

Abstract
It is now recognized that many amyloid-forming proteins can associate into multiple fibril structures. Here, we use two-dimensional infrared spectroscopy to study two fibril polymorphs formed by human islet amyloid polypeptide (hIAPP or amylin), which is associated with type 2 diabetes. The polymorphs exhibit different degrees of structural organization near the loop region of hIAPP fibrils. The relative populations of these polymorphs are systematically altered by the presence of macrocyclic peptides which template β-sheet formation at specific sections of the hIAPP sequence. These experiments are consistent with polymorphs that result from competing pathways for fibril formation and that the macrocycles bias hIAPP aggregation toward one pathway or the other. Another macrocyclic peptide that matches the loop region but extends the lag time leaves the relative populations of the polymorphs unaltered, suggesting that the branching point for structural divergence occurs after the lag phase, when the oligomers convert into seeds that template fibril formation. Thus, we conclude that the structures of the polymorphs stem from restricting oligomers along diverging folding pathways, which has implications for drug inhibition, cytotoxicity, and the free energy landscape of hIAPP aggregation.
Graphical Abstract
The spontaneous self-assembly of soluble proteins into amyloid fibrils is implicated in the pathology of various neurodegenerative and endocrine diseases such as Alzheimer’s, Parkinson’s, and type II diabetes.1–3 As a result, it is imperative to understand the mechanisms by which fibrils form in order to determine how their formation can be prevented. However, such studies are complicated by the intrinsic structural heterogeneity of amyloid fibrils.
Amyloid fibril polymorphs, in which a single protein sequence can self-assemble into different tertiary and quaternary structures, are a common occurrence. A wide range of amyloidogenic proteins have been observed to form multiple fiber morphologies, including amyloid-β (Aβ),4–9 tau,10 human islet amyloid polypeptide (hIAPP),11–14 insulin,15 α-synuclein,16–18 transthyretin,19–21 and immunoglobulin light chains.20,22,23 While most studies of amyloid polymorphs have focused on fibrils formed in vitro, structural heterogeneity has been demonstrated for amyloid fibrils extracted from the diseased tissues of both humans and animals.10,20,22 There is even increasing evidence that different fibril morphologies can have different levels of cytotoxicity17 that may account for varying phenotypes, or strains, within each amyloid disease.6,17,19,24 Such results suggest that the search for effective therapeutics must take one of two approaches: either find specific inhibitors of each individual polymorph, as a single small molecule or peptide inhibitor may not be capable of inhibiting the full range of amyloid polymorphs that form in vivo,25 or identify a species common to all polymorphs in the amyloid aggregation pathway that can be targeted for inhibition.
The structural heterogeneities between amyloid fibril polymorphs can be classified according to three main features: differences in the number of protofilaments that comprise the fiber, differences in the relative arrangement of the protofilaments, and differences in the substructure of the protofilaments themselves (varying polypeptide conformations).26 Fibrils with different morphologies can be observed even in the same preparation,13,15 although their formation is highly sensitive to growth conditions. Often, the predominant fibril morphology can be selected in vitro by adjusting pH, temperature, peptide concentration, or buffer composition, including adding additional salts or metal ions.4,5,7 The first two types of polymorphism can be identified using imaging techniques such as transmission electron microscopy (TEM) and atomic force microscope (AFM), which show individual protofilaments that form larger assemblies with twisted or linear arrangements, different symmetries, and varying sizes7–9,11–13,15,16,21,27 However, the third type of polymorphism is generally not apparent to imaging techniques.7,11,15,16
High resolution structural techniques, such as X-ray crystallography and solid-state nuclear magnetic resonance (ssNMR), have resolved atomic-scale differences in the structures of polymorphs.7–9,16,21,28–30 X-ray diffraction studies revealed that even short hexapeptide fragments can form at least eight distinct fibril structures.31,32 In ssNMR, homogeneous samples are required to obtain a precise structure. Because amyloid polymorphism is self-propagating and it is very difficult for one polymorph to convert into another,7,9 a single polymorph can be prepared by carefully controlling the aggregation conditions and sonicating the resulting fibrils to seed additional fibril growth.7,14 Using this approach, a structural model for one polymorph of hIAPP, the system studied here, was determined, which is shown in Figure 1A.14 The model shows a disordered loop connecting an N-terminal β-sheet extending from residues 8 to 17 and a C-terminal β-sheet from residues 28 to 37. While our understanding of the structural and pathological differences of amyloid fibril polymorphs has increased greatly, as summarized in several excellent review articles,24,33–35 the requirement for static, homogeneous samples has precluded mechanistic studies into the origins of the polymorphs with regards to their secondary structures. Detailed molecular-level aggregation mechanisms are critical if a common species is to be identified for inhibition. Many previous mechanistic studies have used AFM or TEM to study how nanometer-sized units assemble into large supramolecular structures such as multi-stranded cables or twisted ribbons.7,15,36 In the case of Aβ, it has been suggested that secondary nucleation events may play an important role in how oligomers and protofilaments co-operatively aggregate into fibrils.37,38 Thus, formation of polymorphic structures is sensitive to starting aggregation states, i.e., monomer vs oligomer concentration, as well as the solution composition.39 Similar observations have been made for hIAPP using hexafluoroisopropanol (HFIP), which disaggregates amyloid fibrils at high concentrations and accelerates nucleation at lower concentrations.40,41 Most atomic-scale information about polymorph assembly comes from molecular dynamics simulations. One set of simulations identified more than 15 fibril structures that differ by which residues end up in a β-sheet conformation.42 The simulation studies emphasized the importance of side-chain interactions as well as the exposure of individual residues to solvent.42,43 Thus, to further develop mechanistic models of polymorph formation, there is a need for experimental data that precisely probes protein secondary structure and can resolve different polymorph structures.
Figure 1.

Structure and 2D IR spectra of Val17- and Ala25-hIAPP. (A) Structure of hIAPP fibrils derived from ssNMR.16 Spectra and diagonal intensity slices of (B) Val17 and (C) Ala25 fibrils. Spectra of Ala25 fibrils (D) diluted 1:3 with unlabeled hIAPP and (E) formed in 2.5% HFIP. The diagonal slices were fit with pseudo-Voigt functions. The isotope peaks fits are highlighted in red and blue, and the unlabeled peak fits are given in green. The fitting routine and parameters are given in the Supporting Information.
In this report, we use 2D IR spectroscopy, isotope labeling, and macrocyclic β-sheet peptides to investigate polymorph formation in hIAPP. Vibrational couplings and environmental frequency shifts make infrared spectroscopy a sensitive probe of protein secondary structure. 13C–18O labeling of the backbone carbonyl provides residue-level specificity by shifting the frequency of the amide I mode by ~66 cm−1.44 If the labeled residue lies in a region that adopts an in-register conformation such as a parallel β-sheet or stacks of turns,45,46 it will create a linear chain of vibrationally coupled oscillators. The frequency of the coupled linear chain is shifted from the local mode frequency by twice the coupling strength,47,48 and its intensity scales with the number of residues involved in the vibrational mode.49 Moreover, since the coupling strength depends on the exact distance and orientation between residues, peak frequencies are sensitive to structural variations. 2D IR has improved spectral resolution and sensitivity over linear IR spectroscopy because the signal strengths scale more strongly with the transition dipole moment, enhancing signals from isotope labels and suppressing solvent backgrounds.50,51 Previously, we used 2D IR spectroscopy and isotope labeling to study a peptide drug inhibitor,52 deamidation of hIAPP,53 and the kinetics of fibril formation.54–56 In all of these studies, the ability to resolve individual residues provided molecular insights that would be difficult to obtain by other techniques.
MATERIALS AND METHODS
hIAPP Synthesis and Purification.
13C18O-Labeled alanine, glycine, phenylalanine, leucine, and valine were prepared from commercially available 13C-labeled Fmoc-protected amino acids (Cambridge Isotope Laboratories) and 18O water in a procedure previously reported.57 hIAPP was synthesized on a CEM Liberty1 automated microwave peptide synthesizer using PAL–PEG–PS resin to produce an amidated C-terminus and incorporating the labeled amino acid where required.58 Standard trifluoroacetic acid cleavage and deprotection protocols were followed. Crude peptide was dissolved overnight in dimethyl sulfoxide to form the disulfide bond between Cys-2 and Cys-7. The peptide was purified via reverse-phase high-performance liquid chromatography (HPLC) with a Vydac C18 column using 0.125% HCl solution (Buffer A) and 0.125% HCl in an 80/20 acetonitrile/water mixture (Buffer B). HPLC purification was run with a gradient of 1% Buffer B per minute.
hIAPP was dissolved in deuterated HFIP (d-HFIP) to fully disaggregate the peptides and deuterate the amide groups. d-HFIP was removed by lyophilization, and aggregation was initiated by dissolving the dry sample in deuterated buffer (20 mM Tris, pD = 7.4). Experiments were performed in D2O/deuterated buffer to eliminate absorption from the water bend. Additional background subtraction is not necessary due to nonlinear scaling of signal strengths in 2D IR spectra.50 The final total peptide concentration was 0.5 mM for pure hIAPP samples or 1 mM for samples containing equimolar hIAPP and macrocycle.
2D IR Spectroscopy.
The 2D IR experimental setup comprises a single box ultrafast amplifier (Solstice, Spectra-Physics) and a commercial optical parametric amplifier (TOPAS, Light Conversion Ltd.). The signal and idler beams are focused onto a AgGaS2 crystal to generate fs mid-IR laser pulses centered at ~6 μm and subsequently split into pump and probe pulses. The pump pulse travels through a germanium-based acousto-optic modulator (AOM) pulse shaper, enabling shot-to-shot delay scanning and phase cycling.57,59 The beams are focused onto the sample at a cross geometry, and the signal emitted in the probe direction is measured using a mercury cadmium telluride (MCT) array detector. The second frequency axis is obtained by Fourier transform of the time-domain trace measured at each array pixel. More detailed description of the data acquisition process can be found elsewhere.57
RESULTS
Shown in Figure 1B,C are 2D IR spectra of hIAPP 13C18O-labeled at Val17 or Ala25, respectively. The conditions used to collect these spectra were previously reported56 and are reproduced here as a control experiment. Val17 and Ala25 were selected because, according to the ssNMR structural model14 and 2D IR line shape measurements,60 they reside on opposite sides of the loop (Figure 1A). Val17 lies in the N-terminal β-sheet while A25 is at the beginning of the C-terminal sheet, although the NMR model suggests that Ala25 does not adopt a β-sheet structure itself.14
Spectra were collected after at least 5 h of aggregation to ensure that the fibrils had fully formed. The largest features in the 2D IR spectra are the pair of peaks at 1620 cm−1, which are the characteristic β-sheet features created by the unlabeled residues.61 Monitoring aggregation kinetics at 1620 cm−1 is complementary to ThT binding assays (Figure S1) but does not provide site-specific information. The isotope-labeled features appear below 1600 cm−1; their lineshapes and their cross peaks to the unlabeled modes provide information on the solvation and structural heterogeneity of the labeled residue, while their frequencies report on the secondary structure at that position.
Val17 exhibits a single-labeled peak at 1578 cm−1 (Figure 1B). The presence of a single, well-resolved peak for Val17 indicates that this residue resides within a well-ordered region of hIAPP and does not have any structural variations measurable with our technique. In contrast, Ala25 exhibits two labeled modes, which appear at 1585 and 1570 cm−1 (Figure 1C). Isotope dilution experiments, in which fibrils are formed using 25% labeled and 75% unlabeled peptides, have a single peak for Ala25 (Figure 1D). Isotope dilution eliminates the effects of coupling, revealing frequency shifts caused by environmental effects such as hydrogen bonding or solvation.50,55,62,63 Thus, observing a single peak upon dilution indicates that the two peaks are caused by two different magnitudes of coupling. Since couplings are dictated by backbone geometry, the peaks must be caused by polymorphs whose structure differ at Ala25, with the 1585 and 1570 cm−1 peaks created by roughly −5 and −12 cm−1 of coupling. At Val17, the structure is the same to within the resolution of the spectra.
Closer inspection of the Ala25 modes rules out a competing explanation for the double peaks: environmental fluctuations. Mobile water molecules within the fibril core have previously been seen to create multiple closely spaced peaks in 2D IR spectra of Aβ fibrils.28,64 However, the spacing between these peaks was much smaller than the 15 cm−1 difference observed here. Additionally, the formation and breaking of hydrogen bonds with this water on the picosecond time scale also created cross peaks between the isotope peaks, which are not seen here between the 1570 and 1585 cm−1 modes (Figure S3, top), nor do cross peaks become apparent as a function of waiting time (Figure S3, bottom). Moreover, if hydration was the source of the two peaks, they would both still be observed in the dilution studies because hydrogen bonding is independent of coupling. Thus, the only other alternative for these two peaks is ruled out by our experiments.
As stated above, the relative amounts of amyloid polymorphs are often influenced by growth conditions.33,65,66 We find that changing the fibril preparation conditions alters the relative intensities of the two Ala25 peaks but has no effect on Val17. In the presence of 2.5% HFIP (Figure 1E), only the 1585 cm−1 peak appears for Ala25. Thus, we conclude that hIAPP fibrils can adopt two different structures in the region of Ala25, whose relative abundance is influenced by growth conditions. Shown in the Supporting Information are an additional 4 residues that we have labeled for 2D IR in this study (Figure S2). Polymorph structures may also be present at Leu27, which exhibits a weak double peak that has also been observed previously.55 This is not unlikely due to the proximity of Leu27 to Ala25. A very weak second peak may also be present at Gly33. However, in both Leu27 and Gly33, the higher frequency isotope peak is approximately 10× weaker than the lower frequency peak, suggesting that the more coupled structure predominates at these residues. Here, we focus on Ala25, where the polymorph signals are most prominent and appear in nearly equal intensities.
It is not unexpected that hIAPP forms polymorphs; many amyloid-forming proteins have been reported to form structural polymorphs. Indeed, under similar conditions to those used here, TEM images exhibit multiple fibril morphologies that differ by characteristic fibril twists, although the differences in secondary and tertiary structure within those fibrils are not known.12,13 Infrared frequencies are sensitive to structural changes at a single residue or across a few strands, while the TEM images capture periodic twists over tens of nanometers. A correlated 2D IR and TEM study could determine whether Ala25 is responsible for the twist of hIAPP fibrils, but specialized and extensive TEM measurements are needed to reach this conclusion. Ala25 is located at the interface between the disordered loop and the C-terminal β-sheet, according to ssNMR.14 Indeed, two models were found to be consistent with the ssNMR constraints, differing primarily in the structure near Ala25. Although these fibers were formed under very different conditions than those used here, structural differences like these could be the origin of the differences in 2D IR signals.
The results presented so far establish a spectroscopic marker for the identification of hIAPP polymorphs via structural differences at Ala25. To gain mechanistic insights into polymorph formation, we make use of macrocyclic peptides specially designed to recognize and interact with specific fragments of the hIAPP sequence. Macrocyclic peptides, which comprise two antiparallel β-strands connected by δ-linked ornithines that mimic β-turns, were designed to recognize β-sheet segments of hIAPP (Figure 2A). In the presence of monomers, they can template β-sheet formation and thereby act as seeds for fibril formation.56 The residues of the recognition strand were chosen to span key regions of hIAPP fibrils (Figure 2B): two macrocycles target primarily the N-terminal β-sheet (hIAPP11–17 and hIAPP15–21), one targets the partially disordered loop (hIAPP21–27), and two target the C-terminal β-sheet (hIAPP26–32 and hIAPP31–37).
Figure 2.

(A) General design of macrocyclic β-sheet peptides, including the “recognition” β-strand, the “blocking” β-strand (with the Hao unit in blue), and the δ-linked ornithine turns (in green). (B) Sequence of hIAPP-labeled by its secondary structure. The black arrows indicate the β-sheet regions determined by ssNMR,14 while the light gray extension of the C-terminal arrow indicates residues that exhibit β-sheet-like coupling in 2D IR spectra. Sequences targeted by the recognition strand of the macrocycles are listed.
In the top row of Figure 3, we show 2D IR spectra of the isotope-labeled region for fibrils of pure Val17 and Val17 mixed with each of the macrocyclic peptides at a 1:1 ratio. Regardless of which macrocycle is present during aggregation, the frequency of the isotope label is unchanged. As mentioned above, the label frequency depends on many factors, including strength of coupling, structural disorder, and environmental effects.25,33,49,67 Thus, the similarity of the peaks indicates that backbone structure of the amyloid fibrils at the N-terminal β-sheet is invariant at Val17. Moreover, the addition of macrocyclic peptides does not appear to change the macroscopic structure of hIAPP fibrils, as exhibited in our previous work with this system.56
Figure 3.

Macrocyclic peptides do not change fibril structure but systematically alter the distribution of Ala25 polymorphs. 2D IR spectra of Val17-labeled hIAPP (top row) and Ala25-labeled hIAPP (middle row) are mixed with stoichiometric macrocycles. A single Val17 peak is observed regardless of which macrocycle is used to seed fibril formation. Ala25 exhibits two peaks that appear at the same frequencies in all sample preparations, but their relative intensities depend on the macrocycle used to seed fibril formation. Diagonal slices of Ala25 (bottom row) show altered ratios of the two polymorph peaks, highlighted in blue and red, which were fit with pseudo-Voigt functions. The fitting routine and parameters are given in the Supporting Information.
Similarly, the frequencies of the Ala25-labeled modes are unaffected by the addition of macrocyclic peptides. Figure 3G–L shows 2D IR spectra collected for Ala25-labeled peptides, all of which have the same two isotope-labeled peaks. Thus, the backbone structure of each polymorph within the C-terminal β-sheet at Ala25 is also unaffected by the macrocycles. We know that the macrocycles interact with hIAPP during aggregation because every macrocycle alters the kinetics of amyloid formation, as previously reported.56 Therefore, the Ala25 and Val17 results indicate that hIAPP fibrils adopt the same fibril structures, regardless of whether they form in the presence of macrocycles or in buffer alone.
While the macrocycles do not change the polymorph structures, they do alter the relative population of each polymorph that is formed during aggregation. Shown in the bottom row of Figure 3 are diagonal intensity slices that illustrate the relative intensities of the two Ala25 isotope peaks. These slices contain comparable information to linear IR spectra but exhibit improved resolution due to the increased sensitivity of 2D IR signals. The slices are fit to pseudo-Voigt functions,68 and the areas of each isotope peak are used to determine the relative populations of each polymorph. For solutions of pure hIAPP lacking any macrocycles (Figure 3M), the two Ala25 peaks have approximately equal peak areas, with a low-frequency to high-frequency ratio of 0.89. This ratio indicates that there are roughly equal populations of polymorphs with the less coupled polymorph being slightly favored. In contrast, four of the five macrocycles clearly promote the formation of one polymorph over the other. Mac11–17 and Mac31–37 both have low-to-high peak area ratios of 0.48, indicating that formation of the less coupled polymorph is strongly favored in their presence. In contrast, Mac15–21 and Mac26–32 have peak area ratios of 1.34 and 1.90, respectively. Thus, formation of the more strongly coupled polymorph is favored in their presence. Only one macrocycle, Mac21–27, results in polymorph populations that are similar to that pure hIAPP sample.
The areas of each polymorph peak illustrate a correlation in the ratio of polymorphs to the region of the hIAPP sequence that the macrocycles target. Mac15–21 and Mac26–32 target sections of the N- and C-terminal β-sheets, respectively, which lie close to the partially disordered loop. They increase the intensity of the lower frequency peak. Mac11–17 and Mac31–37 target residues farther from the partially disordered loop, near the ends of the N- and C-terminal β-sheets, respectively, and favor the formation of the higher frequency peak. As noted above, Mac21–27, which targets the disordered loop region, does not alter the polymorph ratio.
DISCUSSION
The data presented here is consistent with different aggregation pathways for each polymorph which diverge from a common oligomer species, as shown in Figure 4. The mechanism outlined in Figure 4 draws upon previous experimental and computational studies that have established the presence of a stable oligomeric species. The mechanism of hIAPP aggregation has been studied using ThT, CD, and electron paramagnetic resonance measurements.1,69–73 Through a series of 2D IR studies that include measurements on single-, double-, and penta-labeled peptides, alongside TEM, cross-linking, kinetic modeling, and molecular dynamics studies, we have established that hIAPP aggregation proceeds through an oligomeric intermediate prior to forming amyloid fibrils.56,74–76 Oligomers have a structure that is unique from the fibrils: they have in-register peptides likely to be in a parallel β-sheet, that spans residues 23–27 (Figure 4 “FGAIL intermediate”) and may extend as far as residues 18–32. These oligomers are on-pathway intermediates in the formation of hIAPP fibrils.75 To form the fibrils, the FGAIL β-sheets of the oligomers must rearrange into the disordered loop of the fibrils. Breaking the oligomeric β-sheets is energetically unfavorable and creates a free energy barrier. Concentration dependent studies and kinetic modeling puts this barrier at >3 kcal/mol.75 The barrier causes the oligomers to be long-lived, with near-constant populations during the lag time, as observed in kinetics measurement. The oligomers also have a well-defined secondary structure in region Leu12–Ala13.74 We have suggested that the tertiary fold of the oligomer structure resembles the general fold of leucine rich repeat proteins.74 This oligomer structure is also observed in the presence of vesicles, indicating that membrane catalyzed fibril formation follows a similar pathway.76
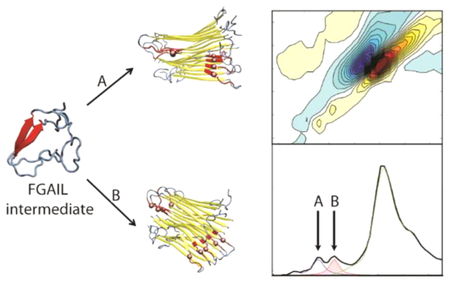
Figure 4.

Schematic mechanism of hIAPP aggregation leading to two different polymorphs. The structures were obtained from REMD simulations of dimeric hIAPP and NMR structures of the fibrils. Folding pathways proceed from disordered monomers through an intermediate with a β-sheet in the FGAIL region (residues 23–27, highlighted in red). The folding pathway diverges after the intermediate, likely via the transition state that nucleates fibril aggregation. In pathway A, the ‡A transition state has the nucleation site close to the FGAIL region (here, residues 15–21, highlighted in yellow). Eventually, fibrils form giving rise to a peak at 1570 cm−1. In pathway B, the ‡B transition state is characterized by the nucleation site far from the FGAIL region (here, residue 31–37) and the FGAIL region in the formed polymorph becomes a part of a partially disordered loop that gives rise to a peak at 1585 cm−1.
Consistent with the experiment, we propose that the oligomer is the branching point from which polymorphs are ultimately formed. Two of four possible pathways for fibril formation are shown in Figure 4. These two pathways diverge starting at the oligomer with each route leading to a different fibril polymorph. Polymorphism is not observed in the structure of the oligomeric intermediate,56,74 indicating that the crucial nucleation steps occur after the intermediate has formed. The difference in the two routes is the location at which the β-sheets of the fibril begin to form: either near the central FGAIL β-sheet, as shown in route A for residues 15–21 (yellow), or near the termini, as shown in route B for residues 31–37. Nucleation at 26–32 and 11–17 (not shown) creates analogous pathways, initially forming short β-sheets either near to, in the case of 26–32, or far from, in the case of 11–17, the FGAIL region.
This model is derived from the macrocycle data, assuming that the macrocycles template β-sheet formation of the residues with which they are designed to interact. The experiments establish that Mac15–21 and Mac26–32, which are designed to template β-sheet formation near the FGAIL region of hIAPP, predominately generate fibrils with the lower frequency peak at 1570 cm−1 (Figure 3O,Q). In contrast, Mac11–17 and Mac31–37 are designed to template β-sheets near the termini of hIAPP and result in fibrils with the higher frequency peak at 1585 cm−1 (Figure 3N,R). When no macrocycles are present, no one pathway is preferred and equal populations of the two polymorphs result.
The proposed mechanism is consistent with previously published kinetics.56 Macrocycles Mac11–17, Mac15–21, Mac26–32, and Mac31–37 increase the rate of fibril formation with a decrease in the lag time. A shorter lag time is consistent with macrocycles initiating β-sheet formation in the oligomer, lowering the transition state free energy, and thereby catalyzing fibril formation. The pathway that is catalyzed is evidenced by increased intensity of the corresponding peak in the 2D IR spectrum. In contrast, macrocycle Mac21–27 slows fibril formation and extends the lag time by 3-times. A longer lag time is consistent with Mac21–27 stabilizing the oligomer to create a higher activation energy. As Mac21–27 overlaps with the FGAIL sequence, it will template and stabilize the oligomer but not the fibrils. Mac21–27 does not alter the ratio of the polymorphs, showing no preference for any of the aggregation pathways. This supports the conclusion that the oligomeric intermediate must be a common step in all four folding pathways and therefore occurs prior to the transition state structures from which the pathways diverge.
The molecular structures of the dimers shown in Figure 4 are taken from replica exchange molecular dynamics (REMD) simulations of hIAPP dimers.56 As described above, the oligomer has been seen experimentally and what is known about its structure mostly matches the illustration. The transition state structure between the oligomer and the fibrils has not been observed experimentally, presumably, because it is present at low concentrations, as is typical for transition state structures. REMD simulations identified potential transition state structures, consisting of a partially or fully disordered FGAIL β-sheet and the beginnings of the β-sheets of the fibril, such as the ones shown in Figure 4. The free energies of these structures were calculated to be higher than both the oligomer and the fibril,75 due to the FGAIL β-sheet being partially disordered and fibril β-sheets not yet fully formed. In our proposed mechanism, the macrocycles seed the formation of one of these transition state structures, thereby lowering the barrier to fibril formation, along that pathway, and catalyzing the formation of a polymorph. Mac21–27 does not match the β-sheets for any of the transition state structures and, so, only acts on the FGAIL oligomer and, thus, slows rather than accelerates fibril formation.
Amyloid fibril formation has historically been considered a nucleation event in which some number of peptides assemble into an oligomer, that once formed, templates the addition of monomers to form fibrils. Models are now being considered that account for long-lived oligomers.37,75 Under the conditions used in our laboratory, standard nucleation models do not fit hIAPP kinetics.75 Formation of the oligomer is consistent with nucleation, but a free energy barrier must be incorporated into the model that represents the structural transformation of the oligomer into the structure of a seed that can template monomers; the oligomer itself cannot seed monomer into fibril. Thus, the oligomer concentrates and aligns the peptides, bringing into proximity the residues that ultimately form the β-sheets found in the fibrils. The length of the lag time is set not by the nucleation of the oligomers themselves but by the subsequent formation of either the N- or C-terminal sheets within the transition state. Thus, the structural transition is more akin to a protein folding event, with a free energy barrier set by the forces between the peptides within the oligomer assembly. The macrocycles enable the structural transformation of the oligomer and thereby lower the free energy of the transition state. The data is not consistent with the macrocycles acting on the monomers directly, because the kinetics are still sigmoidal.3,21,56
It is widely thought that the most cytotoxic species in amyloid formation are oligomeric intermediates.77,78 The oligomeric intermediate discussed here is a probable candidate for cytotoxicity.56 Because hIAPP fibrils are not toxic, the polymorphs themselves are probably not directly responsible for the toxicity of hIAPP. Thus, for hIAPP, we predict that a single inhibitor targeting the oligomer would decrease toxicity and prevent all polymorphs from forming. Other amyloid proteins might diverge directly from the monomer79 in which case each pathway would have a unique oligomeric structure and multiple inhibitors might be needed to prevent fibril formation.
CONCLUSIONS
The critical finding in this Article is that the pathways to different fibril polymorphs diverge from the oligomer state of hIAPP. Polymorphic structures of hIAPP fibrils are known to exist from TEM, NMR, and X-ray experiments,11–14 but our data provides mechanistic insights into the structural origin of fibril polymorphs. We do not know at what point other amyloids diverge, but the strategies provided in this study may find their use in studying polymorphs of other amyloid forming proteins. Aβ has a similar number of residues, a similar fibril fold as hIAPP, a structured intermediate, and polymorphs.7,9,80 2D IR experiments on Aβ similar to those reported here for hIAPP might provide the mechanistic information needed to create a unified theory for its aggregation and cytotoxicity. The branching point of fibril polymorphs have implications for drug inhibition development and cytotoxicity, as discussed above, as well as for probing the free energy landscape that dictates amyloid fibril formation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Randall Massey at the University of Wisconsin Medical School Electron Microscope Facility for help with TEM.
Funding
Support for this research was provided by NIH DK79895 and the NSF CHE-1112188.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00997.
A description of fitting methods, 2D IR kinetics compared to a ThT experiment, 2D IR spectra of different isotope labels, 2D IR spectra of Ala25 measured for different waiting times, and TEM images (PDF)
Notes
The authors declare the following competing financial interest(s): M.T.Z. is co-owner of PhaseTech Spectroscopy, Inc., which sells mid-IR and visible pulse shapers and 2D spectrometers.
REFERENCES
- (1).Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, and Ramamoorthy A (2008) Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc 130, 6424–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Brender JR, Salamekh S, and Ramamoorthy A (2012) Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc. Chem. Res 45, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chiti F, and Dobson CM (2006) Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem 75, 333–366. [DOI] [PubMed] [Google Scholar]
- (4).Klement K, Wieligmann K, Meinhardt J, Hortschansky P, Richter W, and Fändrich M (2007) Effect of Different Salt Ions on the Propensity of Aggregation and on the Structure of Alzheimer’s Aβ(1–40) Amyloid Fibrils. J. Mol. Biol 373, 1321–1333. [DOI] [PubMed] [Google Scholar]
- (5).Kodali R, Williams AD, Chemuru S, and Wetzel R (2010) Aβ(1–40) forms five distinct amyloid structures whose β-sheet contents and fibril stabilities are correlated. J. Mol. Biol 401, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rasmussen J, Mahler J, Beschorner N, Kaeser SA, Häsler LM, Baumann F, Nyström S, Portelius E, Blennow K, Lashley T, Fox NC, Sepulveda-Falla D, Glatzel M, Oblak AL, Ghetti B, Nilsson KPR, Hammarström P, Staufenbiel M, Walker LC, and Jucker M (2017) Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 114, 13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Petkova AT, Leapman RD, Guo Z, Yau W-M, Mattson MP, and Tycko R (2005) Self-Propagating, Molecular-Level Polymorphism in Alzheimer’s β-Amyloid Fibrils. Science 307, 262–265. [DOI] [PubMed] [Google Scholar]
- (8).Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, and Fändrich M (2009) Aβ(1–40) Fibril Polymorphism Implies Diverse Interaction Patterns in Amyloid Fibrils. J. Mol. Biol 386, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Paravastu AK, Leapman RD, Yau W-M, and Tycko R (2008) Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. U. S. A 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).vandenAkker CC, Deckert-Gaudig T, Schleeger M, Velikov KP, Deckert V, Bonn M, and Koenderink GH (2015) Nanoscale Heterogeneity of the Molecular Structure of Individual hIAPP Amyloid Fibrils Revealed with Tip-Enhanced Raman Spectroscopy. Small 11, 4131–4139. [DOI] [PubMed] [Google Scholar]
- (12).Goldsbury CS, Cooper GJS, Goldie KN, Muller SA, Saafi EL, Gruijters WTM, Misur MP, Engel A, Aebi U, and Kistler J (1997) Polymorphic Fibrillar Assembly of Human Amylin. J. Struct. Biol 119, 17–27. [DOI] [PubMed] [Google Scholar]
- (13).Goldsbury C, Goldie K, Pellaud J, Seelig J, Frey P, Müller SA, Kistler J, Cooper GJS, and Aebi U (2000) Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol 130, 352–362. [DOI] [PubMed] [Google Scholar]
- (14).Luca S, Yau WM, Leapman R, and Tycko R (2007) Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid-state NMR. Biochemistry 46, 13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jimenez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM, and Saibil HR (2002) The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. U. S. A 99, 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, and Baldus M (2005) Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. U. S. A 102, 15871–15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, and Melki R (2013) Structural and functional characterization of two alpha-synuclein strains. Nat. Commun 4, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Makky A, Bousset L, Polesel-maris J, and Melki R (2016) Nanomechanical properties of distinct fibrillar polymorphs of the protein α -synuclein. Sci. Rep 6, 37970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bergstrom J, Gustavsson A, Hellman U, Sletten K, Murphy CL, Weiss DT, Solomon A, Olofsson B-O, and Westermark P (2005) Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J. Pathol 206, 224–232. [DOI] [PubMed] [Google Scholar]
- (20).Annamalai K, Gührs K-H, Koehler R, Schmidt M, Michel H, Loos C, Gaffney PM, Sigurdson CJ, Hegenbart U, Schönland S, and Fändrich M (2016) Polymorphism of amyloid fibrils in vivo. Angew. Chem., Int. Ed 55, 4822–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Müller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TPJ, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, and Dobson CM (2013) Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. U. S. A 110, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Annamalai K, Liberta F, Vielberg M, Close W, Lilie H, Guhrs K-H, Schierhorn A, Koehler R, Schmidt A, Haupt C, Hegenbart U, Schonland S, Schmidt M, Groll M, and Fändrich M (2017) Common Fibril Structures Imply Systemically Conserved Protein Misfolding Pathways In Vivo. Angew. Chem., Int. Ed 56, 7510–7514. [DOI] [PubMed] [Google Scholar]
- (23).Close W, Neumann M, Schmidt A, Hora M, Annamalai K, Schmidt M, Reif B, Schmidt V, Grigorieff N, and Fändrich M (2018) Physical basis of amyloid fibril polymorphism. Nat. Commun 9, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Guo JL, and Lee VMY (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med 20, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Duennwald ML, and Shorter J (2010) Countering amyloid polymorphism and drug resistance with minimal drug cocktails. Prion 4, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fändrich M, Meinhardt J, and Grigorieff N (2009) Structural polymorphism of Alzheimer Aβ and other amyloid fibrils. Prion 3, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Green J, Goldsbury C, Mini T, Sunderji S, Frey P, Kistler J, Cooper G, and Aebi U (2003) Full-length rat amylin forms fibrils following substitution of single residues from human amylin. J. Mol. Biol 326, 1147–1156. [DOI] [PubMed] [Google Scholar]
- (28).Ma J, Komatsu H, Kim YS, Liu L, Hochstrasser RM, and Axelsen PH (2013) Intrinsic Structural Heterogeneity and Long-term Maturation of Amyloid β Peptide Fibrils. ACS Chem. Neurosci 4, 1236–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Madine J, Jack E, Stockley PG, Radford SE, Serpell LC, and Middleton DA (2008) Structural insights into the polymorphism of amyloid-like fibrils formed by region 20–29 of amylin revealed by solid-state NMR and X-ray fiber diffraction. J. Am. Chem. Soc 130, 14990–15001. [DOI] [PubMed] [Google Scholar]
- (30).Wu C, Bowers MT, and Shea JE (2010) Molecular structures of quiescently grown and brain-derived polymorphic fibrils of the Alzheimer amyloid Aβ9–40 peptide: A comparison to agitated fibrils. PLoS Comput. Biol 6, e1000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen A, Riekel C, and Eisenberg D (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457. [DOI] [PubMed] [Google Scholar]
- (32).Wiltzius JJW, Landau M, Nelson R, Sawaya MR, Apostol MI, Goldschmidt L, Soriaga AB, Cascio D, Rajashankar K, and Eisenberg D (2009) Molecular mechanisms for protein-encoded inheritance. Nat. Struct. Mol. Biol 16, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tycko R (2014) Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 23, 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tycko R (2015) Amyloid Polymorphism: Structural Basis and Neurobiological Relevance. Neuron 86, 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Adamcik J, and Mezzenga R (2018) Amyloid Polymorphism in the Protein Folding and Aggregation Energy Landscape. Angew. Chem., Int. Ed 57, 8370–8382. [DOI] [PubMed] [Google Scholar]
- (36).Bauer HH, Aebi U, Haner M, Hermann R, Muller M, Arvinte T, and Merkle HP (1995) Architecture and Polymorphism of Fibrillar Supramolecular Assemblies Produced by in vitro Aggregation of Human Calcitonin. J. Struct. Biol 115, 1–15. [DOI] [PubMed] [Google Scholar]
- (37).Auer S (2015) Nucleation of polymorphic amyloid fibrils. Biophys. J 108, 1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jeong JS, Ansaloni A, Mezzenga R, Lashuel HA, and Dietler G (2013) Novel mechanistic insight into the molecular basis of amyloid polymorphism and secondary nucleation during amyloid formation. J. Mol. Biol 425, 1765–1781. [DOI] [PubMed] [Google Scholar]
- (39).Pedersen JS, Andersen CB, and Otzen DE (2010) Amyloid structure - one but not the same: The many levels of fibrillar polymorphism. FEBS J. 277, 4591–4601. [DOI] [PubMed] [Google Scholar]
- (40).Yanagi K, Ashizaki M, Yagi H, Sakurai K, Lee YH, and Goto Y (2011) Hexafluoroisopropanol induces amyloid fibrils of islet amyloid polypeptide by enhancing both hydrophobic and electrostatic interactions. J. Biol. Chem 286, 23959–23966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Padrick SB, and Miranker AD (2002) Islet amyloid: Phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry 41, 4694–4703. [DOI] [PubMed] [Google Scholar]
- (42).Wineman-Fisher V, Atsmon-Raz Y, and Miller Y (2015) Orientations of residues along the β-arch of self-assembled amylin fibril-like structures lead to polymorphism. Biomacromolecules 16, 156–165. [DOI] [PubMed] [Google Scholar]
- (43).Zhao J, Yu X, Liang G, and Zheng J (2011) Structural polymorphism of human islet amyloid polypeptide (hIAPP) oligomers highlights the importance of interfacial residue interactions. Biomacromolecules 12, 210–220. [DOI] [PubMed] [Google Scholar]
- (44).Woys AM, Almeida AM, Wang L, Chiu CC, McGovern M, De Pablo JJ, Skinner JL, Gellman SH, and Zanni MT (2012) Parallel β-sheet vibrational couplings revealed by 2D IR spectroscopy of an isotopically labeled macrocycle: Quantitative benchmark for the interpretation of amyloid and protein infrared spectra. J. Am. Chem. Soc 134, 19118–19128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Colvin MT, Silvers R, Frohm B, Su Y, Linse S, and Griffin RG (2015) High Resolution Structural Characterization of Aβ42 Amyloid Fibrils by Magic Angle Spinning NMR. J. Am. Chem. Soc 137, 7509–7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, and Griffin RG (2016) Atomic Resolution Structure of Monomorphic Aβ 42 Amyloid Fibrils. J. Am. Chem. Soc 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hamm P, and Zanni M (2011) Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, Cambridge. [Google Scholar]
- (48).Buchanan LE, Dunkelberger EB, and Zanni MT (2012) Examining Amyloid Structure and Kinetics with 1D and 2D Infrared Spectroscopy and Isotope Labeling In Protein Folding and Misfolding: Shining Light by Infrared Spectroscopy (Fabian H, and Naumann D, Eds.), pp 217–237, Springer Berlin Heidelberg, Berlin, Heidelberg. [Google Scholar]
- (49).Grechko M, and Zanni MT (2012) Quantification of transition dipole strengths using 1D and 2D spectroscopy for the identification of molecular structures via exciton delocalization: Application to α-helices. J. Chem. Phys 137, 184202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kim YS, and Hochstrasser RM (2009) Applications of 2D IR spectroscopy to peptides, proteins, and hydrogen-bond dynamics. J. Phys. Chem. B 113, 8231–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lomont JP, Ostrander JS, Ho JJ, Petti MK, and Zanni MT (2017) Not All β-Sheets Are the Same: Amyloid Infrared Spectra, Transition Dipole Strengths, and Couplings Investigated by 2D IR Spectroscopy. J. Phys. Chem. B 121, 8935–8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Middleton CT, Marek P, Cao P, Chiu C, Singh S, Woys AM, de Pablo JJ, Raleigh DP, and Zanni MT (2012) Two-dimensional infrared spectroscopy reveals the complex behaviour of an amyloid fibril inhibitor. Nat. Chem 4, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Dunkelberger EB, Buchanan LE, Marek P, Cao P, Raleigh DP, and Zanni MT (2012) Deamidation accelerates amyloid formation and alters amylin fiber structure Deamidation accelerates amyloid formation and alters amylin fiber structure. J. Am. Chem. Soc 134, 12658–12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Strasfeld DB, Ling YL, Shim S-H, and Zanni MT (2008) Tracking Fiber Formation in Human Islet Amyloid Polypeptide with Automated 2D-IR Spectroscopy. J. Am. Chem. Soc 130, 6698–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Shim S-H, Gupta R, Ling YL, Strasfeld DB, Raleigh DP, and Zanni MT (2009) Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc. Natl. Acad. Sci. U. S. A 106, 6614–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Buchanan LE, Dunkelberger EB, Tran HQ, Cheng P-N, Chiu C-C, Cao P, Raleigh DP, de Pablo JJ, Nowick JS, and Zanni MT (2013) Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient β-sheet. Proc. Natl. Acad. Sci. U. S. A 110, 19285–19290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Middleton CT, Woys AM, Mukherjee SS, and Zanni MT (2010) Residue-specific structural kinetics of proteins through the union of isotope labeling, mid-IR pulse shaping, and coherent 2D IR spectroscopy. Methods 52, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Marek P, Woys AM, Sutton K, Zanni MT, and Raleigh DP (2010) Efficient microwave-assisted synthesis of human islet amyloid polypeptide designed to facilitate the specific incorporation of labeled amino acids. Org. Lett 12, 4848–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Karthick Kumar SK, Tamimi A, and Fayer MD (2012) Comparisons of 2D IR measured spectral diffusion in rotating frames using pulse shaping and in the stationary frame using the standard method. J. Chem. Phys 137, 184201. [DOI] [PubMed] [Google Scholar]
- (60).Wang L, Middleton CT, Singh S, Reddy AS, Woys AM, Strasfeld DB, Marek P, Raleigh DP, De Pablo JJ, Zanni MT, and Skinner JL (2011) 2DIR spectroscopy of human amylin fibrils reflects stable β-sheet structure. J. Am. Chem. Soc 133, 16062–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Barth A (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta, Bioenerg. 1767, 1073–1101. [DOI] [PubMed] [Google Scholar]
- (62).Kim YS, Liu L, Axelsen PH, and Hochstrasser RM (2008) Two-dimensional infrared spectra of isotopically diluted amyloid fibrils from Aβ40. Proc. Natl. Acad. Sci. U. S. A 105, 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Petty SA, and Decatur SM (2005) Intersheet rearrangement of polypeptides during nucleation of β-sheet aggregates. Proc. Natl. Acad. Sci. U. S. A 102, 14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Kim YS, Liu L, Axelsen PH, and Hochstrasser RM (2009) 2D IR provides evidence for mobile water molecules in β-amyloid fibrils. Proc. Natl. Acad. Sci. U. S. A 106, 17751–17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Qiang W, Yau WM, and Tycko R (2011) Structural evolution of Iowa mutant β-amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J. Am. Chem. Soc 133, 4018–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Paravastu AK, Petkova AT, and Tycko R (2006) Polymorphic fibril formation by residues 10–40 of the Alzheimer’s β-amyloid peptide. Biophys. J 90, 4618–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Strasfeld DB, Ling YL, Gupta R, Raleigh DP, and Zanni MT (2009) Strategies for extracting structural information from 2D IR spectroscopy of amyloid: Application to islet amyloid polypeptide. J. Phys. Chem. B 113, 15679–15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ida T, Ando M, and Toraya H (2000) Extended pseudo-Voigt function for approximating the Voigt profile. J. Appl. Crystallogr 33, 1311–1316. [Google Scholar]
- (69).Marek P, Abedini A, Song B, Kanungo M, Johnson ME, Gupta R, Zaman W, Wong SS, and Raleigh DP (2007) Aromatic interactions are not required for amyloid fibril formation by islet amyloid polypeptide but do influence the rate of fibril formation and fibril morphology. Biochemistry 46, 3255–3261. [DOI] [PubMed] [Google Scholar]
- (70).Jha S, Sellin D, Seidel R, and Winter R (2009) Amyloidogenic Propensities and Conformational Properties of ProIAPP and IAPP in the Presence of Lipid Bilayer Membranes. J. Mol. Biol 389, 907–920. [DOI] [PubMed] [Google Scholar]
- (71).Jha S, Snell JM, Sheftic SR, Patil SM, Daniels SB, Kolling FW, and Alexandrescu AT (2014) PH dependence of amylin fibrillization. Biochemistry 53, 300–310. [DOI] [PubMed] [Google Scholar]
- (72).Apostolidou M, Jayasinghe SA, and Langen R (2008) Structure of α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem 283, 17205–17210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Konarkowska B, Aitken JF, Kistler J, Zhang S, and Cooper GJS (2006) The aggregation potential of human amylin determines its cytotoxicity towards islet β-cells. FEBS J. 273, 3614–3624. [DOI] [PubMed] [Google Scholar]
- (74).Maj M, Lomont JP, Rich KL, Alperstein AM, and Zanni MT (2018) Site-specific detection of protein secondary structure using 2D IR dihedral indexing: A proposed assembly mechanism of oligomeric hIAPP. Chem. Sci 9, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Serrano AL, Lomont JP, Tu LH, Raleigh DP, and Zanni MT (2017) A Free Energy Barrier Caused by the Refolding of an Oligomeric Intermediate Controls the Lag Time of Amyloid Formation by hIAPP. J. Am. Chem. Soc 139, 16748–16758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Ling YL, Strasfeld DB, Shim S-H, Raleigh DP, and Zanni MT (2009) 2D IR Provides Evidence of an Intermediate in the Membrane-catalyzed Assembly of Diabetic Amyloid. J. Phys. Chem. B 113, 2498–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Butler AE, Janson J, Soeller WC, and Butler PC (2003) Increased β-Cell Apoptosis Prevents Adaptive Increase in β-Cell Mass in Mouse Model of Type 2 Diabetes: Evidence for Role of Islet Amyloid Formation Rather Than Direct Action of Amyloid. Diabetes 52, 2304–2314. [DOI] [PubMed] [Google Scholar]
- (78).Kayed R, Head E, Thompson JL, McIntire T, Milton SC, Cotman CW, and Glabe CG (2003) Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 300, 486–489. [DOI] [PubMed] [Google Scholar]
- (79).Eichner T, and Radford SE (2011) A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Mol. Cell 43, 8–18. [DOI] [PubMed] [Google Scholar]
- (80).Stroud JC, Liu C, Teng PK, and Eisenberg D (2012) Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. U. S. A 109, 7717–7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.