

Abstract
Neurons containing melanin-concentrating hormone (MCH) in the posterior lateral hypothalamus (LH) play an integral role in rapid eye movement sleep (REMs) regulation. As MCH neurons also contain a variety of other neuropeptides [e.g. cocaine- and amphetamine-regulated transcript (CART) and nesfatin-1] and neurotransmitters (e.g. glutamate), the specific neurotransmitter responsible for REMs regulation is not known. We hypothesized that glutamate, the primary fast-acting neurotransmitter in MCH neurons, is necessary for REMs regulation. To test this hypothesis, we deleted vesicular glutamate transporter (Vglut2; necessary for synaptic release of glutamate) specifically from MCH neurons by crossing MCH-Cre mice (expressing Cre recombinase in MCH neurons) with Vglut2flox/flox mice (expressing LoxP-modified alleles of Vglut2), and studied the amounts, architecture and diurnal variation of sleep-wake states during baseline conditions. We then activated the MCH neurons lacking glutamate neurotransmission using chemogenetic methods and tested whether these MCH neurons still promoted REMs. Our results indicate that glutamate in MCH neurons contributes to normal diurnal variability of REMs by regulating the levels of REMs during the dark period, but MCH neurons can promote REMs even in the absence of glutamate.
Keywords: Paradoxical sleep, lateral hypothalamus, conditional knockout, chemogenetics, locomotor activity, body temperature, diurnal rhythms
Introduction
Neurons containing melanin-concentrating hormone (MCH), located in the posterior lateral hypothalamus (LH), play an integral role in sleep-wake regulation (Ferreira et al. 2017; Monti et al. 2013; Torterolo et al. 2011; Yamashita and Yamanaka 2017). MCH neurons express cFos during periods of high rapid eye movement sleep (REMs) following selective REMs deprivation (Verret et al. 2003). The firing rate of MCH neurons is maximal during REMs and minimal during wake suggesting that the MCH neurons may belong to a ‘REMs-on’ type (Hassani et al. 2009). Consistent with these observations, optogenetic and chemogenetic activation of MCH neurons led to substantial increases in REMs without significantly altering other sleep-wake states in mice (Jego et al. 2013; Tsunematsu et al. 2014; Vetrivelan et al. 2016). These evidences strongly suggest that MCH neurons may specifically promote REMs. In addition, loss of MCH neurons produced a significant increase in the diurnal variation of REMs without altering the daily total amounts (Vetrivelan et al. 2016), suggesting that MCH neurons may be necessary for maintaining the diurnal pattern of REMs.
MCH neurons also contain a variety of other neuropeptides including cocaine and amphetamine regulated transcript (CART), nesfatin-1 and a few others (Bittencourt et al. 1992; Cvetkovic et al. 2004; Fujimoto et al. 2017; Hanriot et al. 2007; Jego et al. 2012). In addition to these neuropeptides, almost all (>98%) MCH neurons contain vesicular glutamate transporter 2 (Vglut2), which is necessary for packaging glutamate into synaptic vesicles (Chee et al. 2015a; Mickelsen et al. 2017; Schneeberger et al. 2018). In contrast, none of them express vesicular GABA transporter (Vgat) necessary for packaging GABA (Chee et al. 2015a; Mickelsen et al. 2017) although contrary evidences exist (Jego et al. 2013). Consistent with this, in vitro activation of MCH neurons evokes the release of glutamate but not GABA in the lateral septum, which receives densest input from MCH neurons (Chee et al. 2015a). Thus, MCH neurons may regulate REMs using glutamate or any of the fore-mentioned neuropeptides. We hypothesized that glutamate, the fast-acting neurotransmitter in MCH neurons, might be necessary for REMs regulation. To test this hypothesis, we specifically deleted Vglut2 from MCH neurons and studied the changes in sleep-wake in mice under baseline conditions. We then activated those MCH neurons lacking glutamate neurotransmission and examined if MCH neurons can promote REMs in the absence of glutamate.
In addition to REMs, MCH neurons are also involved in the regulation of spontaneous locomotor activity (LMA) and body temperature (Tb). Loss of MCH neurons induced a robust increase in LMA without increasing the time spent in wake in mice indicating a hyperactivity phenotype (Vetrivelan et al. 2016; Whiddon and Palmiter 2013). Moreover, a moderate hyperthermia (~0.5°C), especially during the dark period, was observed after the loss of MCH neurons (Vetrivelan et al. 2016). Therefore, we studied the changes in LMA and Tb after the deletion of Vglut2 from MCH neurons, as well as after the activation of MCH neurons lacking Vglut2, to assess the potential contribution of glutamate in the regulation of LMA and Tb.
Experimental methods
Generation of transgenic mice lacking Vglut2 in MCH neurons:
We generated MCH-Vglut2KO mice by crossing MCH-Cre mice [expressing Cre recombinase (Cre) specifically in MCH neurons] with Vglut2flox/flox mice (expressing loxP-modified alleles of Vglut2). The F1 offspring heterozygous for Cre and flox-Vglut2 were crossed again with Vglut2flox/flox mice, and only the F2 offspring heterozygous for Cre and homozygous for flox-Vglut2 (MCH-Cre/+;Vglut2flox/flox; henceforth “MCH-Vglut2KO” mice) were used for the experiments. The Cre-mediated recombination event causes deletion of exon2 in the Vglut2 gene, leading to the loss of functional Vglut2 proteins and, thereby, the loss of glutamate release from MCH neurons in the MCH-Vglut2KO mice (Tong et al. 2007).
Both of original lines, MCH-Cre and Vglut2flox/flox mice, have been extensively used in our laboratories. Eutopic expression of Cre in MCH-Cre mice was verified by crossing them with reporter mice (Kong et al. 2010). Selective loss of Vglut2 mRNA and loss of glutamate release from neurons after the Cre-mediated recombination event in the Vglut2flox/flox mice was verified in many previous studies (Krenzer et al. 2011; Tong et al. 2007; Vetrivelan et al. 2009) (Also see Figure 1).
Figure 1: Conditional deletion of Vglut2 in MCH neurons.
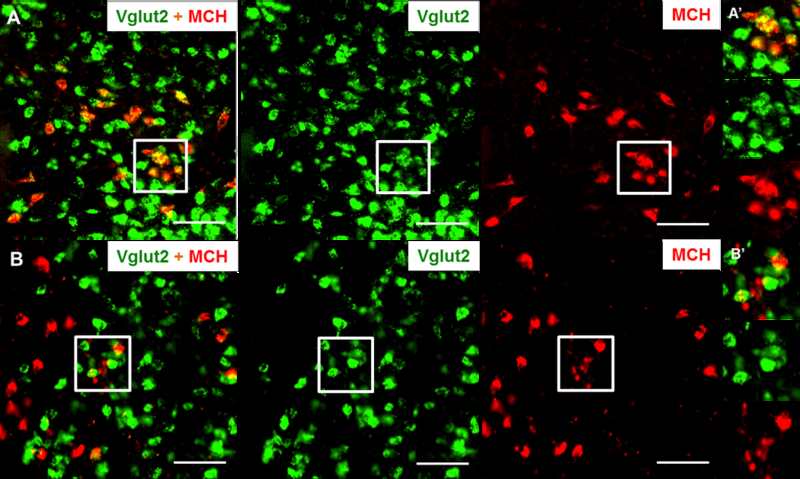
Representative brain sections from a MCH-Cre mouse (A) and a MCH-Vglut2KO mouse (B) labelled for MCH (red) by immunohistochemistry and Vglut2 mRNA (green) by in situ hybridization. More than 90% of the MCH-ir neurons were labelled for Vgult2 mRNA in MCH-Cre mice compared to ~1% in the MCH-Vglut2KO mice. Squares in A and B indicate the region magnified in the A’ and B’. Scale bars - 100 µm.
Animal care:
Adult male MCH-Vglut2KO mice (lacking Vglut2 in MCH neurons) and MCH-Cre mice (controls with intact Vglut2 in MCH neurons) were used in this study. Mice were 10–14 weeks of age and weighed 23–29 g at the time of surgery. All mice were maintained under standard vivarium conditions (including 12:12 h light-dark cycle; lights on at 0700; 150 lux; ambient temperature 22 ± 1°C) with ad libitum access to food and water. Care of the animals met National Institutes of Health standards, as set forth in the Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the BIDMC Institutional Animal Care and Use Committee.
Surgery and recordings:
Under anesthesia (100 mg/kg ketamine + 10 mg/kg xylazine; i.p.), MCH-Vglut2KO (n=11) and MCH-Cre (n=10; controls) mice were stereotaxically injected with AAV8-hSyn-DIO-hM3D(Gq)-mCherry (AAV-hM3Dq; University of North Carolina Vector Core, USA; 260 nl per injections) into the lateral hypothalamic MCH field (anteroposterior: −1.7 mm from bregma, ventral: 4.8 mm from dura, lateral ±0.4 and ±1.1 mm) using a pressure-injection system described previously (Scammell et al. 1998). All mice were then implanted with telemetry transmitters (TLM2-F20EET; Data Sciences International, New Brighton, MN) for recording electroencephalogram (EEG), electromyogram (EMG), Tb and LMA (Vetrivelan et al. 2009; Vetrivelan et al. 2016). Four weeks after the surgery, the mice were transferred to recording chambers and habituated for 5 days. Baseline sleep-wake (EEG/EMG), Tb and LMA were recorded (Dataquest ART 4.1 software; Data Sciences International) for 24 h, following which all mice received i.p. injections of saline (vehicle) or CNO (0.3 mg/kg body weight; MilliporeSigma, USA) at 10:00 AM (3 h after light-onset) and sleep-wake, Tb and LMA were recorded for 9-h post-injections. The order of saline and CNO injections was counterbalanced.
Histology:
After completion of physiological data collection, all mice were injected with CNO (0.3 mg/kg; i.p.) at 10:00AM and were perfused after 3 h with 10% formalin under deep anesthesia (chloral hydrate 700 mg/kg). Harvested brains were post-fixed in 10% formalin overnight and cryoprotected in 20% sucrose solution for 2 days. The brains were then cut on a freezing microtome into 3 series of 40 µm sections.
Two series of sections were processed for double-labelling of Vglut2 mRNA by in situ hybridization and MCH or mCherry (to label hM3Dq-expressing neurons) by immunochemistry. For this, brain sections were mounted on Superfrost Plus slides in RNAse-free conditions and RNAScope hybridization was first performed on those slides using RNAScope Multiplex Fluorescent Reagent Kit V2 (Catalog # 323100, Advanced Cell Diagnostics, Hayward, CA) as described in the company-provided procedure. Briefly, the sections were pretreated with hydrogen peroxide for 20 minutes in room temperature and target retrieval was done by placing the slides in a streamer (Temperature >99°C) for 5 minutes. Then sections were dehydrated in 100% alcohol and air-dried for 5 minutes. Sections were then treated with protease reagent (Protease III, RNAScope) for 30 minutes at 40°C. After rinsing in sterile water sections were incubated in an RNAScope probe for Vglut2-C2 (Mm-Slc17a6-C2, Catalog #319171-C2, Advanced Cell Diagnostics) for 2 hours at 40°C for hybridization. Sections were then subjected 3 amplification steps at 40°C (AMP1-FL and AMP2-FL: 30 mins each; AMP3-FL: 15 mins) followed by incubation in HRP blocker for 15 minutes. Sections were incubated in TSA plus Fluorescein fluorophore (Catalog # NEL741001, Perkin Elmer, Waltham, MA) for 30 mins for visualization (Channel 2 at 488nm) of Vglut2 mRNA. Following Vglut2 mRNA labeling, the sections were processed for immunolabelling. Sections were incubated in Rabbit anti-MCH (Gift from Dr. Eleftheria Maratos-Flier, BIDMC; 1:7500 dilution) or Rabbit anti-DsRed (for labeling mCherry, Clontech, USA; catalog # 632496; 1:5,000 dilution) overnight at 4°C, washed in PBS (2×2 minutes) and incubated in secondary antibody (Alexa fluro555 Donkey anti-Rabbit, Life Technologies, Catalog # A-31572) for 2 h at room temperature. After another round of washes, slides were dried and cover-slipped with Vectashield (Catalog # h-1400, Vector laboratories, Burlingame, CA).
Third series of sections were double-labelled for cFos (neuronal activation) and mCherry (to label the hM3Dq-expressing neurons) by immunohistochemistry (Vetrivelan et al. 2016). Briefly, the brain sections were washed in phosphate buffered saline (PBS; 3× 5 min washes) and incubated overnight in primary antibody, 2.5% Triton-X, Sodium azide and 1% normal horse serum (NHS) in PBS. On the next day, sections were washed again in PBS (3× 5 min washes) and were incubated in appropriate secondary antibody for 2 h. After another round of washes, sections were incubated in avidin-biotin complex (ABC) solution for 1 h and visualized using a 3,3-diaminobenzidine tetrahydrochloride (DAB; 0.06% solution) solution with (to label cFos in black) or without Ni-Co (to label mCherry in brown). All sections were then mounted, dehydrated and coverslipped using Permamount. The following antibodies were used: rabbit anti-DsRed (1:10,000; catalog #632496, Clontech, Mountain View, CA; for labeling mCherry), rabbit anti-cFos (1:30000; catalog #4188, Oncogene Sciences, Uniondale, NY) and biotin-SP-conjugated donkey rabbit IgG (1:1000; catalog #711-065-152, Jackson ImmunoResearch, West Grove, PA). The specificity of MCH and DsRed antibodies was confirmed by the absence of staining in negative controls [in mice with MCH neuron deletion and in wild type mice receiving AAV-hM3Dq respectively (Vetrivelan et al. 2016)].
Data Analysis:
Sleep-wake (EEG/EMG) recordings were manually scored in 12 s epochs as wake, NREMs or REMs using SleepSign software (Kissei Comtec, Nagano, Japan) (Lu et al. 2000). For baseline data, percentages of individual sleep-wake stages (wake, NREMs and REMs) and the number and average duration of their bouts during the 12 h light and 12 h dark periods, independently, as well as, during the entire 24 h were calculated. In addition, percentages of sleep-wake stages in 1-h and 3-h bins were calculated. Total LMA and mean Tb were also calculated for similar time intervals. Diurnal variation index (DVI; referred to as circadian index in previous publications) for all these variables was then calculated using the formula [(Datadark period – Datalight period) /Data24h] as described previously (Vetrivelan et al. 2016). The term ‘circadian index’ was used in the previous studies (Chou et al. 2003; Vetrivelan et al. 2016) for similar quantification of diurnal variation of physiological parameters even though the animals were maintained on entrained conditions (light-dark cycle) during the recordings. Hence, the term ‘diurnal variability index’ used in the current study is more appropriate than ‘circadian index’ to quantify the amplitude of light-dark variation in sleep-wake or other variables.
For chemogenetic experiments, data from the first 9 h after i.p. saline/CNO injections were divided into three 3-h bins, and percentage of time spent, bout numbers and average duration of individual sleep-wake states in each bin were calculated. Additionally, total LMA and mean Tb in these 3-h bins were calculated. Finally, NREMs and REMs latencies were calculated as the time to that stage from saline/CNO injections.
Statistical analysis:
Baseline sleep-wake, LMA and Tb data from MCH-Vglut2KO mice were compared to those from the MCH-Cre mice using an unpaired t-test. In addition, post-saline and post-CNO data from control and MCH-Vglut2KO mice were compared using two-way ANOVA followed by Sidak’s post-hoc tests. All statistics were performed using GraphPad Prism version 7.0 (GraphPad Software, La Jolla, CA) and statistical significance was set at P<0.05.
Results
Histology
We first surveyed the brain sections from MCH-Cre and MCH-Vglut2KO mice for the presence of Vglut2 mRNA in MCH neurons. In the MCH-Cre mice, 90.0±2.8% of MCH-immunoreactive (MCH-ir) neurons were positive for Vglut2 mRNA confirming the presence of Vglut2 in most MCH neurons (Fig. 1A) reported by previous studies (Chee et al. 2015a; Schneeberger et al. 2018). In contrast, virtually no MCH-ir neurons (1.6 ±0.5%) in the MCH-Vglut2KO mice were positive for Vglut2 mRNA (Fig. 1B). Vglut2 expression in the adjacent neurons remained unaffected in the MCH-Vglut2KO mice and numerous Vglut2-labelled neurons were found within the LH as well as in the surrounding regions (e.g. ventromedial hypothalamus). These observations demonstrate that the loss of Vglut2 mRNA in MCH-Vglut2KO mice was primarily restricted to MCH neurons and rule out any non-specific knockdown in other neurons.
Stereotaxic injections of AAV-hM3Dq into the LH of MCH-Cre as well as MCH-Vglut2KO mice (Fig. 2A) resulted in expression of hM3Dq in MCH neurons (65.1±6.9% and 61.3±4.7% of MCH-ir neurons within LH were positive for mCherry in MCH-Cre and MCH-Vglut2KO mice, respectively). Moreover, 92.5±1.8% and 91.9±1.8% of mCherry-ir neurons were positive for MCH in MCH-Cre and MCH-Vglut2KO mice, respectively, indicating high specificity of the AAV-hM3Dq. The mCherry-ir neurons in the MCH-Vglut2KO mice were not labeled for Vglut2 mRNA further confirming the absence of Vglut2 in the MCH neurons transfected with hM3Dq (Fig. 2B). Importantly, i.p. administration of CNO, 3 h prior to perfusion, induced significant cFos expression in all mCherry-ir MCH neurons (Fig. 2C). It is important to note that MCH neurons do not express cFos during baseline conditions and therefore robust cFos observed in all mCherry-ir neurons is a clear indication of MCH neuron activation after CNO.
Figure 2: Chemoactivation of MCH neurons by CNO in MCH-Cre and MCH-Vglut2KO mice.
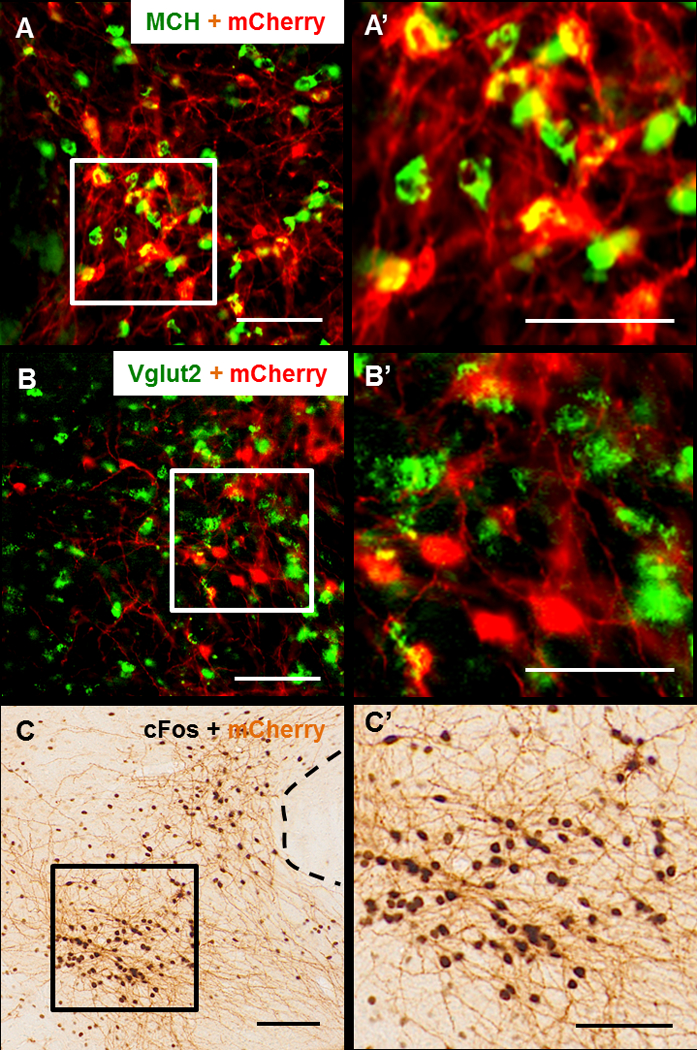
(A, B) Representative brain sections, labelled for mCherry (red) and MCH (green in A, A’) or Vglut2 mRNA (green in B, B’) from a MCH-Vglut2KO mouse injected with AAV-hM3Dq into the LH. AAV injections resulted in specific expression of hM3Dq in MCH neurons (>90% of hM3Dq-mcherry expressing neurons were positive for MCH). These mCherry-ir neurons did not contain Vglut2 mRNA further indicating that hM3Dq was expressed in the MCH neurons lacking Vglut2. (C, C’) Representative brain section from a MCH-Vglut2KO mouse labelled for cFos (black) and mCherry (brown) using DAB immunohistochemistry. This mouse was intraperitoneally injected with CNO (0.3 mg/kg) and sacrificed after 3 h. CNO induced cFos in almost all the mCherry+ neurons indicating their activation. Squares in A, B and C indicate the region magnified in the A’, B’ and C’ respectively. Scale bars - 100 µm.
Sleep-wake changes in MCH-Vglut2KO mice
We first examined whether specific elimination of glutamate neurotransmission from MCH neurons altered spontaneous sleep-wake amounts and architecture in MCH-Vglut2KO mice. We found that MCH-Vglut2KO mice exhibited alterations in the distribution of sleep-wake across light and dark periods although their total sleep and wake amounts remained unchanged. Percentages of wake, NREMs or REMs in the MCH-Vglut2KO mice during the entire 24-h day or specifically during light and dark periods were not significantly different from MCH-Cre mice with intact glutamate neurotransmission (Fig. 3A). Also, bout number and average bout duration of individual sleep-wake states in MCH-Vglut2KO mice during these periods were comparable to MCH-Cre mice (Table 1). However, diurnal variation of REMs, expressed as DVI, was significantly higher in MCH-Vglut2KO mice (142.0±11.92% of MCH-Cre mice; Mann-Whitney U test; p=0.018: Fig. 3B). In addition, we observed that individual MCH-Vglut2KO mice displayed periods of unusually high or low REMs across the 24-h day. Many MCH-Vglut2KO mice displayed complete absence of REMs for 4–6 h during the dark period which was not commonly observed in MCH-Cre mice (Fig. 3C), indicating deficits in nocturnal REMs control in these mice. We therefore analyzed the sleep-wake data from all mice in 1 and 3 h bins to identify these specific periods of REMs dysregulation. We found that the REMs amounts were significantly decreased during the middle of the dark period (between ZT16-21, i.e. 4–9 h from lights-off) (ZT16-18: 1.64±0.34 vs.3.18±0.54 in MCH-Cre mice, ZT19-21: 3.41±0.62 vs. 5.41±0.60 in MCH-Cre, Mann-Whitney U test; P=0.026 and 0.044 respectively: Fig. 3D). On the other hand, the REMs was increased during the 1 h just prior to lights-on (ZT24) along with a corresponding increase in NREMs and a decrease in wake (REMs: 7.09±1.37 vs. 3.31±0.66 in MCH-Cre mice, P=0.029; NREMs: 58.05±5.49 vs. 37.75±4.67 in MCH-Cre mice, P=0.012; Wake: 34.86±6.72 vs. 58.93±4.95 in MCH-Cre mice, P=0.0085; Holm-Sidak Multiple comparisons: Fig 3E). While the percentages of REMs in MCH-Vglut2KO mice were higher during the first 6 h of light period, they did not reach statistical significance (Fig. 3D).
Figure 3: Specific elimination of glutamate neurotransmission from MCH neurons alters diurnal variation of REM sleep.
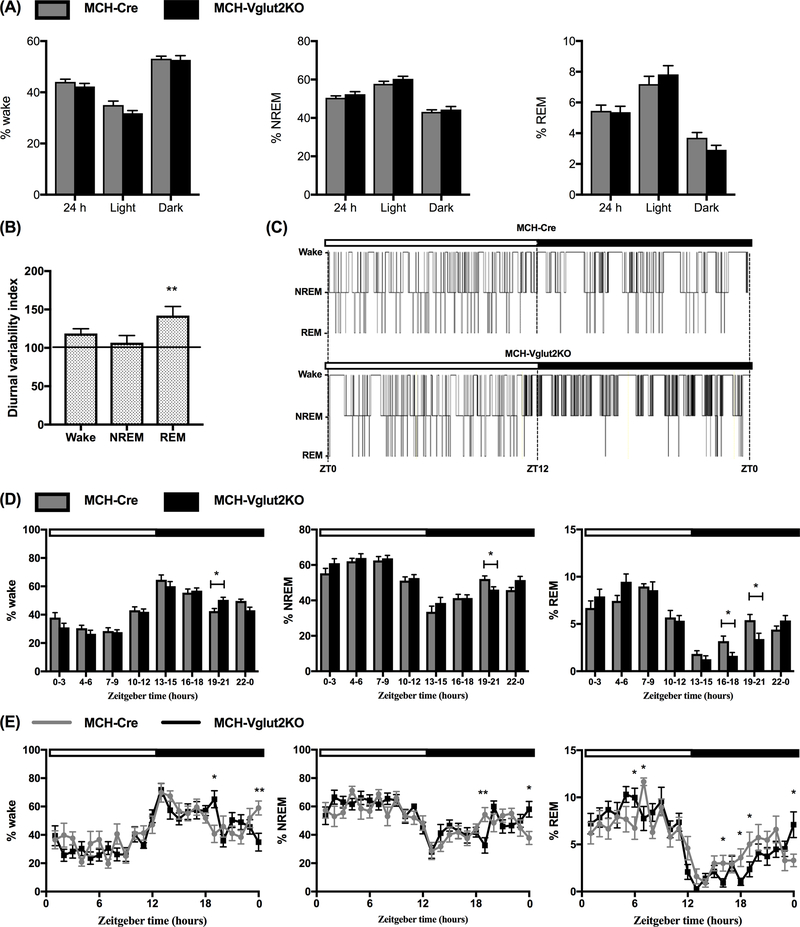
(A) Percentages of wake, non-rapid eye movement sleep (NREMs) and rapid eye movement sleep (REMs) in MCH Cre (n=10; gray bars) and MCH-Vglut2KO mice (n=12; black bars) during the entire 24-h or specifically during the light and dark periods. (B) Diurnal variability index, a measure of diurnal amplitude of wake, NREMs and REMs in MCH-Vglut2KO mice normalized to control data (100%) from MCH-Cre mice (horizontal line) (Mann-Whitney U test; **p < 0.001. (C) Representative 24-h hypnograms from one MCH-Cre (controls, upper trace) and one MCH-Vglut2KO mouse (lower trace). MCH-Vglut2KO mice displayed significant REMs suppression during the dark period compared to MCH-Cre mice. (D) and (E) Percentages of wake, NREMs and REMs in 3-h bins (D) and 1-h bins (E) in MCH-Cre (gray lines or bars) and MCH-Vglut2KO mice (black lines or bars) *p < 0.05; **p < 0.01; Multiple t-test with Holm-Sidak method. In C, D and E, white horizontal bar indicates the light period (ZT0-12) and the black horizontal bar depicts the dark period (ZT12-0). All data are mean ± SEM.
Table 1: Sleep-wake architecture in MCH-Cre and MCH-Vglut2KO mice.
Number of bouts and average bout duration of individual sleep-wake states in MCH-Cre (n=10) and MCH-Vglut2KO mice (n=11). All data are mean ± SEM.
| MCH-Cre (n = 10) | MCH-Vglut2KO (n = 12) | ||
|---|---|---|---|
| Number of bouts | |||
| Wake | Light | 145.50 ± 12.76 | 136.75 ± 11.47 |
| Dark | 141.80 ± 12.54 | 123.83 ± 12.04 | |
| 24 h | 287.70 ± 24.50 | 255.50 ± 19.74 | |
| NREM | Light | 150.70 ± 12.09 | 141.83 ± 10.73 |
| Dark | 144.20 ± 12.33 | 125.08 ± 11.85 | |
| 24 h | 294.90 ± 23.74 | 256.00 ± 17.36 | |
| REM | Light | 46.50 ± 4.27 | 45.92 ± 4.45 |
| Dark | 24.80 ± 3.97 | 17.50 ± 2.69 | |
| 24 h | 71.20 ± 7.57 | 63.42 ± 6.36 | |
| Mean bout duration(s) | |||
| Wake | Light | 104.20 ± 9.17 | 105.33 ± 9.78 |
| Dark | 162.90 ± 14.60 | 190.50 ± 15.56 | |
| 24 h | 134.40 ± 11.45 | 147.25 ± 11.40 | |
| NREM | Light | 174.90 ± 15.26 | 195.92 ± 18.66 |
| Dark | 135.80 ± 9.94 | 167.92 ± 17.88 | |
| 24 h | 155.40 ± 12.13 | 186.83 ± 16.80 | |
| REM | Light | 68.90 ± 4.68 | 76.58 ± 4.33 |
| Dark | 68.40 ± 4.93 | 79.08 ± 6.01 | |
| 24 h | 67.80 ± 2.94 | 76.25 ± 3.90 | |
In contrast to REMs changes, MCH-Vglut2KO mice did not display any major alterations in LMA or Tb. Total LMA and mean Tb either during entire 24-h day or during the individual light- and dark- periods in MCH-Vglut2KO mice were comparable to MCH-Cre mice (Fig. 4). Consistently, the DVI of LMA and Tb also remained unaltered in MCH-Vglut2-KO mice (LMA: 92.40±0.037 and Tb: 87.71±0.21; Mann-Whitney U test; P=0.27 and 0.86, respectively).
Figure 4: Specific elimination of glutamate neurotransmission from MCH neurons did not alter the locomotor activity and body temperature.
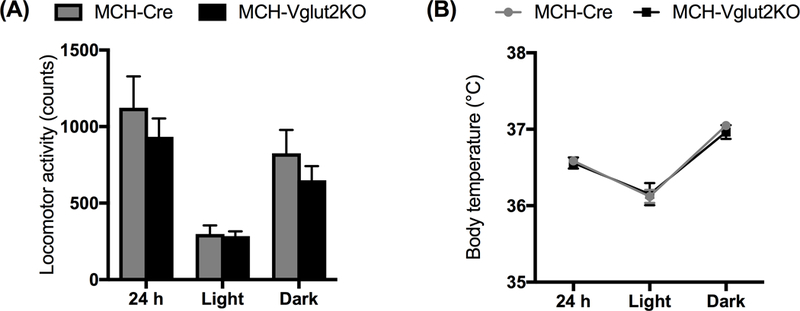
Locomotor activity counts (A) and mean body temperature (B) in MCH-Cre (black bars or lines) and MCH-Vglut2KO (grey bars or lines) during entire 24- h or specifically during the light and dark periods. All data are mean ± SEM.
Collectively, specific deletion of Vglut2 from MCH neurons led to deficits in diurnal variation of REMs by reducing REMs during the dark period but did not alter the daily amounts of REMs or other sleep-wake states.
Activation of MCH neurons lacking glutamatergic neurotransmission
We then tested whether REMs promotion by MCH neurons (Vetrivelan et al. 2016) depended upon glutamate release by activating the MCH neurons in MCH-Vglut2KO mice. Chemoactivation of MCH neurons in MCH-Cre as well as in MCH-Vglut2KO mice significantly increased REMs without altering NREMs or wake. When compared to saline, CNO (0.3 mg/kg; i.p.; 10:00 AM) increased REMs by 81% in MCH-Cre mice and 158% in MCH-Vglut2KO mice during the first 3 h after the injections (two-way ANOVA followed by Sidak’s multiple comparisons test, p=0.0020 and p<0.0001 in MCH-Cre mice and MCH-Vglut2KO, respectively; Fig. 5). REMs during the 4–6 h period after CNO was comparable to saline in MCH-Cre mice, whereas, a significant (83%) increase was observed in MCH-Vglut2KO mice even during this period (two-way ANOVA followed by Sidak’s multiple comparisons test, p=0.037). However, REMs amounts after CNO in MCH-Vglut2KO mice were not significantly different from those after CNO in MCH-Cre mice (Fig 5). The increase in REMs in both sets of mice was due to an increase in the number of REMs bouts and a decrease in REMs latency (Table 2). REM bout durations, on the other hand, were not altered after CNO in either set of mice (Table 2). Finally, neither the total LMA nor the mean Tb was significantly altered after CNO injections in MCH-Cre as well as MCH-Vglut2KO mice (Table 3).
Figure 5: Chemoactivation of MCH neurons increase REM sleep in both MCH-Cre and MCH-Vglut2KO mice.
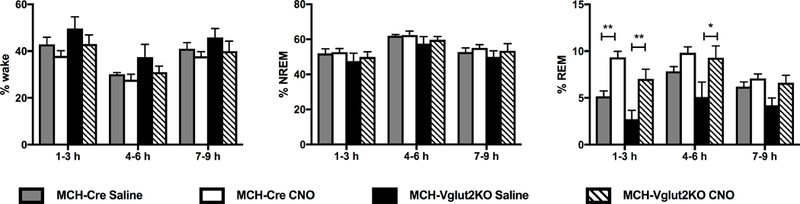
Percentages of wake, NREMs and REMs in 3 h bins after intraperitoneal administration of vehicle (saline) or CNO (0.3 mg/kg, i.p., 10:00 AM) in MCH-Cre (n=10) and MCH-Vglut2KO (n=11) that were injected with AAV-hM3Dq into the lateral hypothalamus. Two-way ANOVA between gene and treatment (1–3 h: interaction F(1,31)=0.0069, p=0.93; treatment F(1,31)=28.65, p<0.0001; gene F(1, 31)=8.98, p=0.0053. 4–6 h: interaction F(1,31)=1.30; treatment F(1,31)=10.00, p=0.0035; gene F(1,31)=2.80, p=0.10) followed by Sidak’s Multiple Comparison test; * p<0.05, ** p<0.01. All data are mean ± SEM.
Table 2: Changes in sleep-wake architecture after chemogenetic activation of MCH neurons in MCH-Cre and MCH-Vglut2KO mice.
Number of bouts and average bout duration of individual sleep-wake states in 3 h bins following intraperitoneal administration of saline or CNO in MCH-Cre mice (n=10) or MCH-Vglut2KO mice (n=8) injected with AAV-hM3Dq into the lateral hypothalamus. All data are mean ± SEM.
| Number of bouts | Mean bout duration(s) | ||||
|---|---|---|---|---|---|
| Post-saline | Post-CNO | Post-saline | Post-CNO | ||
| MCH-Cre (n = 10) | |||||
| Wake | 1–3 h | 30.80 ± 2.60 | 33.80 ± 2.66 | 162.60 ± 21.10 | 129.00 ± 13.93 |
| 4–6 h | 31.70 ± 2.43 | 32.50 ± 2.70 | 107.20 ± 7.46 | 104.70 ± 13.16 | |
| 7–9 h | 33.60 ± 2.08 | 38.60 ± 3.05 | 137.80 ± 14.69 | 107.00 ± 8.01 | |
| NREM | 1–3 h | 31.10 ± 2.53 | 35.60 ± 2.73 | 186.90 ± 11.85 | 165.90 ± 10.45 |
| 4–6 h | 33.40 ± 2.26 | 34.90 ± 2.64 | 207.90 ± 12.94 | 196.10 ± 12.48 | |
| 7–9 h | 34.60 ± 1.99 | 40.70 ± 2.96 | 168.70 ± 10.95 | 155.90 ± 13.42 | |
| REM | 1–3 h | 7.70 ± 1.05 | 13.30 ± 0.75 * | 75.80 ± 5.15 | 74.90 ± 3.70 |
| 4–6 h | 12.20 ± 0.59 | 13.00 ± 1.18 | 69.70 ± 4.25 | 82.00 ± 6.60 | |
| 7–9 h | 9.20 ± 0.73 | 10.50 ± 1.18 | 74.40 ± 5.44 | 78.80 ± 7.26 | |
| MCH-Vglut2KO (n = 8) | |||||
| Wake | 1–3 h | 36.00 ± 3.18 | 26.75 ± 2.54 | 151.71 ± 10.26 | 199.63 ± 46.12 |
| 4–6 h | 43.00 ± 7.86 | 29.63 ± 3.23 | 123.86 ± 37.36 | 146.13 ± 46.22 | |
| 7–9 h | 41.29 ± 5.72 | 29.13 ± 2.85 | 150.86 ± 35.55 | 160.63 ± 26.97 | |
| NREM | 1–3 h | 36.00 ± 2.98 | 28.25 ± 2.58 | 159.43 ± 31.65 | 199.00 ± 15.84 |
| 4–6 h | 45.14 ± 7.30 | 32.63 ± 2.99 | 176.00 ± 38.81 | 213.63 ± 27.30 | |
| 7–9 h | 42.14 ± 5.56 | 30.88 ± 2.81 | 152.00 ± 33.64 | 194.63 ± 20.85 | |
| REM | 1–3 h | 3.57 ± 1.22 | 10.00 ± 1.86 * | 62.57 ± 16.71 | 85.63 ± 8.13 |
| 4–6 h | 7.29 ± 2.28 | 11.63 ± 1.68 | 69.86 ± 13.77 | 89.13 ± 4.70 | |
| 7–9 h | 7.14 ± 1.35 | 9.13 ± 0.90 | 68.71 ± 8.19 | 78.00 ± 4.98 | |
p < 0.05
Mann-Whitney U-test.
Table 3: Changes of locomotor activity and body temperature after chemogenetic activation of MCH neurons in MCH-Cre and MCH-Vglut2KO mice.
Locomotor activity (LMA) and body temperature (Tb) of MCH-Cre (n=10) and MCH-Vglut2KO mice (n=8) in 3 h bins following intraperitoneal administration of saline or CNO in MCH-Cre mice or MCH-Vglut2KO mice injected with AAV-M3Dq into the lateral hypothalamus. All data are mean ± SEM.
| MCH-Cre (n = 10) | MCH-Vglut2KO (n = 8) | ||||
|---|---|---|---|---|---|
| Post-saline | Post-CNO | Post-saline | Post-CNO | ||
| Total LMA (counts) |
1–3 h | 134.59 ± 20.14 | 156.70 ± 15.15 | 107.49 ± 6.96 | 117.17 ± 10.52 |
| 4–6 h | 82.23 ± 10.70 | 72.10 ± 11.70 | 61.43 ± 11.64 | 58.12 ± 9.75 | |
| 7–9 h | 135.83 ± 10.74 | 91.88 ± 9.96 | 87.24 ± 8.87 | 75.24 ± 10.96 | |
| Mean Tb (°C) |
1–3 h | 36.12 ± 0.13 | 36.22 ± 0.10 | 36.47 ± 0.15 | 36.37 ± 0.092 |
| 4–6 h | 35.63 ± 0.089 | 35.66 ± 0.078 | 35.92 ± 0.10 | 35.98 ± 0.11 | |
| 7–9 h | 35.95 ± 0.13 | 35.89 ± 0.087 | 36.24 ± 0.22 | 36.53 ± 0.33 | |
Collectively, chemoactivation of MCH neurons lacking glutamate neurotransmission increased REMs similar to chemoactivation of intact MCH neurons.
Discussion
Using a combination of conditional knockout and chemogenetic activation approaches, we investigated the role of glutamate neurotransmission from MCH neurons in sleep-wake. We found that intact glutamate neurotransmission is crucial for nocturnal REMs control and thereby normal expression of diurnal variation in REMs in mice, but MCH neuron activation can increase REMs even in the absence of glutamate.
Technical considerations
Although the Cre-loxP based conditional knockout and chemogenetic activation methods have been extensively used in many previous studies, combining them was novel and was absolutely necessary to determine the precise physiological role of glutamate in MCH neurons. While selective elimination of glutamate from MCH neurons alone can reveal its functional importance, it is also necessary to activate MCH neurons missing glutamate because MCH neurons may be strongly activated only under specific conditions but not under baseline conditions (Verret et al. 2003). We were able to express the excitatory DREADD, hM3Dq, selectively in MCH neurons in MCH-Vglut2KO mice as Cre expression persists in the MCH-Vglut2KO mice. AAV injections produced expression of hM3Dq in MCH neurons and CNO i.p injections induced robust cFos expression in MCH-Vglut2KO mice (Fig. 2) similar to MCH-Cre mice. Thus, chemogenetic activation of MCH neurons in MCH-Vglut2KO mice is expected to be similar to their activation in MCH-Cre mice except that glutamate would not be released in the former. Although we have not verified the loss of glutamate release, we confirmed the absence of Vglut2 mRNA in MCH neurons in MCH-Vglut2KO mice. However, previous studies from our lab and other labs confirmed that Cre-mediated elimination of Vglut2 mRNA led to loss of glutamate release in various neuronal populations in the hypothalamus and brainstem (Krenzer et al. 2011; Tong et al. 2007; Vetrivelan et al. 2009). Importantly, most MCH neurons express only Vglut2, but not other vesicular glutamate transporters viz. Vglut1 or Vglut3 (only a very small percentage of MCH neurons express Vglut3) (Mickelsen et al. 2017) and therefore loss of Vglut2 was expected to completely abolish glutamate release from these neurons.
Loss of glutamate in MCH neurons leads to deficits in nocturnal REMs control
Our present results in MCH-Vglut2KO mice demonstrate that the loss of glutamate neurotransmission does not alter daily amounts of sleep-wake states, but reduces REMs during the middle of dark period and increases the diurnal variability (DVI) of REMs. Similar changes in REMs were observed after the complete deletion of MCH neurons (Vetrivelan et al. 2016). While MCH neuronal loss caused a significant reduction in REMs during the entire dark period, loss of glutamate decreased REMs for only for 4–6 h. Nevertheless, increase in DVI in MCH-Vglut2-KO mice was comparable to that observed after MCH neuron deletions. Thus, glutamate release from MCH neurons may primarily contribute to the diurnal variability of REMs by decreasing REMs during the dark period. In addition, an increase in wake and decrease in NREM sleep was also observed between ZT19-21, but a rebound increase in NREMs occurred by the end of dark period leading to unaltered total wake and NREM amounts during the overall dark period. These NREMs perturbations in MCH-Vglut2KO mice could be related to the dysregulation of nocturnal REMs because MCH neurons regulate REMs by increasing the depth and adjusting the dynamics of NREMs (Varin et al. 2018). In contrast, MCH neurons have also been implicated in NREMs promotion (Benedetto et al. 2013; Konadhode et al. 2013; Monti et al. 2013; Torterolo et al. 2011).
In addition to increasing DVI of REMs, MCH neuronal loss also produced a robust increase in LMA (ca. 120% increase) and a moderate increase in Tb (0.5°C) in mice, especially during the dark period (Vetrivelan et al. 2016), but these changes were not observed in the MCH-Vglut2KO mice. Thus, glutamatergic neurotransmission from MCH neurons may contribute to the nocturnal REMs control, but not to LMA and Tb regulation. On the other hand, hyperactivity was observed in mice that lack the MCH peptide or MCH-receptors (Chee et al. 2015b; Willie et al. 2008; Zhou et al. 2005). Similarly, mice lacking MCH receptor (MCH-R1) exhibit higher Tb during baseline than wild-type littermates (Ahnaou et al. 2011) and chronic intracerebroventicular (i.c.v.) infusion of MCH induce hypothermia in mice (Glick et al. 2009). Thus, hyperactivity and hyperthermia observed after the loss of MCH neurons could be due to the loss of the MCH peptide but not glutamate.
Is glutamate necessary for the REMs increase following MCH neuron activation?
Although the loss of glutamate transmission from MCH neurons induced deficits in nocturnal REMs, chemoactivation of MCH neurons lacking Vglut2 was still able to increase REMs, indeed more strongly and for longer duration (6 h in MCH-Vglut2KO mice vs. 3 h in MCH-Cre mice). However, the absolute amounts of REMs after CNO in both sets of mice were comparable and thus significant increase in REMs during 4–6 h after CNO was probably contributed by lower REMs after saline in MCH-Vglut2KO mice. Lower REMs after saline injections could be due to altered stress-reactivity in these mice (stress associated with handling and injections) although this needs to be systematically investigated. Nevertheless, REMs increase after chemoactivation of MCH neurons in MCH-Vglut2KO mice indicates that glutamate may be dispensable for REMs promotion by MCH neurons. In addition, previous studies have indicated that global loss of the MCH receptor 1 (only receptor in rodents) cannot prevent the REMs increase following MCH neuron activation (Jego et al. 2013), thus questioning the importance of the MCH peptide in REMs regulation. It is surprising that removal of any one of the two major neuromodulators, which are present in almost all MCH neurons, had no effect on REMs amounts. Nevertheless, MCH neurons contain nesfatin-1, CART and other neuromodulators that could be responsible for the REMs increase. A subpopulation of MCH neurons containing CART expressed higher cFos during REMs rebound compared to the CART-negative MCH neurons suggesting a potential role for CART in REMs promotion (Hanriot et al. 2007; Kitka et al. 2011). However, the CART-positive MCH neurons do not have descending projections to the brainstem REM regulatory structures (Brischoux et al. 2002; Cvetkovic et al. 2003a; Cvetkovic et al. 2004; Cvetkovic et al. 2003b), and so, their activity may not be related to REMs generation but to cortical activation and theta activity during this state. Moreover, i.c.v. administration of CART increased wake rather than REMs (Keating et al. 2010) although this can’t be taken against the role of CART in MCH neurons, as CART-positive neurons are also present in other regions. Similarly, the role nesfatin-1 in MCH neurons is largely unclear as both nesfatin-1 and its antiserum produced significant REMs suppression (Jego et al. 2012; Vas et al. 2013).
In addition to these neuropeptides, MCH neurons are also thought to be GABAergic as the majority of MCH neurons express glutamate acid decarboxylase 1 (GAD 1, also known as GAD 67), an enzyme necessary for GABA synthesis (Sapin et al. 2010). Jego and colleagues reported that optogenetic stimulation of MCH terminals evoked inhibitory postsynaptic currents (IPSCs) in TMN neurons which could be blocked by bicuculline (Jego et al. 2013). These IPSCs persisted even in mice lacking MCH receptors, and inhibition of MCH terminals increased REMs bout durations (Jego et al. 2013). These evidences indicate that MCH neurons may extend REMs bouts by GABA-mediated inhibition of TMN neurons. In contrast, we recently found that MCH neurons may not contain vesicular GABA transporter (Vgat) necessary for packaging GABA into the vesicles for synaptic release (Chee et al. 2015a). While vesicular monoamine transporters (VMAT 1 and 2) can mediate GABAergic neurotransmission, MCH neurons are also devoid of these transporters (Mickelsen et al. 2017). Consistent with this, optogenetic stimulation of MCH neurons did not release GABA in the lateral septum, the brain region that receives the densest MCH projections (Chee et al. 2015a). On the contrary, MCH neurons released glutamate but inhibited the lateral septal neurons by feed forward inhibition i.e. by evoking GABA release from other inputs to the lateral septum (Chee et al. 2015a). Our current findings indicate that loss of glutamate cannot prevent the REMs increase following MCH neuron activation. It is likely that each of these neuromodulators may be involved in REMs promotion and thus loss of one of them is compensated by others. In support of this hypothesis, MCH-R1 KO mice had higher REMs even at baseline conditions (Adamantidis et al. 2008) indicating a compensatory response. However, it will be ultimately necessary to study the REMs changes after acute elimination of glutamate and other neuromodulators from MCH neurons in adult stage in order to further understand the complex interplay between them.
Limitations of the study
We did not investigate homeostatic control of REMs in MCH-Vglut2KO mice. Considering the high cFos expression in MCH neurons during REMs rebound (Verret et al. 2003), it is possible that glutamate in these neurons may contribute to REMs homeostasis. However, increased REMs after chemoactivation of MCH neurons lacking glutamate in the current study suggest the contrary. Nevertheless, chemoactivation may not entirely mimic the activity pattern of MCH neurons during REMs rebound and therefore REMs homeostasis in MCH-Vglut2KO mice needs to be investigated in future studies.
Conclusions
Our current results indicate that glutamate in the MCH neurons may regulate nocturnal REMs and thereby contribute to the normal day-night variation in REMs, but it is not absolutely necessary for REMs induction by MCH neurons. Taken together with previous studies, these data suggest that REMs regulation by MCH neurons may not be solely dependent upon any single neurotransmitter or peptide, but each of them may contribute and play synergistic roles in this function.
Acknowledgements
We thank Quan Ha, Minh Ha and Celia Gagliardi for excellent technical assistance and Dr. Daniel Kroeger for his help with histology imaging. We also thank Dr. Eleftheria Maratos-Flier (Department of Medicine, Beth Israel Deaconess Medical Center, Boston) for providing the rabbit anti-MCH antibody.
Funding
This work was supported by National Institutes of Health Grants [R21-NS074205, R01- NS088482 (to RV)].
Footnotes
Compliance with Ethical Standards
Conflict of Interest
This was not an industry-supported study. The authors declare that they have no competing interests.
Research involving Human Participants and/or Animals
Animals
Statement on the animal welfare and Ethical approval
Care of the animals met National Institutes of Health standards, as set forth in the Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the BIDMC Institutional Animal Care and Use Committee.
Informed consent
Not applicable
References
- Adamantidis A et al. (2008) Sleep architecture of the melanin-concentrating hormone receptor 1-knockout mice Eur J Neurosci 27:1793–1800 doi: 10.1111/j.1460-9568.2008.06129.x [DOI] [PubMed] [Google Scholar]
- Ahnaou A, Dautzenberg FM, Huysmans H, Steckler T, Drinkenburg WH (2011) Contribution of melanin-concentrating hormone (MCH1) receptor to thermoregulation and sleep stabilization: evidence from MCH1 (−/−) mice Behav Brain Res 218:42–50 doi: 10.1016/j.bbr.2010.11.019 [DOI] [PubMed] [Google Scholar]
- Benedetto L, Rodriguez-Servetti Z, Lagos P, D’Almeida V, Monti JM, Torterolo P (2013) Microinjection of melanin concentrating hormone into the lateral preoptic area promotes non-REM sleep in the rat Peptides 39:11–15 doi: 10.1016/j.peptides.2012.10.005 [DOI] [PubMed] [Google Scholar]
- Bittencourt JC et al. (1992) The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization J Comp Neurol 319:218–245 doi: 10.1002/cne.903190204 [DOI] [PubMed] [Google Scholar]
- Brischoux F, Cvetkovic V, Griffond B, Fellmann D, Risold PY (2002) Time of genesis determines projection and neurokinin-3 expression patterns of diencephalic neurons containing melanin-concentrating hormone Eur J Neurosci 16:1672–1680 [DOI] [PubMed] [Google Scholar]
- Chee MJ, Arrigoni E, Maratos-Flier E (2015a) Melanin-concentrating hormone neurons release glutamate for feedforward inhibition of the lateral septum J Neurosci 35:3644–3651 doi: 10.1523/JNEUROSCI.4187-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee MJ et al. (2015b) Melanin-concentrating hormone is necessary for olanzapine-inhibited locomotor activity in male mice Eur Neuropsychopharmacol 25:1808–1816 doi: 10.1016/j.euroneuro.2015.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J (2003) Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms J Neurosci 23:10691–10702 doi:23/33/10691 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetkovic V, Brischoux F, Griffond B, Bernard G, Jacquemard C, Fellmann D, Risold PY (2003a) Evidence of melanin-concentrating hormone-containing neurons supplying both cortical and neuroendocrine projections Neuroscience 116:31–35 [DOI] [PubMed] [Google Scholar]
- Cvetkovic V, Brischoux F, Jacquemard C, Fellmann D, Griffond B, Risold PY (2004) Characterization of subpopulations of neurons producing melanin-concentrating hormone in the rat ventral diencephalon J Neurochem 91:911–919 doi: 10.1111/j.1471-4159.2004.02776.x [DOI] [PubMed] [Google Scholar]
- Cvetkovic V, Poncet F, Fellmann D, Griffond B, Risold PY (2003b) Diencephalic neurons producing melanin-concentrating hormone are influenced by local and multiple extra-hypothalamic tachykininergic projections through the neurokinin 3 receptor Neuroscience 119:1113–1145 [DOI] [PubMed] [Google Scholar]
- Ferreira JGP, Bittencourt JC, Adamantidis A (2017) Melanin-concentrating hormone and sleep Curr Opin Neurobiol 44:152–158 doi: 10.1016/j.conb.2017.04.008 [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Fukuda S, Sakamoto H, Takata J, Sawamura S (2017) Neuropeptide glutamic acid-isoleucine (NEI)-induced paradoxical sleep in rats Peptides 87:28–33 doi: 10.1016/j.peptides.2016.11.007 [DOI] [PubMed] [Google Scholar]
- Glick M, Segal-Lieberman G, Cohen R, Kronfeld-Schor N (2009) Chronic MCH infusion causes a decrease in energy expenditure and body temperature, and an increase in serum IGF-1 levels in mice Endocrine 36:479–485 doi: 10.1007/s12020-009-9252-5 [DOI] [PubMed] [Google Scholar]
- Hanriot L, Camargo N, Courau AC, Leger L, Luppi PH, Peyron C (2007) Characterization of the melanin-concentrating hormone neurons activated during paradoxical sleep hypersomnia in rats J Comp Neurol 505:147–157 doi: 10.1002/cne.21482 [DOI] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Jones BE (2009) Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle Proc Natl Acad Sci U S A 106:2418–2422 doi: 10.1073/pnas.0811400106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego S et al. (2013) Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus Nat Neurosci 16:1637–1643 doi: 10.1038/nn.3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego S, Salvert D, Renouard L, Mori M, Goutagny R, Luppi PH, Fort P (2012) Tuberal hypothalamic neurons secreting the satiety molecule Nesfatin-1 are critically involved in paradoxical (REM) sleep homeostasis PLoS One 7:e52525 doi: 10.1371/journal.pone.0052525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating GL, Kuhar MJ, Bliwise DL, Rye DB (2010) Wake promoting effects of cocaine and amphetamine-regulated transcript (CART) Neuropeptides 44:241–246 doi: 10.1016/j.npep.2009.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitka T et al. (2011) Association between the activation of MCH and orexin immunorective neurons and REM sleep architecture during REM rebound after a three day long REM deprivation Neurochem Int 59:686–694 doi: 10.1016/j.neuint.2011.06.015 [DOI] [PubMed] [Google Scholar]
- Konadhode RR et al. (2013) Optogenetic stimulation of MCH neurons increases sleep J Neurosci 33:10257–10263 doi: 10.1523/JNEUROSCI.1225-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D et al. (2010) Glucose stimulation of hypothalamic MCH neurons involves K(ATP) channels, is modulated by UCP2, and regulates peripheral glucose homeostasis Cell Metab 12:545–552 doi: 10.1016/j.cmet.2010.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenzer M et al. (2011) Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia PLoS One 6:e24998 doi: 10.1371/journal.pone.0024998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Greco MA, Shiromani P, Saper CB (2000) Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep J Neurosci 20:3830–3842 doi:20/10/3830 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelsen LE et al. (2017) Neurochemical Heterogeneity Among Lateral Hypothalamic Hypocretin/Orexin and Melanin-Concentrating Hormone Neurons Identified Through Single-Cell Gene Expression Analysis eNeuro 4 doi: 10.1523/ENEURO.0013-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti JM, Torterolo P, Lagos P (2013) Melanin-concentrating hormone control of sleep-wake behavior Sleep Med Rev 17:293–298 doi: 10.1016/j.smrv.2012.10.002 [DOI] [PubMed] [Google Scholar]
- Sapin E, Berod A, Leger L, Herman PA, Luppi PH, Peyron C (2010) A very large number of GABAergic neurons are activated in the tuberal hypothalamus during paradoxical (REM) sleep hypersomnia PLoS One 5:e11766 doi: 10.1371/journal.pone.0011766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell T, Gerashchenko D, Urade Y, Onoe H, Saper C, Hayaishi O (1998) Activation of ventrolateral preoptic neurons by the somnogen prostaglandin D2 Proc Natl Acad Sci U S A 95:7754–7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger M et al. (2018) Functional analysis reveals differential effects of glutamate and MCH neuropeptide in MCH neurons Mol Metab 13:83–89 doi: 10.1016/j.molmet.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q et al. (2007) Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia Cell Metab 5:383–393 doi: 10.1016/j.cmet.2007.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torterolo P, Lagos P, Monti JM (2011) Melanin-concentrating hormone: a new sleep factor? Front Neurol 2:14 doi: 10.3389/fneur.2011.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu T et al. (2014) Optogenetic manipulation of activity and temporally controlled cell-specific ablation reveal a role for MCH neurons in sleep/wake regulation J Neurosci 34:6896–6909 doi: 10.1523/JNEUROSCI.5344-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varin C, Luppi PH, Fort P (2018) Melanin-concentrating hormone-expressing neurons adjust slow-wave sleep dynamics to catalyze paradoxical (REM) sleep Sleep 41 doi: 10.1093/sleep/zsy068 [DOI] [PubMed] [Google Scholar]
- Vas S et al. (2013) Nesfatin-1/NUCB2 as a potential new element of sleep regulation in rats PLoS One 8:e59809 doi: 10.1371/journal.pone.0059809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L et al. (2003) A role of melanin-concentrating hormone producing neurons in the central regulation of paradoxical sleep BMC Neurosci 4:19 doi: 10.1186/1471-2202-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivelan R, Fuller PM, Tong Q, Lu J (2009) Medullary circuitry regulating rapid eye movement sleep and motor atonia J Neurosci 29:9361–9369 doi: 10.1523/JNEUROSCI.0737-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivelan R et al. (2016) Melanin-concentrating hormone neurons specifically promote rapid eye movement sleep in mice Neuroscience 336:102–113 doi: 10.1016/j.neuroscience.2016.08.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiddon BB, Palmiter RD (2013) Ablation of neurons expressing melanin-concentrating hormone (MCH) in adult mice improves glucose tolerance independent of MCH signaling J Neurosci 33:2009–2016 doi: 10.1523/JNEUROSCI.3921-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie JT, Sinton CM, Maratos-Flier E, Yanagisawa M (2008) Abnormal response of melanin-concentrating hormone deficient mice to fasting: hyperactivity and rapid eye movement sleep suppression Neuroscience 156:819–829 doi: 10.1016/j.neuroscience.2008.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Yamanaka A (2017) Lateral hypothalamic circuits for sleep-wake control Curr Opin Neurobiol 44:94–100 doi: 10.1016/j.conb.2017.03.020 [DOI] [PubMed] [Google Scholar]
- Zhou D, Shen Z, Strack AM, Marsh DJ, Shearman LP (2005) Enhanced running wheel activity of both Mch1r- and Pmch-deficient mice Regul Pept 124:53–63 doi: 10.1016/j.regpep.2004.06.026 [DOI] [PubMed] [Google Scholar]