

Abstract
BACKGROUND & AIMS:
Little is known about the signaling pathways that initiate and promote acute pancreatitis (AP). The pathogenesis of AP has been associated with abnormal increases in cytosolic Ca2+, mitochondrial dysfunction, impaired autophagy, and endoplasmic reticulum (ER) stress. We analyzed the mechanisms of these dysfunctions and their relationships, and how these contribute to development of AP in mice and rats.
METHODS:
Pancreatitis was induced in C57BL/6J mice (control) and mice deficient in peptidylprolyl isomerase D (cyclophilin D, encoded by Ppid) by administration of L-arginine (also in rats), caerulein, bile acid, or an AP-inducing diet. Parameters of pancreatitis, mitochondrial function, autophagy, ER stress, and lipid metabolism were measured in pancreatic tissue, acinar cells, and isolated mitochondria. Some mice with AP were given trehalose to enhance autophagic efficiency. Human pancreatitis tissues were analyzed by immunofluorescence.
RESULTS:
Mitochondrial dysfunction in pancreas of mice with AP was induced by either mitochondrial Ca2+ overload or through a Ca2+ overload-independent pathway that involved reduced activity of ATP synthase (80% inhibition in pancreatic mitochondria isolated from rats or mice given L-arginine). Both pathways were mediated by cyclophilin D and led to mitochondrial depolarization and fragmentation. Mitochondrial dysfunction caused pancreatic ER stress, impaired autophagy, and deregulation of lipid metabolism. These pathologic responses were abrogated in cyclophilin D-knockout mice. Administration of trehalose largely prevented trypsinogen activation, necrosis, and other parameters of pancreatic injury in mice with L-arginine AP. Tissues from patients with pancreatitis had markers of mitochondrial damage and impaired autophagy, compared with normal pancreas.
CONCLUSIONS:
In different animal models, we find a central role for mitochondrial dysfunction, and for impaired autophagy as its principal downstream effector, in development of AP. In particular, the pathway involving enhanced interaction of cyclophilin D with ATP synthase mediates L-arginine–induced pancreatitis, a model of severe AP the pathogenesis of which has remained unknown. Strategies to restore mitochondrial and/or autophagic function might be developed for treatment of AP.
Keywords: Pancreas, Inflammatory Response, Acinar Cell, Lamellar Bodies
Graphical Abstract
The pathogenic mechanism of acute pancreatitis (AP), a common and sometimes fatal disease, is incompletely understood, and no specific/effective treatment is available.1,2 Because of limited access to human tissue, the knowledge on molecular/cellular pathways initiating and driving pancreatitis comes mainly from experimental models that appear to reproduce key responses of human disease.3 There are significant gaps in our understanding of these pathways, in particular the role of mitochondrial dysfunction. It has been recently shown4 that persistent opening of permeability transition pore (PTP) caused by mitochondrial Ca2+ overload is an early event in experimental AP models associated with pathologic (global and sustained) increases in free cytosolic Ca2+ ([Ca2+]i), such as those induced by caerulein (CER-AP) or pancreatic ductal infusion of taurolithocholic acid sulfate (TLCS-AP). PTP is a non-specific channel traversing both the outer and inner mitochondrial membranes, persistent opening of which causes depolarization and, ultimately, adenosine triphosphate (ATP) drop.5 In those models, PTP blockade by genetic or pharmacologic knockdown of its major regulator peptidylprolyl isomerase D (cyclophilin D) markedly ameliorated disease.4 However, it remains unknown whether mitochondrial dysfunction drives AP in models where massive increases in [Ca2+]i have not been reported (such as that induced by L-arginine (Arg-AP) or by genetic modifications6); and if so, what the underlying mechanisms are.
The paradigm on PTP molecular nature has dramatically changed, with recent findings showing that the 15-subunit ATP synthase is a central PTP component.5 Under certain conditions, ATP synthase undergoes cyclophilin D-dependent conformational transition, forming the PTP channel. The detailed mechanism is a matter of intense investigation, but it is believed that the multisubunit structure of ATP synthase restricts the switch to PTP conformation and this restriction is removed by cyclophilin D.5,7 Whether such mechanism operates in disease has not been shown.
Very little is known about the mechanisms linking mitochondrial dysfunction to AP responses. Recent studies revealed, in particular, that impaired autophagy is a major pathologic event prominent in both experimental and human pancreatitis.8,9 It is implicated in disease initiation because genetic modifications to inhibit or impair autophagy cause pancreatitis.10–12 Endoplasmic reticulum (ER) stress is another pathway believed to be involved in AP.13,14 However, whether disordering of pancreatic autophagy or activation of ER stress are linked to mitochondrial dysfunction remains unclear. Furthermore, it is not known whether normalizing these pathways alleviates AP. Hypertriglyceridemia is an established risk factor for human pancreatitis15,16; however, it is not known whether pancreatitis alters acinar cell lipid metabolism and whether mitochondria and/or autophagy play a role in these alterations.
To examine these issues, we primarily utilized the Arg-AP model. Although this noninvasive model of severe AP was introduced decades ago and is being increasingly used,17,18 its pathogenic mechanism remains largely unknown.
We find that mitochondrial dysfunction in dissimilar AP models involves both Ca2+-overload–dependent and –independent mechanisms, converging on cyclophilin D-mediated perturbation of ATP synthase. It causes impaired autophagy, ER stress, and deregulates acinar cell lipid metabolism. Normalizing either the mitochondrial function (by cyclophilin D knockout) or autophagy (with the disaccharide trehalose) greatly ameliorates AP.
Materials and Methods
Detailed description of materials and methods is provided in the Supplementary Materials.
In Vivo Pancreatitis Models
Arg-AP was induced in rats by 2 hourly intraperitoneal (i.p.) injections of 3 g/kg Arg; and in mice, by 3 hourly i.p. injections of 3.3 g/kg Arg. CER-AP was induced in mice by 7 hourly injections of 50 μg/kg cerulein. The TLCS-AP and choline deficient, ethionine-supplemented diet (CDE)-AP models are described in the Supplementary Materials. In experiments aimed to restore efficient autophagy, 2 g/kg trehalose was given to mice by daily i.p. injections for 12 days followed by induction of Arg-AP or CER-AP.
Human Pancreas Specimens
Briefly, human tissue specimens of normal pancreas and pancreatitis were evaluated for the presence of acinar compartment and provided de-identified as formalin-fixed paraffin-embedded samples.
Lipidomics
Pancreatic levels of tri- and diacylglycerols, free fatty acids (FFAs), and total phosphatidylcholine and phosphatidylethanolamine were analyzed by liquid chromatography-mass spectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS) at the UCSD LIPID MAPS Lipidomics Core (http://www.ucsd-lipidmaps.org/).
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical analysis was performed with Prizm5 software (https://www.graphpad.com/scientific-software/prism/) using 2-tailed Student’s t-test. P < .05 was considered statistically significant.
Results
Mitochondrial Dysfunction in Experimental Pancreatitis is Mediated Through Ca2+-overload Independent and Dependent Mechanisms
We analyzed mitochondrial alterations in Arg-AP in comparison with other models of AP, particularly those (CER-AP and TLCS-AP) associated with pathologic increases in acinar cell [Ca2+]i.19 Mitochondria were isolated from pancreas (as well as liver) at different times after Arg-AP induction (Figure 1), and their function assessed by measuring changes in the mitochondrial membrane potential (ΔΨm), both basal and in response to exogenous adenosine diphosphate (ADP). Pancreatic mitochondria are markedly sensitive to Ca2+-induced depolarization20 and lose ΔΨm when exposed to even low-micromolar Ca2+. Therefore, in experiments in Figure 1A-D, mitochondria were isolated and assayed in a Ca2+-free medium with EGTA, thus preventing mitochondrial Ca2+ overload. As expected, in mitochondria from control animals, ADP-stimulated oxidative phosphorylation caused ΔΨm drop, which was restored to basal after ADP is converted to ATP (Figure 1A,C). The ΔΨm recovery after ADP addition was inhibited in mitochondria from mice and rats with Arg-AP, compared with control (Figure 1A-D). This effect was evident as early as at 2 hours, and the recovery was completely lost after 24 hours of Arg-AP. Another effect of Arg-AP on mitochondria was progressive decrease in basal ΔΨm (Figure 1A-D). Both effects were prevented by cyclophilin D knockout (Figure 1C,D), indicating that Arg-induced mitochondrial depolarization is because of sustained PTP opening.
Figure 1.

Mitochondrial dysfunction occurs early in Arg-AP; it is mediated by cyclophilin D but not by Ca2+ overload. Rats (A,B) and wild-type (wt) or cyclophilin D knockout (CypD KO) mice (C-E) were subjected to Arg-AP, as detailed in Methods, and killed at indicated times. Pancreatic mitochondria were isolated and assayed either in Ca2+-free medium containing 1 mmol/L EGTA (A-D) or at indicated free Ca2+ concentrations maintained with Ca2+/EGTA buffers (E). ΔΨm was measured with tetraphenyl phosphonium ion (TPP+) electrode and quantified as the difference between TPP+ levels in mitochondria suspension before and after addition of 5 μmol/L DNP, a mitochondrial uncoupler [illustrated in (A)]. The rate of ΔΨm recovery after ADP (20 μmol/L) -induced drop was measured as a slope of ΔΨm increase back to steady-state level [shown in (A) in red]. Both parameters were normalized to mitochondria from control animals. Shown are the individual values and means ± SEM (n = 6–10 per group). *P < .05 vs control (saline-treated) animals; #P < .05 vs CypD KO mice with the same treatment. (F). Wt mouse acinar cells were loaded with 25 μmol/L BAPTA-AM (where noted) and incubated with 20 μmol/L Arg for indicated times. ΔΨm was measured using the dye TMRM (1 μmol/L). The difference between TMRM fluorescence in control cells before and after addition of the mitochondrial uncoupler FCCP (10 μmol/L) was taken as 100% ΔΨm. Values are mean ± SEM from 3–4 cell preparations. *P < .05 vs control cells.
These results indicate that Arg-AP causes an “irreversible” damage of pancreatic mitochondria that persists in conditions precluding Ca2+ overload. To further examine the role of Ca2+, mitochondria isolated from mice with Arg-AP were subjected to low-micromolar Ca2+ concentrations (Figure 1E, Supplementary Figure 1A). The observed Ca2+-dose–dependent depolarization demonstrates that mitochondria retain their sensitivity to Ca2+ overload. However, same as at “zero” Ca2+, mitochondria from mice with Arg-AP at all Ca2+ concentrations had approximately 50% lower ΔΨm than those from control mice (Figure 1E, Supplementary Figure 1A), indicating that Ca2+ and Arg-AP cause depolarization through additive pathways.
In accord with the results on isolated mitochondria, incubation of mouse pancreatic acinar cells with Arg (20–40 mmol/L) caused time-dependent decrease in ΔΨm (Figure 1F). Notably, acinar cell mitochondrial depolarization persisted in the presence of Ca2+ chelator BAPTA (Figure 1F), which prevents Ca2+ overload.20 Pre-incubation with Arg did not abrogate cholecystokinin-8 (CCK)-induced depolarization in acinar cells (Supplementary Figure 1B). Furthermore, Arg did not affect the basal [Ca2+]i, and did not block CCK-induced [Ca2+]i increase (Supplementary Figure 1C), indicating no depletion of intracellular Ca2+ stores by Arg.
In stark contrast with Arg-AP, mitochondria isolated from mice with CER-AP or TLCS-AP were not depolarized and showed complete ΔΨm recovery after ADP-induced drop when assayed in the absence of Ca2+ (Supplementary Figure 2A-D). Moreover, when assayed at different low-micromolar Ca2+ concentrations, mitochondria from mice with CER-AP or TLCS-AP displayed the same extent of Ca2+-dependent depolarization as the mitochondria from control animals (Supplementary Figure 2B,D). These results on isolated mitochondria are in accord with the data on acinar cells, showing that depolarization induced by CCK or TLCS is caused by mitochondrial Ca2+ overload.4,21 Of note, BAPTA completely prevents CCK- or TLCS-induced depolarization in acinar cells4,21; and Ca2+ chelators reverse Ca2+-induced depolarization in isolated mitochondria.20
We next examined mitochondrial dysfunction in the AP model induced with choline deficient, ethionine-supplemented diet (CDE-AP), in which massive increases in [Ca2+]i have not been documented.22 Pancreatic mitochondria from mice fed CDE diet displayed characteristics similar to those in Arg-AP, that is, marked depolarization in the absence of Ca2+ and Inhibition of ΔΨm recovery after addition of ADP (Supplementary Figure 2E,F). These data indicate that, similar to Arg-AP, mitochondrial damage in CDE-AP is “irreversible” and persists in conditions precluding Ca2+ overload.
Collectively, the above results indicate that mitochondrial dysfunction in Arg-AP, as well as CDE-AP, involves mechanisms independent of Ca2+ overload. In contrast, mitochondrial damage induced by CER/CCK or TLCS in in vivo or ex vivo AP models is mediated by Ca2+ overload and is completely reversible upon removal of Ca2+.
Mechanism of Mitochondrial Dysfunction in Arg-AP
Arg-AP dramatically increases free Arg level in pancreatic mitochondria.
We asked whether Arginduced mitochondrial damage could be because of increased intramitochondrial Arg. Mice subjected to Arg-AP displayed dramatic increases in the intrapancreatic level of free Arg, with peak at a 25-fold increase at 4 hours (Figure 2A). Arg level in pancreatic mitochondria from mice with Arg-AP was approximately 140-fold greater than that of control mice (Figure 2A), indicating that Arg preferentially accumulates in the mitochondria. Of note, Arg accumulation causes mitochondrial damage in neurons.23
Figure 2.

Mitochondrial dysfunction in Arg-AP is caused by cyclophilin D-mediated perturbation of ATP synthase. Mice (A,C-H) and rats (B,H) were subjected to Arg-AP or CER-AP. Pancreatic levels of free Arg (A) were measured with LC-MS in whole tissue and isolated mitochondria; ATP synthase enzymatic activity (B,C), in isolated mitochondria; and ATP levels (D), in tissue homogenates. Shown are the individual values and means ± SEM (n = 3–9 per group). *P < .05 vs control animals; # P < .05 vs CypD KO mice. (E-G). ATP synthase and cyclophilin D were immunoprecipitated (IP) from pancreatic mitochondria isolated from mice at indicated conditions, and their levels in the immunoprecipitates were measured by IB. (H). Pathologic alterations in mitochondrial ultrastructure in pancreas of rats and mice with Arg-AP (EM). Scale bars: 0.5 μm. Larger fields from these micrographs are shown in Supplementary Figure 5.
Arg-induced depolarization is not mediated through nitric oxide pathway; but L-ornithine, another Arg metabolite, depolarizes pancreatic mitochondria.
We next tested the role of Arg metabolism in its effects on mitochondria. Arg is primarily metabolized by nitric oxide synthase (generating nitric oxide) and arginase, which converts it to L-ornithine. Nitric oxide synthase inhibitor L-NMMA had no effect on Arg-induced depolarization (Supplementary Figure 3A), indicating the nitric oxide pathway is not involved. Differently, L-ornithine caused ΔΨm decrease in mouse acinar cells; and it did not block CCK-induced depolarization (Supplementary Figure 3B) – same as observed for Arg (Supplementary Figure 1B). Arginase-II isoform, residing in mitochondria, is abundant in acinar cells24; thus, it is possible that L-ornithine mediates the effect of Arg on ΔΨm. Of note, L-ornithine causes pancreatitis responses similar to Arg.18
Accumulation of Arg in pancreatic mitochondria is associated with cyclophilin D-dependent perturbation of ATP synthase.
The impaired ΔΨm recovery after ADP-induced drop observed in Arg-AP (Figure 1A-D) suggested dysregulation of mitochondrial ATP synthesis because of Arg accumulation in mitochondria. Indeed, we found progressive, up to approximately 80% inhibition of ATP synthase activity in pancreatic mitochondria isolated from rats or mice with Arg-AP (Figure 2B,C). The inhibition was pronounced within 2 hours after Arg-AP induction, and was followed by decreased pancreatic ATP (Figure 2D). Both effects were prevented in cyclophilin D knockout mice (Figure 2C,D). We next examined whether Arg-AP affects cyclophilin D interaction with ATP synthase by using co-immunoprecipitation (Figure 2E-G). Independent of whether immunoprecipitation was done through cyclophilin D or ATP synthase, complex formation between these proteins markedly increased in Arg-AP (Figure 2E,G). The increased binding of cyclophilin D to ATP synthase is in accord with the effects of Arg-AP on pancreatic ATP synthase activity (Figure 2B,C) and PTP opening (Figure 1). These data are consistent with the recently proposed paradigm that cyclophilin D promotes ATP synthase transition to PTP channel conformation.5,7
In striking contrast, CER-AP did not increase the amount of cyclophilin D in complex with ATP synthase (Figure 2F,G). As stated earlier, CER/CCK, as well as TLCS, causes mitochondrial Ca2+ overload in acinar cells4; and Ca2+ overload was shown to facilitate ATP synthase transition to PTP channel conformation.5,7 Thus, our data indicate that mitochondrial dysfunction in AP is caused by cyclophilin D-mediated transition of ATP synthase to PTP channel conformation, but the underlying mechanisms differ between AP models. Whereas PTP opening in Arg-AP is driven by enhanced complex formation between cyclophilin D and ATP synthase, in CER-AP and TLCS-AP it is mediated by mitochondrial Ca2+ overload. Cyclophilin D knockout prevents mitochondrial dysfunction mediated through both mechanisms (Figures 1, 2; and Ref.4).
Arg-AP does not damage liver mitochondria, nor does it increase their Arg level.
The basal level of free Arg in liver of control mice (0.11 ± 0.03 μmol/mg, n = 9) was much lower than that in pancreas (1 ± 0.13 μmol/mg, n = 8), and it only increased approximately 4-fold in Arg-AP (Supplementary Figure 4A). Furthermore, in contrast to pancreatic mitochondria (Figure 2A), Arg level did not increase in liver mitochondria isolated from mice with Arg-AP (Supplementary Figure 4A). Congruently, in liver mitochondria Arg-AP did not induce complex formation between cyclophilin D and ATP synthase, did not inhibit ATP synthase activity and ΔΨm recovery after ADP addition, and did not decrease ΔΨm (Supplementary Figure 4B-D). These data provide an explanation as to why Arg administration does not cause liver damage. The observed differences in the effects of Arg administration on pancreas and liver are likely because of different rates of Arg metabolism and/or Arg uptake by liver and pancreatic mitochondria.
Cyclophilin D Knockout Prevents Pancreatitis-induced Alterations in Mitochondrial Ultrastructure and Dynamics
Changes in mitochondrial ultrastructure occurred early in Arg-AP, manifested by the appearance of swollen mitochondria with flocculent matrix and loss of cristae, and mitochondria with condensed matrix and dilated cristae (Figure 2H, Supplementary Figure 5). Mitochondrial swelling results from persistent PTP opening that allows water to enter the matrix,25 whereas mitochondrial condensation is indicative of impaired ATP synthesis and ADP excess (respiratory state III).26 Cyclophilin D knockout essentially prevented these pathologic alterations (Figure 2H, Supplementary Figure 5).
Mitochondria continuously undergo fission and fusion, which is necessary for energy production and cell survival.27 Deranged dynamics is often associated with mitochondrial dysfunction and is a characteristic feature of various disorders27; however, it has not been assessed in pancreatitis. We examined changes in the mitochondrial network by morphologic localization of mitochondrial resident proteins Tom20 and VDAC1. Both markers revealed interconnected tubular mitochondrial network in control pancreas (Supplementary Figure 6A,C). Arg-AP caused dramatic disorganization of the network, already prominent at 12 hours (Supplementary Figure 6A,C). In accord with morphologic evidence of mitochondrial fragmentation, immunoblot (IB) analysis showed decreases in the fusion effectors OPA1, MFN1, and MFN2, whereas the fission mediator, Fis1, was up-regulated in Arg-AP (Supplementary Figure 6B,D-E). CER-AP also caused dramatic mitochondrial fragmentation and Fis1 up-regulation (Supplementary Figure 6F,G); however, different from Arg-AP, OPA1 and other fusion markers were not affected (Supplementary Figure 6F; and data not shown). Cyclophilin D ablation largely restored the tubular mitochondrial network in Arg-AP (Supplementary Figure 6A,C), as well as the levels of fusion mediators (Supplementary Figure 6B,D,E).
Notably, we found prominent mitochondrial fragmentation in human pancreatitis assessed by VDAC1 immunolocalization (Supplementary Figure 6H).
Restoring Mitochondrial Function With Cyclophilin D Knockout Greatly Improves Arg-AP
Cyclophilin D knockout greatly improved pancreas histology and blocked (or markedly diminished) all key pathologic responses of Arg-AP, ie, increases in serum amylase and lipase, acinar cell vacuolization, trypsinogen activation, necrosis, and apoptosis (Figure 3A,B). Compared with wildtype (wt), cyclophilin D knockout mice with Arg-AP displayed less neutrophilic and macrophage infiltration (Figure 3C,D; Supplementary Figure 7). The transcription factor nuclear factor kappa B mediates inflammation in AP. Its activation in Arg-AP was marked by increases in p65/RelA phosphorylation and increased DNA binding activity; both were reduced in cyclophilin D knockout mice (Figure 3E,F). Another mechanism of the inflammatory response in AP is through damage-associated molecular patterns (DAMPs) released by injured cells.28 Arg-AP caused a dramatic release into the cytosol of nuclear HMGB1, a prototypical DAMP (Figure 3G,H). This response was blocked by cyclophilin D ablation (Figure 3G,H).
Figure 3.

Normalizing mitochondrial function by cyclophilin D genetic ablation improves Arg-AP. Mice were subjected to Arg-AP, killed at 72 hours (A), 24 hours (C,E-G), or as indicated (B); and histopathologic changes (A; H&E staining) and pancreatitis responses (B-H) were measured. (C,D). Inflammatory infiltration was measured on pancreatic tissue immunostained for the neutrophil marker myeloperoxidase (MPO) and macrophage marker F4/80 (see also Supplementary Figure 7). (E,F). Pancreatic levels of phosphorylated and total p65/RelA (E) were measured by IB, and NF-κB DNA binding activity (F), by EMSA. In this and other figures, ERK1/2 or GAPDH are loading controls, and each lane on IB represents an individual animal. The narrow white space on EMSA gel indicates the lanes are on the same gel but not contiguous. (G,H). Subcellular localization of HMGB1 in pancreas was analyzed by IF using Volocity software. Scale bars: 10 μm. Values are mean ± SEM (n = 3–4). *P < .05 vs wt saline-treated mice; #P < .05 vs same treatment in CypD KO mice; $P < .05 vs saline-treated CypD KO mice.
Pancreatitis Causes Dramatic Changes in Acinar Cell Lipid Metabolism, Which Are Largely Prevented by Cyclophilin D Knockout
Massive accumulation of small/”empty” vacuole-like structures in acinar cells is an early histopathologic response of rat Arg-AP,13,29,30 which we also observed in mouse Arg-AP (Figure 4A). Although the previous studies13,29,30 concluded these vacuoles were autophagic, their nature was not investigated. [H&E-stained pancreatic tissue sections from rat and mouse Arg-AP also display large, morphologically different, cargo-containing, vacuoles (Figure 3A; and Ref.8), identified with electron microscopy (EM) as autolysosomes (see below, Figure 5C and Supplementary Figure 9; and Ref.8)]. EM analysis revealed (Figure 4B,C; Supplementary Figure 8A) that the small/”empty” vacuoles have features of lamellar bodies (LBs), cellular organelles that are composed of concentric membrane layers and function in lipid storage and secretion.31 LBs play physiologic roles in skin and lung, but their formation in other organs is usually caused by defects in lipid degradation; in particular, LBs accumulate in lysosomal storage diseases with defective lipid turnover.32 EM also showed the appearance of lipid droplets (LDs) in Arg-AP (Figure 4D), organelles that store fatty acids in the form of triacylglycerols (TAGs).33 Supporting the EM data, IB showed up-regulation of perilipin 2 (PLIN2), a marker of lipid storage organelles,33 in pancreas of mice with Arg-AP (Figure 4E) and CER-AP (data not shown). We did not detect PLIN1, a specific marker of adipocyte LDs,33 in either normal pancreas or pancreatitis tissue, indicating no contamination with adipocytes.
Figure 4.

Arg-AP profoundly disturbs pancreatic lipid metabolism, which is prevented by cyclophilin D genetic ablation. Mice were subjected to 24 hours Arg-AP. H&E staining (A) shows multiple small vacuoles in pancreas of wt animals with Arg-AP, identified by EM as lamellar bodies (B,C) and lipid droplets (D). Boxed area in (B), showing accumulation of lamellar bodies, is enlarged in (C). Scale bars: 10 μm (A), 6.7 μm (B), and 1.3 μm (C,D). (E). IB analysis of perilipin 2 (PLIN2), a marker of lipid droplets. The narrow white space indicates the lanes are on the same blot but not contiguous. (F-L). Pancreatic tissue triacylglycerols (TAG) and free fatty acids (FFA) were measured with LC-MS and GC-MS as detailed in Methods. Total, saturated, and unsaturated TAG and FFA levels were quantified based on their detailed profiles (see Supplementary Figure 8C,D), normalized per mg protein, and further normalized to wt control mice. (I-L). Effect of Arg-AP on individual FFAs (the numbers of carbons and of double bonds are indicated). Values are mean ± SEM (n = 3). *P < .05 vs wt saline-treated mice; #P < .05 vs same treatment in CypD KO mice.
Figure 5.

Cyclophilin D genetic ablation alleviates ER stress and normalizes autophagy in Arg-AP. Mice were subjected to Arg-AP and killed at indicated times (A,B) or at 24 hours (C-I). Markers/mediators of (A,B) ER stress, (D-F) autophagic/lysosomal pathways, and (G) mitophagy were analyzed in pancreatic tissue by IB. Ub, ubiquitin; CatB, cathepsin B. The IB in (F) shows the intermediate/single-chain (sc) CatB form and the heavy chain of mature/double-chain (dc) form. (C). EM showing abnormally large autolysosome with poorly degraded material in pancreas of Arg-AP mouse. The boxed area is enlarged in Supplementary Figure 10. Scale bar: 1.7 μm. (H,I). Pancreatic acinar cells were isolated from mice of indicated groups, incubated for 2 hours in the presence or absence of lysosomal protease inhibitors, E64D + pepstatin A (20 μmol/L each), and LC3-I/II levels measured by IB. In (B) and (I), densitometric band intensities for specified proteins were normalized to that of ERK in the same sample, and the mean ratios further normalized to that in wt control group. Values are mean ± SEM (n = 3). *P < .05 vs wt saline-treated mice; #P < .05 vs same treatment in CypD KO mice; $P < .05 vs saline-treated CypD KO mice.
To confirm that pancreatitis, indeed, causes deregulation of lipid metabolism, we assessed the effect of Arg-AP on pancreatic levels of TAGs and FFAs using LC-MS and GC-MS. Arg-AP decreased total pancreatic TAG content (Figure 4F) by decreasing TAGs containing unsaturated fatty acids, without changing saturated TAGs (Figure 4F; Supplementary Figure 8B). It caused profound and selective changes in the composition and levels of FFAs in pancreas (Figure 4G-L; Supplementary Figure 8C-E). Arg-AP markedly decreased the levels of saturated FFAs, such as myristic, palmitic, and stearic acids; increased the levels of long-chain polyunsaturated fatty acids (PUFAs), eg, arachidonic and eicosadienoic acids; but had no effect on medium-chain unsaturated FFAs, eg, linoleic acid. Because the majority of FFAs are saturated (Supplementary Figure 8C), these changes resulted in a decrease in total pancreatic FFAs in Arg-AP (Figure 4G) and a shift of FFA profile toward unsaturated fatty acids. Thus, the ratio of total saturated FFAs to PUFAs decreased from approximately 2.5 in control pancreas to approximately 1.0 in Arg-AP (Figure 4H). As an example, the ratio of the most abundant saturated palmitic acid to unsaturated arachidonic acid decreased 5.2 times in Arg-AP.
Remarkably, cyclophilin D knockout prevented Arginduced accumulation of LBs seen with H&E staining and EM, as well as the up-regulation of PLIN2 (Figure 4E). It greatly attenuated pancreatitis-induced changes in the levels and composition of TAGs and FFAs (Figure 4F-L; Supplementary Figure 8E), and largely restored the ratio of saturated FFAs to PUFAs in Arg-AP (Figure 4H).
These data indicate an important role of mitochondria in maintaining exocrine pancreas lipid homeostasis.
Cyclophilin D Knockout Alleviates ER Stress and Enhances Autophagic Flux in Pancreatitis
Early activation of ER stress is a prominent feature of Arg-AP.13 It is manifest by up-regulation of ER stress markers CHOP, GRP78, and phosphorylated (p)-IRE1, and down-regulation of spliced XBP1 (sXBP1) that plays a protective role in pancreatitis.14 ER stress in Arg-AP was already evident at 6 hours, and further increased by 24 hours (Figure 5A,B). Cyclophilin D knockout largely prevented ER stress in Arg-AP, evidenced by delayed induction and decreased protein expression of CHOP, GRP78, and p-IRE1, and by sustained level of sXBP1 (Figure 5A,B).
Impaired autophagy is a characteristic feature of AP in various models, caused by reduced ability of lysosomes to degrade cargo and a concomitant increase in autophagosome formation.8,9 Indeed, acinar cells in Arg-AP showed accumulation of abnormally large autolysosomes containing poorly degraded cargo (Figure 5C, Supplementary Figure 9). The pancreas also exhibited increased levels of the autophagic vacuole marker LC3-II and autophagy substrate p62/SQSTM1, and accumulation of ubiquitinated proteins (Figure 5D,E), all consistent with reduced autophagic flux in Arg-AP. Importantly, we find massive increase in LC3-positive puncta in pancreatic tissue from patients with pancreatitis (Supplementary Figure 10), suggesting that autophagic flux is impaired in human disease. Indeed, EM of human pancreatitis prominently shows large autophagic vacuoles with poorly degraded material.8,9
One characteristic of decreased lysosomal proteolytic function in experimental AP is defective processing/ maturation of cathepsins, a major class of lysosomal hydrolases.8,9 In particular, the key cathepsin B (CatB) is mainly present in control pancreas as fully mature (doublechain) form; whereas in Arg-AP the level of its intermediate, partially processed (single-chain) form markedly increases while the mature form is reduced (Figure 5F). Cyclophilin D knockout restored CatB processing in Arg-AP (Figure 5F), indicating that mitochondria regulate the endolysosomal pathway. Interestingly, EM showed that defective/condensed mitochondria wrap around the abnormally large autolysosomes in acinar cells, suggesting a direct interaction between these organelles in AP (Figure 5C, Supplementary Figure 9). In correlation with improved CatB processing, cyclophilin D knockout normalized autophagic flux in Arg-AP, resulting in decreased levels of LC3-II, p62, and ubiquitinated proteins (Figure 5D,E) and markedly less acinar cell vacuolization (Figure 3B). To further confirm the role of mitochondria in maintaining pancreatic autophagy, we used the compound GFP-LC3;Ppid−/− mice generated by crossing cyclophilin D knockout mice and GFP-LC3 mice that express LC3 conjugated with green fluorescent protein. Cyclophilin D ablation in GFP-LC3 mice almost completely prevented accumulation of both the endogenous LC3-II and GFP-LC3-II induced by Arg-AP (Supplementary Figure 11).
To more specifically assess the role of mitochondria in autophagy induction and autophagic flux, we isolated acinar cells from wt and cyclophilin D knockout mice subjected to Arg-AP, and incubated them with inhibitors of lysosomal proteolytic degradation, E64D plus pepstatin A (Figure 5H,I). In this type of analysis, when lysosomal degradation is blocked, changes in LC3-II (ie, those induced by Arg-AP) solely reflect autophagosome formation.34 In the presence of lysosomal inhibitors, acinar cells from wt mice with Arg-AP displayed greater levels of LC3-II than cells isolated from control mice, indicating autophagy induction (ie, increased autophagosome formation) in Arg-AP. The same analysis on acinar cells isolated from cyclophilin D knockout mice showed almost no autophagy induction by Arg-AP (Figure 5H,I). The data also indicate that Arg-AP activates mitophagy, a selective autophagy of mitochondria,35 which is prevented by cyclophilin D knockout. Indeed, Parkin1, a key protein initiating mitophagy,35 was up-regulated by Arg-AP in wt but not in cyclophilin D knockout mice (Figure 5G).
Thus, the results indicate that Arg-AP both activates pancreatic autophagy (in particular, mitophagy) and causes impaired autophagic flux. We found a similar situation in CER-AP.8,9 Cyclophilin D ablation normalizes autophagy (Figure 5), revealing the link between mitochondrial and autophagic dysfunctions.
Enhancing Autophagic Activity With Trehalose Improves AP
Genetic alterations that specifically target autophagic pathways induce pancreatitis-like injury.6,10–12,36 We therefore examined whether the toxic effects of mitochondrial dysfunction on pancreas are mediated, at least in part, by impaired autophagy. To normalize autophagy in pancreatitis, wt mice were treated with trehalose, a disaccharide recently shown to stimulate autophagic flux and clearance of autophagic vacuoles.37 Trehalose enhanced autophagic activity in Arg-AP, which was manifest by decreases in LC3-II puncta (Supplementary Figure 12A) and IB levels of LC3-II, p62, and ubiquitinated proteins (Figure 6A,B); and by normalized CatB processing (Figure 6B). The histopathologic alterations caused by Arg-AP were greatly improved in trehalose-treated mice (Figure 6C); and EM showed no accumulation of abnormally large autolysosomes with poorly degraded cargo, typical of pancreatitis (Figure 6D).
Figure 6.

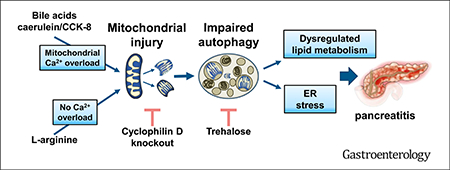
Enhancement of autophagic activity with trehalose improves Arg-AP. Wt mice received daily i.p. injections of trehalose (or vehicle) for 2 weeks, then subjected to Arg-AP and killed 24 hours later. (A,B,F). Effects of trehalose on markers/mediators of autophagic and lysosomal pathways (LC3, p62, ubiquitin, CatB), ER stress (CHOP, sXBP1), lipid droplets (PLIN2), and inflammation (phospho-p65/RelA) were analyzed by IB. The IB in (B) shows the intermediate, single-chain (sc) and the mature, double-chain (dc) forms of CatB. (C,D). H&E staining and EM demonstrate the absence of abnormally large autolysosomes and lamellar bodies in trehalose-treated mice with Arg-AP. Scale bars: 10 μm (C), 2 μm (D). (E,G). Effects of trehalose on Arg-AP responses (E) and pancreatic FFA levels (G), measured as in Figure 5. *P < .05 vs control (saline-treated) mice without trehalose; #P < .05 vs trehalose-treated mice with Arg-AP; $P < .05 vs control (saline-treated) mice with trehalose. (H). Schematic illustrating the pathways of mitochondrial dysfunction and their links to pancreatitis.
Restoring autophagy with trehalose markedly alleviated pancreatitis responses (Figure 6E,F). In particular, pancreatic necrosis was decreased several fold and trypsinogen activation completely prevented, as was HMGB1 release from nuclei (Figure 6E, Supplementary Figure 12B). There was no increase in p65/RelA phosphorylation (Figure 6F) and less neutrophilic infiltration as determined by MPO staining (Figure 6E; Supplementary Figure 12C). Trehalose attenuated ER stress in Arg-AP, manifest by downregulation of CHOP and up-regulation of sXBP1 (Figure 6F); prevented formation of LBs and LDs, as evidenced with EM and light microscopy (Figure 6C,D) and by PLIN2 down-regulation (Figure 6F); and it restored the levels of saturated FFAs (Figure 6G). Thus, trehalose’s effects of improving pancreatitis were similar (albeit of lesser extent) to those of cyclophilin D knockout, indicating impaired autophagy as a key downstream effector of mitochondrial dysfunction in AP. However, the mechanisms of the protective effects of cyclophilin D ablation and trehalose treatment do not completely overlap. For example, cyclophilin D ablation reduced necrosis in Arg-AP to a greater extent than trehalose (Figures 3B and 6E).
To determine whether trehalose treatment is also protective in AP model associated with [Ca2+]i increase, we examined its effects on CER-AP. In this model as well, trehalose normalized autophagic flux, as reflected by decreased LC3-II and p62 levels (Supplementary Figure 13A); and improved other parameters of AP including histology, trypsinogen activation, and CHOP up-regulation (Supplementary Figure 13B,C).
Trehalose could be expected to improve mitochondrial “health status” by enhancing autophagic elimination of damaged mitochondria. Indeed, we found that trehalose treatment partially restored tubular mitochondrial network in pancreas of mice with Arg-AP and CER-AP (Supplementary Figure 14A). However, trehalose did not restore ΔΨm recovery after ADP-induced drop in isolated mitochondria (Supplementary Figure 14B), and did not prevent CCK-induced loss of ΔΨm in acinar cells (Supplementary Figure 14C). Thus, in accord with findings in other cells,38,39 mitochondria in pancreas are not directly targeted by trehalose. In contrast, cyclophilin D knockout improves pancreatitis through directly preventing mitochondrial damage (Figures 1, 2; Supplementary Figures 5, 6).
Discussion
Our study aimed to elucidate the mechanisms of mitochondrial dysfunction in AP and pathways linking mitochondrial damage to pancreatitis pathologies. We find that mitochondrial dysfunction drives Arg-AP, a widely used model of severe AP, the pathogenic mechanism of which has largely remained unknown. Arg-AP early and dramatically increased Arg level in pancreatic mitochondria, and promoted complex formation between cyclophilin D and ATP synthase resulting in decreased ATP synthase activity and PTP opening. Thus, we find that this mechanism operates in a disease model, validating the recent paradigm.5,7 Of note, Arg does not cause liver mitochondria damage.
We compared the mechanisms of mitochondrial dysfunction in Arg-AP with other models. Arg-AP, as well as CDE-AP, causes “irreversible” damage of pancreatic mitochondria that persists in conditions precluding Ca2+ overload. In contrast, mitochondrial damage induced by CER/ CCK or TLCS in in vivo or ex vivo AP models is mediated by Ca2+ overload and is reversible upon removal of Ca2+. The differences in parameters of mitochondrial dysfunction are summarized in Supplementary Table 1. Our results, together with Mukherjee et al4 and Shalbueva et al,40 indicate that both mechanisms converge on cyclophilin D-dependent PTP opening, causing depolarization, ATP drop, and mitochondrial fragmentation. Hence, cyclophilin D ablation prevents mitochondrial dysfunction in all models tested (this study4,40).
The finding of a Ca2+-overload independent mechanism of mitochondrial dysfunction does not imply there is no aberrant Ca2+ signaling or that Ca2+ is not involved in Arg-AP pathogenesis. It is well established in other cells that mitochondria control Ca2+ signaling; in particular, fragmentation of mitochondria disrupts their contacts with ER that regulate ER Ca2+ transport.41 In acinar cells, mitochondrial inhibitors convert the physiologic (oscillatory/transient) [Ca2+]i response to neurohormones into pathologic (peak-and-plateau) signal characteristic of pancreatitis.19 Mitochondrial dysfunction in Arg-AP, as well as in other models, likely perturbs Ca2+ signaling, thus contributing to pancreatitis pathologies.
We find that mitochondrial dysfunction mediates key pathologic responses of Arg-AP: hyperamylasemia, trypsinogen activation, necrosis, vacuolization, and inflammation. We further find dramatic and specific changes in exocrine pancreas lipid metabolism. Recent studies have elucidated the injurious role of peripancreatic fat necrosis in pancreatitis15,16; however, alterations in acinar cell lipid metabolism have not been investigated. Our data show that Arg-AP causes accumulation in acinar cells of lipid storage organelles, LBs and LDs, thus illuminating the nature of copious small vacuoles appearing in this model. LD accumulation was reported in pancreatitis patients42; and in rat AP induced by L-lysine.43 Arg-AP alters the levels and composition of pancreatic TAGs and FFAs, shifting the FFA profile toward PUFAs. These changes could be very detrimental because acinar cell function critically relies on efficient membrane trafficking and retrieval. Furthermore, PUFAs, eg, arachidonic acid, damage acinar cells and worsen pancreatitis severity.15,16 Remarkably, cyclophilin D knockout largely prevented lipid metabolism abnormalities, revealing the role of mitochondria in maintaining pancreatic lipid homeostasis.
Deregulation of acinar cell lipid metabolism is a previously unrecognized pathology of AP. Certainly, more detailed studies are needed to examine the effects of pancreatitis on various classes of lipids in acinar cells, the underlying mechanisms, and role in disease pathogenesis.
Acinar cells have among the highest rates of protein synthesis, making them highly susceptible to ER perturbations. ER dysfunction triggers the protective unfolded protein response, which, if unsuccessful, leads to ER stress. Manifestations of the latter are present in pancreatitis models,13,14,44 particularly Arg-AP.13 ER stress was markedly reduced by cyclophilin D ablation in both Arg-AP and CER-AP, indicating a role for mitochondrial dysfunction in this response. Several mechanisms may link mitochondrial dysfunction to ER stress in pancreatitis: ATP decrease could compromise unfolded protein response; critical ER–mitochondria contacts could be disrupted41; or autophagy impairment overwhelmed unfolded protein response capacity to eliminate protein aggregates.6,10,12
Impaired autophagy is emerging as a key pathogenic event in pancreatitis4,6,8–12,36; it is implicated in disease initiation because genetic modifications to impair or block autophagy cause pancreatitis.6,10–12 The data indicate autophagy is activated in experimental pancreatitis, but its completion (lysosomal degradation) is inhibited. One mechanism underlying deficient lysosomal activity in pancreatitis could be defective/incomplete cathepsin (eg, CatB) processing.8,9,11 Notably, cyclophilin D genetic ablation normalized CatB processing in Arg-AP, demonstrating that mitochondria regulate endolysosomal and autophagic pathways in exocrine pancreas. Our results indicate that pancreatitis also activates mitophagy, the selective autophagy of mitochondria. In particular, Arg-AP displayed all the preconditions for mitophagy activation: mitochondrial depolarization and fragmentation, and increase in Parkin1, a protein that initiates mitophagy by decorating depolarized mitochondria.35 Cyclophilin D ablation cancelled all these effects.
To test whether autophagy mediates toxic effects of mitochondrial damage in AP, we used trehalose, a disaccharide that enhances autophagic flux and clearance of protein aggregates.37 Trehalose enhanced autophagic activity in both Arg-AP and CER-AP (manifested by reduced accumulation of LC3-II, p62, and ubiquitinated proteins), largely normalized lipid metabolism, and alleviated pancreatitis responses. Notably, in both Arg-AP and CER-AP trypsinogen activation was completely prevented, providing further evidence for a critical role of defective autophagy in this signature AP response.8,9,11 Trehalose has no direct effect on mitochondria in acinar cells, in accord with findings in other cells.38 Thus, the results with cyclophilin D ablation identify mitochondrial dysfunction as the primary defect driving Arg-AP, whereas the results with trehalose indicate that key pathologic responses caused by mitochondrial damage are mediated through impaired autophagy.
Importantly, we find that pancreatic tissue from patients with pancreatitis displays mitochondrial fragmentation and increase in LC3 puncta, the same manifestations of mitochondrial and autophagic dysfunctions as observed in AP models.
In sum, our results show that mitochondrial dysfunction in AP is induced by not only Ca2+ overload but also through enhanced complex formation between cyclophilin D and ATP synthase, a mechanism that does not necessitate Ca2+ overload. We find, in particular, that this mechanism drives Arg-AP. Cyclophilin D ablation normalizes mitochondrial function and greatly improves AP in models both driven and not by Ca2+ overload. We find deregulation of acinar cell lipid metabolism in AP. The impaired autophagy, ER stress, and deregulated lipid metabolism are all mediated by mitochondrial dysfunction and normalized by cyclophilin D knockout (Figure 6H). Thus, our results, together with previous findings,4,40 reveal a central role for mitochondrial dysfunction in AP pathogenesis. Further, restoring efficient autophagy with trehalose markedly ameliorates pancreatic injury, indicating impaired autophagy as a major downstream effector of mitochondrial damage in AP. Both mitochondrial and autophagic dysfunctions are prominent in human disease; thus, approaches to normalize these pathways could be promising for disease treatment.
Supplementary Material
EDITOR’S NOTES.
BACKGROUND AND CONTEXT
Acinar cell mitochondrial dysfunction and impaired autophagy are implicated in the pathogenesis of pancreatitis, a disease with no effective treatment. Mechanisms of these dysfunctions, their interrelations, and links to acute pancreatitis pathology are poorly understood.
NEW FINDINGS
Mitochondrial dysfunction in experimental acute pancreatitis is mediated by cyclophilin D and involves Ca2+-overload-dependent or -independent pathways. The latter mediates L-arginine pancreatitis, a severe model with unknown pathogenesis. Mitochondrial dysfunction causes impaired autophagy, ER stress and deregulated lipid metabolism, which are normalized by cyclophilin D knockout.
LIMITATIONS
There is limited information on the mechanisms of mitochondrial dysfunction and impaired autophagy in human disease.
IMPACT
Restoration of mitochondrial function and/or efficient autophagy greatly ameliorates pancreatitis in mouse models, and thus offers potential treatment approaches for this disease.
Acknowledgments
The authors thank Dr Oswald Quehenberger (LIPID MAPS at the University of California San Diego [UCSD]) for advice on lipidomics analyses and Dr Alexander Andreyev (UCSD) for helpful discussion of the manuscript.
Funding
This work was supported by NIH grant no. P01DK098108 and the US Veterans Administration Merit Review award (both, to A.S.G.), and the Rosztoczy Foundation fellowship (to G.B. and E.T.V.).
Abbreviations used in this paper:
- ADP
adenosine diphosphate
- AP
acute pancreatitis
- Arg
L-arginine
- Arg-AP
L-arginine-induced acute pancreatitis
- ATP
adenosine triphosphate
- CatB
cathepsin B
- CCK
cholecystokinin-8
- CDE-AP
pancreatitis induced with choline deficient, ethionine-supplemented diet
- CER-AP
cerulein-induced acute pancreatitis
- CypD
cyclophilin D
- [Ca2+]i
free cytosolic Ca2+ concentration
- ΔΨm
mitochondrial membrane potential
- EM
electron microscopy
- EMSA
electrophoretic mobility shift assay
- ER
endoplasmic reticulum
- FFA
free fatty acid
- GC-MS
gas chromatography-mass spectrometry
- IB
immunoblot
- IF
immunofluorescence
- IHC
immunohistochemistry
- i.p.
intraperitoneal
- KO
knockout
- LB
lamellar body
- LC-MS
liquid chromatography-mass spectrometry
- LD
lipid droplet
- L-MMNA
NG-monomethyl-L-arginine acetate
- MPO
myeloperoxidase
- PLIN
perilipin
- PTP
permeability transition pore
- PUFA
polyunsaturated fatty acid
- SEM
standard error of the mean
- TAG
triacylglycerol
- TLCS-AP
pancreatitis induced by taurolithocholic acid sulfate infusion
- wt
wild type
Footnotes
Conflicts of interest
The authors disclose no conflicts.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2017.10.012.
References
- 1.Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology 2012;143:1179–1187. e1-e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pandol SJ, Saluja AK, Imrie CW, et al. Acute pancreatitis: bench to the bedside. Gastroenterology 2007;132: 1127–1151. Erratum: 2008;133:1056. [DOI] [PubMed] [Google Scholar]
- 3.Lerch MM, Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology 2013;144:1180–1193. [DOI] [PubMed] [Google Scholar]
- 4.Mukherjee R, Mareninova OA, Odinokova IV, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 2016;65:1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernardi P, Rasola A, Forte M, et al. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev 2015;95:1111–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li N, Wu X, Holzer RG, et al. Loss of acinar cell IKKalpha triggers spontaneous pancreatitis in mice. J Clin Invest 2013;123:2231–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giorgio V, von Stockum S, Antoniel M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A 2013;110: 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mareninova OA, Hermann K, French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 2009;119:3340–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gukovskaya AS, Gukovsky I. Autophagy and pancreatitis. Am J Physiol Gastrointest Liver Physiol 2012; 303:G993–G1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diakopoulos KN, Lesina M, Wormann S, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 2015;148:626–638.e17. [DOI] [PubMed] [Google Scholar]
- 11.Mareninova OA, Sendler M, Malla SR, et al. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell Mol Gastroenterol Hepatol 2015;1: 678–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonucci L, Fagman JB, Kim JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A 2015;112:E6166–E6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubisch CH, Sans MD, Arumugam T, et al. Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 2006;291:G238–G245. [DOI] [PubMed] [Google Scholar]
- 14.Lugea A, Tischler D, Nguyen J, et al. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2011;140:987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noel P, Patel K, Durgampudi C, et al. Peripancreatic fat necrosis worsens acute pancreatitis independent of pancreatic necrosis via unsaturated fatty acids increased in human pancreatic necrosis collections. Gut 2016; 65:100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang YT, Chang MC, Tung CC, et al. Distinctive roles of unsaturated and saturated fatty acids in hyperlipidemic pancreatitis. World J Gastroenterol 2015; 21:9534–9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawra R, Sharif R, Phillips P, et al. Development of a new mouse model of acute pancreatitis induced by administration of L-arginine. Am J Physiol Gastrointest Liver Physiol 2007;292:G1009–G1018. [DOI] [PubMed] [Google Scholar]
- 18.Kui B, Balla Z, Vegh ET, et al. Recent advances in the investigation of pancreatic inflammation induced by large doses of basic amino acids in rodents. Lab Invest 2014; 94:138–149. [DOI] [PubMed] [Google Scholar]
- 19.Gerasimenko JV, Gerasimenko OV, Petersen OH. The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 2014;592:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odinokova IV, Sung KF, Mareninova OA, et al. Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut 2009;58:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voronina SG, Barrow SL, Gerasimenko OV, et al. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem 2004;279:27327–27338. [DOI] [PubMed] [Google Scholar]
- 22.Okada N, Ohshio G, Manabe T, et al. Intracellular Ca2+ dynamics and in vitro secretory response in acute pancreatitis induced by a choline-deficient, ethionine-supplemented diet in mice. Digestion 1995; 56:502–508. [DOI] [PubMed] [Google Scholar]
- 23.Braun RJ, Sommer C, Leibiger C, et al. Accumulation of basic amino acids at mitochondria dictates the cytotoxicity of aberrant ubiquitin. Cell Rep 2015;10: 1557–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiong Y, Yepuri G, Necetin S, et al. Arginase-II promotes tumor necrosis factor-alpha release from pancreatic acinar cells causing beta-cell apoptosis in aging. Diabetes 2017;66:1636–1649. [DOI] [PubMed] [Google Scholar]
- 25.Kaasik A, Safiulina D, Zharkovsky A, et al. Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol 2007;292:C157–C163. [DOI] [PubMed] [Google Scholar]
- 26.Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 1966; 30:269–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biala AK, Dhingra R, Kirshenbaum LA. Mitochondrial dynamics: orchestrating the journey to advanced age. J Mol Cell Cardiol 2015;83:37–43. [DOI] [PubMed] [Google Scholar]
- 28.Kang R, Chen R, Zhang Q, et al. HMGB1 in health and disease. Mol Aspects Med 2014;40:1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaccaro MI, Grasso D, Ropolo A, et al. VMP1 expression correlates with acinar cell cytoplasmic vacuolization in arginine-induced acute pancreatitis. Pancreatology 2003;3:69–74. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Rouse RL. Histopathology and pathogenesis of caerulein-, duct ligation-, and arginine-induced acute pancreatitis in Sprague-Dawley rats and C57BL6 mice. Histol Histopathol 2014;29:1135–1152. [DOI] [PubMed] [Google Scholar]
- 31.Schmitz G, Muller G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J Lipid Res 1991;32: 1539–1570. [PubMed] [Google Scholar]
- 32.Anderson N, Borlak J. Drug-induced phospholipidosis. FEBS Lett 2006;580:5533–5540. [DOI] [PubMed] [Google Scholar]
- 33.Barbosa AD, Savage DB, Siniossoglou S. Lipid dropletorganelle interactions: emerging roles in lipid metabolism. Curr Opin Cell Biol 2015;35:91–97. [DOI] [PubMed] [Google Scholar]
- 34.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shirihai OS, Song M, Dorn GW 2nd. How mitochondrial dynamism orchestrates mitophagy. Circ Res 2015; 116:1835–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gukovsky I, Gukovskaya AS. Impaired autophagy triggers chronic pancreatitis: lessons from pancreas-specific atg5 knockout mice. Gastroenterology 2015;148: 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubinsztein DC, Bento CF, Deretic V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J Exp Med 2015;212:979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007;282:5641–5652. [DOI] [PubMed] [Google Scholar]
- 39.Wu F, Xu HD, Guan JJ, et al. Rotenone impairs autophagic flux and lysosomal functions in Parkinson’s disease. Neuroscience 2015;284:900–911. [DOI] [PubMed] [Google Scholar]
- 40.Shalbueva N, Mareninova OA, Gerloff A, et al. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 2013; 144:437–446.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raffaello A, Mammucari C, Gherardi G, et al. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci 2016;41:1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aho HJ, Nevalainen TJ, Havia VT, et al. Human acute pancreatitis: a light and electron microscopic study. Acta Pathol Microbiol Immunol Scand A 1982;90: 367–373. [PubMed] [Google Scholar]
- 43.Biczo G, Hegyi P, Dosa S, et al. The crucial role of early mitochondrial injury in L-lysine-induced acute pancreatitis. Antioxid Redox Signal 2011;15:2669–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sah RP, Garg SK, Dixit AK, et al. Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J Biol Chem 2014;289:27551–27561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.