

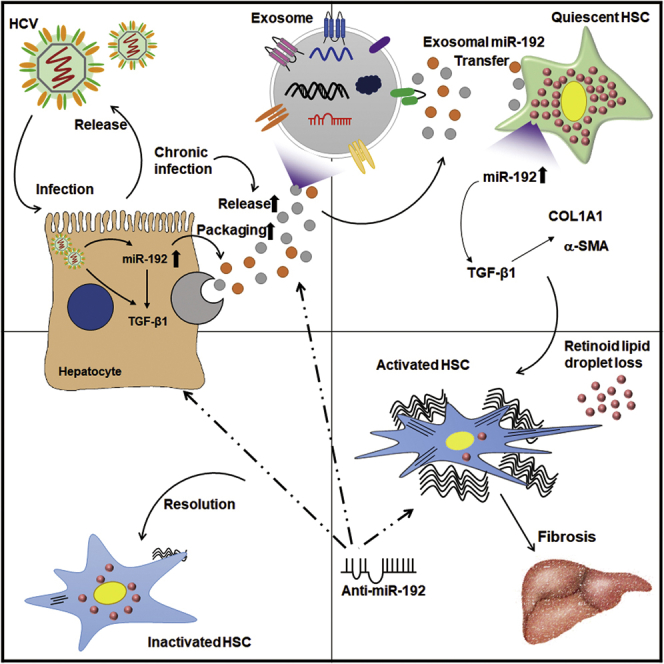
Abstract
The mechanism by which hepatitis C virus (HCV) causes fibrosis and other chronic liver diseases remains poorly understood. Previously, we observed that HCV infection induces microRNA-192 (miR-192) expression, which in turn upregulates transforming growth factor β1 (TGF-β1) in hepatocytes. In this study, we aimed to determine the roles and mechanisms of HCV-induced miR-192 expression during chronic liver injury and fibrosis and to identify potential target of the liver disease. Noticeably, miR-192 is secreted and transmitted through exosomes from HCV-replicating hepatocytes into hepatic stellate cells (HSCs). Exosomal transferred miR-192 upregulated fibrogenic markers in HSCs through TGF-β1 upregulation, resulting in the activation and transdifferentiation of HSCs into myofibroblasts. Anti-miR-192 treatment of HCV-replicating hepatocytes efficiently reduced miR-192 levels in exosomes, downregulated miR-192 and fibrogenic marker levels in HSCs, and impeded transdifferentiation of the cells. In contrast, miR-192 mimic RNA treatment significantly increased miR-192 levels in exosomes from naive hepatocytes, increased miR-192 and fibrogenic marker expression in HSCs, and induced transdifferentiation of the cells. Notably, transdifferentiation of exosome-exposed HSCs was reversed following treatment with anti-miR-192 into the HSCs. This study revealed a novel mechanism of HCV-induced liver fibrosis and identified exosomal miR-192 as a major regulator and potential treatment target for HCV-mediated hepatic fibrosis.
Keywords: hepatitis C virus, liver fibrosis, hepatic stellate cells, hepatocytes, exosome, microRNA, miR-192, transdifferentiation, myofibroblasts, fibrogenic marker
Graphical Abstract
Introduction
Hepatitis C virus (HCV) is an enveloped, positive-sense, single-stranded RNA virus belonging to the family Flaviviridae. The virus annually infects 170 million people worldwide.1 Chronic HCV infection carries an approximately 15%–30% risk of liver cirrhosis,1 and HCV-related cirrhosis is the major risk factor for hepatocellular carcinoma.2, 3 Numerous growth factors, chemokines, and cytokines are activated in virus-infected hepatocytes, thereby causing chronic hepatic inflammation, which is the major driver of fibrogenesis.2 Chronic liver injury results in both increased extracellular matrix (ECM) levels and alterations of ECM composition,4 which are typical findings of liver fibrosis. The advanced stage of fibrosis, termed cirrhosis, distorts the normal architecture and vasculature of the liver.5
Hepatic stellate cell (HSC) activation is the central event leading to the development of liver fibrosis.5 Under homeostatic conditions, HSCs store large amounts of retinoids and exhibit a quiescent phenotype.6 Following chronic liver injury, quiescent HSCs are activated, after which they transdifferentiate into a myofibroblast-like activated phenotype.5 HSC activation consists of two major phases: initiation and perpetuation.6 Once activated, HSCs secrete inflammatory cytokines and upregulate the gene expression of fibrogenic markers, such as α-smooth muscle actin (α-SMA) and ECM components, including fibronectin and collagens, particularly types I and III, leading to liver fibrosis.7 Of note, attenuation of the source of injury results in either reversion of the HSCs to an inactivated state with an intermediate quiescent phenotype or apoptosis and clearance of activated HSCs.8, 9 Therefore, reversion of HSC activation or the induction of apoptosis represents attractive targets for hepatic fibrosis modulation.
HCV core protein, as well as NS3 and NS5 proteins, indirectly triggers HSC activation by stimulating pro-mitogenic or pro-inflammatory pathways.10 Moreover, HCV core protein and subgenomic replicons induce oxidative stress, increasing reactive oxygen species production and in turn activating transforming growth factor β1 (TGF-β1) expression and HSCs.11, 12, 13 However, the mechanism of the progression and pathogenesis of liver fibrosis induced by HCV infection remains unclear.
Meanwhile, TGF-β participates in the initiation and maintenance of fibrogenesis as a powerful activator of HSCs.14 Among three major isoforms of TGF-β (1–3), TGF-β1 is the predominant fibrogenic cytokine in the liver.6, 15, 16 TGF-β1 induces the transcription of pro-fibrotic molecules, including α-SMA and collagen type I (COL1), by stimulating SMAD or non-SMAD-associated pathways, such as p38/mitogen-activated protein kinase (MAPK), Akt, and JNK signaling, resulting in ECM formation.17, 18 In addition, TGF-β1 blocks ECM degradation by matrix metalloproteinases (MMPs) through protease inhibitors, such as tissue inhibitors of MMPs (TIMPs).15, 18 The TIMP versus MMP balance has a powerful influence on the development and resolution of liver fibrosis.8, 19
Exosomes are extracellular vesicles of 30–100 nm in diameter that originate from multivesicular endosomes (MVEs) and that are released via exocytosis when MVEs fuse with the plasma membrane, in contrast to microvesicles that directly bud from the plasma membrane.20, 21 Exosomes are released by various cell types, and they can be detected in local or circulating body fluids.20 Exosomes play important roles in many biological processes, including immune responses; the progression of various diseases, such as cancer; and virus transmission,21, 22 through mediating intracellular communication via the transfer of proteins, mRNAs, and non-coding RNAs, such as microRNAs (miRNAs)23 without direct cell-to-cell contact. Replication-competent HCV RNA can also be detected in exosomes isolated from the plasma of HCV-infected patients.24 Moreover, HCV infection can be transmitted by exosomal HCV genomic RNA, and the exosomes may cause a productive infection in hepatocytes.25 However, the exact role of exosomes in HCV infection and the components in exosomes that influence the recipient cells are unclear.
We previously observed that HCV infection and replication increases TGF-β1 mRNA expression through microRNA-192 (miR-192) upregulation in hepatocytes.26 Thus, in this study, we investigated whether and how HCV-induced miR-192 influences liver fibrosis. To this end, we tested whether upregulated miR-192 in HCV-replicating hepatocytes affects HSC activation and determined the mechanism by which HCV-induced miR-192 regulates HSC activation. Furthermore, we confirmed the roles of miR-192 in HSC activation by treating miR-192 mimic RNA or anti-miR-192.
Results
Chronic HCV Replication Leads to HSC Activation through miR-192 Regulation
We initially tested whether HCV replication affects HSC activation, the key event in liver fibrosis. In this aim, JFH-1-stably replicating (JFH-1 stable cells) or naive Huh-7 cells were co-cultured with human HSC line LX-2 (Figure 1A). Then, RNA expressions of TGF-β1 and fibrosis markers, such as α-SMA and collagen type I alpha I (COL1A1), were increased in LX-2 cells when co-cultured with JFH-1 stable cells (Figure 1B), indicating that HCV replication activates HSCs. miR-192 levels were increased in both JFH-1-infected Huh-7 cells and JFH-1 stable cells, compared with naive Huh-7 cells (Figure S1A), as observed in our previous study.26 TGF-β1 mRNA levels as well as miR-192 levels were also elevated in co-cultured JFH-1 stable cells, and HCV genome RNA was detected in these cells (Figures S1B and S1C). However, no HCV RNA was found using real-time qPCR in LX-2 cells co-cultured with JFH-1 stable cells (Figure S1C). Notably, miR-192 expression was increased in LX-2 cells by co-culture with JFH-1 stable cells (Figure 1B). Direct transfection of miR-192 mimic RNA into LX-2 cells increased the expression of TGF-β1, COL1A1, and α-SMA (Figures 1C and S1D), indicating that miR-192 directly affects the expression of TGF-β1 and fibrosis markers in these cells. In addition, RNA levels of TGF-β1 and fibrosis markers, as well as those of miR-192, were decreased in LX-2 cells after co-culturing with JFH-1 stable cells that were treated with anti-miR-192 (Figure 1D), indicating that downregulation of miR-192 in HCV-replicating hepatocytes could directly or indirectly affect LX-2 cells. miR-192 and TGF-β1 mRNA levels in JFH-1 stable cells were also depressed by anti-miR-192 treatment (Figure S1E). These results suggest that miR-192 levels in HCV-replicating hepatocytes are linked to those of TGF-β1, fibrogenic markers, and miR-192 in LX-2 cells.
Figure 1.

Effects of HCV Replication and HCV-Induced miR-192 on HSC Activation
(A) Scheme of co-culture of LX-2 cells with naive Huh-7 or HCV (JFH-1) stable cells. (B) RNA levels of two activation markers (COL1A1 and α-SMA) and TGF-β1, as well as miR-192 levels, in co-cultured LX-2 cells are shown. (C) Effects of miR-192 on HSC activation are shown. LX-2 cells were transfected with miR-192 mimic RNA or negative-control miRNA (siNTC). RNA level of COL1A1, α-SMA, and TGF-β1 was quantified relative to those in siNTC-transfected LX-2 cells. (D) Effects of miR-192 depletion on HCV stable cells are shown. Experimental scheme is shown (left). JFH-1 stable cells were transfected with anti-miR-192 RNA or scramble siRNA (scr) and co-cultured with LX-2 cells starting 6 h after transfection. RNA levels of COL1A1, α-SMA, TGF-β1, and miR-192 in co-cultured LX-2 cells were quantified relative to those in cells co-cultured with scr-transfected JFH-1 stable cells (right). All data are the means of at least three independent experiments, each performed in triplicate. Error bars represent the SEM. p values were determined via a one-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001.
miR-192 Is Transferred to LX-2 Cells through the Supernatant
No HCV RNA in LX-2 cells co-cultured with HCV-producing JFH-1 stable cells (Figure S1C) and no transgene expression in LX-2 cells infected with HCV pseudoparticles carrying the luciferase gene (data not shown), as demonstrated previously,27 suggest a lack of HCV infection and replication in LX-2 cells. Nevertheless, exposure to supernatant from JFH-1 stable cells increased the expression of fibrogenic markers, TGF-β1, and miR-192 in LX-2 cells (Figure 2A). In contrast to the concentration-dependent effects of TGF-β1 treatment on TGF-β1, α-SMA, and COL1A1 mRNA expression in LX-2 cells, miR-192 expression was not affected by exposure to TGF-β1 (Figure 2B). These results imply that factors other than TGF-β1 and HCV particles in supernatant from HCV-replicating hepatocytes influence miR-192 expression in LX-2 cells. Notably, the expression level of fibrogenic markers, TGF-β1, and miR-192 was not affected in LX-2 cells exposed to supernatant from naive Huh-7 cells, which is human hepatoma cell lines, compared with control media, suggesting no effects of any components of supernatant from naive liver cancer cells on HSC activation (Figure S2A). miR-192 levels were elevated in supernatant from JFH-1 stable cells (Figure S2B), indicating that miR-192 is secreted into the supernatant of HCV-replicating hepatocytes. miR-192 levels were also increased in supernatant from miR-192 mimic-transfected hepatocytes and in LX-2 cells treated with the supernatant (Figure 2C). Contrarily, miR-192 levels were decreased in supernatant from anti-miR-192-transfected hepatocytes and in LX-2 cells treated with this supernatant. Moreover, TGF-β1 and fibrogenic marker expression in LX-2 cells was affected by the supernatant of hepatocytes transfected with miR-192 mimic or anti-miR-192 (Figure S2C). These results suggest that miR-192 secreted into the supernatant of HCV-replicating hepatocytes is linked to the accumulation of miR-192 and HSC activation in LX-2 cells.
Figure 2.

Effects of Supernatant from HCV-Replicating Cells on miR-192 Expression in HSCs
(A) Experimental scheme (left). LX-2 cells were treated with supernatant from naive Huh-7 or JFH-1 stable cells for 3 days. The RNA levels of COL1A1, α-SMA, and TGF-β1 and miR-192 levels in LX-2 cells treated with JFH-1 supernatant were quantified relative to those in cells treated with naive Huh-7 supernatant (right). (B) Effects of TGF-β1 on miR-192 expression in HSCs are shown. LX-2 cells were cultured with serum-free medium for 16 h and then treated with different concentrations of recombinant TGF-β1 protein for 72 h. The mRNA levels of COL1A1, α-SMA, and TGF-β1 and miR-192 levels in LX-2 cells were assessed relative to those in control PBS-treated cells. (C) Effects of miR-192 introduction or depletion on Huh-7 cells are shown. Experimental scheme is shown (left). Huh-7 cells were transfected with miR-192 mimic RNA or anti-miR-192 for 48 h and then the supernatant was collected and used to treat LX-2 cells for 72 h. The miR-192 copy number in each supernatant was quantified (middle). miR-192 levels in LX-2 cells treated with each type of supernatant were assessed relative to those in cells treated with supernatant from negative-control miRNA (siNTC)- or scramble RNA (scr)-treated Huh-7 cells (right). All data are presented as the means of at least three independent experiments, each performed in triplicate. Error bars represent the SEM. p values were determined via a one-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., non-significant.
Exosomes Facilitate HCV Replication-Induced HSC Activation
We examined whether miR-192 secreted from HCV-replicating hepatocytes is transferred into LX-2 cells through exosomes. Exosomes were purified from supernatant of Huh-7 or JFH-1 stable cells, and nanoparticles trafficking analysis (NTA) was used for measuring the particle size and concentration of exosomes (Figure S3A). The levels of exosomal markers, such as CD63, CD81, HSP70, and LAMP2, were significantly higher in exosomes isolated from JFH-1 stable cell-free supernatant (JFH-1 exosome) than in control exosomes isolated from naive Huh-7 cell-free supernatant (Huh-7 exosome; Figure 3A). Calnexin, GM130, cytochrome C, and NUP98 were not detected in both JFH-1 exosome and Huh-7 exosome, indicating that the isolated exosomes were not contaminated by other organelles, including the endoplasmic reticulum, Golgi, mitochondria, and nucleoporin, respectively (Figure 3B). Moreover, NTA showed that the number of exosomes derived from JFH-1 exosome was approximately 2-fold higher than that isolated from Huh-7 exosome (Figure S3A), indicating that HCV replication increases exosome release. Notably, miR-192 and miR-122 levels were higher in JFH-1 exosome than in Huh-7 exosome (Figure 3C). HCV-positive genome RNA was also detected in JFH-1 exosome, as revealed in previous studies24, 28 (Figure 3D). As recently reported for subgenomic HCV RNA,29 miR-19-3p was more abundant in JFH-1 exosome despite its lower copy number relative to the findings for miR-192 and miR-122 (Figure S3B).
Figure 3.

Effects of HCV Replication on Exosome Release, Exosomal RNA Level, and HSC Activation
(A) Comparison of exosome release from naive Huh-7 (Huh-7 exo) and JFH-1 stable cells (JFH-1 exo). Immunoblot analysis of exosome marker proteins (CD63, LAMP2, HSP70, and CD81) in increasing volume of exosomes purified from each cell type is shown (left). The level of each exosome marker protein was quantified and presented as a percentage of that in Huh-7 exo (middle). Exosome total proteins were quantified using a bicinchoninic acid assay (BCA) (right). (B) Immunoblot analysis of total cell lysates (lysate) and isolated exosomes (exo) of naive Huh-7 or JFH-1 stable cells is shown. (C and D) Quantification of miRNAs and HCV RNA within exosomes from naive Huh-7 or JFH-1 cells is shown. After isolating exosomes, we re-suspended the exosomes in 100 μL PBS buffer and used 2 μL or 20 μL of the solution for the analysis of (C) the RNA levels of miR-192 (left: Huh-7 exo, 3.03 ± 2.69 × 105 copies per 2 μL and 3.09 ± 2.26 × 106 copies per 20 μL; JFH-1 exo, 7.20 ± 4.74 × 105 copies per 2 μL and 8.80 ± 7.89 × 106 copies per 20 μL), miR-122 (right: Huh-7 exo, 2.25 ± 1.80 × 105 copies per 2 μL and 1.60 ± 1.43 × 106 copies per 20 μL; JFH-1 exo, 4.43 ± 2.29 × 105 copies per 2 μL and 5.06 ± 4.90 × 106 copies per 20 μL), and (D) HCV RNA (JFH-1 exo, 2.42 ± 3.07 × 105 copies per 2 μL and 2.15 ± 2.79 × 106 copies per 20 μL). (E and F) Quantification of fibrotic marker RNAs and proteins in LX-2 cells treated with each type of exosomes is shown. (E) Relative intracellular mRNA levels of fibrosis markers (left) and copy numbers of miR-192 (right) in LX-2 cells treated with each type of exosomes are shown. Copy numbers of miR-192 were 3.30 ± 1.67 × 105 and 6.18 ± 2.78 × 105 per 100 ng of total RNA in Huh-7 exo-treated and JFH-1 exo-treated LX-2 cells, respectively. (F) Immunoblot analysis of the fibrosis markers COL1A1 and α-SMA, as well as TGF-β1, in LX-2 cells treated with each type of exosome (left) and quantification of their levels relative to those of tubulin (right) are shown. (G) Effects of exosomes on HSC activation are shown. LX-2 cells were treated with each exosome type for 72 h, fixed, stained, and visualized using fluorescence microscopy. Representative images (left) of α-SMA and DAPI staining in LX-2 cells are shown. Scale bars represent 50 μm. α-SMA protein levels (right) in LX-2 cells treated with each type of exosomes were quantified from more than five fields of three independent experiments and shown relative to the levels in cells treated with Huh-7 exo. Data are also shown in Figure S4. All data are presented as the means of at least three independent experiments, each performed in triplicate. Error bars represent the SEM. p values were determined using a one-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Exposure of Huh-7 exosome showed little effects on protein and RNA levels in LX-2 cells, compared with media (Figure S3C). By sharp contrast, miR-192 levels and the mRNA levels of fibrosis markers and TGF-β1 were increased in JFH-1 exosome-treated LX-2 cells (Figure 3E). Protein levels of fibrosis markers, including COL1A1 and α-SMA, as well as TGF-β1 were also induced in JFH-1 exosome-treated LX-2 cells (Figure 3F). Moreover, LX-2 cells treated with JFH-1 exosome were more efficiently activated and transdifferentiated into a myofibroblast-like phenotype than cells treated with Huh-7 exosome, and treatment with JFH-1 exosome was also associated with elevated α-SMA expression (Figures 3G and S4). However, little HCV RNA was found in LX-2 cells 48 h post-exposure of JFH-1 exosome, even though HCV RNA was detected in the cells at 24 h post-exposure (Figure S3D), indicating transfer of HCV RNA into LX-2 cells through JFH-1 exosome but no HCV RNA replication in the cells. These results suggest that LX-2 cell activation is not caused by the delivered HCV genomic RNA.
We confirmed our findings in human primary HSCs. Exposure of JFH-1 exosome showed increase in RNA levels of fibrosis markers and TGF-β1 as well as miR-192 and miR-122 levels, but no effects in miR-19-3p, in the primary HSCs (Figures 4A and 4B). Furthermore, exposure of JFH-1 exosome induced high levels of fibrosis marker proteins and activation of primary HSCs (Figures 4C, 4D, and S5). These results indicate that the number of miR-192-containing exosomes is increased by HCV replication, and they are released from the virus-replicating hepatocytes, after which they are transmitted to HSCs to activate and induce differentiation of the cells.
Figure 4.

Primary HSCs Activation by Exosome Isolated from HCV-Replicating Hepatocytes
(A–C) Quantification of fibrotic marker RNAs and proteins in primary HSCs treated with each type of exosomes. (A) Relative intracellular mRNA levels of fibrosis markers and (B) copy numbers of miR-192, miR-122, and miR-19-3p in primary HSCs treated with each type of exosome are shown. (C) Immunoblot analysis of the fibrosis markers COL1A1 and α-SMA, as well as TGF-β1, in primary HSCs treated with each type of exosome (left) and quantification of their levels relative to those of tubulin (right) are shown. (D) Effects of exosomes on primary HSCs activation are shown. Primary HSCs were treated with each exosome type for 72 h, fixed, stained, and visualized using fluorescence microscopy. Representative images (left) of α-SMA and DAPI staining in primary HSCs are shown. Scale bars represent 50 μm. α-SMA protein levels (right) in primary HSCs treated with each type of exosomes were quantified from more than five fields and shown relative to the levels in cells treated with Huh-7 exo. Data are also shown in Figure S5. All data are presented as the means of at least three independent experiments, each performed in triplicate. Error bars represent the SEM. p values were determined using a one-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., non-significant.
Transferred Exosomes and Exosomal miR-192 Affect LX-2 Activation
We tested whether exosome-induced LX-2 cell activation was directly caused by exosomes. JFH-1 stable cells were treated with GW4869, a known inhibitor of exosome release,28, 30 which led to decreased levels of the exosome marker CD63 and total exosome proteins in JFH-1 exosome (Figure 5A). The suppression of exosome release following GW4869 treatment was accompanied by reductions of miR-192, miR-122, and HCV genome RNA levels within the exosomes (Figure 5B). Treatment with exosomes isolated from GW4869-treated JFH-1 cells (GW4869 exosome) decreased the expression of HSC activation markers (COL1A1, α-SMA, and TGF-β1) of LX-2 cells (Figure 5C). Furthermore, treatment with GW4869 exosome effectively suppressed the transdifferentiation of LX-2 cells and reduced α-SMA accumulation in these cells (Figures 5D and S6). Similar to its reduction within GW4869 exosomes, miR-192 levels were also decreased in LX-2 cells by GW4969 exosome treatment (Figure 5E).
Figure 5.

Effects of Blocking Exosome Release on HSCs
Effects of exosomes from JFH-1 stable cells treated with the exosome release inhibitor GW4869 (10 μM) (GW4869 exo). Control exosomes from untreated JFH-1 stable cells are referred as DMSO exo. (A) Comparison of CD63 (left) or exosome total protein levels using a BCA assay (right) within each exosome is shown. (B) Levels of miR-192 (left: 2.18 ± 1.62 × 106 and 7.82 ± 8.46 × 105 copies per 20 μL of control DMSO exo and GW4869 exo, respectively), miR-122 (middle: 1.98 ± 0.99 × 106 and 8.96 ± 4.68 × 105 copies per 20 μL of DMSO exo and GW4869 exo, respectively), and HCV RNA (right: 4.33 ± 3.21 × 105 and 7.73 ± 8.51 × 104 per 20 μL of DMSO exo and GW4869 exo, respectively) within each type of exosomes were quantified. (C) Immunoblot analysis of the fibrogenic markers COL1A1 and α-SMA, as well as TGF-β1, in LX-2 cells treated with each exosome type (left) is shown. Protein levels were quantified relative to those of tubulin (right). (D) LX-2 cells were treated with each exosome type for 72 h, fixed, stained, and visualized via fluorescence microscopy. Representative images of α-SMA and DAPI staining in each cell are shown (left). Scale bars represent 50 μm. Intensity of α-SMA staining was quantified from more than five fields in three independent experiments (right). Data are also shown in Figure S6. (E) Intracellular miR-192 levels in LX-2 cells treated with each exosome type were quantified. All data are presented as the means of at least three independent experiments, each performed in triplicate. Error bars represent the SEM. p values were determined using a one-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we determined the effects of exosomal miR-192. Exosomes isolated from JFH-1 stable cells treated with anti-miR-192 (anti-miR-192 exosome) had no effects on CD63 or total exosome protein levels, indicating that exosome release was not affected by miR-192 downregulation (Figure 6A). However, miR-192 copy numbers were decreased in these exosomes. By contrast, miR-122 and HCV genome RNA levels in JFH-1 exosomes were not affected by anti-miR-192 treatment (Figure 6B). Expression of the fibrosis markers COL1A1 and α-SMA as well as that of TGF-β1 in LX-2 cells was diminished by anti-miR-192 exosome treatment (Figure 6C). In addition, treatment with anti-miR-192 exosome significantly decreased the transdifferentiation of LX-2 cells and reduced α-SMA accumulation in these cells (Figures 6D and S7). miR-192 levels in LX-2 cells were also decreased by anti-miR-192 exosome treatment (Figure 6E). These results suggest that HCV-exposed exosomes alter the activation of LX-2 cells and that exosomal miR-192 directly or indirectly influences this activation.
Figure 6.

Effects of Blocking Exosomal miR-192 Level on HSCs
Effects of exosomes from JFH-1 stable cells treated with anti-miR-192 (anti-miR-192 exo). Control exosomes from scramble-treated JFH-1 stable cells are referred to as scr exo. (A) Comparison of CD63 (left) and exosome total protein levels (right) using a BCA assay is shown. (B) Levels of miR-192 (left: 3.27 ± 1.85 × 106 and 1.24 ± 0.62 × 106 copies per 20 μL of control scr exo and anti-miR-192 exo, respectively), miR-122 (middle: 2.56 ± 1.71 × 106 and 2.49 ± 1.32 × 106 copies per 20 μL of scr exo and anti-miR-192 exo, respectively), and HCV RNA (right: 1.67 ± 1.03 × 106 and 1.54 ± 1.15 × 106 copies per 20 μL of scr exo and anti-miR-192 exo, respectively) within each exosome type were quantified. (C) Immunoblot analysis of the fibrogenic markers COL1A1 and α-SMA, as well as TGF-β1, in LX-2 cells treated with each exosome type (left) is shown. Protein levels were quantified relative to those of tubulin (right). (D) LX-2 cells were treated with each exosome type for 72 h, fixed, stained, and visualized via fluorescence microscopy. Representative images of α-SMA and DAPI staining in each cell are shown (left). Scale bars represent 50 μm. Intensity of α-SMA staining was quantified from more than five fields in three independent experiments (right). Data are also shown in Figure S7. (E) Intracellular miR-192 levels in LX-2 cells treated with each exosome type were quantified. All data are presented as the means of at least three independent experiments, each performed in triplicate. Error bars represent the SEM. p values were determined using a one-tailed unpaired Student’s t test. *p < 0.05; ***p < 0.001; n.s., non-significant.
Exosomal miR-192 Directly Influences HSC Activation
We tested whether miR-192 within exosomes can activate HSCs independently of HCV replication (Figure 7A). No changes in CD63 and total exosome protein levels were noted in exosomes isolated from naive Huh-7 cells transfected with miR-192 mimic RNA (miR-192 exosome; Figure 7B), indicating that exosome numbers were not affected by the treatment, as observed for anti-miR-192 transfection in JFH-1 cells in Figure 6A. However, miR-192 levels within miR-192 exosome were significantly increased, whereas those of miR-122 remained unchanged (Figure 7C). Treatment with miR-192 exosome increased the transdifferentiation of LX-2 cells and expression of COL1A1, α-SMA, and TGF-β1 in the cells (Figures 7D, 7E, and S8A). Moreover, LX-2 cells treated with miR-192 exosome displayed elevated miR-192 levels (Figure 7F).
Figure 7.

Effects of Exosomes from miR-192-Transfected Hepatocytes or Exosomes Packaged with miR-192
(A) Experimental scheme: negative-control miRNA (siNTC) or miR-192 mimic RNA was transfected into Huh-7 cells, and exosomes were purified from the transfected cells (siNTC and miR-192 exo, respectively) and used to treat LX-2 cells. (B) Comparisons of protein levels of the exosome markers LAMP2 and CD63 using western blotting (left) and exosome total protein levels using a BCA assay (right) within exosomes from each transfected cell type are shown. (C) Quantification of miR-192 (left: 3.98 ± 1.71 × 106 and 2.08 ± 2.09 × 108 copies per 20 μL of control siNTC exo and miR-192 exo, respectively) and miR-122 levels (right: 2.04 ± 1.48 × 106 and 1.88 ± 1.27 × 106 copies per 20 μL of siNTC exo and miR-192 exo, respectively) in exosomes from each transfected cell type is shown. (D) Immunoblot analysis of the fibrosis markers COL1A1 and α-SMA, as well as TGF-β1, in LX-2 cells treated with each exosome type is shown. (E) LX-2 cells were treated with each type of exosomes for 72 h, fixed, stained, and visualized via fluorescence microscopy. Representative images of α-SMA and DAPI staining in each cell type (left). Scale bars represent 50 μm. The intensity of α-SMA staining was quantified using more than five fields from at least three independent experiments (right). Data are also shown in Figure S8A. (F) Intracellular miR-192 levels in LX-2 cells treated with each type of exosomes were quantified. (G) Experimental scheme: exosomes were first purified from naive Huh-7 cells and then packaged with siNTC or miR-192 (siNTC- and miR-192-packaged exosomes, respectively). (H) Immunoblot analysis of COL1A1, α-SMA, and TGF-β1 expression in LX-2 cells treated with each packaged exosome type is shown (left). Protein levels were quantified relative to those of tubulin (right). (I) LX-2 cells were treated with each packaged exosome type for 72 h, fixed, stained, and visualized using fluorescence microscopy. Representative images of α-SMA and DAPI staining in each cell are shown (left). Scale bars represent 50 μm. Intensity of α-SMA staining was quantified using more than five fields from at least three independent experiments is shown (right). Data are also shown in Figure S8B. (J) Intracellular miR-192 levels in LX-2 cells treated with each packaged exosome type were quantified. All data are presented as the means of at least three independent experiments, each performed in triplicate. Error bars represent SEM. p values were determined using a one-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; n.s., non-significant.
Next, we more directly tested the effects of miR-192 by initially isolating exosomes from naive Huh-7 cells and then packaging miR-192 directly into these exosomes (Figure 7G). Treatment of LX-2 cells with miR-192-packaged exosomes induced transdifferentiation of the cells and increased levels of COL1A1, α-SMA, and TGF-β1 (Figures 7H, 7I, and S8B). Higher miR-192 levels were also found in LX-2 cells treated with miR-192-packaged exosomes (Figure 7J). These results suggest that LX-2 cell activation is directly influenced by miR-192 within exosomes.
Exosomal miR-192 Released from Hepatocytes Activates LX-2 Cells
We investigated whether miR-192 in hepatocytes could translocate into LX-2 cells through exosomes and activate LX-2 cells. To visualize the transport of extracellular miR-192 derived from hepatocytes, exosomes were isolated from Huh-7 cells transfected with Cy3-labeled miR-192 and then incubated with LX-2 cells (Figure 8A). Cellular uptake of Cy3-miR-192 was observed in LX-2 cells (Figure 8B), indicating the transmission of miR-192 derived from Huh-7 cells into LX-2 cells via exosomes. Moreover, LX-2 cells that accumulated exosomal miR-192 exhibited transdifferentiation and increased α-SMA expression. Loss of lipid droplets and intracellular retinoids is a distinguished feature of HSC activation, although it remains unclear whether these changes are required for HSC activation.31 Meanwhile, inactive HSCs reverted from an activated state display a quiescent-like phenotype characterized by the presence of intracellular retinoid lipid droplets.32 Treatment of JFH-1 exosome-exposed LX-2 cells with anti-miR-192 significantly rescued the presence of retinoid lipid droplets in the cells and decreased levels of COL1A1, α-SMA, and TGF-β1 (Figures 8C and 8D). Levels of miR-192 were much more downregulated in exosome-exposed LX-2 cells treated with anti-miR-192 than even those in PBS-treated LX-2 cells (Figure 8E). These results indicate that downregulating miR-192 can reverse HSC activation caused by HCV replication through decreasing TGF-β1 signal.
Figure 8.

Transmission of Exosomal miR-192 Released from Hepatocytes into HSCs and Effects of miR-192 Downregulation on Activated HSCs
(A) Experimental scheme: Huh-7 cells were transfected with Cy3-labeled miR-192, and exosomes were purified and used to treat LX-2 cells. (B) Confocal microscopy image of Cy3-labeled miR-192 (red), α-SMA, and DAPI staining in exosome-treated LX-2 cells is shown. An image of two representative fields is shown, and the scale bars represent 50 μm. (C) LX-2 cells were exposed to PBS or JFH-1 exosomes and then treated with scrambled RNA (scr) or anti-miR-192. Oil Red O staining image of LX-2 cells using a light microscope (left) and quantification of Oil-Red-O-stained cells using a microplate reader after destaining are shown (right). The results are representative of at least three independent experiments. (D) Immunoblot analysis of COL1A1, α-SMA, and TGF-β1 expression in exosome-exposed LX-2 cells treated with each RNA is shown. (E) Intracellular miR-192 levels in exosome-exposed LX-2 cells treated with each RNA were quantified and presented as the means of at least three independent experiments, each performed in triplicate. Error bars represent the SEM. p values were determined using a one-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
miRNAs are key regulators of HCV-induced liver diseases, and the expression patterns of miRNAs can be changed during HSC activation.33, 34, 35 However, the mechanism by which miRNAs mediate HCV-induced liver diseases, including fibrosis, is unclear. We previously observed that HCV infection and replication increases the levels of miR-192 through HCV core protein in hepatocytes and stimulates TGF-β1 transcription in the cells through miR-192-mediated ZEB1 downregulation.26 In this study, we observed the fibrotic activation of HSCs by HCV-induced miR-192 when co-cultured with HCV-replicating hepatocytes. Notably, miR-192 accumulated in HSCs through exosomes released from HCV-replicating hepatocytes, but its accumulation was not induced by either TGF-β1 or HCV particles. The transmitted exosomal miR-192 stimulated the transdifferentiation of not only LX-2 hepatic stellate cell lines but also human primary HSCs into myofibroblasts through TGF-β1 stimulation.
Exosomes from HCV-infected patients contain replication-competent viral RNA as well as miR-122.36 In this study, we demonstrated that HCV replication increased exosome release and exosomal miR-192 levels in addition to those of exosomal HCV genome RNA and miR-122. Recently, Devhare et al.29 reported that exosomes released from HCV-infected hepatocytes promoted HSC activation, which was related to the exosomal transmission of miR-19a. We also examined that miR-19a (7.99 ± 4.39 × 105 copies per 20 μL of JFH-1 exosome) was packaged in the exosomes but at a much lower copy number than miR-192 (8.80 ± 7.89 × 106 copies per 20 μL of JFH-1 exosome) and miR-122 (5.06 ± 4.90 × 106 copies per 20 μL of JFH-1 exosome). However, the mechanism by which HCV infection increases exosomal miR-19a levels is unclear. Moreover, it is uncertain whether HCV replication induces miR-19a expression or enhances miR-19a packaging into exosomes. Contrarily, the observation of a significant reduction of miR-192 levels within GW4869 or anti-miR-192 exosome in this study and the previous finding of miR-192 induction in HCV-infected hepatocytes26 strongly indicate that HCV-induced exosomal miR-192 level is mainly due to HCV-mediated increases in miR-192 expressions in the infected cells combined with an HCV-induced increment of the number of exosomes. Notably, we observed efficient transmission of miR-192, but not miR-19-3p, into not only LX-2 cell lines but also primary HSCs by JFH-1 exosome. In addition, effective suppression of HSC activation by treatment with anti-miR-192 exosome was observed in this study, strongly indicating that exosomal miR-192 is the major inducer of the development of HCV-induced liver fibrosis. Moreover, whether exosome-packaged miR-19a solely activates HSCs independently of HCV infection and replication is unclear. Contrarily, the observation of efficient HSC activation and transdifferentiation following treatment with exosomes from miR-192 mimic RNA-transfected hepatocytes or with miR-192-packaged exosomes in this study strongly suggests that exosome-packaged miR-192 alone can induce HSC activation.
The most widely known functions of miR-192 are inducing renal fibrosis through TGF-β1 upregulation in the kidneys37 and downregulating target transcripts, including several genes associated with cell cycle regulation in colon cancer and myeloma.38, 39 Concerning liver diseases, miR-192 has emerged as a new biomarker of acetaminophen-induced acute liver injury.40 Moreover, liver injury is associated with miR-122 and miR-192 levels in the sera of patients with chronic hepatitis C.41 In contrast, miR-192 levels tend to be lower in cirrhotic livers of patients and were downregulated in HSCs during progression of hepatic fibrosis in mouse models established by treatment of bile duct ligation surgery or chronic CCl4, and miR-192 may play a role in maintaining the quiescence state of HSCs.42 However, HSCs were clearly activated following transfection with miR-192 mimic RNA in this study. Moreover, HSCs were efficiently transdifferentiated into myofibroblasts following treatment with miR-192-containing exosomes released from HCV-replicating or miR-192-transfected hepatocytes or with miR-192-packaged exosomes. Furthermore, HSC activation was significantly suppressed when exosomal miR-192 transmission into the cells was reduced by anti-miR-192 treatment of HCV-replicating hepatocytes. Most importantly, HCV-activated HSCs reverted to an inactivated state after treatment with anti-miR-192. Taken together, the results illustrate that miR-192 is critical for the initiation and/or perpetuation of HSC activation following HCV replication. Components released from naive cancerous cell lines used in this study may not be related with HSC activation due to no observation of activation in LX-2 cells exposed to supernatant from Huh-7 cells. The miRNA expression profile in patients with liver diseases or HSCs exhibits a number of differences, and heterogeneity between studies has been noted.43, 44 These discordances might be due to differences in the causes of hepatic injury, methods of HSC activation, or species studied.
Unlike liver cirrhosis, liver fibrosis is known as a reversible disease.8 Therefore, diagnosis and treatment are considered attractive goals in this stage. In particular, HSC activation is a critical step in the development of fibrosis; hence, resolution of HSC activation through reversion or apoptosis is an attractive target for hepatic fibrosis regulation. miR-192 levels are increased in the sera of patients with fibrosis caused by nonalcoholic liver disease, thus making it a potential biomarker.34, 45 In addition, our study suggests exosomal miR-192 as a potential HCV-related fibrosis marker, given its important role in HSC activation through TGF-β1 stimulation. Moreover, our study reveals that downregulation of HCV-induced miR-192 in hepatocytes reduced miR-192 levels in exosomes and impeded the development of hepatic fibrosis. More importantly, downregulating miR-192 in HCV-activated HSCs was sufficient to reverse the transdifferentiation of these cells through the downregulation of the major fibrogenic trigger TGF-β1. Therefore, miR-192 antagonism should be a useful therapeutic approach against HCV-induced fibrosis.
In conclusion, we identified exosomal miR-192 as a major regulator of HCV-mediated hepatic fibrosis. HCV replication increases the level of miR-192 in hepatocytes and the release of miR-192-packaged exosomes. HCV-induced exosomal miR-192 is transmitted into HSCs and increases the production of TGF-β1 and liver fibrosis markers in the cells, resulting in HSC activation and transdifferentiation into myofibroblasts. Downregulating miR-192 in either HCV-replicating hepatocytes or exosome-activated HSCs efficiently suppresses HSC activation or leads to reversion of activated HSCs. These findings suggest that miR-192 is useful as a diagnostic and therapeutic target for HCV-mediated liver fibrosis. Further studies are required to determine whether exosomal miR-192 levels are associated with hepatic fibrosis of chronic hepatitis C patients and whether any additional exosomal miRNAs have functional roles in HCV replication progression of HSC activation through coordination with miR-192.
Materials and Methods
Cells and HCV
An infectious, replication-competent JFH-1 virus (HCV genotype 2a) was generated as previously described.46, 47 The human hepatoma cell line Huh-7 (JCRB Cell Bank, JCRB0403, Osaka, Japan) and JFH-1 stably replicating Huh-7 cells (JFH-1 stable cells) were cultured and generated as previously described.48, 49 Briefly, Huh-7 and JFH-1 stable cells were maintained in DMEM with high glucose (GE Healthcare Hyclone, Logan, Utah) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (GE Healthcare Hyclone) and 1% penicillin and streptomycin (p/s) at 37°C. The human HSC line LX-2 was purchased from EMD Millipore (SCC064, Ontario, Canada) and maintained in DMEM with high glucose supplemented with 2% heat-inactivated FBS and 1% p/s at 37°C. The passage number of cell culture was less than five for Huh-7 and JFH-1 stable cells and less than three in LX-2 cells for all experiments in this study. The human primary HSCs were purchased from iXCells Biotechnologies (10HU-210, San Diego, CA) and maintained in Stellate Cell Growth Medium (cat. no. MD-0014, iXCells Biotechnologies, San Diego, CA, USA) at 37°C. After thawing of frozen cells, the primary HSCs cells were used immediately when the cells reached 80% confluence rather than sub-cultured.
Co-culture of LX-2 Cells with Naive Huh-7 or HCV Stable Cells
Co-culture experiments were conducted using Transwell Permeable Supports 0.4-μm membranes (Corning, Corning, NY, USA). Naive Huh-7 or JFH-1 stable cells (1 × 105) were seeded into 0.4-μm Transwell membranes overnight. Naive Huh-7 cells or JFH-1 stable cells were co-incubated with LX-2 cells at a density of 2 × 105 cells in 37°C in a CO2 incubator for 72 h and harvested for subsequent experimentation.
Transfection of miRNA Mimic RNA and Anti-miRNA
Cells (2 × 105) were seeded in 35-mm culture dishes and transfected with 50 pmol of miRNA mimic RNA or anti-miRNA twice using Lipofectamine 2000 reagent (Thermo Fisher Scientific, Waltham, MA, USA). The sequences are listed in Table S1. siNTC and anti-scramble were used as a negative control for miRNA and anti-miRNA, respectively. Cells were washed with 1× PBS, and the medium was changed to penicillin and streptomycin-free medium at 6 h post-transfection followed by incubation for 48 h for subsequent experimentation.
Total RNA Isolation and Quantification of miRNA, mRNA, and HCV RNA
Cells were washed once with 1× PBS, and total RNA was extracted using Tri-reagent (MRC, Cincinnati, OH) according to the manufacturer’s protocol. After RNA isolation, total RNA was treated with RNase-free DNase I (Roche, Penzberg, Germany) and reverse transcribed, and real-time qPCR was performed. Reverse transcription was performed using two different methods. First, cDNA of mRNA and HCV genome RNA was synthesized from 1 μg of total RNA using reverse transcriptase (ABM, Richmond, Canada) and amplified using Power SYBR Green Master Mix (Applied Biosystems, Posters, CA, USA). Second, miRNA (100 ng) was reverse transcribed into cDNA using a reverse transcription kit (no. 4366596; Applied Biosystems). Quantification was performed using the TaqMan microRNA Assay and TaqMan PCR Master Mix (no. 4324018; Applied Biosystems). Real-time PCR was performed in triplicate wells for each condition using the Step One Plus Real-Time PCR System (Applied Biosystems) or Rotor-Gene Real-Time System (QIAGEN, Hilden, Germany). The copy numbers of HCV-positive-strand genomes and miRNAs were calculated in comparison to a standard curve of full-length in vitro transcribed HCV RNA and miRNA mimic RNAs, respectively. RNA levels were normalized to those of 18S rRNA or GAPDH mRNA in each sample, but not for exosome samples. The primer sequences for real-time PCR are listed in Table S2. All data are the means of at least three independent experiments, each performed in triplicate.
TGF-β1 Treatment
Serum-starved LX-2 cells (3 × 105) were seeded in 6-well plates. After 16 h of culture, TGF-β1 recombinant protein (GF111, EMD Millipore, Darmstadt, Germany; concentration 1–25 ng/mL) was treated with DMEM supplemented with 2% FBS.
Analysis and Treatment of Cell-free Supernatant
The supernatant of cultured cells was harvested and centrifuged at 2,000 rpm for 10 min to remove cells and debris. The supernatant was analyzed by real-time qPCR to detect miRNAs and HCV genome RNA. Supernatant from Huh-7 cells or JFH-1 stable cells (1 mL) was used to treat LX-2 cells.
Isolation and Treatment of Exosome
Exosomes from cell culture supernatants were isolated using ExoQuick-TC (System Bioscience, Palo Alto, CA) according to the manufacturer’s protocol. Specifically, the same numbers of Huh-7 and JFH-1 stable cells were incubated for 3 days. To inhibit exosome release, cells were treated for 48 h with 10 μM GW4869 (Sigma Aldrich, St. Louis, MO, USA) dissolved in DMSO (Sigma Aldrich). To assess the effects of miR-192, cells were transfected with miR-192 mimic RNA or anti-miR-192. siNTC or scramble RNA was used as a control. Supernatant from each cell type was collected and centrifuged at 3,000 rpm for 15 min to remove cells and debris. The supernatant (5 mL) was added to ExoQuick-TC (1 mL) and mixed well by inverting. After overnight culture at 4°C, the mixture was centrifuged at 1,500 × g for 30 min at 4°C. The supernatant was then aspirated and centrifuged at 1,500 × g for 5 min. The whole-exosome pellet was re-suspended in 100 μL of 1× PBS, separated into 20 μL aliquots, and stored at −80°C. For RNA analysis, total RNA was extracted from the re-suspended exosomes using Tri-reagent (MRC) and real-time qPCR was performed.
Immunoblot Analysis
Cells or isolated exosomes were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 2 mM EDTA) supplemented with a protease inhibitor cocktail (Invitrogen, Carlsbad, CA, USA) and then maintained by constant agitation for 30 min at 4°C. Lysates were harvested by centrifugation at 4°C. Proteins quantified using the SMART bicinchoninic acid Protein Assay (iNtRON Biotechnology, Gyeonggi-do, Republic of Korea) were separated by SDS-PAGE and electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes (EMD Millipore) in 2 mM Tris-192 mM glycine buffer. The membrane was blocked with 5% blocking reagent (Amersham ECL Prime Blocking Reagent, GE Healthcare Life Sciences, Buckinghamshire, UK) in Tris-buffered saline with 0.1% Tween 20 (TBS-T) and incubated with the following primary antibodies overnight at 4°C: anti-CD63 (1:1,000 dilution; EXOAB-CD63A-1; System Bioscience or sc-5275; Santa Cruz Biotechnology, Dallas, TX, USA); anti-LAMP2 (1:2,000 dilution; sc-18822; Santa Cruz Biotechnology); anti-HSP70 (1:1,000 dilution; EXOAB-HSP70A-1; System Biosciences); anti-CD81 (1:1,000 dilution; EXOAB-SD81A-1; System Biosciences); anti-α-SMA (1:1,000 dilution; ab7817; Abcam, Cambridge, UK); anti-COL1A1 (1:1,000 dilution; ab34710; Abcam); anti-TGFβ1 (1:1,000 dilution; ab92486; Abcam); anti-cytochrome C (1:2,000; no. 11940; Cell Signaling Technology, Danvers, MA, USA); anti-Calnexin (1:2,000; no. 2679; Cell Signaling Technology); anti-NUP98 (1:2,000; no. 2598; Cell Signaling Technology); anti-GM130 (1:2,000; no. 12480; Cell Signaling Technology); or anti-α-tubulin (1:1,000 dilution; PM054; BioMax, Seoul, Republic of Korea). After washing, membranes were incubated with horseradish peroxidase-coupled secondary antibody for 1 h at 25°C. The secondary antibodies were goat anti-mouse immunoglobulin G (IgG)-horseradish peroxidase (HRP) (1:6,000; sc-2005; Santa Cruz Biotechnology) and goat anti-rabbit IgG-HRP (1:3,000; sc-2004; Santa Cruz Biotechnology or TSG101; System Biosciences). The protein bands were visualized using an enhanced chemiluminescence protocol (Advansta, San Jose, CA, USA) and exposed onto Kodak X-ray film. The densitometric intensities of the immunoreactive bands were quantified using ImageJ (NIH, Rockville, MD, USA) or Image Studio software (LI-COR Biosciences, Lincoln, NE, USA).
Immunofluorescence Microscopy
Cells were washed three times in 1× PBS, fixed in 3% formaldehyde (DUKSAN, Gyeonggi-do, Republic of Korea) in cold PBS at 4°C for 30 min, rinsed three times in 1× PBS, and permeabilized with 0.5% Triton X-100 in 1× PBS at room temperature for 10 min. Cells were further rinsed three times in 1× PBS for 5 min each, blocked with 5% BSA in PBS at room temperature for 1 h, and stained with α-SMA antibody (1:200; ab7817; Abcam) for 3 h at room temperature followed by five rinses in 1× PBS for 5 min each. The cells were incubated with goat anti-mouse IgG conjugated to Alexa 488 (1:1,000; A11001; Invitrogen) for 1 h at room temperature in the dark and rinsed in 1× PBS five times for 5 min each. Coverslips were mounted using SlowFade Gold antifade reagent with DAPI (S36938; Invitrogen). Fluorescence images were visualized under an optical microscope (Ni-SS; Nikon Instruments, Melville, NY, USA). Positive immunofluorescence was quantified on the basis of optical intensity using NIS Elements BR software. Representative images of α-SMA (Alexa 488; excited at 488 nm; green) and DAPI (nuclei; excited at 405 nm; blue) staining were taken, and the average intensity in more than five fields from at least three independent experiments was graphed.
Cellular Uptake of miRNAs
The cellular uptake of Cy3-labeled miR-192 was examined using confocal microscopy. Huh-7 cells were transfected with Cy3-labeled miR-192. After 72 h of transfection, exosomes were isolated using ExoQuick-TC (System Bioscience). LX-2 cells were incubated with the exosomes for 72 h, fixed, and stained with α-SMA antibody (1:200; ab7817; Abcam) as described in the Immunofluorescence Microscopy section. Confocal fluorescence images were obtained using a laser-scanning confocal microscope (A1-Rsi; Nikon Instruments). Representative images of miR-192 (red; excited at 543 nm), α-SMA (Alexa 488; excited at 488 nm; green), and DAPI (nuclei; excited at 405 nm; blue) staining were taken, and the average intensity of five fields from at least three independent experiments was graphed.
Packaging of miRNA in Exosomes
Exo-Fect (System Bioscience) was used to package miRNA into isolated exosomes according to the manufacturer’s protocol. Briefly, isolated exosomes from Huh-7 cell-free supernatant (5 mL) were re-suspended in 100 μL of 1× PBS and then the components, consisting of 10 μL of Exo-Fect solution, 20 μL of miR-192 or non-template control siRNA (siNTC) (20 pmol), 100 μL of 1× PBS, and 20 μL of purified exosomes, were mixed by inversion three times. The component mixture was incubated at 37°C in a shaker for 10 min and then immediately placed on ice. To stop the reaction, 30 μL of ExoQuick-TC reagents were added to the component mixture and mixed by inverting six times. miRNA-packaged exosomes were placed on ice for 30 min and then centrifuged for 3 min at 13,000 rpm. The supernatant was removed, and the packaged exosome pellet was re-suspended in 200 μL of 1× PBS.
Oil Red O Staining and Destaining
After incubating LX-2 cells with exosomes derived from JFH-1 cell supernatant for 72 h, cells were transfected with scramble or anti-miR-192 RNA for 48 h. Cells were washed with 1× PBS three times and fixed in 10% formaldehyde at room temperature for 20 min. The cells were then washed three times in 1× PBS and stained with a freshly prepared Oil Red O (Sigma-Aldrich) working solution (diluted in water and filtered through two-layer Whatman filter paper) for 20 min at room temperature. After removing the Oil Red O solution, cells were washed with 1× PBS five times, and the red-stained lipid droplets were observed via light microscopy. To quantify staining, Oil Red O was extracted from cells using 100% isopropanol and transferred to a 96-well plate, and the optical density was measured at 540 nm using a microplate reader (Epoch; BioTek, Winooski, VT, USA).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Student’s unpaired t test and one-way ANOVA were used for statistical analyses of the data. Results were expressed as the mean ± SE (SEM; n > 3).
Author Contributions
S.-W.L. designed the experiments; J.H.K. and C.H.L. conducted the experiments; and J.H.K. and S.-W.L. analyzed data and wrote the manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Acknowledgments
We thank Takaji Wakita (National Institute of Infectious Diseases, Japan) for supplying HCV genotype 2a JFH-1 constructs. This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2013R1A2A2A01004649, 2012M3A9B6055200, 2017R1A2B2003368, and 2017M3A9C8031781).
Footnotes
Supplemental Information includes eight figures and two tables and can be found with this article online at https://doi.org/10.1016/j.omtn.2019.01.006.
Supplemental Information
References
- 1.Perz J.F., Armstrong G.L., Farrington L.A., Hutin Y.J., Bell B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006;45:529–538. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Manns M.P., Buti M., Gane E., Pawlotsky J.M., Razavi H., Terrault N., Younossi Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers. 2017;3:17006. doi: 10.1038/nrdp.2017.6. [DOI] [PubMed] [Google Scholar]
- 3.Yamane D., McGivern D.R., Masaki T., Lemon S.M. Liver injury and disease pathogenesis in chronic hepatitis C. Curr. Top. Microbiol. Immunol. 2013;369:263–288. doi: 10.1007/978-3-642-27340-7_11. [DOI] [PubMed] [Google Scholar]
- 4.Friedman S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 5.Iwaisako K., Brenner D.A., Kisseleva T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012;27(Suppl 2):65–68. doi: 10.1111/j.1440-1746.2011.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee U.E., Friedman S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambrecht J., Mannaerts I., van Grunsven L.A. The role of miRNAs in stress-responsive hepatic stellate cells during liver fibrosis. Front. Physiol. 2015;6:209. doi: 10.3389/fphys.2015.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellicoro A., Ramachandran P., Iredale J.P., Fallowfield J.A. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014;14:181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 9.Troeger J.S., Mederacke I., Gwak G.Y., Dapito D.H., Mu X., Hsu C.C., Pradere J.P., Friedman R.A., Schwabe R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–1083.e22. doi: 10.1053/j.gastro.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bataller R., Paik Y.H., Lindquist J.N., Lemasters J.J., Brenner D.A. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology. 2004;126:529–540. doi: 10.1053/j.gastro.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 11.Lin W., Tsai W.L., Shao R.X., Wu G., Peng L.F., Barlow L.L., Chung W.J., Zhang L., Zhao H., Jang J.Y. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology. 2010;138:2509–2518.e1. doi: 10.1053/j.gastro.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miura K., Taura K., Kodama Y., Schnabl B., Brenner D.A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48:1420–1429. doi: 10.1002/hep.22486. [DOI] [PubMed] [Google Scholar]
- 13.Okuda M., Li K., Beard M.R., Showalter L.A., Scholle F., Lemon S.M., Weinman S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 14.Bauer M., Schuppan D. TGFbeta1 in liver fibrosis: time to change paradigms? FEBS Lett. 2001;502:1–3. doi: 10.1016/s0014-5793(01)02655-2. [DOI] [PubMed] [Google Scholar]
- 15.Friedman S.L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 18.Xu F., Liu C., Zhou D., Zhang L. TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J. Histochem. Cytochem. 2016;64:157–167. doi: 10.1369/0022155415627681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons C.J., Bradford B.U., Pan C.Q., Cheung E., Schauer M., Knorr A., Krebs B., Kraft S., Zahn S., Brocks B. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology. 2004;40:1106–1115. doi: 10.1002/hep.20425. [DOI] [PubMed] [Google Scholar]
- 20.Schorey J.S., Cheng Y., Singh P.P., Smith V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015;16:24–43. doi: 10.15252/embr.201439363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Théry C., Zitvogel L., Amigorena S. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 22.Shen J., Huang C.K., Yu H., Shen B., Zhang Y., Liang Y., Li Z., Feng X., Zhao J., Duan L., Cai X. The role of exosomes in hepatitis, liver cirrhosis and hepatocellular carcinoma. J. Cell. Mol. Med. 2017;21:986–992. doi: 10.1111/jcmm.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raposo G., Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masciopinto F., Giovani C., Campagnoli S., Galli-Stampino L., Colombatto P., Brunetto M., Yen T.S., Houghton M., Pileri P., Abrignani S. Association of hepatitis C virus envelope proteins with exosomes. Eur. J. Immunol. 2004;34:2834–2842. doi: 10.1002/eji.200424887. [DOI] [PubMed] [Google Scholar]
- 25.Ramakrishnaiah V., Thumann C., Fofana I., Habersetzer F., Pan Q., de Ruiter P.E., Willemsen R., Demmers J.A., Stalin Raj V., Jenster G. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA. 2013;110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J.H., Lee C.H., Lee S.W. Hepatitis C virus infection stimulates transforming growth factor-β1 expression through up-regulating miR-192. J. Microbiol. 2016;54:520–526. doi: 10.1007/s12275-016-6240-3. [DOI] [PubMed] [Google Scholar]
- 27.Florimond A., Chouteau P., Bruscella P., Le Seyec J., Mérour E., Ahnou N., Mallat A., Lotersztajn S., Pawlotsky J.M. Human hepatic stellate cells are not permissive for hepatitis C virus entry and replication. Gut. 2015;64:957–965. doi: 10.1136/gutjnl-2013-305634. [DOI] [PubMed] [Google Scholar]
- 28.Dreux M., Garaigorta U., Boyd B., Décembre E., Chung J., Whitten-Bauer C., Wieland S., Chisari F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe. 2012;12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devhare P.B., Sasaki R., Shrivastava S., Di Bisceglie A.M., Ray R., Ray R.B. Exosome-mediated intercellular communication between hepatitis C virus-infected hepatocytes and hepatic stellate cells. J. Virol. 2017;91 doi: 10.1128/JVI.02225-16. e02225-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J., Liu K., Liu Y., Xu Y., Zhang F., Yang H., Liu J., Pan T., Chen J., Wu M. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 2013;14:793–803. doi: 10.1038/ni.2647. [DOI] [PubMed] [Google Scholar]
- 31.Blaner W.S., O’Byrne S.M., Wongsiriroj N., Kluwe J., D’Ambrosio D.M., Jiang H., Schwabe R.F., Hillman E.M., Piantedosi R., Libien J. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta. 2009;1791:467–473. doi: 10.1016/j.bbalip.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El Taghdouini A., Najimi M., Sancho-Bru P., Sokal E., van Grunsven L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenesis Tissue Repair. 2015;8:14. doi: 10.1186/s13069-015-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee C.H., Kim J.H., Lee S.W. The role of microRNA in pathogenesis and as markers of HCV chronic infection. Curr. Drug Targets. 2017;18:756–765. doi: 10.2174/1389450117666160401125213. [DOI] [PubMed] [Google Scholar]
- 34.Pirola C.J., Fernández Gianotti T., Castaño G.O., Mallardi P., San Martino J., Mora Gonzalez Lopez Ledesma M., Flichman D., Mirshahi F., Sanyal A.J., Sookoian S. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64:800–812. doi: 10.1136/gutjnl-2014-306996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayes C.N., Chayama K. MicroRNAs as biomarkers for liver disease and hepatocellular carcinoma. Int. J. Mol. Sci. 2016;17:280. doi: 10.3390/ijms17030280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bukong T.N., Momen-Heravi F., Kodys K., Bala S., Szabo G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014;10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung A.C., Huang X.R., Meng X., Lan H.Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 2010;21:1317–1325. doi: 10.1681/ASN.2010020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braun C.J., Zhang X., Savelyeva I., Wolff S., Moll U.M., Schepeler T., Ørntoft T.F., Andersen C.L., Dobbelstein M. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008;68:10094–10104. doi: 10.1158/0008-5472.CAN-08-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pichiorri F., Suh S.S., Rocci A., De Luca L., Taccioli C., Santhanam R., Zhou W., Benson D.M., Jr., Hofmainster C., Alder H. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18:367–381. doi: 10.1016/j.ccr.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Beger R.D., Bhattacharyya S., Yang X., Gill P.S., Schnackenberg L.K., Sun J., James L.P. Translational biomarkers of acetaminophen-induced acute liver injury. Arch. Toxicol. 2015;89:1497–1522. doi: 10.1007/s00204-015-1519-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Meer A.J., Farid W.R., Sonneveld M.J., de Ruiter P.E., Boonstra A., van Vuuren A.J., Verheij J., Hansen B.E., de Knegt R.J., van der Laan L.J., Janssen H.L. Sensitive detection of hepatocellular injury in chronic hepatitis C patients with circulating hepatocyte-derived microRNA-122. J. Viral Hepat. 2013;20:158–166. doi: 10.1111/jvh.12001. [DOI] [PubMed] [Google Scholar]
- 42.Coll M., El Taghdouini A., Perea L., Mannaerts I., Vila-Casadesús M., Blaya D., Rodrigo-Torres D., Affò S., Morales-Ibanez O., Graupera I. Integrative miRNA and gene expression profiling analysis of human quiescent hepatic stellate cells. Sci. Rep. 2015;5:11549. doi: 10.1038/srep11549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitano M., Bloomston P.M. Hepatic stellate cells and microRNAs in pathogenesis of liver fibrosis. J. Clin. Med. 2016;5:E38. doi: 10.3390/jcm5030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leti F., Malenica I., Doshi M., Courtright A., Van Keuren-Jensen K., Legendre C., Still C.D., Gerhard G.S., DiStefano J.K. High-throughput sequencing reveals altered expression of hepatic microRNAs in nonalcoholic fatty liver disease-related fibrosis. Transl. Res. 2015;166:304–314. doi: 10.1016/j.trsl.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker P.P., Rau M., Schmitt J., Malsch C., Hammer C., Bantel H., Müllhaupt B., Geier A. Performance of serum microRNAs -122, -192 and -21 as biomarkers in patients with non-alcoholic steatohepatitis. PLoS ONE. 2015;10:e0142661. doi: 10.1371/journal.pone.0142661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lohmann V., Körner F., Koch J., Herian U., Theilmann L., Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 47.Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H.G., Mizokami M. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blight K.J., McKeating J.A., Rice C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee C.H., Lee Y.J., Kim J.H., Lim J.H., Kim J.H., Han W., Lee S.H., Noh G.J., Lee S.W. Inhibition of hepatitis C virus (HCV) replication by specific RNA aptamers against HCV NS5B RNA replicase. J. Virol. 2013;87:7064–7074. doi: 10.1128/JVI.00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.