

Abstract
Barth syndrome (BTHS) is a rare mitochondrial disease that causes severe cardiomyopathy and has no disease-modifying therapy. It is caused by recessive mutations in the gene tafazzin (TAZ), which encodes tafazzin—an acyltransferase that remodels the inner mitochondrial membrane lipid cardiolipin. To identify novel mechanistic pathways involved in BTHS and evaluate the effects of gene therapy on proteomic profiles, we performed a multiplex tandem mass tagging (TMT) quantitative proteomics analysis to compare protein expression profiles from heart lysates isolated from BTHS, healthy wild-type (WT), and BTHS treated with adeno-associated virus serotype 9 (AAV9)-TAZ gene replacement as neonates or adults. 197 proteins with ≥2 unique peptides were identified. Of these, 91 proteins were significantly differentially expressed in BTHS compared to WT controls. Cause-effect relationships between tafazzin deficiency and altered protein profiles were confirmed through demonstrated significant improvements in expression levels following administration of AAV9-TAZ. The importance of TMEM65 in Cx43 localization to cardiac intercalated discs was revealed as a novel consequence of tafazzin deficiency that was improved following gene therapy. This study identifies novel mechanistic pathways involved in the pathophysiology of BTHS, demonstrates the ability of gene delivery to improve protein expression profiles, and provides support for clinical translation of AAV9-TAZ gene therapy.
Keywords: Barth syndrome, BTHS, TMT proteomics, mitochondrial disease, cardiac gene therapy, AAV9, TMEM65, tafazzin, Cx43, gap junction localization
Graphical Abstract
Introduction
Barth syndrome (BTHS) is a rare mitochondrial disorder caused by X-linked, recessive loss-of-function mutations in Tafazzin (TAZ), which encodes tafazzin. Tafazzin is a nuclear-encoded acyltransferase that is trafficked to the inner mitochondrial membrane, where it remodels monolysocardiolipin (MLCL) back to its fully functional form, cardiolipin (CL). CL is an inner mitochondrial membrane lipid that is important for stabilization of electron transport chain (ETC) proteins. In BTHS, this cycle is impaired and the accumulation of MLCL causes destabilization of the inner mitochondrial membrane, abnormal cristae structure, and inefficient ETC-mediated ATP production.1, 2, 3, 4, 5, 6, 7, 8, 9
The timing and clinical presentation of BTHS is variable; however, typical symptoms include impaired muscle bioenergetics; decreased cardiac reserve; and diminished muscle O2 utilization, neutropenia, and 3-methylglutaconic aciduria, with cardiomyopathy being the primary cause of death.10, 11, 12, 13 BTHS standard of care currently includes administration of medications, such as diuretics and angiotensin-converting enzyme (ACE) inhibitors—there is currently no disease-modifying therapy for this disorder.12 In addition, previous studies have indicated that cardiac TAZ mRNA expression is diminished in several heart failure animal models, chronic human heart failure patients, and human patients with dilated cardiomyopathy, suggesting that tafazzin expression influences both BTHS and common heart failure.3, 14, 15, 16, 17, 18
We recently demonstrated that intravenous administration of adeno-associated virus serotype 9 (AAV9)-Des-TAZ to either neonatal or adult BTHS mice significantly improves mitochondrial structure and function, cardiac function, and whole-body activity levels.19 Previously, an elegant investigation into the proteomic profile of BTHS was performed by other investigators using two-dimensional differential gel electrophoresis (2D-DIGE) and isobaric tags for relative and absolute quantification (iTRAQ) analyses on mitochondrial lysates isolated from the hearts of BTHS and wild-type (WT) mice.20 The study identified a total of 26 differentially expressed proteins in BTHS cardiac mitochondria as compared to WT controls. In so doing, several novel mechanisms and compensatory pathways involved in BTHS pathogenesis were revealed. Specifically, this study demonstrated disruptions between fatty acid oxidation and ETC interactions and provided further evidence for ETC supercomplex destabilization. To expand upon previous BTHS proteomics studies, our present study provides a deeper investigation into the consequences of tafazzin deficiency in total heart lysates using a multi-plex tandem mass tagging (TMT) isobaric labeling quantitative approach. In addition, cause-effect relationships between tafazzin deficiency and proteomic profiles are confirmed through our clinically relevant gene-replacement strategy.19
Although the general utility of TMT multiplex labeling has been demonstrated in a variety of previously published characterization studies, our data reveal the full scope of how this technique can be also be applied to evaluations to determine the broad efficacy of molecular medicines.21, 22 Specifically, we observed successful normalization of the vast majority of dysregulated protein expression profiles in the hearts of AAV9-TAZ-treated BTHS mice, regardless of their treatment age. Our data also identify several novel mechanistic pathways involved in BTHS that will be interesting to explore in other cardiac disorders in which tafazzin expression levels are diminished. In sum, we present pre-clinical data that provide strong support for further translation of BTHS gene therapy into the clinical realm and identify novel proteins that contribute to the pathophysiology of this disorder.
Results
TMT-Based Proteomics Isobaric Multiplex Labeling and LC-MS/MS Analysis
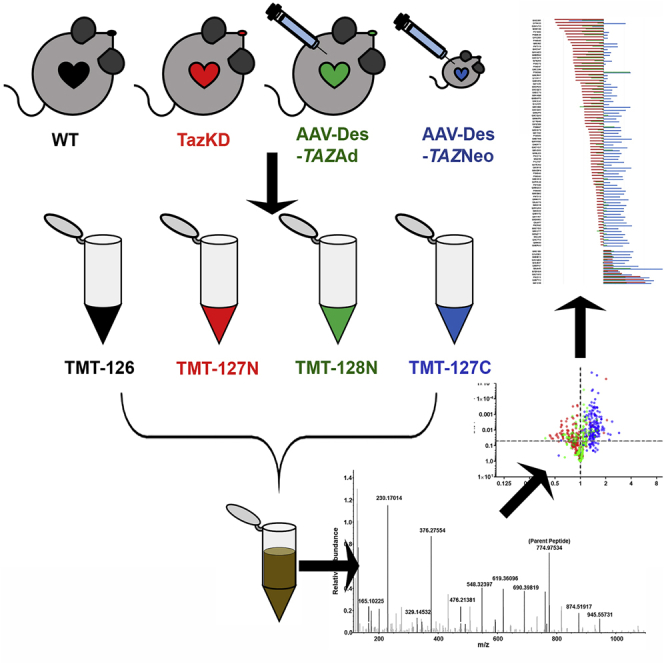
Multiplex tandem mass tagging (TMT) isobaric-label-based quantitative proteomics comparisons were performed to identify proteins significantly dysregulated in BTHS heart lysates as compared to healthy WT controls. AAV9-TAZ gene replacement in adult and neonatal BTHS mice was also performed to confirm cause-effect relationships between tafazzin deficiency and aberrant protein expression profiles and to evaluate the potential for gene therapy to broadly improve cardiac proteomic alterations in BTHS. We labeled heart lysates from 5-month-old healthy WT, untreated BTHS, BTHS mice treated as adults (AAV9-TAZAd), and BTHS mice treated as neonates (AAV9-TAZNeo; Figures 1A–1C). Lysates were combined and purified for QE Orbitrap liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (Figures 1D–1F). The results were then searched against a Mus musculus protein database for identification.
Figure 1.

TMT Experimental Workflow
(A) AAV9-TAZ was administered to BTHS mice as adults (3 months) or neonates (1 or 2 days). (B) Hearts from WT, untreated BTHS, and treated BTHS mice were collected at 5 months of age. (C) Heart lysates were labeled with unique isobaric labels. (D) The labeled samples were combined and purified (E). (F) LC-MS/MS was performed on the purified sample, and data were analyzed against the Mus musculus protein database for peptide identification.
Identification of Significantly Differentially Expressed Proteins in BTHS
In total, 197 proteins were identified by TMT-based proteomics as being represented by ≥2 unique peptides (Figure 2A). Among those protein, 91 were identified as being significantly differentially expressed in untreated BTHS samples as compared to healthy WT control heart samples (Table S1). A volcano plot was generated to depict the distribution of the 106 unchanged, 12 upregulated, and 79 downregulated proteins BTHS heart lysate (Figure 2B). A previous study using 2D-DIGE and iTRAQ proteomic analysis to compare mitochondria isolated from WT and BTHS mouse hearts identified 26 differentially expressed proteins.20 Two of the 26 proteins identified in that study were also identified in our study (cytochrome b-c1 complex subunit Rieske and myoglobin), 24 were only found in the previous study, and 89 novel proteins were identified by this TMT study (Figure 2C).
Figure 2.

Proteins Differentially Expressed in BTHS
(A) Flowchart depicting the study screening process. (B) A volcano plot displaying log 2 fold change ratios of untreated BTHS heart proteins as compared to healthy WT controls. All proteins above the horizontal dashed line (p = 0.05) display significantly altered expression levels. (C) A Venn diagram comparison of proteins identified as being differentially expressed in a previous study using isolated heart mitochondrial lysates and this study using whole-heart lysates.
The full list of 91 differentially expressed proteins identified by TMT-based proteomics was assessed using the Protein Analysis trough Evolutionary Relationships (PANTHER) ontology classification system.23 In total, 78 of the 79 proteins determined to be significantly downregulated in BTHS and 11 of the 12 proteins determined to be significantly upregulated in BTHS were recognized by PANTHER and categorized based upon molecular function, involvement in biological processes, or specific protein classes (Figure S1). Based upon this classification system, highly impacted protein classes in both upregulated and downregulated datasets include nucleic acid binding, oxidoreductases, transferase, and hydrolase classes (Figure S1A). By far, the two most highly impacted molecular functions in BTHS are catalytic and binding activities (Figure S1B). Finally, the biological processes that are most impacted by BTHS are metabolic, cellular, and cell organization and/or biogenesis (Figure S1C).
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) analysis (online database of known and predicted protein-protein interactions) enabled generation of a string diagram depiction of associations between 41 of the proteins that were differentially expressed (Figure S2), downregulated (Figure S3), and upregulated (Figure S4) between BTHS and WT mice. These revealed several clusters, including multiple proteins, including those directly or indirectly involved in mitochondrial electron transport chain function and those involved in various aspects of transcription or translation. Further STRING assessment identified multiple functional enrichments within the set of differentially expressed proteins. The strongest hit among biological pathways was the oxidative-reduction process (Table S2). The top molecular function was oxidoreductase activity. In addition to the unsurprising links between BTHS and mitochondrial electron transport chain function, STRING molecular function analyses also identified strong associations between proteins in the dataset involved in ion binding, transition metal ion binding, and small-molecule binding (Table S3). The strongest Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway identified was oxidative phosphorylation (Table S4). Several specific diseases known to have mitochondrial impairment that were listed among KEGG pathways as being heavily associated with this dataset included Alzheimer’s, Parkinson’s, Huntington’s, and non-alcoholic fatty liver disease.
Although the vast majority of proteins identified in this study were downregulated, several were also found to be upregulated in BTHS (Table S5). Although the mechanistic functions associated with several of these proteins point toward an upregulation of various metabolic processes and may suggest the involvement of compensatory mechanisms, there were no obvious links tying these proteins together to reveal clear upregulation of a precise mechanistic pathway. Furthermore, there were a total of 9 proteins that were not rescued following AAV9-TAZ treatment at either age. UniProt investigations as well as STRING and PANTHER analyses did not reveal any apparent connections between these proteins to reveal a particular mechanistic pathway that remains uncorrected (Table S6).24
TMT-Based Proteomics Data Demonstrate the Widespread Benefits of AAV-Mediated Rescue and further Confirm Cause-Effect Relationships between Tafazzin Deficiency and Cardioskeletal Myopathy
To confirm that deficient tafazzin expression was the root cause of the aberrant protein expression profiles observed by TMT-based proteomics and to demonstrate the ability of AAV-mediated TAZ gene delivery to correct these profiles, we assessed TMT-based proteomics data generated from the hearts of mice that had been administered AAV9-TAZ either as neonates or adults. Log2 ratios were calculated to quantify fold change in expression between untreated BTHS, BTHS mice treated as adults (AAV9-TAZAd), and BTHS mice treated as neonates (AAV9-TAZNeo), as compared to healthy WT controls for each of the 91 differentially expressed proteins (Figure 3). When plotted against −log10 (p values) in the y axis, volcano plots demonstrate global improvement in expression in both AAV treatment groups as compared to untreated controls with an overall shift toward significantly increased expression levels (Figure 3A). A horizontal line diagram depiction of log2 fold change for each sample as compared to WT controls for each of the 91 proteins further demonstrates the dramatic improvements observed in AAV9-TAZ-treated BTHS mice (Figure 3B). In all, the improvements observed in AAV9-TAZ-treated BTHS mice protein profiles complement the significant improvements in function observed in our previously published study.19
Figure 3.

Relative Distribution of Proteins from BTHS, AAV9-TAZAd, and AAV9-TAZNeo Mouse Hearts
(A) Volcano plot showing an overlay of log2 ratio fold changes in protein expression levels between untreated BTHS mice, AAV9-TAZneo, and AAV9-TAZad-treated cohorts as compared to healthy WT control levels clearly demonstrates improved expression in treatment groups. (B) Log2 fold change line diagram displaying relative expression levels for each protein identified as significantly differentially expressed in BTHS (red) and corresponding AAV9-TAZAd (green) and AAV9-TAZNeo (blue) levels as compared to that of healthy WT controls.
Further evidence demonstrating a significant improvement in protein expression profiles was shown through volcano plots of protein log2 ratios from healthy WT controls, AAV9-TAZAd, and AAV9-TAZNeo samples as compared to untreated BTHS expression levels (Figure 4). An overlay of all three samples (WT, AAV9-TAZAd, and AAV9-TAZNeo) reveals a highly similar distribution between the adult treatment group and healthy WT controls (Figure 4A). Individual plots from mice treated as adults or neonates reveals more dramatically increased upregulation of differentially expressed proteins in mice treated as neonates as compared to those treated as adults (Figures 4B and 4C). Individually, 85% (for AAV9-TAZAd treated) and 92% (for AAV9-TAZNeo treated) of the proteins identified as being significantly differentially expressed in BTHS showed improved expression levels. 82% (75) of the differentially expressed proteins demonstrated significantly increased expression levels in both neonatal and adult treatment groups (Figure 4D). Evaluations of those proteins only corrected in mice from either the adult treatment age 3% or the neonatal treatment age 10% did not reveal closely related pathways or processes (Table S7).
Figure 4.

AAV9-TAZ Administration Improves Aberrant Protein Expression Profiles
(A) Volcano plots of log2 fold change ratios of proteins from healthy WT (black), AAV9-TAZAd-treated (green), and AAV9-TAZNeo-treated (blue) heart lysate samples as compared to untreated BTHS. (B) The AAV9-TAZAd-treated profile is very similar to that of healthy WT controls. (C) The AAV9-TAZNeo-treated profile shows the overall highest expression levels. (D) A Venn diagram showing the total numbers of proteins found to be differentially expressed in BTHS that display significant improvement in expression levels in the treated groups.
TMT-Based Proteomics Analysis Reveals Proteins Involved in Heart Failure, Cardiac Development, Carnitine Biosynthesis, Transcription, and Translation that Are Impacted by BTHS
As previous BTHS studies have demonstrated differential expression of proteins important for mitochondrial metabolism, we focused further analyses on 6 novel proteins that were highly downregulated, improved with gene therapy, and were not previously identified as being involved in BTHS (Figure 5). Several proteins newly identified by our study as being differentially expressed in BTHS have been previously found to be involved in heart failure and cardiac development: transmembrane protein 65 (Tmem65) (UniProt: Q4VAE3), lumican (Lum) (UniProt: P51885), and the 4 1/2 LIM 2 protein (Fhl2) (UniProt: O70433) were all downregulated in BTHS hearts but displayed increased expression levels following AAV treatment. The first enzyme in the L-carnitine biosynthetic pathway, trimethyllysine dioxygenase (Tmlhe) (UniProt: Q91ZE0), also displayed a significantly reduced expression level in BTHS that was improved with gene therapy.
Figure 5.

MS2 Spectra for Representative Proteins Identified by TMT
Reporter ions for each are located on the right-hand side of each spectra.
Two other interesting proteins that were discovered as being involved in BTHS and upregulated following gene therapy include (1) the fat-storage-inducing transmembrane protein 2 (Fitm2) (UniProt: P59266), which is a multi-pass transmembrane protein localized to the endoplasmic reticulum, where it plays an important role in lipid droplet accumulation, the regulation of cell morphology, and cytoskeletal organization, and (2) the heat shock protein 70 2B (Hspa12b) (UniProt: Q9CZJ2), which has been shown to protect against cardiac dysfunction following myocardial infarction.
Real-time PCR gene expression analyses on transcripts for these 6 hits (Lum, Tmlhe, Hspa12b, Fitm2, Fhl2, and Tmem65) revealed significantly decreased RNA transcription levels in BTHS samples as compared to healthy WT controls (Figure 6). TAZ gene delivery ameliorated the deficiencies in transcript expression levels following at least one if not both administration ages for each of the proteins evaluated (Figures 6A–6F).
Figure 6.

Relative Fold mRNA Transcript Levels for 6 Highly Interesting Proteins Identified by TMT as Being Significantly Differentially Expressed in BTHS
(A) Lumican (Lum), (B) Trimethyllysine dioxygenase (Tmlhe), (C) heat shock 70 kDa protein 12B (Hspa12b), (D) fat-storage-inducing transmembrane protein 2 (Fitm2), (E) four and a half LIM domains protein 2 (Fhl2), and (F) transmembrane protein 65 (Tmem65) (all data are presented as ± SE; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
One of the significantly differentially expressed proteins that we found particularly interesting and relevant to the arrhythmogenic cardiomyopathy observed in BTHS was TMEM65. Western blot evaluation of TMEM65 protein expression confirmed TMT-based proteomics and transcription results and showed downregulation in untreated BTHS hearts as compared to healthy WT controls as well as AAV9-TAZAd- and AAV9-TAZNeo-treated BTHS mice (Figure 7).
Figure 7.

Tmem65 Expression Levels
(A) Western blot (WB) analysis confirms decreased TMEM65 expression in BTHS that is improved with AAV9-TAZ treatment. (B) Graphical representation of TMEM65 as compared to GAPDH expression by WB (n = 5). (C) MS2 spectra identified by TMT with reporter ions located on the right-hand side (all data are presented as ± SE; *p ≤ 0.05; ***p ≤ 0.001).
As TMEM65 has been shown to bind to Cx43 and play a key role in the trafficking of Cx43 to the intercalated discs of cardiomyocytes, we performed immunofluorescence (IF) analyses on mouse heart tissue sections to evaluate whether there were differences in Cx43 localization. IF microscopy revealed distinct localization of Cx43 to the intercalated discs of healthy WT control cardiomyocytes. In contrast, disorganized, cytoplasmic Cx43 expression was observed in untreated BTHS heart sections, indicating deficient trafficking to the intercalated discs (Figures 8A and 8B). Both AAV9-TAZ treatment groups showed normalization of this phenomenon (Figures 8C and 8D). A cartoon depiction comparing correct and incorrect Cx43 trafficking on cardiomyocytes is provided (Figures 8E and 8F).
Figure 8.

Cx43 Localization Is Impaired in Untreated BTHS Mice
(A–D) IF staining showing DAPI staining of nuclei (blue), MTCO2 expression localized to mitochondria (green), and Cx43 expression (red) in (A) healthy WT control (scale bar = 25 μm), (B) untreated BTHS, (C) BTHS AAV9-TAZ adult, and (D) BTHS AAV9-TAZ neonatal treated heart tissues. (E) A depiction of how Cx43 properly localizes to the polar ends of healthy cardiomyocytes. (F) A depiction of how Cx43 is mislocalized to the longitudinal sides and more clumped in untreated BTHS cardiomyocytes.
Discussion
This manuscript describes the use of TMT-based proteomics multiplex labeling to identify differentially expressed proteins in the hearts of a BTHS mouse model. Confirmation of the cause-effect relationship between tafazzin deficiency and aberrant proteomic profiles was demonstrated through significant improvement in the proteomic profiles of mice treated with AAV9-Des-TAZ at two different ages. As AAV9 is the most naturally cardiotropic serotype available to date and, in general, AAV provides stable gene transfer without disruption of genes by insertional mutagenesis, we are developing this capsid for future clinical translation of BTHS gene therapy.25, 26, 27, 28, 29
It was important to perform comparative analyses between the dataset generated by this TMT-based proteomics study and that generated by a previously described study on BTHS mouse hearts using 2D-DIGE and iTRAQ analyses because different proteomic approaches can yield varying results.20 Even so, both studies identified two of the exact same proteins as being differentially expressed in BTHS, which further validates the TMT-based proteomics approach. Although we feel that both studies provide valuable information and greatly complement one another, it is important to consider several differences in how they were designed that led to the differences in proteins identified. One key point is that, although our study evaluated whole-heart lysates, the previous study was performed on purified mitochondrial samples. Thus, any mitochondrial proteins identified in our study were found among the broad melee of non-mitochondrial proteins that would be present in whole-heart lysate. Another important difference is the difference in ages of the mice evaluated. Although the mice in our study were all 5 months of age, those used in the previous study were 3 months old and at a less advanced disease state.
Finally, both studies would have been impacted by the effect of BTHS on mitochondrial numbers due to extensive fragmentation. Although the outer mitochondrial membrane in BTHS samples remains intact and any proteins that exist in the mitochondrial matrix will have a greater amount of space upon which to exist, the cristae or inner mitochondrial membrane structures are greatly decreased in BTHS samples. This likely impacts proteomic results from BTHS mitochondrial samples, which may skew toward an increase in outer membrane or matrix proteins and a decrease in inner membrane proteins, even though the total number of mitochondrial proteins being compared to WT samples may be the equal.
Among those proteins identified specifically in our study, several interesting protein associations were revealed. One major cluster highlighted through STRING analysis consisted of various subunits of ETC proteins. As BTHS has long been known to be associated with instability of the inner mitochondrial membrane and a subsequent decrease in ETC supercomplex formation, this was not a surprising result. Other clusters of differentially expressed proteins (in the context of BTHS) consisted of those involved in beta-oxidation (Hadh and Decr1), mRNA processing (Prpf6, Lsm3, Prpf19, and Cpsf7), proteasome-mediated degradation (Ngly1, Psmc1, and Psmb1), and protein translation (Eif3l, Rps29, and Rpl38), suggesting that these functions may play pivotal roles in the manifestation of BTHS. These data support the pursuit of future investigations into TMT-based proteomics profiling of other forms of cardiomyopathy to distinguish BTHS-specific effects versus those that occur throughout more common forms of heart disease.
Evaluations into specific proteins found by TMT-based proteomics analysis as being differentially expressed in BTHS revealed mechanistic links between well-documented BTHS phenotypes and decreased function at the protein level. One example is downregulation of the first enzyme in the L-carnitine biosynthetic pathway, trimethyllysine dioxygenase (Tmlhe). Carnitine levels are low failing in hearts in general, and a previous BTHS study of patient plasma samples revealed carnitine and acylcarnitines as being among metabolites that reveal clear distinctions between BTHS patients and healthy controls using broad-spectrum NMR metabolomics and targeted metabolomics.30, 31, 32
Previously published reports have described somewhat disparate functions for one of the novel proteins identified in our study as being significantly downregulated in BTHS—TMEM65. Originally identified as a protein of unknown function that was upregulated by the steroid receptor RNA activator (SRA) ncRNA, TMEM65 has more recently been shown (in separate studies) to be able to localize to the mitochondrial inner membrane, play a role in mitochondrial function through maintenance of mtDNA copy numbers, and bind to connexin 43 (Cx43) and participate in Cx43 localization to the intercalated discs of cardiomyocytes.33, 34, 35, 36 SRA has also been shown to have disease association with human dilated cardiomyopathy and results in dilated cardiomyopathy when genetically knocked down in zebrafish.37 One study evaluating TMEM65 knockdown in fibroblasts revealed its role as an inner mitochondrial membrane protein and showed that a particular mutation in TMEM65 resulted in a mitochondrial myopathy with severe neurological manifestations.34 A separate investigation evaluated TMEM65 function using lentiviral short hairpin RNA (shRNA)-mediated knockdown in mouse cardiomyocytes and morpholino-based knockdown in zebrafish. This study demonstrated that TMEM65 is an intercalated disc protein required for correct localization of connexin 43 (Cx43), as its deletion resulted in Cx43 internalization and deficient cardiac development. Due to its importance for both Cx43 localization and mitochondrial function as well as its activation by SRA RNA, TMEM65’s involvement in the development of cardiomyopathy represents an interesting potential new therapeutic target for a variety of disorders, which we look forward to investigating in future studies.
Multiple proteins identified as being downregulated in BTHS hearts are known to be involved in more common forms of heart failure and/or in cardiac development. Lumican is a proteoglycan that localizes to the extracellular matrix (ECM) and has been shown to play a role in alterations of the cardiac ECM during development of heart failure in mouse models and human patients.38, 39 Other studies suggested that lumican controls cardiomyocyte growth through regulation of the pericellular ECM and showed that mouse hearts deficient in lumican display an increase in myocardial tissue without a significant increase in cell proliferation as well as an increased susceptibility to aging and isoproterenol-induced myocardiac fibrosis.40, 41 Another differentially expressed protein in our data—the 41/2 LIM 2 protein (Fhl2) has been shown to act as a repressor of pathological cardiac growth through suppression of stress-induced calcineurin activation.42 Fhl2 also interacts with the basic-helix-loop-helix (bHLH) factor Hand1 to repress Hand1/E12 heterodimer-induced transcription during healthy cardiac development.43
Of note, the vast majority of proteins that were identified as differentially expressed between WT and BTHS mouse hearts by our TMT-based proteomics analysis were downregulated. It is possible that this is partly a consequence of a generalized decrease in translational activity, as several ribosomal proteins important for translation of either nuclear or mitochondrial encoded genes were found to be downregulated.
To both further confirm cause-effect relationships between tafazzin and TMT-based proteomics results and generate strong pre-clinical data supporting the potential for AAV9-TAZ gene therapy as a treatment option for BTHS patients, we compared TMT-based proteomics protein expression profiles from WT and BTHS mice to those from mice treated with AAV9-TAZ as adults or neonates and found dramatic improvements. The breadth of proteomic information obtained through TMT-based proteomics comparisons demonstrates the widespread, positive impact that AAV-mediated gene therapy can have on BTHS. As TAZ deficiency has been associated with both BTHS and a variety of non-BTHS heart diseases and CL abnormalities have been detected in a range of other disorders (cardiac ischemia, diabetes, cancer, and Parkinson’s and Alzheimer’s diseases), the identification of a broad range of proteins involved in this disorder provides an important contribution to BTHS and more common maladies.3, 14, 15, 16, 44, 45, 46
Our results reveal a multitude of proteins not previously identified as being involved in BTHS pathophysiology, identify a protein directly involved in BTHS arrhythmogenic cardiomyopathy (TMEM65), demonstrate the broadly impactful ability of gene delivery to improve proteomic profiles, and provide substantial support for a potential definitive treatment that significantly improves cardiac protein expression profiles in BTHS whether delivered to very young or fully grown mice. Further research comparing BTHS proteomic profiles to those of other forms of heart disease may reveal novel therapeutic targets (pharmaceutical, molecular, or otherwise) relevant to a wide variety of cardiomyopathies. As a multitude of AAV-mediated gene delivery strategies are currently being evaluated for clinical efficacy in humans to treat a range of genetically inherited disorders, a strong precedent exists, and our data support this as a strategy to be successfully translated into the clinic.47, 48
Materials and Methods
BTHS Mice
The Institutional Animal Care and Use Committee from University of Florida approved all animal studies. WT C57BL/6J female mice were mated to transgenic males (ROSA26 H1/TetO-shRNA:taz) CB57BL/129S6 (previously characterized BTHS mouse model) for 5 days.49, 50, 51, 52 Females were then separated from males and placed on a doxycycline (dox) diet containing 200 mg of dox/kg of chow (TD98186, Envigo). Transgenic pups were identified by PCR genotyping of tail genomic DNA and maintained on the dox diet throughout their lives (BTHS). Non-transgenic WT littermates were also fed the dox diet and used as controls.
AAV Vector Design and Administration
The AAV used in this study was cloned into a previously described double-stranded (ds) AAV plasmid sequence kindly provided by Dr. Xiao Xiao (UNC).53 A previously described desmin (Des) promoter drove expression of the full-length human TAZ transgene cDNA (CCDS14748.1).54 The dsAAV-Des-TAZ plasmid was packaged into recombinant AAV9 capsids, which were generated at the University of Florida Vector Core facility. AAV9-TAZ was administered intravenously to BTHS mice at a dose of 1 × 1013 vg/kg. Adult injections were administered through the jugular vein, and neonatal injections were administered through the superficial temporal vein as previously described.27 At 5 months of age, mice were euthanized and necropsies were performed. Hearts were collected and stored at −80°C.
Protein Extraction, Digestion, TMT Labeling, and LC-MS/MS
Four hearts were used for each sample. A piece of each heart was lysed by Fast Prep FP120 Cell Disrupter using 2-mL tubes filled with approximately 200 μL of zirconia/silica (1 mm) beads and 500 μL of radioimmunoprecipitation assay (RIPA) buffer. The lysate was centrifuged at 16,000 × g for 10 min at 4°C, the supernatant transferred to a new tube, quantified, and then 25 μg from each heart were combined to generate 100 μg of heart protein lysate to a final volume of 100 μL in 100 mM triethyl ammonium bicarbonate (TEAB) as per previous TMT analyses.55 For each: WT, BTHS untreated, BTHS AAV9-TAZ treated (neonate), and BTHS AAV9-TAZ treated (adult) mice. Each lysate was incubated with 5 μL of 200 mM Bond-Breaker TCEP Solution (Thermo Fisher) at 55°C for 1 h, 5 μL of 375 mM iodoacetamide at room temperature for 30 min protected from light, and precipitation of protein was performed overnight using previous described methanol and chloroform protocol.56 The samples were centrifuged at 8,000 × g for 10 min at 4°C, and the tubes were inverted to decant the methanol and chloroform and dry the protein pellet. Each protein pellet was resuspended with 100 μL of 100 mM (TEAB) and digested overnight with 2.5 μg of trypsin per 100 μg of protein. The peptide labeling was performed following TMT Multiplex (Thermo Fisher) manufacturer’s instructions. There were four biological replicates pooled together per cohort as demonstrated in previous studies.55 Each treatment group and internal control lysate sample was tagged with a unique isobaric label (126, 127N, 127C, and 128N) and submitted to the UF Proteomics Core for purification using C18 spin columns and analysis using high-resolution Q Exactive hybrid quadrupole-Orbitrap LC-MS/MS (Thermo Fisher Scientific, Bremen, Germany). WT lysate was labeled with 126, BTHS untreated lysate was labeled with 127N, BTHS AAV9-TAZ treated (neonate) was labeled with 127C, and BTHS AAV9-TAZ treated (adult) mice was labeled with 128C. The labeled samples were mixed in 1:1:1:1 ratio. Labeled peptides were desalted with C18-solid phase extraction and dissolved in strong cation exchange (SCX) solvent A (25% [v/v] acetonitrile, 10 mM ammonium formate, and 0.1% [v/v] formic acid [pH 2.8]). The peptides were fractionated using an Agilent high-performance liquid chromatographer (HPLC) 1260 with a polysulfethyl A column (2.1 × 100 mm2; 5 μm; 300 Å; PolyLC, Columbia, MD, USA). Peptides were eluted with a linear gradient of 0%–20% solvent B (25% [v/v] acetonitrile and 500 mM ammonium formate [pH 6.8]) over 50 min followed by ramping up to 100% solvent B in 5 min. The absorbance at 280 nm was monitored, and fractions were collected. The fractions were lyophilized and resuspended in LC solvent A (0.1% formic acid in 97% water [v/v] and 3% acetonitrile [v/v]). High-energy collision dissociation (HCD) was used in each MS and MS/MS cycle. The instrument was run in data-dependent mode with a full MS (300–5,000 m/z) resolution of 70,000 and five MS/MS experiments (HCD NCE = 28%; isolation width = 3 Th; first mass = 105 Th; 5% underfill ratio; peptide match set to “preferred” and an AGC target of 1e6). Dynamic exclusion for 10 s was used to prevent repeated analysis of the same peptides, and a lock mass of m/z 445.12003 (polysiloxane ion) was used for real-time internal calibration. The MS system was interfaced with an automated Easy-nLC 1000 system (Thermo Fisher Scientific, Bremen, Germany). Each sample fraction was loaded onto an Acclaim Pepmap 100 precolumn (20 mm × 75 μm; 3 μm-C18) and separated on a PepMap RSLC analytical column (250 mm × 75 μm; 2 μm-C18) at a flow rate at 300 nL/min during a linear gradient from solvent A (0.1% formic acid [v/v]) to 25% solvent B (0.1% formic acid [v/v] for 80 min and to 99.9% acetonitrile [v/v]) for an additional 15 min.
Proteomics Data Processing
The UF Proteomics core facility processed RAW files using Proteome Discoverer (PD version 1.4.0.288; Thermo Fisher Scientific, Bremen, Germany) with the SEQUEST algorithm.57 Spectrum Properties filter min. precursor mass: 300 Da and max. precursor mass: 5,000 Da was used with total intensity threshold: 0 and minimum peak count: 1. Scan event filters of min. collision energy: 0 and max. collision energy: 1,000 was used. Peak filters: with S/N threshold (FT-only): 1.5 was used. Precursor mass tolerance: 10 ppm and max. RT difference (min): 1.1 was set to filter. SEQUEST processing of protein database: uniprot-mouse_20170905.fasta with full tryptic peptides and maximum missed cleavage sites: 2 was allowed. Peptide scoring options of maximum peptides considered: 500, maximum peptides output: 10, calculate probability scores: false, absolute XCorr threshold: 0.4, fragment ion cutoff percentage: 0.1, and peptide without protein XCorr threshold: 1.5 were used. Protein scoring options of maximum protein references per peptide: 100, protein relevance threshold: 1.5, and peptide relevance factor: 0.4 was applied for identified proteins. Precursor mass tolerance 10 ppm and fragment mass tolerance 0.6 Da was used. Neutral loss of a ions, b ions, and y ions were used with weight of a ions: 0, weight of b ions: 1, weight of c ions: 0, weight of x ions: 0, weight of y ions: 1, and weight of z ions: 0 used for ion series. N-terminal modification: TMT10plex/+229.163 Da (any N terminus) and dynamic modification: TMT10plex/+229.163 Da (K) with max. modifications per peptide: 4 were allowed. Static modifications of carbamidomethyl/+57.021 Da (C) were implemented. All MS/MS spectra were searched by SEQUEST combined with the Percolator algorithm (version 2.0) for peptide spectra matched (PSM) search optimization.58 Input data search with maximum Delta Cn: 0.05 was allowed. Decoy database search with target false discovery rate (FDR) (Strict): 0.01–0.05 and target FDR (relaxed): 0.05 with validation based on: q-Value was used.
For quantification of reporter ion, peak integration tolerance of 20 ppm and integration method being most confident centroid was used. Scan event filters of mass analyzer: FTMS, MS order: MS2, activation type: HCD with min. collision energy: 0 and max. collision energy: 1,000 was used. Mass precision of 2 ppm and S/N threshold of 1 was used for event detection. Fold change threshold for up-/downregulation was set at 2 with a maximum allowed fold change being 100. Only unique peptides were considered for protein quantification. There was no experimental bias. Quantification channels like residue modification TMT10plex/+229.163 Da (K) and any N-terminal modification: TMT10plex/+229.163 Da was used with 126: monoisotopic m/z = 126.12773 Da, average m/z = 126.21930 Da; 127_N: monoisotopic m/z = 127.12476 Da, average m/z = 127.21270 Da; 127_C: monoisotopic m/z = 127.13108 Da, average m/z = 127.21140 Da; and 128_N: monoisotopic m/z = 128.12812 Da, average m/z = 128.20400 Da. Ratio calculations were performed with minimum and maximum quan value being 384.4. Ratios reported were 126/126, 127_C/126, 127_N/126, and 128_N/126 as well as 127_C/127_N, 127_N/127_N, 128_C/127_N, and 126/127_N.
For processing results, peptide grouping options like true peptide groups with mass and sequence was used. Shared peptides are discarded. Protein grouping options like considering leucine and isoleucine as equal, considering only PSMs with confidence and delta Cn better than: 0.15, was implemented. Strict maximum parsimony principle was applied. No filters were applied for data reduction. Result filters like peptide score (peptide score: SEQUEST [XCorr]; score threshold: 1), peptide rank (maximum rank: 1), peptide length (lowest peptide length: 6; highest peptide length: 27), peptide rank (maximum rank: 1), peptide Delta Cn (maximum delta Cn: 0.1), peptides per protein (minimal number of peptides: 1; count peptide only in top scored proteins: true) were used. Distinct proteins with precursor mass (lowest precursor mass: 600 Da; highest precursor mass: 3,500 Da) and peptides per protein (minimal number of peptides: 2 with peptide only in top scored proteins) were considered for quantification to generate a final list of 91 proteins identified as being differentially expressed between BTHS and WT mouse hearts. BTHS and AAV-treated (neo or adult) peptide quantities were normalized to WT and expressed as log2 fold change, and later, AAV-treated adult and neonatal proteins were normalized to BTHS to visualize the corrected proteins. Peptide matches that pass the filter associated with the strict FDR (0.01) possessing a green confidence indicator were shown in Figures 6 and S3. Volcano plots were generated in GraphPad Prism.59, 60
Common Contaminants
The following proteins were regarded as common contaminants on the basis of their occurrence and ambiguity in all the samples. These proteins are trypsin (XP_094996); keratin 1; keratin 2a; keratin 5; keratin, type II cytoskeletal 6F; keratin 9; similar to keratin, type I cytoskeletal 10; keratin 10; keratin 14; and keratin 16. They were confirmed by running through Contaminant Repository for Affinity Purification (http://www.crapome.org) against H. sapiens.61, 62
Proteomics Analyses
STRING version 10.5 was used to identify protein-protein interactions between those hits identified in our TMT study.63 All differentially expressed proteins were included in the assessment. The minimum required interaction score was set to 0.400 (medium confidence), and disconnected nodes (proteins with no known interactions) were not included in the view. PANTHER version 13.1 ontology classification software was used to group differentially expressed proteins based upon molecular functions, biological processes, and protein classes.23, 64
Statistical Analyses
GraphPad Prism (version 7) was used for all data analysis. For the number of proteins quantified by reporter ion ratios with FDR 0.05 and FDR 0.01 threshold with validation based on q values, p values were calculated based on Student’s t tests and p ≤ 0.05 was considered significant.
Real-Time PCR Gene Expression Analyses
Total RNA was isolated using the RNA Extraction kit (Zymo Research). cDNA was synthesized using the High Capacity RNA-to-cDNA kit (Applied Biosystems), and real-time qPCR was performed using TaqMan Master Mix (Thermo Scientific) and specific primers (Table S8; Thermo Scientific) on a StepOnePlus Real-Time PCR System (Applied Biosystems). All relative fold change expression levels were calculated using the ΔΔCt method.
Protein Quantification and Western Blot Analyses
Tissues were homogenized in RIPA buffer, and protein lysate concentrations were determined using the DC Protein Assay kit (Bio-Rad). Samples were prepared in 6× loading buffer, boiled for 5 min, and resolved on SDS-PAGE (12% bis-tris polyacrylamide) denaturing gels. Proteins were transferred to nitrocellulose membranes. Membranes were blocked and then incubated with primary antibodies (Table S9) overnight at 4°C. On the following day, membranes were washed with 1× tris-buffered saline + Tween (TBST), incubated with secondary antibodies conjugated to horseradish peroxidase (1:1,000) for 1 h, and washed again with 1× TBST. Bands were detected using the Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Science).
Immunofluorescence on Formalin-Fixed Paraffin-Embedded Tissue
Freshly excised tissue samples were fixed in 10% neutral-buffered formalin overnight at 4°C. Following fixation, tissues were dehydrated, cleared, and embedded in paraffin wax. 50-μM tissue sections were cut from embedded blocks, floated onto a water bath at 42°C, and placed on microscope slides. Slides are dried overnight at 37°C and then dewaxed, rehydrated, and subjected to antigen retrieval using trypsin-EDTA. The slides were blocked in PBS + 5% BSA for 1 h at room temperature then incubated in primary antibodies diluted in PBS + 5% BSA overnight at 4°C. Following PBS washes, slides were incubated in secondary antibodies diluted (1:200) in PBS + 5% BSA for 1 h at room temperature. Finally, slides were incubated in PBS + 1 μg/mL DAPI (Sigma-Aldrich) for 5 min, rinsed in 1× PBS, mounted in VectaMount (Vector Laboratories), and stored at 4°C. IF images were acquired within 24 h of immunolabeling.
Author Contributions
The manuscript was written through contributions from all authors. S.S.-H., M.S.S., and C.A.P. designed the study and contributed acquisition and analysis of data. M.S. designed, generated, and tested vector. P.B.K. contributed acquisition and analysis of data. B.J.B. and W.T.C. contributed to analysis of data. All authors have given approval to the final version of the manuscript.
Conflicts of Interest
No competing financial interests exist.
Acknowledgments
Barth Syndrome Foundation: Association Barth France and the Will McCurdy Fund for the Advancement of Therapies for Barth Syndrome AGR DTD 03-06-2015 (C.A.P.), Barth Syndrome Foundation: Barth Syndrome Foundation of Canada AGR DTD 7-14-2017 (C.A.P.), the American Heart Association-Scientist Development Grant no. 17SDG33410467 (C.A.P.), the Children’s Miracle Network: UF Pediatrics Pilot Project (C.A.P.), the NIH R01 HL136759-01A1 (C.A.P.), and NIH R01 HL107406-01A1 (W.T.C.) all provided funding support for this study. We would also like to acknowledge Dr. Xiao Xiao (UNC) for sharing the dsAAV vector, the UF Vector Core facility for AAV production, and the UF Proteomics ICBR core for protein purification and RAW data assessment.
Footnotes
Supplemental Information includes nine tables and four figures and can be found with this article online at https://doi.org/10.1016/j.omtm.2019.01.007.
Supplemental Information
References
- 1.Houtkooper R.H., Turkenburg M., Poll-The B.T., Karall D., Pérez-Cerdá C., Morrone A., Malvagia S., Wanders R.J., Kulik W., Vaz F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta. 2009;1788:2003–2014. doi: 10.1016/j.bbamem.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Neuwald A.F. Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 1997;7:R465–R466. doi: 10.1016/s0960-9822(06)00237-5. [DOI] [PubMed] [Google Scholar]
- 3.Saini-Chohan H.K., Holmes M.G., Chicco A.J., Taylor W.A., Moore R.L., McCune S.A., Hickson-Bick D.L., Hatch G.M., Sparagna G.C. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 2009;50:1600–1608. doi: 10.1194/jlr.M800561-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 2008;49:1607–1620. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlame M., Kelley R.I., Feigenbaum A., Towbin J.A., Heerdt P.M., Schieble T., Wanders R.J., DiMauro S., Blanck T.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Schlame M., Ren M., Xu Y., Greenberg M.L., Haller I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids. 2005;138:38–49. doi: 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Valianpour F., Mitsakos V., Schlemmer D., Towbin J.A., Taylor J.M., Ekert P.G., Thorburn D.R., Munnich A., Wanders R.J., Barth P.G., Vaz F.M. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 2005;46:1182–1195. doi: 10.1194/jlr.M500056-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Wang G., McCain M.L., Yang L., He A., Pasqualini F.S., Agarwal A., Yuan H., Jiang D., Zhang D., Zangi L. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014;20:616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y., Kelley R.I., Blanck T.J., Schlame M. Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 2003;278:51380–51385. doi: 10.1074/jbc.M307382200. [DOI] [PubMed] [Google Scholar]
- 10.Bashir A., Bohnert K.L., Reeds D.N., Peterson L.R., Bittel A.J., de Las Fuentes L., Pacak C.A., Byrne B.J., Cade W.T. Impaired cardiac and skeletal muscle bioenergetics in children, adolescents, and young adults with Barth syndrome. Physiol. Rep. 2017;5:e13130. doi: 10.14814/phy2.13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cade W.T., Spencer C.T., Reeds D.N., Waggoner A.D., O’Connor R., Maisenbacher M., Crowley J.R., Byrne B.J., Peterson L.R. Substrate metabolism during basal and hyperinsulinemic conditions in adolescents and young-adults with Barth syndrome. J. Inherit. Metab. Dis. 2013;36:91–101. doi: 10.1007/s10545-012-9486-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spencer C.T., Bryant R.M., Day J., Gonzalez I.L., Colan S.D., Thompson W.R., Berthy J., Redfearn S.P., Byrne B.J. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 2006;118:e337–e346. doi: 10.1542/peds.2005-2667. [DOI] [PubMed] [Google Scholar]
- 13.Spencer C.T., Byrne B.J., Bryant R.M., Margossian R., Maisenbacher M., Breitenger P., Benni P.B., Redfearn S., Marcus E., Cade W.T. Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H2122–H2129. doi: 10.1152/ajpheart.00479.2010. [DOI] [PubMed] [Google Scholar]
- 14.Brooks W.W., Shen S.S., Conrad C.H., Goldstein R.H., Bing O.H. Transition from compensated hypertrophy to systolic heart failure in the spontaneously hypertensive rat: Structure, function, and transcript analysis. Genomics. 2010;95:84–92. doi: 10.1016/j.ygeno.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Cappuzzello C., Napolitano M., Arcelli D., Melillo G., Melchionna R., Di Vito L., Carlini D., Silvestri L., Brugaletta S., Liuzzo G. Gene expression profiles in peripheral blood mononuclear cells of chronic heart failure patients. Physiol. Genomics. 2009;38:233–240. doi: 10.1152/physiolgenomics.90364.2008. [DOI] [PubMed] [Google Scholar]
- 16.Rowell J., Koitabashi N., Kass D.A., Barth A.S. Dynamic gene expression patterns in animal models of early and late heart failure reveal biphasic-bidirectional transcriptional activation of signaling pathways. Physiol. Genomics. 2014;46:779–787. doi: 10.1152/physiolgenomics.00054.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tulacz D., Mackiewicz U., Maczewski M., Maciejak A., Gora M., Burzynska B. Transcriptional profiling of left ventricle and peripheral blood mononuclear cells in a rat model of postinfarction heart failure. BMC Med. Genomics. 2013;6:49. doi: 10.1186/1755-8794-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y., Malhotra A., Claypool S.M., Ren M., Schlame M. Tafazzins from Drosophila and mammalian cells assemble in large protein complexes with a short half-life. Mitochondrion. 2015;21:27–32. doi: 10.1016/j.mito.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki-Hatano S., Saha M., Rizzo S.A., Witko R.L., Gosiker B.J., Ramanathan M., Soustek M.S., Jones M.D., Kang P.B., Byrne B.J. AAV-mediated TAZ gene replacement restores mitochondrial and cardioskeletal function in Barth syndrome. Hum. Gene Ther. 2018 doi: 10.1089/hum.2018.020. Published online October 3, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y., Powers C., Madala S.K., Greis K.D., Haffey W.D., Towbin J.A., Purevjav E., Javadov S., Strauss A.W., Khuchua Z. Cardiac metabolic pathways affected in the mouse model of barth syndrome. PLoS ONE. 2015;10:e0128561. doi: 10.1371/journal.pone.0128561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raso C., Cosentino C., Gaspari M., Malara N., Han X., McClatchy D., Park S.K., Renne M., Vadalà N., Prati U. Characterization of breast cancer interstitial fluids by TmT labeling, LTQ-Orbitrap Velos mass spectrometry, and pathway analysis. J. Proteome Res. 2012;11:3199–3210. doi: 10.1021/pr2012347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rauniyar N., Gao B., McClatchy D.B., Yates J.R., 3rd Comparison of protein expression ratios observed by sixplex and duplex TMT labeling method. J. Proteome Res. 2013;12:1031–1039. doi: 10.1021/pr3008896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45(D1):D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bish L.T., Morine K., Sleeper M.M., Sanmiguel J., Wu D., Gao G., Wilson J.M., Sweeney H.L. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum. Gene Ther. 2008;19:1359–1368. doi: 10.1089/hum.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inagaki K., Fuess S., Storm T.A., Gibson G.A., Mctiernan C.F., Kay M.A., Nakai H. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pacak C.A., Mah C.S., Thattaliyath B.D., Conlon T.J., Lewis M.A., Cloutier D.E., Zolotukhin I., Tarantal A.F., Byrne B.J. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- 28.Piras B.A., Tian Y., Xu Y., Thomas N.A., O’Connor D.M., French B.A. Systemic injection of AAV9 carrying a periostin promoter targets gene expression to a myofibroblast-like lineage in mouse hearts after reperfused myocardial infarction. Gene Ther. 2016;23:469–478. doi: 10.1038/gt.2016.20. [DOI] [PubMed] [Google Scholar]
- 29.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 30.Ferrari R., Merli E., Cicchitelli G., Mele D., Fucili A., Ceconi C. Therapeutic effects of L-carnitine and propionyl-L-carnitine on cardiovascular diseases: a review. Ann. N Y Acad. Sci. 2004;1033:79–91. doi: 10.1196/annals.1320.007. [DOI] [PubMed] [Google Scholar]
- 31.Sethi R., Wang X., Ferrari R., Dhalla N.S. Improvement of cardiac function and beta-adrenergic signal transduction by propionyl L-carnitine in congestive heart failure due to myocardial infarction. Coron. Artery Dis. 2004;15:65–71. doi: 10.1097/00019501-200402000-00010. [DOI] [PubMed] [Google Scholar]
- 32.Sandlers Y., Mercier K., Pathmasiri W., Carlson J., McRitchie S., Sumner S., Vernon H.J. Metabolomics reveals new mechanisms for pathogenesis in Barth syndrome and introduces novel roles for cardiolipin in cellular function. PLoS ONE. 2016;11:e0151802. doi: 10.1371/journal.pone.0151802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foulds C.E., Tsimelzon A., Long W., Le A., Tsai S.Y., Tsai M.J., O’Malley B.W. Research resource: expression profiling reveals unexpected targets and functions of the human steroid receptor RNA activator (SRA) gene. Mol. Endocrinol. 2010;24:1090–1105. doi: 10.1210/me.2009-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nazli A., Safdar A., Saleem A., Akhtar M., Brady L.I., Schwartzentruber J., Tarnopolsky M.A. A mutation in the TMEM65 gene results in mitochondrial myopathy with severe neurological manifestations. Eur. J. Hum. Genet. 2017;25:744–751. doi: 10.1038/ejhg.2017.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishimura N., Gotoh T., Oike Y., Yano M. TMEM65 is a mitochondrial inner-membrane protein. PeerJ. 2014;2:e349. doi: 10.7717/peerj.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma P., Abbasi C., Lazic S., Teng A.C., Wang D., Dubois N., Ignatchenko V., Wong V., Liu J., Araki T. Evolutionarily conserved intercalated disc protein Tmem65 regulates cardiac conduction and connexin 43 function. Nat. Commun. 2015;6:8391. doi: 10.1038/ncomms9391. [DOI] [PubMed] [Google Scholar]
- 37.Friedrichs F., Zugck C., Rauch G.J., Ivandic B., Weichenhan D., Müller-Bardorff M., Meder B., El Mokhtari N.E., Regitz-Zagrosek V., Hetzer R. HBEGF, SRA1, and IK: three cosegregating genes as determinants of cardiomyopathy. Genome Res. 2009;19:395–403. doi: 10.1101/gr.076653.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engebretsen K.V., Lunde I.G., Strand M.E., Waehre A., Sjaastad I., Marstein H.S., Skrbic B., Dahl C.P., Askevold E.T., Christensen G. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J. 2013;280:2382–2398. doi: 10.1111/febs.12235. [DOI] [PubMed] [Google Scholar]
- 39.Engebretsen K.V., Waehre A., Bjørnstad J.L., Skrbic B., Sjaastad I., Behmen D., Marstein H.S., Yndestad A., Aukrust P., Christensen G. Decorin, lumican, and their GAG chain-synthesizing enzymes are regulated in myocardial remodeling and reverse remodeling in the mouse. J. Appl. Physiol. 2013;114:988–997. doi: 10.1152/japplphysiol.00793.2012. [DOI] [PubMed] [Google Scholar]
- 40.Dupuis L.E., Berger M.G., Feldman S., Doucette L., Fowlkes V., Chakravarti S., Thibaudeau S., Alcala N.E., Bradshaw A.D., Kern C.B. Lumican deficiency results in cardiomyocyte hypertrophy with altered collagen assembly. J. Mol. Cell. Cardiol. 2015;84:70–80. doi: 10.1016/j.yjmcc.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen S.W., Tung Y.C., Jung S.M., Chu Y., Lin P.J., Kao W.W., Chu P.H. Lumican-null mice are susceptible to aging and isoproterenol-induced myocardial fibrosis. Biochem. Biophys. Res. Commun. 2017;482:1304–1311. doi: 10.1016/j.bbrc.2016.12.033. [DOI] [PubMed] [Google Scholar]
- 42.Hojayev B., Rothermel B.A., Gillette T.G., Hill J.A. FHL2 binds calcineurin and represses pathological cardiac growth. Mol. Cell. Biol. 2012;32:4025–4034. doi: 10.1128/MCB.05948-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill A.A., Riley P.R. Differential regulation of Hand1 homodimer and Hand1-E12 heterodimer activity by the cofactor FHL2. Mol. Cell. Biol. 2004;24:9835–9847. doi: 10.1128/MCB.24.22.9835-9847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barth P.G., Van den Bogert C., Bolhuis P.A., Scholte H.R., van Gennip A.H., Schutgens R.B., Ketel A.G. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratory-chain abnormalities in cultured fibroblasts. J. Inherit. Metab. Dis. 1996;19:157–160. doi: 10.1007/BF01799418. [DOI] [PubMed] [Google Scholar]
- 45.Chicco A.J., Sparagna G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007;292:C33–C44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- 46.He Q. Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H210–H216. doi: 10.1152/ajpheart.00098.2010. [DOI] [PubMed] [Google Scholar]
- 47.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 49.Phoon C.K., Acehan D., Schlame M., Stokes D.L., Edelman-Novemsky I., Yu D., Xu Y., Viswanathan N., Ren M. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J. Am. Heart Assoc. 2012;1 doi: 10.1161/JAHA.111.000455. jah3-e000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soustek M.S., Baligand C., Falk D.J., Walter G.A., Lewin A.S., Byrne B.J. Endurance training ameliorates complex 3 deficiency in a mouse model of Barth syndrome. J. Inherit. Metab. Dis. 2015;38:915–922. doi: 10.1007/s10545-015-9834-8. [DOI] [PubMed] [Google Scholar]
- 51.Soustek M.S., Falk D.J., Mah C.S., Toth M.J., Schlame M., Lewin A.S., Byrne B.J. Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene Ther. 2011;22:865–871. doi: 10.1089/hum.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiebish M.A., Yang K., Liu X., Mancuso D.J., Guan S., Zhao Z., Sims H.F., Cerqua R., Cade W.T., Han X., Gross R.W. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013;54:1312–1325. doi: 10.1194/jlr.M034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Z., Ma H.I., Li J., Sun L., Zhang J., Xiao X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003;10:2105–2111. doi: 10.1038/sj.gt.3302133. [DOI] [PubMed] [Google Scholar]
- 54.Pacak C.A., Sakai Y., Thattaliyath B.D., Mah C.S., Byrne B.J. Tissue specific promoters improve specificity of AAV9 mediated transgene expression following intra-vascular gene delivery in neonatal mice. Genet. Vaccines Ther. 2008;6:13. doi: 10.1186/1479-0556-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zakirova Z., Reed J., Crynen G., Horne L., Hassan S., Mathura V., Mullan M., Crawford F., Ait-Ghezala G. Complementary proteomic approaches reveal mitochondrial dysfunction, immune and inflammatory dysregulation in a mouse model of Gulf War Illness. Proteomics Clin. Appl. 2017;11:1600190. doi: 10.1002/prca.201600190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wessel D., Flügge U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 57.Eng J.K., McCormack A.L., Yates J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 58.Käll L., Canterbury J.D., Weston J., Noble W.S., MacCoss M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 59.Oberg A.L., Mahoney D.W. Statistical methods for quantitative mass spectrometry proteomic experiments with labeling. BMC Bioinformatics. 2012;13(Suppl 16):S7. doi: 10.1186/1471-2105-13-S16-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paulo J.A., Kadiyala V., Banks P.A., Conwell D.L., Steen H. Mass spectrometry-based quantitative proteomic profiling of human pancreatic and hepatic stellate cell lines. Genomics Proteomics Bioinformatics. 2013;11:105–113. doi: 10.1016/j.gpb.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hodge K., Have S.T., Hutton L., Lamond A.I. Cleaning up the masses: exclusion lists to reduce contamination with HPLC-MS/MS. J. Proteomics. 2013;88:92–103. doi: 10.1016/j.jprot.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jensen L.J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mi H., Muruganujan A., Casagrande J.T., Thomas P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.