- [100].Borner C, Monney L, Apoptosis without caspases: an inefficient molecular guillotine?, Cell Death Differ 6 (1999) 497–507. doi: 10.1038/sj.cdd.4400525. [DOI] [PubMed] [Google Scholar]
- [101].Conrad M, Transgenic mouse models for the vital selenoenzymes cytosolic thioredoxin reductase, mitochondrial thioredoxin reductase and glutathione peroxidase 4, Biochim. Biophys. Acta BBA - Gen. Subj 1790 (2009) 1575–1585. doi: 10.1016/j.bbagen.2009.05.001. [DOI] [PubMed] [Google Scholar]
- [102].Fiers W, Beyaert R, Declercq W, Vandenabeele P, More than one way to die: apoptosis, necrosis and reactive oxygen damage, Oncogene 18 (1999) 7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- [103].Loscalzo J, Membrane Redox State and Apoptosis: Death by Peroxide, Cell Metab 8 (2008) 182–183. doi: 10.1016/j.cmet.2008.08.004. [DOI] [PubMed] [Google Scholar]
- [104].Kim WH, Choi CH, Kang SK, Kwon CH, Kim YK, Ceramide Induces Non-Apoptotic Cell Death in Human Glioma Cells, Neurochem. Res 30 (2005) 969–979. doi: 10.1007/s11064-005-6223-y. [DOI] [PubMed] [Google Scholar]
- [105].Mochizuki T, Asai A, Saito N, Tanaka S, Katagiri H, Asano T, Nakane M, Tamura A, Kuchino Y, Kitanaka C, Kirino T, Akt protein kinase inhibits non-apoptotic programmed cell death induced by ceramide, J. Biol. Chem 277 (2002) 2790–2797. doi: 10.1074/jbc.M106361200. [DOI] [PubMed] [Google Scholar]
- [106].Sawada M, Nakashima S, Kiyono T, Nakagawa M, Yamada J, Yamakawa H, Banno Y, Shinoda J, Nishimura Y, Nozawa Y, Sakai N, p53 regulates ceramide formation by neutral sphingomyelinase through reactive oxygen species in human glioma cells, Oncogene 20 (2001) 1368–1378. doi: 10.1038/sj.onc.1204207. [DOI] [PubMed] [Google Scholar]
- [107].Novgorodov SA, Voltin JR, Gooz MA, Li L, Lemasters JJ, Gudz TI, Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis, J. Lipid Res 59 (2018) 312–329. doi: 10.1194/jlr.M080374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Dolma S, Lessnick SL, Hahn WC, Stockwell BR, Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells, Cancer Cell 3 (2003) 285–96. [DOI] [PubMed] [Google Scholar]
- [109].Yang WS, Stockwell BR, Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells, Chem. Biol 15 (2008) 234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Vigil D, Cherfils J, Rossman KL, Der CJ, Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy?, Nat. Rev. Cancer 10 (2010) 842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR, RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels, Nature 447 (2007) 864–8. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR, Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis, Elife 3 (2014) e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gout PW, Buckley AR, Simms CR, Bruchovsky N, Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug, Leukemia 15 (2001) 1633–1640. [DOI] [PubMed] [Google Scholar]
- [114].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR, Regulation of ferroptotic cancer cell death by GPX4, Cell 156 (2014) 317–31. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Pe’er D, Shaik F, Bulinski JC, Stockwell BR, Modulatory profiling identifies mechanisms of small molecule-induced cell death, Proc. Natl. Acad. Sci. U. S. A 108 (2011) E771–E780. doi: 10.1073/pnas.1106149108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Gao M, Monian P, Jiang X, Metabolism and iron signaling in ferroptotic cell death, Oncotarget 6 (2015) 35145–6. doi: 10.18632/oncotarget.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Gao M, Monian P, Quadri N, Ramasamy R, Jiang X, Glutaminolysis and Transferrin Regulate Ferroptosis, Mol Cell 59 (2015) 298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Yang WS, Stockwell BR, Ferroptosis: Death by Lipid Peroxidation, Trends Cell Biol 26 (2016) 165–76. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Fenton HJH, LXXIII.—Oxidation of tartaric acid in presence of iron, J. Chem. Soc. Trans 65 (1894) 899–910. doi: 10.1039/CT8946500899. [DOI] [Google Scholar]
- [120].Dunford HB, Oxidations of iron(II)/(III) by hydrogen peroxide: from aquo to enzyme, Coord. Chem. Rev 233–234 (2002) 311–318. doi: 10.1016/S0010-8545(02)00024-3. [DOI] [Google Scholar]
- [121].Golberg L, Martin LE, Batchelor A, Biochemical changes in the tissues of animals injected with iron. 3. Lipid peroxidation, Biochem. J 83 (1962) 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Dixon SJ, Stockwell BR, The role of iron and reactive oxygen species in cell death, Nat. Chem. Biol 10 (2014) 9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- [123].Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, Yamada K, An essential role for functional lysosomes in ferroptosis of cancer cells, Biochem. J 473 (2016) 769–777. doi: 10.1042/BJ20150658. [DOI] [PubMed] [Google Scholar]
- [124].Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR, Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis, Proc Natl Acad Sci U A 113 (2016) E4966–75. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, Li G, Yang L, Liu W, Min J, Wang F, Characterization of ferroptosis in murine models of hemochromatosis, Hepatol. Baltim. Md 66 (2017) 449–465. doi: 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Cheng Z, Li Y, What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update, Chem. Rev 107 (2007) 748–766. doi: 10.1021/cr040077w. [DOI] [PubMed] [Google Scholar]
- [127].Andrews NC, Schmidt PJ, Iron homeostasis, Annu. Rev. Physiol 69 (2007) 69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- [128].Latunde-Dada GO, Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy, Biochim. Biophys. Acta 1861 (2017) 1893–1900. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- [129].Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X, Ferroptosis is an autophagic cell death process, Cell Res 26 (2016) 1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, Kang R, Tang D, Autophagy promotes ferroptosis by degradation of ferritin, Autophagy 12 (2016) 1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Barradas Manuel A., Jeremy Jamie Y., Kontoghiorghes George J., Mikhailidis Dimitri P., Hoflbrand A Victor, Dandona Paresh, Iron chelators inhibit human platelet aggregation, thromboxane A2 synthesis and lipoxygenase activity, FEBS Lett 245 (2001) 105–109. doi: 10.1016/0014-5793(89)80201-7. [DOI] [PubMed] [Google Scholar]
- [132].Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D, Ferroptosis: process and function, Cell Death Differ 23 (2016) 369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Krainz T, Gaschler MM, Lim C, Sacher JR, Stockwell BR, Wipf P, A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis, ACS Cent. Sci 2 (2016) 653–659. doi: 10.1021/acscentsci.6b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ji J, Baart S, Vikulina AS, Clark RS, Anthonymuthu TS, Tyurin VA, Du L, St Croix CM, Tyurina YY, Lewis J, Skoda EM, Kline AE, Kochanek PM, Wipf P, Kagan VE, Bayır H, Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion, J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 35 (2015) 319–328. doi: 10.1038/jcbfm.2014.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR, Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis, ACS Chem. Biol 13 (2018) 1013–1020. doi: 10.1021/acschembio.8b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Schubert D, Kimura H, Maher P, Growth factors and vitamin E modify neuronal glutamate toxicity., Proc. Natl. Acad. Sci. U. S. A 89 (1992) 8264–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Tan S, Schubert D, Maher P, Oxytosis: A novel form of programmed cell death, Curr. Top. Med. Chem 1 (2001) 497–506. [DOI] [PubMed] [Google Scholar]
- [138].Cao JY, Dixon SJ, Mechanisms of ferroptosis, Cell. Mol. Life Sci 73 (2016) 2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, O’Connell D, Zhang P, Li Y, Gao T, Ren W, Yang Y, miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma, Cell Death Differ (2018). doi: 10.1038/s41418-017-0053-8. [DOI] [PMC free article] [PubMed]
- [140].Cassago A, Ferreira APS, Ferreira IM, Fornezari C, Gomes ERM, Greene KS, Pereira HM, Garratt RC, Dias SMG, Ambrosio ALB, Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism, Proc. Natl. Acad. Sci 109 (2012) 1092–1097. doi: 10.1073/pnas.1112495109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Jin L, Alesi GN, Kang S, Glutaminolysis as a target for cancer therapy, Oncogene 35 (2016) 3619–3625. doi: 10.1038/onc.2015.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Robison WG, Kuwabara T, Bieri JG, The roles of vitamin E and unsaturated fatty acids in the visual process, Retina Phila. Pa 2 (1982) 263–281. [PubMed] [Google Scholar]
- [143].Li Y, Maher P, Schubert D, A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion, Neuron 19 (1997) 453–463. [DOI] [PubMed] [Google Scholar]
- [144].Angeli JPF, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Rådmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Förster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M, Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice, Nat. Cell Biol 16 (2014) 1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR, Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models, J Am Chem Soc 136 (2014) 4551–6. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR, Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death, ACS Chem. Biol 10 (2015) 1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trümbach D, Mao G, Qu F, Bayir H, Füllekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JPF, Conrad M, ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition, Nat. Chem. Biol 13 (2017) 91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Agmon E, Solon J, Bassereau P, Stockwell BR, Modeling the effects of lipid peroxidation during ferroptosis on membrane properties, Sci. Rep 8 (2018) 5155. doi: 10.1038/s41598-018-23408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Sato H, Tamba M, Ishii T, Bannai S, Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins, J. Biol. Chem (1999) 11455–11458. [DOI] [PubMed]
- [150].Ishii T, Bannai S, Sugita Y, Mechanism of growth stimulation of L1210 cells by 2-mercaptoethanol in vitro. Role of the mixed disulfide of 2-mercaptoethanol and cysteine, J. Biol. Chem 256 (1981) 12387–12392. [PubMed] [Google Scholar]
- [151].Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR, Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation, Cell Death Differ 23 (2016) 270–8. doi: 10.1038/cdd.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD, Dual roles of Nrf2 in cancer, Pharmacol. Res 58 (2008) 262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ikeda H, Nishi S, Sakai M, Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis, Biochem. J 380 (2004) 515–521. doi: 10.1042/BJ20031948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Yu H, Guo P, Xie X, Wang Y, Chen G, Ferroptosis, a new form of cell death, and its relationships with tumourous diseases, J. Cell. Mol. Med 21 (2017) 648–657. doi: 10.1111/jcmm.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D, Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells, Hepatol. Baltim. Md 63 (2016) 173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Fan Z, Wirth A-K, Chen D, Wruck CJ, Rauh M, Buchfelder M, Savaskan N, Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis, Oncogenesis 6 (2017) e371. doi: 10.1038/oncsis.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Wang S-J, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W, Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression, Cell Rep 17 (2016) 366–373. doi: 10.1016/j.celrep.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, Chauffert B, Galmiche A, Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors, Anticancer Res 34 (2014) 6417–6422. [PubMed] [Google Scholar]
- [159].Louandre C, Ezzoukhry Z, Godin C, Barbare J-C, Mazière J-C, Chauffert B, Galmiche A, Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib, Int. J. Cancer 133 (2013) 1732–1742. doi: 10.1002/ijc.28159. [DOI] [PubMed] [Google Scholar]
- [160].Panka DJ, Wang W, Atkins MB, Mier JW, The Raf inhibitor BAY 43–9006 (Sorafenib) induces caspase-independent apoptosis in melanoma cells, Cancer Res 66 (2006) 1611–1619. doi: 10.1158/0008-5472.CAN-05-0808. [DOI] [PubMed] [Google Scholar]
- [161].Coriat R, Nicco C, Chéreau C, Mir O, Alexandre J, Ropert S, Weill B, Chaussade S, Goldwasser F, Batteux F, Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo, Mol. Cancer Ther 11 (2012) 2284–2293. doi: 10.1158/1535-7163.MCT-12-0093. [DOI] [PubMed] [Google Scholar]
- [162].Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C, Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5, Cancer Res 66 (2006) 11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- [163].Sauzay C, Louandre C, Bodeau S, Anglade F, Godin C, Saidak Z, Fontaine J-X, Usureau C, Martin N, Molinie R, Pascal J, Mesnard F, Pluquet O, Galmiche A, Protein biosynthesis, a target of sorafenib, interferes with the unfolded protein response (UPR) and ferroptosis in hepatocellular carcinoma cells, Oncotarget 9 (2018) 8400–8414. doi: 10.18632/oncotarget.23843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Larraufie M-H, Yang WS, Jiang E, Thomas AG, Slusher BS, Stockwell BR, Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility, Bioorg. Med. Chem. Lett 25 (2015) 4787–4792. doi: 10.1016/j.bmcl.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, Rodríguez Martínez M, López G, Mattioli M, Realubit R, Karan C, Stockwell BR, Bansal M, Califano A, Elucidating Compound Mechanism of Action by Network Perturbation Analysis, Cell 162 (2015) 441–451. doi: 10.1016/j.cell.2015.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Zilka O, Shah R, Li B, Friedmann Angeli J.P., Griesser M, Conrad M, Pratt DA, On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death, ACS Cent. Sci 3 (2017) 232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ingold KU, Pratt DA, Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective, Chem. Rev 114 (2014) 9022–9046. doi: 10.1021/cr500226n. [DOI] [PubMed] [Google Scholar]
- [168].Evans HM, Bishop KS, On the Existence of a Hitherto Unrecognized Dietary Factor Essential for Reproduction, Science 56 (1922) 650–651. doi: 10.1126/science.56.1458.650.43 [DOI] [PubMed] [Google Scholar]
- [169].Burton GW, Ingold KU, Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function, Acc. Chem. Res 19 (1986) 194–21. doi: 10.1021/ar00127a001. [DOI] [Google Scholar]
- [170].Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL, Conrad M, Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration, Redox Biol 9 (2016) 22–31. doi: 10.1016/j.redox.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M, T cell lipid peroxidation induces ferroptosis and prevents immunity to infection, J. Exp. Med 221 (2015) 555–568. doi: 10.1084/jem.20140857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR, Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis, Nat Chem Biol 12 (2016) 497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Wortmann M, Schneider M, Pircher J, Hellfritsch J, Aichler M, Vegi N, Kölle P, Kuhlencordt P, Walch A, Pohl U, Bornkamm GW, Conrad M, Beck H, Combined Deficiency in Glutathione Peroxidase 4 and Vitamin E Causes Multiorgan Thrombus Formation and Early Death in MiceNovelty and Significance, Circ. Res 113 (2013) 408–471 doi: 10.1161/CIRCRESAHA.113.279984. [DOI] [PubMed] [Google Scholar]
- [174].Angeli JPF, Shah R, Pratt DA, Conrad M, Ferroptosis Inhibition: Mechanisms and Opportunities, Trends Pharmacol. Sci 38 (2017) 489–498. doi: 10.1016/j.tips.2017.02.005. [DOI] [PubMed] [Google Scholar]
- [175].Ulatowski LM, Manor D, Vitamin E and neurodegeneration, Neurobiol. Dis 84 (2015) 78–83. doi: 10.1016/j.nbd.2015.04.002. [DOI] [PubMed] [Google Scholar]
- [176].Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, Kölle P, Tschoep K, Issels RD, Daniel PT, Conrad M, Bornkamm GW, The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death, Oncogene 27 (2008) 1618–1628. doi: 10.1038/sj.onc.1210796. [DOI] [PubMed] [Google Scholar]
- [177].Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, Elia A, Berger T, Cescon DW, Adeoye A, Brüstle A, Molyneux SD, Mason JM, Li WY, Yamamoto K, Wakeham A, Berman HK, Khokha R, Done SJ, Kavanagh TJ, Lam C-W, Mak TW, Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression, Cancer Cell 27 (2015) 211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- [178].Mandal PK, Seiler A, Perisic T, Kölle P, Banjac Canak A., Förster H, Weiss N, Kremmer E, Lieberman MW, Bannai S, Kuhlencordt P, Sato H, Bornkamm GW, Conrad M, System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency, J. Biol. Chem 285 (2010) 22244–22253. doi: 10.1074/jbc.M110.121327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].May JM, Morrow JD, Burk RF, Thioredoxin reductase reduces lipid hydroperoxides and spares alpha-tocopherol, Biochem. Biophys. Res. Commun 292 (2002) 45–49. [DOI] [PubMed] [Google Scholar]
- [180].Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, Ye LF, Tyurina YY, Lin AJ, Shchepinov MS, Chan AY, Peguero-Pereira E, Fomich MA, Daniels JD, Bekish AV, Shmanai VV, Kagan VE, Mahal LK, Woerpel KA, Stockwell BR, FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation, Nat. Chem. Biol 14 (2018) 507–515. doi: 10.1038/s41589-018-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Bentinger M, Brismar K, Dallner G, The antioxidant role of coenzyme Q, Mitochondrion 7 Suppl (2007) S41–50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- [182].Ernster L, Dallner G, Biochemical, physiological and medical aspects of ubiquinone function, Biochim Biophys Acta 1271 (1995) 195–204. [DOI] [PubMed] [Google Scholar]
- [183].Villalba JM, Navas P, Plasma membrane redox system in the control of stress-induced apoptosis, Antioxid Redox Signal 2 (2000) 213–30. doi: 10.1089/ars.2000.2.2-213. [DOI] [PubMed] [Google Scholar]
- [184].Adams JD, Odunze IN, Oxygen free radicals and Parkinson’s disease, Free Radic. Biol. Med 10 (1991) 161–169. [DOI] [PubMed] [Google Scholar]
- [185].Hall ED, Novel inhibitors of iron-dependent lipid peroxidation for neurodegenerative disorders, Ann. Neurol 32 Suppl (1992) S137–142. [DOI] [PubMed] [Google Scholar]
- [186].Praticò D, MY Lee V, Trojanowski JQ, Rokach J, Fitzgerald GA, Increased F2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 12 (1998) 1777–1783. [DOI] [PubMed] [Google Scholar]
- [187].Honig LS, Rosenberg RN, Apoptosis and neurologic disease, Am. J. Med 108 (2000) 317–330. [DOI] [PubMed] [Google Scholar]
- [188].Belaidi AA, Bush AI, Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics, J. Neurochem 139 Suppl 1 (2016) 179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- [189].Do Van B, Gouel F, Jonneaux A, Timmerman K, Gelé P, Pétrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian J-C, Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC, Neurobiol. Dis 94 (2016) 169–178. doi: 10.1016/j.nbd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- [190].Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S, Ferroptosis and cell death mechanisms in Parkinson’s disease, Neurochem. Int 104 (2017) 34–48. doi: 10.1016/j.neuint.2017.01.004. [DOI] [PubMed] [Google Scholar]
- [191].Morris G, Berk M, Carvalho AF, Maes M, Walker AJ, Puri BK, Why should neuroscientists worry about iron? The emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases, Behav. Brain Res 341 (2018) 154–175. doi: 10.1016/j.bbr.2017.12.036. [DOI] [PubMed] [Google Scholar]
- [192].Faux NG, Rembach A, Wiley J, Ellis KA, Ames D, Fowler CJ, Martins RN, Pertile KK, Rumble RL, Trounson B, Masters CL, AIBL Research Group AI Bush, An anemia of Alzheimer’s disease, Mol. Psychiatry 19 (2014) 1227–1234. doi: 10.1038/mp.2013.178. [DOI] [PubMed] [Google Scholar]
- [193].Gangania MK, Batra J, Kushwaha S, Agarwal R, Role of Iron and Copper in the Pathogenesis of Parkinson’s Disease, Indian J. Clin. Biochem. IJCB 32 (2017) 353–356. doi: 10.1007/s12291-016-0614-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Wang J-F, Shao L, Sun X, Young LT, Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia, Bipolar Disord 11 (2009) 523– 529. doi: 10.1111/j.1399-5618.2009.00717.x. [DOI] [PubMed] [Google Scholar]
- [195].Medina-Hernández V, Ramos-Loyo J, Luquin S, Sánchez LFC, García-Estrada J, Navarro-Ruiz A, Increased lipid peroxidation and neuron specific enolase in treatment refractory schizophrenics, J. Psychiatr. Res 41 (2007) 652–658. doi: 10.1016/j.jpsychires.2006.02.010. [DOI] [PubMed] [Google Scholar]
- [196].Selley ML, Increased (E)-4-hydroxy-2-nonenal and asymmetric dimethylarginine concentrations and decreased nitric oxide concentrations in the plasma of patients with major depression, J. Affect. Disord 80 (2004) 249–256. doi: 10.1016/S0165-0327(03)00135-6. [DOI] [PubMed] [Google Scholar]
- [197].Romano A, Serviddio G, Calcagnini S, Villani R, Giudetti AM, Cassano T, Gaetani S, Linking lipid peroxidation and neuropsychiatric disorders: focus on 4-hydroxy-2-nonenal, Free Radic. Biol. Med 111 (2017) 281–293. doi: 10.1016/j.freeradbiomed.2016.12.046. [DOI] [PubMed] [Google Scholar]
- [198].Ross CA, Tabrizi SJ, Huntington’s disease: from molecular pathogenesis to clinical treatment, Lancet Neurol 10 (2011) 83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- [199].Johnson WM, Wilson-Delfosse AL, Mieyal JJ, Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases, Nutrients 4 (2012) 1399–1440. doi: 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Muller M, Leavitt BR, Iron dysregulation in Huntington’s disease, J. Neurochem 130 (n.d.) 328–350. doi: 10.1111/jnc.12739. [DOI] [PubMed] [Google Scholar]
- [201].Lavados M, Guillón M, Mujica MC, Rojo LE, Fuentes P, Maccioni RB, Mild Cognitive Impairment and Alzheimer Patients Display Different Levels of Redox-Active CSF Iron, J. Alzheimers Dis 13 (2008) 225–232. doi: 10.3233/JAD-2008-13211. [DOI] [PubMed] [Google Scholar]
- [202].Qin Y, Zhu W, Zhan C, Zhao L, Wang J, Tian Q, Wang W, Investigation on positive correlation of increased brain iron deposition with cognitive impairment in Alzheimer disease by using quantitative MR R2$ mapping, J. Huazhong Univ. Sci. Technolog. Med. Sci 31 (2011) 578. doi: 10.1007/s11596-011-0493-1. [DOI] [PubMed] [Google Scholar]
- [203].Smith MA, Zhu X, Tabaton M, Liu G, Jr M, W D, Cohen ML, Wang X, Siedlak SL, Dwyer BE, Hayashi T, Nakamura M, Nunomura A, Perry G, Increased Iron and Free Radical Generation in Preclinical Alzheimer Disease and Mild Cognitive Impairment, J. Alzheimers Dis 19 (2010) 363–372. doi: 10.3233/JAD-2010-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Zhu W, Zhong W, Wang W, Zhan C, Wang C, Qi J, Wang J, Lei T, Quantitative MR Phase-corrected Imaging to Investigate Increased Brain Iron Deposition of Patients with Alzheimer Disease, Radiology 253 (2009) 497–504. doi: 10.1148/radiol.2532082324. [DOI] [PubMed] [Google Scholar]
- [205].Butterfield DA, Bader Lange M.L., Sultana R, Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease, Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 1801 (2010) 924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD, Lipid peroxidation in aging brain and Alzheimer’s disease1,2 1Guest Editors: Mark A. Smith and George Perry 2This article is part of a series of reviews on “Causes and Consequences of Oxidative Stress in Alzheimer’s Disease.” The full list of papers may be found on the homepage of the journal., Free Radic. Biol. Med 33 (2002) 620–626. doi: 10.1016/S0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- [207].Sultana R, Perluigi M, Allan Butterfield D., Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain, Free Radic. Biol. Med 62 (2013) 157–169. doi: 10.1016/j.freeradbiomed.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Hofmans S, Berghe TV, Devisscher L, Hassannia B, Lyssens S, Joossens J, Van Der Veken P, Vandenabeele P, Augustyns K, Novel Ferroptosis Inhibitors with Improved Potency and ADME Properties, J. Med. Chem 59 (2016) 2041–2053. doi: 10.1021/acs.jmedchem.5b01641. [DOI] [PubMed] [Google Scholar]
- [209].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144 (2011) 646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [210].Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S, Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis, Free Radic. Biol. Med 108 (2017) 610–626. doi: 10.1016/j.freeradbiomed.2017.04.024. [DOI] [PubMed] [Google Scholar]
- [211].Toyokuni S, Role of iron in carcinogenesis: Cancer as a ferrotoxic disease, Cancer Sci 100 (2009) 9–16. doi: 10.1111/j.1349-7006.2008.01001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Toyokuni S, Okamoto K, Yodoi J, Hiai H, Persistent oxidative stress in cancer, FEBS Lett 358 (1995) 1–3. doi: 10.1016/0014-5793(94)01368-B. [DOI] [PubMed] [Google Scholar]
- [213].Kansanen E, Kuosmanen SM, Leinonen H, Levonen A-L, The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer, Redox Biol 1 (2013) 45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C, Role of Glutathione in Cancer Progression and Chemoresistance, Oxid. Med. Cell. Longev (2013). doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed]
- [215].Arnér ESJ, Holmgren A, The thioredoxin system in cancer, Semin. Cancer Biol 16 (2006) 420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- [216].Schott C, Graab U, Cuvelier N, Hahn H, Fulda S, Oncogenic RAS Mutants Confer Resistance of RMS13 Rhabdomyosarcoma Cells to Oxidative Stress-Induced Ferroptotic Cell Death, Front. Oncol 5 (2015) 131. doi: 10.3389/fonc.2015.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Conrad M, Angeli JPF, Vandenabeele P, Stockwell BR, Regulated necrosis: disease relevance and therapeutic opportunities, Nat. Rev. Drug Discov 15 (2016) 348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B, The Role of Ferroptosis in Cancer Development and Treatment Response, Front. Pharmacol 8 (2018). doi: 10.3389/fphar.2017.00992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X, Emerging Strategies of Cancer Therapy Based on Ferroptosis, Adv. Mater 30 (n.d.) 1704007. doi: 10.1002/adma.201704007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Torti SV, Torti FM, Iron and cancer: more ore to be mined, Nat. Rev. Cancer 13 (2013) 342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG, Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition, ELife 6 (2017). doi: 10.7554/eLife.27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, Hu M, Chan DA, Ethier SP, van ‘t Veer LJ, Polyak K, McCormick F, Gray JW, Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target, Cancer Cell 24 (2013) 450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Huang Y, Dai Z, Barbacioru C, Sadée W, Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance, Cancer Res 65 (2005) 7446–7454. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- [224].Liu X-X, Li X-J, Zhang B, Liang Y-J, Zhou C-X, Cao D-X, He M, Chen G-Q, He J-R, Zhao Q, MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11, FEBS Lett 585 (2011) 1363–1367. doi: 10.1016/j.febslet.2011.04.018. [DOI] [PubMed] [Google Scholar]
- [225].Duffy MJ, Synnott NC, McGowan PM, Crown J, O’Connor D, Gallagher WM, p53 as a target for the treatment of cancer, Cancer Treat. Rev 40 (2014) 1153–1160. doi: 10.1016/j.ctrv.2014.10.004. [DOI] [PubMed] [Google Scholar]
- [226].Bieging KT, Mello SS, Attardi LD, Unravelling mechanisms of p53-mediated tumour suppression, Nat. Rev. Cancer 14 (2014) 359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Valente LJ, Gray DHD, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A, p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa, Cell Rep 3 (2013) 1339–1345. doi: 10.1016/j.celrep.2013.04.012. [DOI] [PubMed] [Google Scholar]
- [228].Cui X, Reactive Oxygen Species: The Achilles’ Heel of Cancer Cells?, Antioxid. Redox Signal 16 (2012) 1212–1214. doi: 10.1089/ars.2012.4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Maillet A, Pervaiz S, Redox Regulation of p53, Redox Effectors Regulated by p53: A Subtle Balance, Antioxid. Redox Signal 16 (2011) 1285–1294. doi: 10.1089/ars.2011.4434. [DOI] [PubMed] [Google Scholar]
- [230].Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W, Ferroptosis as a p53-mediated activity during tumour suppression, Nature 520 (2015) 57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Guo W, Zhao Y, Zhang Z, Tan N, Zhao F, Ge C, Liang L, Jia D, Chen T, Yao M, Li J, He X, Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma, Cancer Lett 312 (2011) 55–61. doi: 10.1016/j.canlet.2011.07.024. [DOI] [PubMed] [Google Scholar]
- [232].Clarke PG, Developmental cell death: morphological diversity and multiple mechanisms, Anat. Embryol. (Berl.) 181 (1990) 195–213. [DOI] [PubMed] [Google Scholar]
- [233].Esposito F, Ammendola R, Faraonio R, Russo T, Cimino F, Redox Control of Signal Transduction, Gene Expression and Cellular Senescence, Neurochem. Res 29 (2004) 617– 628. doi: 10.1023/B:NERE.0000014832.78725.1a. [DOI] [PubMed] [Google Scholar]
- [234].Jabs T, Reactive oxygen intermediates as mediators of programmed cell death in plants and animals, Biochem. Pharmacol 57 (1999) 231–245. doi: 10.1016/S0006-2952(98)00227-5. [DOI] [PubMed] [Google Scholar]
- [235].Ufer C, Wang CC, The Roles of Glutathione Peroxidases during Embryo Development, Front. Mol. Neurosci 4 (2011) 12. doi: 10.3389/fnmol.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Salas-Vidal E, Valencia C, Covarrubias L, Differential tissue growth and patterns of cell death in mouse limb autopod morphogenesis, Dev. Dyn 220 (n.d.) 295–306. doi: 10.1002/dvdy.1108. [DOI] [PubMed] [Google Scholar]
- [237].Schnabel D, Salas-Vidal E, Narváez V, del Rayo Sánchez-Carbente M, Hernández-García D, Cuervo R, Covarrubias L, Expression and regulation of antioxidant enzymes in the developing limb support a function of ROS in interdigital cell death, Dev. Biol 291 (2006) 291–299. doi: 10.1016/j.ydbio.2005.12.023. [DOI] [PubMed] [Google Scholar]
- [238].Distéfano AM, Martin MV, Córdoba JP, Bellido AM, D’Ippólito S, Colman SL, Soto D, Roldán JA, Bartoli CG, Zabaleta EJ, Fiol DF, Stockwell BR, Dixon SJ, Pagnussat GC, Heat stress induces ferroptosis-like cell death in plants, J. Cell Biol 216 (2017) 463–476. doi: 10.1083/jcb.201605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Mushegian AA, Ferroptosis-like cell death in plants, Sci. Signal 10 (2017). doi: 10.1126/scisignal.aan0450. [DOI] [PubMed] [Google Scholar]
- [240].Kuhn H, Banthiya S, van Leyen K, Mammalian lipoxygenases and their biological relevance, Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 1851 (2015) 308–330. doi: 10.1016/j.bbalip.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR, Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways, J. Neurosci. Off. J. Soc. Neurosci 29 (2009) 8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Burczynski ME, Sridhar GR, Palackal NT, Penning TM, The reactive oxygen species-- and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the alpha,beta-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene, J. Biol. Chem 276 (2001) 2890–2897. doi: 10.1074/jbc.M006655200. [DOI] [PubMed] [Google Scholar]
- [243].Lou H, Du S, Ji Q, Stolz A, Induction of AKR1C2 by Phase II Inducers: Identification of a Distal Consensus Antioxidant Response Element Regulated by NRF2, Mol. Pharmacol 69 (2006) 1662–1672. doi: 10.1124/mol.105.019794. [DOI] [PubMed] [Google Scholar]
- [244].Kwon M-Y, Park E, Lee S-J, Chung SW, Kwon M-Y, Park E, Lee S-J, Chung SW, Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death, Oncotarget 6 (2015) 24393–24403. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB, Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism, J Pharmacol Exp Ther 187 (1973) 185–94. [PubMed] [Google Scholar]
- [246].Geiger PG, Thomas JP, Girotti AW, Lethal damage to murine L1210 cells by exogenous lipid hydroperoxides: Protective role of glutathione-dependent selenoperoxidases, Arch. Biochem. Biophys 288 (1991) 671–680. doi: 10.1016/0003-9861(91)90250-M. [DOI] [PubMed] [Google Scholar]
- [247].Wolpaw AJ, Stockwell BR, Chapter Eleven - Multidimensional Profiling in the Investigation of Small-Molecule-Induced Cell Death, in: Ashkenazi A, Wells JA, Yuan J (Eds.), Methods Enzymol, Academic Press, 2014: pp. 265–302. doi: 10.1016/B978-0-12-801430-1.00011-1. [DOI] [PubMed] [Google Scholar]