

Abstract
Targeting the “undruggable” proteome remains one of the big challenges in drug discovery. Recent innovations in the field of targeted protein degradation and manipulation of the ubiquitin-proteasome system open up new therapeutic approaches for disorders that cannot be targeted with conventional inhibitor paradigms. Proteolysis targeting chimeras (PROTACs) are bivalent ligands in which a compound that binds to the protein target of interest is connected to a second molecule that binds an E3 ligase via a linker. The E3 protein is usually either Cereblon or Von Hippel-Lindau. Several examples of selective PROTAC molecules with potent effect in cells and in vivo models have been reported. The degradation of specific proteins via these bivalent molecules is already allowing for the study of biochemical pathways and cell biology with more specificity than was possible with inhibitor compounds. In this review, we provide a comprehensive overview of recent developments in the field of small molecule mediated protein degradation, including transcription factors, kinases and nuclear receptors. We discuss the potential benefits of protein degradation over inhibition as well as the challenges that need to be overcome.
Keywords: PROTAC, Protein degradation, Degrader, Proteasome, Chimera, Bivalent ligand
Abbreviations: ABCB1, ATP-binding cassette sub-family B member 1; AD, Alzheimer's disease; AHR, aryl hydrogen receptor; ALK, anaplastic lymphoma kinase; Aβ, amyloid-β; Bcl6, B-cell lymphoma 6; BET, bromodomain and extra-terminal; Brd4, bromodomain 4; BTK, Bruton's tyrosine kinase; CDK9, cyclin dependent kinase 9; cIAP1, cellular inhibitor of apoptosis protein; CK2, Casein kinase 2; CLIPTAC, click-formed proteolysis targeting chimera; CRBN, Cereblon; DC50, the compound concentration that results in 50% target protein degradation; DHODH, Dihydroorotate dehydrogenase; ERK1, extracellular signal-regulated kinase 1; ERRα, estrogen-related receptor alpha; ERα, estrogen receptor alpha; EZH2, enhancer of zeste homolog 2; FLT3, FMS-like tyrosine kinase-3; FRS2, fibroblast growth factor receptor substrate 2; GCN5, general control nonderepressible 5; GPCR, G-protein coupled receptor; GST, glutathione S-transferase; HDAC, histone deacetylase; HTS, high-throughput screening; MDM2, mouse double-minute 2 homolog; MetAP-2, methionine aminopeptidase-2; PCAF, P300/CBP-associated factor; PEG, polyethylene glycol; PI3K, phosphatidylinositol-3-kinase; PLK-1, polo-like kinase 1; POI, protein of interest; PROTAC, proteolysis targeting chimeras; RAR, retinoic acid receptor; RIPK2, receptor-interacting serine/threonine-protein kinase 2; RTK, receptor tyrosine kinase; SARM, selective androgen receptor modulator; SNIPER, specific and non-genetic IAP-dependent protein eraser; TBK1, TANK-Binding kinase 1; TRIM24, tripartite motif-containing 24 (also known as TIF1α); VHL, Von Hippel-Lindau
Graphical Abstract

1. Introduction
During the last two decades, genetic methods for modulating in vivo protein expression such as CRISPR/Cas9, antisense oligonucleotides and RNA interference have proven to be powerful for target validation and hold promise for therapeutic interventions, especially for proteins which are difficult to target with small molecules. However, clinical application of these techniques require the administration of large biomolecules, which poses significant challenges in terms of bioavailability, stability and delivery to the tissue of interest [1,2]. Conventional small molecules suffer less from these complications, which is why PROTACs have recently received significant attention as a new modality for therapeutic intervention based on modulation of protein levels [3]. The technology is based on the creation of bivalent molecules that bring together an E3 ligase and a target protein that is to be degraded. These bivalent molecules therefore usually consist of an E3 ligase ligand connected with a defined linker to a specific ligand that binds the target protein. A successful PROTAC ligand positions the E3 ligase at the appropriate distance and orientation to the target protein, allowing the latter to be ubiquitinated. The ubiquitinated target protein is subsequently recognized by the proteasome, where it is degraded [4,5]. Or to put it more poetically, a PROTAC molecule is the instigator of the interaction by which an E3 ligase is in the position to give its kiss of death to the target protein.
Potentially, a mode of action whereby a target protein is degraded has several advantages over a more traditional therapeutic approach based on inhibition. First, a PROTAC compound only needs to bring the E3 protein close to the target of interest. Once that target protein has been ubiquitinated, the PROTAC ligand can dissociate and diffuse to the next target copy, to repeat its action. As such, the PROTAC ligand acts like a catalyst, and even sub-stoichiometric amounts of a PROTAC therapeutic can be expected to achieve (near) complete protein degradation [6]. In theory, much lower concentrations would be needed compared to traditional inhibitors, which obtain their effect from long-term binding at concentrations that need to exceed the affinity constant for the interaction. Second, and related to this, since PROTACs can be effective at low occupancies, lower-affinity ligands may be sufficient to achieve pharmacological effects [7,8]. This may open the way for low-ligandable proteins that are often out of reach for traditional inhibitors [9]. Third, and again related to the catalytic potential of PROTAC ligands, one may expect significant duration of pharmacodynamic effects. When the initial pool of target protein has been depleted, the PROTAC only needs to sweep up the newly synthesized protein molecules. And even after the inevitable metabolic degradation of the PROTAC itself, it may take a significant amount of time for the cells to achieve pharmacologically relevant levels of the target protein. Fourth, as it is hypothesized that PROTAC efficiency depends strongly on the protein-protein interactions between the protein of interest (POI) and the E3 ligase [10], and therefore largely on surface residues of the POI, a PROTAC may display increased selectivity between closely related proteins, compared to a compound that relies only on active-site binding. Furthermore, PROTACs may be more effective in countering feedback mechanisms in which target protein expression is upregulated as a result of inhibitory intervention. Finally, since PROTACs do not rely on inhibiting a protein's activity, but merely need to bind to the POI, they can neutralize any target that contains an area or pocket where adequate affinity can be found. This makes PROTACs particularly attractive to address scaffolding proteins, pseudokinases and transcription factors. A large number of studies have recently been published demonstrating potent in vitro efficacy of PROTAC-induced protein degradation in relevant cell lines. Efficient in vivo target degradation has been demonstrated in mouse xenograft models (see below), and the first PROTAC molecules are expected to enter clinical trials early 2019. These promises have propelled PROTACs into the center of attention of present-day drug discovery.
Many excellent reviews on PROTACs have appeared over the past years [[11], [12], [13], [14], [15], [16]], and many of them provide a historic perspective on PROTAC discovery. In the current review, we focus on the more recent developments and aim to provide a comprehensive overview of the current state of PROTAC research, including the role of ternary complex formation in PROTAC design. The majority of PROTACs have been designed against epigenetic targets, kinases and nuclear hormone receptors and we chose to cluster and discuss them in these target groups. Finally, we discuss the latest advances of Brd4 degradation, which is the area of biology most extensively explored by PROTACs.
1.1. Prequel
The concept of using a bivalent ligand to modulate protein levels by hijacking the ubiquitination pathway dates back to a patent application in 1999 by Kenten & Roberts from Proteinix Inc. [17]. This application became a granted patent in 2001, with the single claim of: “a method of generating a compound for activating ubiquitination of a target protein which comprises covalently linking a target protein binding element able to bind specifically to said protein to a ubiquitination element.” Specific examples in the application are largely based on peptidic ubiquitination recognition elements: small peptides like Arg-Ala-caproic acid-Cys, and also larger motives like PEST [18] and Destruction box [19]. The patent describes synthesis routes in which these recognition elements are directly connected to a set of ligands for various target proteins – no spacer moiety was incorporated to connect the two pieces. Described in most detail are bivalent ligands containing derivatives of L-cichoric acid for degradation of HIV integrase, glutathione for degradation of GST, and fluorescein-5-maleimide for degradation of an anti-fluorescein antibody [17]. A125I radiography assay is described to measure protein levels, and data is shown for lysozyme and GST degradation, but unfortunately there is no discussion as to the effect of the molecules. Meanwhile, this patent has expired due to nonpayment of maintenance fees.
A different piece of work from the 1990s involving a bivalent inducer of heterodimerization of proteins was published by the Schreiber lab [20] who extended the concept of cell-permeable chemical inducers of dimerization [21] to promote heterodimerization. These bivalent inducers were envisioned to alter the subcellular localization of signalling proteins, thereby modulating a variety of transcriptional and translational events.
1.2. Proteolysis-Targeting Chimeras
The term PROTAC was first introduced in a seminal paper by Sakamoto and coworkers in 2001 [22], that describes how the Skp1-Cullin-F box E3 ligase complex can be employed to degrade MetAP-2. This was done through a chimeric compound where the IκBα peptide and the angiogenesis inhibitor ovalicin were linked through a suberate-based linker. The IκBα peptide is recognized by the F-box protein, and ovalicin binds covalently to MetAP-2. This construct was shown to both ubiquitinate and degrade MetAP-2. While the authors proposed PROTACs to be useful research tools to study cellular phenotypes, they also recognized that upon replacing the IκBα peptide with a small, cell-permeable molecule, PROTACs would have the potential to become therapeutic agents.
In the absence of small-molecule E3 binders, other peptidic PROTACs were developed that were sufficiently small to be cell permeable. Several of these were based on a 7-amino acid sequence derived from Hypoxia-inducible factor 1, which is the key binding moiety to the VHL E3 ligase complex. Examples of these were shown to target a FKBP12 mutant [23], ERα [24] and AHR [25], and these are described in more detail below.
These peptidic PROTAC molecules were unlikely to have the potential to become therapeutic modalities, because of their low biostability and limited bioavailability. It was not until all-small-molecule PROTACs were developed that the targeted protein degradation field started to flourish. Initially, these were based on small-molecule inhibitors of MDM2 [26] and cIAP1 [[27], [28], [29]]. These PROTACs were still not very effective, and high concentrations of ligand were required to achieve significant target protein degradation. This is probably attributable to (1) the MDM2 and cIAP1 ligands not being potent enough when incorporated in PROTACs, and (2) MDM2 and cIAP1 themselves lacking sufficient ubiquitination activity. The field really accelerated when Cullin RING E3 ligase complexes could be addressed with small molecules, in particular VHL and CRBN. The vast majority of PROTACs that have been disclosed in the literature over the last two-three years are based on a selective inhibitor of VHL or on CRBN inhibitors (thalidomide, lenalidomide, pomalidomide and derivatives thereof) – see Fig. 1. Recently, new E3 ligases have been reported that can be hijacked for protein degradation: DCAF16 [30] and RNF114 [31,32].
Fig. 1.

E3 ligase ligands used for PROTACs: thalidomide derivatives targeting Cereblon. Bestatin and compound 7 are ligands of cIAP [34], nutlin is a ligand of MDM2 [26]. The VHL ligand 9 was optimized starting from a peptide using structure guided design [35]. The asterisk shows the attachment point for the linker.
Most of these published PROTACs target epigenetic proteins, nuclear hormone receptors, and a variety of kinases. An overview of these PROTAC molecules constitutes the body of this review, with the focus on more recent publications. Over the last years, other terms have been introduced to describe molecules or platforms for targeted protein degradation. The general expression ‘target protein degradation’ or simply ‘degraders’ is common. Researchers at C4 Therapeutics and at the Dana Farber Institute refer to their degradation technology as the Degronimid platform, which was further optimized and designated the dTag system [33]. Degradation inducers based on cIAP1 are called SNIPERs, for specific and non-genetic IAP-dependent protein erasers [27]. However, PROTAC is the most commonly used term in the literature, and we will use it in this review for any type of chimeric protein degrader.
1.3. Mode of Action of PROTACs
The basic mode of action of PROTAC molecules is visualized in Fig. 2. The intention of the bifunctional molecule is to bring together an E2/E3 ligase complex with the protein that is to be degraded (the POI). In most cases, the E3 ligase has not evolved to bind to the POI, so there is no natural complementarity of surfaces or other features that promote the ligase and POI to interact. The interaction therefore critically depends on the bridging molecule, and consequently, this bifunctional ligand needs to have adequate affinity for both the POI and the E3 ligase. No functional activity is required towards either the POI or the E3 proteins. The sole purpose of the ligand is to position the E2/E3 ligase complex so that it can efficiently (poly)ubiquitinate the POI. The ubiquitinated POI will be recognized by the proteasome as an entity that needs to be degraded, which leads to a depletion of the POI over time. The PROTAC molecule, once dissociated from the proteins, will be able to repeat its action on the next POI copy, and essentially act as a catalyst for POI degradation. The fact that the POI and the chosen E3 ligase are no natural binding partners leads to a number of implications that deviate from conventional drug discovery paradigms [36].
-
•
Once the POI has been ubiquitinated, there is no need for the PROTAC to continue to bind to the POI. In fact, it would be more advantageous for it to dissociate and find a fresh target to be ubiquitinated. Very tight binding of the PROTAC to the POI (i.e. slow off-rate) may even reduce the overall efficiency of the ligand. In enzymology terms, the residence time of the PROTAC ligand on the POI will affect its catalytic turnover number. This effect may well have a bell-shaped dependency: too short residence time (low affinity) and the E3 will not have enough time to catalyse the transfer of ubiquitin from the E2 to the POI. A residence time, on the other hand, that is too long may slow down the traveling of the PROTAC ligand between different POI copies. Of course, once the POI is being degraded, the (non-covalent) PROTAC molecule will be released so it will always be able to achieve a base level of catalysis. This suggests there is no need for exquisite affinity of the bivalent ligand for the POI. Indeed, an elegant and meticulous study reported by the Crews group [10] clearly shows that very potent kinase binders are not necessarily effective degraders. This observation can be explained by the phenomenon outlined directly below.
-
•
Given that the E3 ligase chosen to be targeted with the bivalent molecule has no natural interaction surface with the POI, it is not obvious that a PROTAC will succeed in positioning the E2/E3 ligase complex so that it can transfer ubiquitin molecules from the E2 to the POI. That is, a PROTAC molecule may have very good potency for both the POI and E3 ligase, but if these two proteins cannot form a complex that allows for ubiquitination, there will be no degradation. This is very apparent from the aforementioned study by the Crews lab [10]. They systematically determined the amount of PROTAC-induced degradation for two different PROTACS against a panel of kinases. Both constructs were based on the promiscuous kinase ligand foretinib, one targeting CRBN and the other VHL. In a first pass, they looked for global proteomic degradation and found that 86 proteins were downregulated by one of the two PROTACs, while only 12 were degraded by both. Perhaps even more strikingly, when the authors studied 54 kinases in more detail, they found no correlation between the affinity of the PROTAC molecule for the kinases and the extent of induced degradation. Clearly, the affinity of foretinib for the protein is not a determining factor here, and instead it is the formation of the ternary complex that governs the efficiency.
-
•
This, in turn, means that the linker plays an important role. As the POI and E3 ligase are not meant to interact, the linker will to a large extent determine the relative orientation of the two proteins in the ternary complex. An effective linker will position the proteins in such a way that some surface complementarity arises (with POI lysines accessible to the E3 ligase), and a local interaction minimum is formed. This will yield the ternary complex with sufficient stability for the ubiquitination to occur. Besides linker length, the attachment of the linker to the individual POI and E3 ligands plays a role. For example, in a study of PROTACs for BTK, different degradation efficiencies were observed when the same 8-atom linker was connected to the C5 compared to C4 atom on the pomalidomide phthalimide ring [37].
-
•
Finally, in conventional inhibitor discovery, selectivity between members within a target family (such as kinases, GPCRs) or between isoforms is usually obtained by exploring differences in the active site of proteins. However, in the case of PROTACs, several examples have been reported where a non-selective ligand produced selective degradation effects once incorporated in a PROTAC. For example, when the pan-BET inhibitor JQ1 was conjugated to VHL ligand 9, the resulting PROTAC molecule selectively degraded Brd4 over Brd2 and Brd3 [38]. Likewise, when a pan-HDAC inhibitor was tethered to thalidomide-type ligands with various linkers, one of the PROTAC molecules showed selective degradation of HDAC6 over the other HDACs tested [39]. This selectivity is a result of different interaction surface complementarities of the E3 ligase with the Brd and HDAC proteins, respectively. It is a direct consequence of the mode of action of these bifunctional molecules.
Fig. 2.

Schematic representation of the PROTAC mode of action. POI: protein of interest – the protein that is meant to be degraded. E3 denotes the E2/E3 ligase complex, commonly VHL, CRBN, cIAP1 or MDM2. The bifunctional molecule links together a ligand for the POI, and an E3 ligase binder. The induced proximity between the ligase and protein of interest leads to poly-ubiquitination (Ub) of the POI, which triggers its proteasomal degradation. The released PROTAC can exert its function again, which gives rise to a catalytic mode of action.
Another aspect of the mode of action of PROTAC molecules is the so-called hook or prozone effect. This is a type of negative interference commonly observed in sandwich (immunometric) assays, and refers to the observation that when a bivalent molecule acts as a linker between two proteins, the amount of fully formed ternary complex actually decreases with high enough concentrations of that molecule. It is a delicate source of false negatives in sandwich immunoassays, particularly well-known in home pregnancy tests [40]. These types of bell-shaped dose response curves are also frequently observed with PROTACs, and is simply the observation that high doses of bivalent molecules will saturate the individual proteins, as opposed to linking them together [41]. It is an intrinsic property of the PROTAC mode of action, and has significant impact on dosing strategies. However, computational methods suggest that cooperativity of binding can mitigate or reduce the hook effect [42,43]. In the context of PROTAC, cooperativity means that binding of the bivalent molecule to one protein increases the affinity for the second protein, skewing the equilibrium from the formation of binary complexes to the desired ternary complex. The MT-820 PROTAC developed for BTK does not show an observable hook effect at concentrations ten times above maximal observed degradation, which is attributed to positive cooperativity of binding [37].
1.4. Challenges and Limitations
Despite the continuous and significant improvements that were made over the last years, several fundamental challenges limiting the therapeutic applications of PROTACs remain to be addressed. First, the bivalent ligands have high molecular weight and polar surface areas, factors that are commonly associated with poor cell permeability, bioavailability and tissue distribution. These are issues that have troubled peptide and oligonucleotide drug discovery efforts for many decades, and have stopped many such molecules from becoming therapeutics. While it is encouraging that many groups have reported activity of PROTAC molecules in cell systems and of in vivo protein depletion [7,44,45], it remains to be seen how effective these compounds will be in a therapeutic setting.
Second, as was mentioned above, the size, orientation and composition of the linker plays an important role in PROTAC efficiency, and we are only starting to learn how these principles can be used in a rational way. Until then, PROTAC development will be very much a hit and miss exercise. Indeed, some PROTAC patent applications contain hundreds of pages listing rather diverse linkers, eg [45,46]. Protein-protein docking may help addressing this, and assist in the design of optimized linkers.
Furthermore, the published PROTAC molecules to date all rely on hijacking one of only a handful different E3 ligases. Variable target specificity and efficiency has been demonstrated for the different E3 ligases [47,48], most likely dominated by the nature of the protein-protein interactions between the ligase and the POI. Since an estimated 600 E3 ligases are encoded in the human genome [49], it is not unconceivable that E3 ligases exist with degradation efficacies vastly superior to the ones in use today. Hopefully, efforts will concentrate on finding small-molecule binders to these alternative E3 ligases. Finally, it is fair to say that the PROTACs that have been described to date, are based on previously known protein inhibitors. Maybe the biggest challenge, but also opportunity, for the PROTAC field is to degrade proteins for which no inhibitors can be found, such as scaffolding proteins or transcription factors.
2. Overview of Published PROTAC Molecules
Protein destabilization tools and other approaches to influence protein levels using the ubiquitination system have been reported over the last two decades. Successful applications include the use of heat-shock protein inhibitors [50], and of non-natural fusion proteins incorporating destabilizing domains [51]. The first bivalent molecules came in the form of hydrophobic tags, mimicking the (partially) unfolded state of proteins. These tags are believed to recruit chaperones, which mediate proteosomal degradation. The second-generation bifunctional molecules are instead mediated by E3 ligases, and these are generally referred to as PROTACs. These molecules consist of a distinct and selective target protein binding moiety, connected to an E3 ligase-binding molecule via a linker. Connection of the linker should not result in a loss of binding affinity of the target ligand to the protein of interest and for successful degradation the proximity and orientation must allow for target ubiquitination. Initially, small peptide chains were used to target VHL mediated degradation. These molecules have been extensively reviewed by Crews [52,53] and Chopra [54].
The seven amino-acid peptide ALAPYIP is recognized by VHL when the center proline is hydroxylated, and this entity was incorporated in the first cell permeable bifunctional molecule. A poly-D-arginine was fused to the peptide to increase cell permeability and stability. As a proof of principle, small degraders of FKBP12 and the AR were prepared, which were effective in selective protein degradation at 25 μM concentrations [23]. Following a similar approach, PROTACs were shown to hijack the VHL-CUL2 complex to degrade ERα at a concentration of 2 μM. Estradiol was linked via an n-hexyl linker to a shortened LAPOHYI pentapeptide with a hydroxylated proline, and in this case the poly-D-Arg tag was not required [24,55]. A two-headed approach for the degradation of ERα using the same penta-peptide was also shown to be effective in ERα degradation but a clear loss in solubility was observed for this molecule [56]. The same pentapeptide sequence was used to convert the small molecule apigenin into a PROTAC targeting the aryl hydrocarbon receptor. Despite a loss in binding affinity of the PROTAC compared to apigenin, the PROTAC was found to degrade AHR at 10 μM concentrations after 12 h of incubation [25]. Examples of PROTACs using two conjugated peptide sequences targeting the E3 ligase and the protein of interest were used to target FRS2α and PI3K [57]. The degradation of FRS2α and PI3K depends on intracellular phosphorylation of the proline in the VHL targeting peptide sequence and the concentration giving maximal target degradation was 60 μM. More recently, the Smad3 receptor, a critical signalling protein associated with renal fibrosis was subjected to virtual screening with the intention to construct a PROTAC molecule. Indeed, suitable binders were identified from the screen which were linked to the aforementioned pentapeptide and resulted in a PROTAC that decreased Smad3 protein levels in a dose-dependent manner in relevant cellular systems [58].
Not surprisingly, these peptidic PROTAC molecules are not particularly druglike, and their poor pharmacokinetic properties (notably absorption/distribution, solubility and stability) likely prevent the progression of these compounds into therapeutic modalities. Therefore, the discovery of small-molecule ligands for several E3 ligases (Fig. 1) was an important milestone for the PROTAC field, as this opened the door to the development of more druglike molecules. Below we will give an overview of the small-molecule PROTACs described to date. The majority of these have been designed against epigenetic targets, kinases or nuclear hormone receptors. We chose to cluster and discuss them accordingly.
2.1. PROTACs Targeting Epigenetic Processes
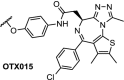
There is a large demand for therapeutic molecules that can effectively modulate transcriptional activity. The development of small-molecule inhibitors for transcription factors is challenging due to the lack of discrete cavities that allow for modulation of functional activity with ligands [59,60]. However, gene expression is also modulated indirectly by epigenetic processes such as DNA methylation and histone modification. As it turns out, many of the enzymes involved in these epigenetic processes can be targeted with small-molecule ligands, and many research groups have developed inhibitors of epigenetic proteins to modulate protein transcription [[61], [62], [63]]. Not surprisingly, those epigenetic targets were amongst the first to be explored with PROTACs. Degradation of bromodomain proteins, especially Brd4, has been researched extensively using the JQ1 inhibitor or the closely related OTX015, resulting in a variety of BET degraders. Some of the Brd4 molecules have become commercially available, putting PROTACs in the hands of any biologist who wants to study the effect of knocking out the Brd4 protein. A specific paragraph will be devoted to Brd4 PROTACs in this review.
The bromodomain-containing protein Brd9, a subunit of the human BAF nucleosome remodelling complex, has been implicated in acute myeloid leukemia [64] and several medicinal chemistry efforts targeting Brd9 have been reported [65,66]. Degraders of Brd9 demonstrated enhanced potency towards the parental ligands (Table 1, entry 6). Efficient degradation of Brd9 was shown at concentrations starting at 5 nM in MOLM-13 cells and Brd9 was the only protein with an altered abundance compared to 7326 proteins [67].
Table 1.
E3 protein ligand, structure of the linker and POI ligand with a description of the potency and efficacy against the specific epigenetic target.
| Table entry | E3 protein (ligand) | Linker | POI ligand | POI | Potency/efficacy | Reference |
|---|---|---|---|---|---|---|
| 1. | CRBN (pomalidomide) | 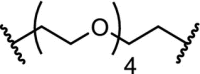 |
 |
Brd4 | Near complete degradation at 10nM within 6 hours, in all tested Burkitt’s lymphoma cell lines. | ARV-825 Lu [82] |
| 2. | CRBN (thalidomide, lenalidomide and pomalidomide) |
n-butyl |
 |
Brd4 | Near complete degradation at 100 nM in a human AML cell line. | dBET1 Winter [6] |
| 3. | VHL (9) | 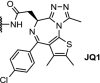 |
Brd4 | Selective degradation of Brd4 over Brd2 and Brd3 at low concentrations. | MZ1 Zengerle [38] | |
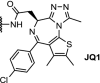
| 4. | VHL (9) | 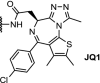 |
Brd4 | Suppression of both AR signaling and AR levels and tumor regression in a CRPC mouse xenograft model | ARV-771 Raina [83] | |
| 5 | CRBN (thalidomide) | 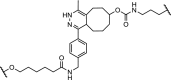 |
 |
Brd4 | In-cell click reaction with complete degradation of Brd4 at 10 and 3 μM and partial degradation at 1 and 0.3 μM | Lebraud [84] |
| 6. | CRBN (Lenalidomide, pomalidomide and 5) | 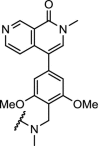 |
Brd9 | Selective degradation of Brd9 over Brd4 and Brd7. Efficient and fast degradation in the concentration range of 5 to 50 nM. | Remillard [67] | |
| 7. | CRBN (thalidomide) | 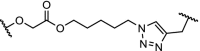 |
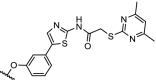 |
Sirt2 | Selective over Sirt1 and Sirt3. Degradation of Sirt2 observed in the range of 0.05 to 5 μM | Schiedel [71] |
| 8. | CRBN (thalidomide) and VHL (9) | 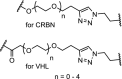 |
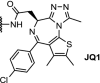 |
Brd4 | Longest linker was the most active, resulting in a DC50 of 0.20 μM for the CRBN PROTACs. | Wurz [47] |
| 9. | CRBN (thalidomide) | Extensive linker optimization, highest potency was obtained for the n-pentyl linker | 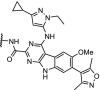 |
Brd4 | Effective Brd4 degradation at 30 pM in RS4-11 leukemia cells. IC50 51 pM in inhibition of RS4-11 cell growth. Induced rapid tumor regression in vivo against xenograft tumors | Zhou [85] |
| 10. | VHL (9) | 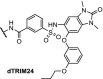 |
TRIM24 | dTRIM24 induced rapid, selective and sustained proteasomal degradation of TRIM24. Dependence of acute leukemia on TRIM24 was shown | Gechijian [9] | |
| 11. | CRBN (pomalidomide) | n-pentyl | 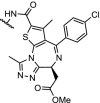 |
Brd4 | Selective Brd4 degradation over Brd2/3. Off-target degradation can be tuned by the linker composition | ZXH-3-26 Nowak [86] |
| 12. | CRBN (pomalidomide) | 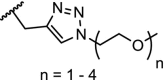 |
 |
HDAC6 | DC50: 34 nM with a maximum HDAC6 degradation of 70% in MCF-7 cells. | Yang [39] |
| 13. | CRBN (thalidomide) | 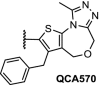 |
Brd4 | Effective degradation at low pM concentrations in human leukemia cell lines. QCA570 achieves tumor regression in both the MV4-11 and RS4-11 acute leukemia xenograft models | QCA570 Qin [87] | |
| 14. | CRBN (thalidomide) |  |
 |
PCAF/ GCN5 | Degradation of PCAF and GCN5 in THP1 cells, DC50: 1.5 nM and 3 nM, inhibiting the differentiation of monocytes into macrophages | Bassi [79] |
| 15. | CRBN (thalidomide) | 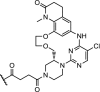 |
Bcl6 | No significant phenotypic response. Incomplete Bcl6 degradation despite sufficient cellular concentration, excellent selectivity and target engagement. | McCoull [81] |
The structure of the CRBN ligands and the VHL ligand (9) are shown in Fig. 2.
Sirtuins are proteins that possess either mono-ADP-ribosyltransferase or deacetylase activity. Sirtuins influence a range of cellular processes such as inflammation, aging and apoptosis. The human genome encodes seven isotypes of sirtuins (Sirt1–7). Sirt2 has been shown to be a pivotal regulator of cell cycle regulation [68] and dysregulation of Sirt2 has been associated with neurodegenerative diseases [69] and cancer [70]. Investigations into the role of Sirt2 as epigenetic regulator protein has been hampered by the lack of compounds that have sufficient isotype selectivity and good pharmacokinetic properties. To investigate the effects of Sirt2-dependent deacetylation and downstream signalling, selective Sirt2 degraders were designed. Effective degradation of Sirt2 was shown in the concentration range of 0.05 to 5 μM. At higher concentrations the efficacy of the PROTAC dropped, which is likely due to the hook effect [71].
TRIM24, also known as TIF1α, is a multidomain transcriptional co-regulator involved in nuclear receptor signalling [72,73]. Potent and selective inhibitors of the TRIM24 bromodomain have not been able to demonstrate efficacious effects on cancer proliferation [74]. The potent TRIM24 inhibitor IACS-9571 [75] was converted into a heterobifunctional degrader hijacking VHL, which achieved efficient, selective and sustained degradation of TRIM24 (Table 1, entry 10). Furthermore, TRIM24 dependency in acute leukemia was demonstrated [9].
HDACs are epigenetic erasers, which control gene transcription via the removal of epigenetic markers, that play crucial roles in many diseases [76]. In an effort to create degraders for this protein family, a non-selective HDAC inhibitor was connected to pomalidomide via click chemistry. Four different length PEG linkers were tested, and the bifunctional molecules were observed to non-specifically inhibit HDACs. However, the most potent bifunctional molecule selectively degraded HDAC6 over other members of the family. The concentration at which half-maximal degradation was achieved (DC50) was 34 nM, and the maximum percentage of degradation was 70% [39]. Degradation of HDAC6 may negate the effects of overexpression of HDAC6 in cancer by reducing the protein amount to physiological levels [77,78].
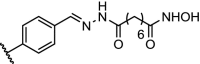
The epigenetic proteins PCAF and GCN5 each contain an acetyltransferase domain and a bromodomain. The small molecule GSK-4027 (Table 1, entry 14) is a potent inhibitor of the bromodomains of PCAF and GCN5, but inhibition alone was found to be insufficient to disrupt the immunomodulatory functions of these proteins. To explore whether PCAF or GCN5 degradation would be more successful, a PROTAC was designed from GSK-4027. The resulting compound had a DC50 (the compound concentration that results in 50% target protein degradation) of 1.5 nM and 3 nM in THP1 cells for PCAF and GCN5, respectively. Degradation was observed rapidly, with 80% reduction of protein levels after 10 min at a concentration of 30 nM. This PROTAC was shown to potently modulate the expression of multiple inflammatory mediators in lipopolysaccharide-stimulated macrophages and dendritic cells and affect the differentiation of monocytes. This is a compelling study in which protein degradation and not inhibition provides therapeutic opportunities [79].
Bcl6 inhibition is a promising strategy for the treatment of diffuse large B-cell lymphoma cancers. Recently, potent and selective macrocyclic Bcl6 inhibitors were discovered, amongst others via a fragment-based drug discovery program [80]. In an effort to develop high-quality chemical probes to evaluate the therapeutic potential of Bcl6, the macrocycles were converted into PROTACs to engage the ubiquitination pathway. Despite efficient target engagement and effective cellular concentrations, the PROTAC failed to induce a significant phenotypic response in B-cell lymphoma cell lines. Detailed mass analysis showed a small residual Bcl6 population across all subcellular fractions [81].
2.2. PROTACs Targeting Kinases
GPCRs and kinases are the two biggest target classes in drug discovery. And while there are many GPCR-targeting drugs on the market, covering many indications with great efficacy and safety [88], kinase inhibitors are largely developed for oncology indications only [89]. They often suffer from the occurrence of resistance [90] and the lack of kinase selectivity, translating into side effects. Not surprisingly, the kinase field was early and eager to test whether depletion of target kinases would be beneficial over inhibition. Since PROTAC efficacy appears to depend to a large extent on the interface between the E3 ligase and the POI, and less on specific interactions at a protein's active site, it was hypothesized that PROTACs will suffer less from inter-kinase cross-reactivity. Also, it has been rationalized that resistance through upregulation of the kinase can be more successfully countered with a protein degrader as opposed to an inhibitor [91,92].
Inhibitors of RIPK2 are of interest for the treatment of inflammatory diseases, because RIPK2 plays a central role in the innate immune response and the production of inflammatory cytokines. RIPK2 also has a pro-inflammatory scaffolding function [93], which makes degradation of RIPK2 an attractive therapeutic strategy. A RIPK2 modulator appended with a linker and a VHL recruiting ligand (9, Fig. 1) showed catalytic degradation of RIPK2 with high specificity and a DC50 of 1.4 nM (Table 2, entry 1) [7].
Table 2.
E3 protein ligand, structure of the linker and POI ligand with a description of the properties for kinases.
| Table entry | E3 protein (ligand) | Linker | POI ligand | POI | Potency/efficacy | Reference |
|---|---|---|---|---|---|---|
| 1. | VHL (9) |  |
 |
RIPK2 | 50% RIPK2 degradation at 1.4 nM after 1 hour. Dmax of >95% at 10 nM | Bondeson [7] |
| 2. | VHL and CRBN (9 and pomalidomide) |  |
imatinib, bosutinib, and dasatinib | Tyrosine kinase: BCR-ABL | >60% degradation at 1 μM for the dasatinib-CRBN PROTAC. No degradation for the VHL-based PROTACs. | Lai [48] |
| 3. | CRBN (thalidomide) | 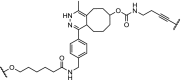 |
 |
ERK1 and ERK2 | In-cell click reaction where ERK1/2 degradation is observed partially after 4 h and is complete after 16 h at 10 μM | Lebraud [84] |
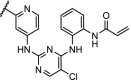
| 4. | CRBN (thalidomide) | n-pentyl | 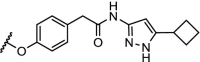 |
CDK9 | Selective for CDK9. 56% degradation of CDK9 at 10 μM. | Robb [95] |
| 5. | VHL (9) | Diethylene glycol | Lapatinib, gefitinib, afatinib | RTKs: EGFR, HER2 and c-MET | Efficient degradation of transmembrane RTKs with superior outcome over RTK inhibitors. | Burslem [98] |
| 6. | CRBN (pomalidomide) | 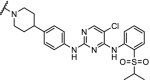 |
Multi- kinase degrader | Quantitative proteomics showed degradation of 28 kinases including BTK, FLT3 and nine members of the CDK family | Huang [91] | |
| 7. | CRBN (pomalidomide) | 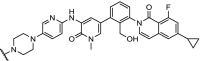 |
BTK | Most efficient degradation at 100 nM concentration. In addition, a bosutinib-based degrader was reported | Huang [91] | |
| 8. | CRBN (pomalidomide) | 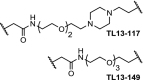 |
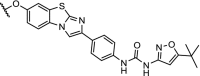 |
FLT3 | Concentrations between 10 and 100 nM resulted in the most efficient degradation of FLT3. | Huang [91] |
| 9. | VHL (9) | 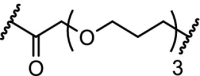 |
TBK1 | Extensive linker optimization. DC50: 12 nM with Dmax: 96%. | Crew [99] | |
| 10. | CRBN (pomalidomide) | 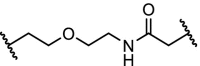 |
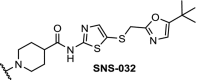 |
CDK9 | The multi target inhibitor SNS-032 became selective for CDK9 as PROTAC with near complete degradation at <250 nM concentration | Olson [96] |
| 11. | CRBN (pomalidomide) | Six different linkers were used. The best results were obtained with: 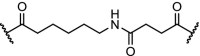
|
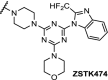 |
PI3K | IC50 for PI3K: 24 nM. time-/concentration-dependent degradation PI3K protein observed. Inhibition of HepG2 cell growth via autophagy | Li [100] |
| 12. | CRBN (pomalidomide) | 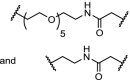 |
 |
ALK | DC50: 3 and 11 nM respectively after 16 h in SU-DHL-1 cells. Potent inhibition of proliferation of SU-DHL-1 cells. Both linkers effective | Zhang [101] |
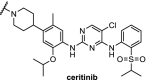
| 13. | CRBN (pomalidomide) | Ceritinib and TAE684[108] | ALK | DC50 of 10 nM for both PROTACs in H3122 cells. ABCB1 was responsible for efflux. | Powel [102] | |
| 14. | CRBN (pomalidomide) | 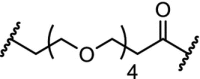 |
 |
CDK8 | CDK8 IC50: 159 nM. Significant degradation of CDK8 in Jurkat cells after treatment for 24 hr at 1 μM concentration | Hatcher [103] |
| 15. | CRBN (pomalidomide) | 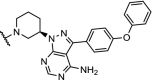 |
BTK | Efficiently degradation of BTK-WT. Induced degradation of ibrutinib-resistant BTK-C481S (50% degradation efficiency at 30 nM) | Sun [104] | |
| 16. | CRBN (5) | 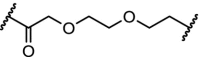 |
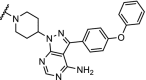 |
BTK | >99% degradation of mutated C481S and wildtype BTK at nM concentrations. Enhanced selectivity over ibrutinib | Buhimschi [37] |
| 17. | CRBN (pomalidomide) | 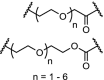 |
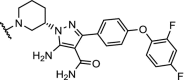 |
BTK | For n = 4 – 6 DC50’s between 1 and 40 nM. n = 1 – 3 are ineffective in degradation of BTK. | Zorba [41] |
| 18. | CRBN (pomalidomide) | 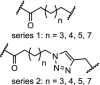 |
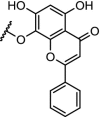 |
CDK9 | Serie 2 (n = 5) showed the most efficient CDK9 degradation in the concentration range of 1 to 30 μM. Inhibition of MCF cell proliferation with an IC50 of 17 μM | Bian [97] |
| 19. | CRBN (pomalidomide) | 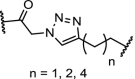 |
 |
CK2 | Most promising results for n = 2. Degradation of CK2 in a dose and time-dependent manner resulting in downstream reduced phosphorylation of Akt | Chen [107] |
| 20. | VHL (9) | 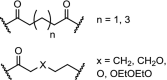 |
 |
ALK | n = 1 displayed the best properties. 90% ALK degradation at 1 μM after 16 h. in SU-DHL-1 cells. Excellent efficacy in tumor xenograft mice. | Kang [100] |
The structure of the CRBN ligands and the VHL ligand (9) are shown in Fig. 2.
The marketed kinase inhibitors imatinib, bosutinib and dasatinib were conjugated to either a VHL or CRBN ligand with a range of linkers to demonstrate the degradation of c-ABL and BCR-ABL. Effective protein degradation depended on the recruited E3 ligase and linker length [48].
The MAP kinase signalling cascade with the centrally placed ERK kinases is involved in a large variety of cellular and physiological processes [94]. A click-chemistry approach showed efficient in-cell assembly of a covalent PROTAC (Table 2, entry 3), followed by complete and time-dependent degradation of ERK1/2 [84]. The main potential advantage of PROTAC assembly in cell via click chemistry is a significant reduction of the molecular weight and polar surface area of the separate reaction partners compared to the pre-assembled PROTAC molecule. The main difficulty, on the other hand, for this approach is that the two click partners may undergo their reaction outside the cell.
Enhanced selectivity by converting kinase inhibitors into PROTACs was demonstrated for CDK9. Development of selective CDK inhibitors is challenging because of the structural similarity of the ATP binding sites between the CDKs. However, the surfaces of the CDKs differ and this offers the opportunity to develop selective CDK degraders. Three independent research groups have described selective CDK9 degraders (Table 2, entries 4, 10 and 18). A pan CDK inhibitor was appended with a n-pentyl linker and converted into a PROTAC hijacking cereblon. Selective degradation of CDK9 over other CDKs was shown in the concentration range of 5 to 25 μM [95]. Following this success another PROTAC was designed which was also more selective in degrading CDK9 over several other CDKs compared to the parent molecule SNS-032. At 250 nM, the PROTAC did not achieve measurable occupancy of any other CDK target of SNS-032 in cells, even though these targets are engaged in cell lysates. This suggests that the selectivity was induced by the ability of the PROTAC construct to act catalytically, at sub-stochiometric concentrations in the cell [96], and/or due to the fact that PROTACs do induce a protein-protein interaction between the protein and the ligase, leading to a more selective action. Finally, a series of PROTACs based on the natural product wogonin was effective in selective downregulation of CDK9 in MCF-7 cells in the concentration range of 10 to 30 μM [97].
The degradation of receptor tyrosine kinases (RTKs) was first demonstrated using the kinase inhibitors lapatinib, gefitinib and afatinib. The PROTAC molecules capable of degrading the intended RTKs inhibited downstream signalling and cell proliferation at lower concentrations than the parent compounds achieved through inhibition only. Resistance through mutations and rewiring of the kinome, which is common with kinase inhibitors could be avoided [98].
Through a chemoproteomic approach, a multi-kinase degrader was found, that targeted 28 kinases including the drug targets BTK, FLT3 and nine members of the CDK family. This study showed that the degraded set of 28 kinases represented only a fraction of the targets to which the degrader bound, indicating that successful degradation depends on more than only target engagement. Selective degraders for BTK and FLT3 were found that were efficacious at concentrations of 100 nM [91]. Selective and effective TBK1 degraders were generated via systematic optimization of the linker and the TBK1 binding ligand. Greater potency and selectivity was achieved than anticipated from the individual components [99].
PI3K inhibitors have been advanced for the treatment of cancer, but their clinical application is hindered by acquired drug resistance. This prompted the design of PI3K targeting PROTAC molecules, through conjugation of pomalidomide with the pan class-I PI3K inhibitor ZSTK474, using six different linkers (Table 2, entry 11). Four of the linkers demonstrated time- and concentration-dependent degradation of PI3K and mediated expression of downstream proteins. Interestingly, further study on a representative compound indicated the construct inhibited cancer cell growth by autophagy and not by the induction of apoptosis or by cell cycle arrest, suggesting that degradation can lead to a different pharmacological phenotype compared to inhibition [100].
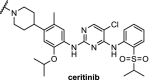
The kinase ALK is an interesting target for degradation because absence of ALK protein is not toxic in mammals, and the four FDA-approved inhibitors used against ALK-positive non-small cell lung cancers suffer from drug resistance. Three independent research groups have reported PROTACs based on the selective ALK inhibitor ceritinib using different linkers and employing either CRBN or VHL (Table 2, entries 12, 13 and 20). The PROTACs hijacking the cereblon ligase all demonstrate low nM DC50s in various cell lines. Moreover, these PROTACs showed beneficial downstream effects and displayed good plasma exposure in mice [101]. In addition, for the first time a drug transporter protein (ABCB1) was identified to be responsible for efflux of a PROTAC [102]. Utilizing the VHL E3 ligase, the PROTAC showed 90% degradation of ALK at 1 μM concentration in SU-DHL-1 cells and demonstrated excellent efficacy in an in vivo xenograft mouse model [100].
In attempts to simplify the CDK8 ligand cortistatin A, a steroid scaffold was developed which exhibited very high selectivity and only a slight reduction in potency against CDK8 compared to cortistatin A. Further investigations revealed that this scaffold could be converted into a proteasomal degrader of CDK8. Significant degradation of CDK8 was observed after 24 h at 1 μM concentration. The degrader was used to confirm target engagement and will be used to investigate the pharmacological consequences of degrading CDK8 [103].
Inhibition of BTK with the irreversible inhibitor ibrutinib has emerged as a treatment for patients with chronic lymphocytic leukemia, but a large population of patients develop resistance through a mutation of cysteine 481 to a serine (C481S). Three recent independent studies report efficient and highly potent BTK PROTACs capable of degrading both BTK wildtype and the C481S mutant at low nM concentrations [37,41,104].
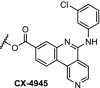
CK2 is a constitutively active serine/threonine kinase that can phosphorylate over 300 substrates [105]. Overexpression of this kinase has been reported in several cancers and different kinds of tumors. CK2 function can be reduced by downregulation of the protein levels or inhibition by small molecules [106]. The CK2 inhibitor CX-4945 (Table 2, entry 19) was appended to pomalidomide via click chemistry, and was found to degrade CK2 in a time- and dose-dependent manner, albeit with relatively high concentrations in the range of 10 μM. Degradation of CK2 resulted in downstream changes, such as reduced phosphorylation of Akt and upregulation of the P53 tumor suppressor [107].
In summary, a large number of potent and efficient PROTACs have been described for kinases. While none of these have been extensively tested in a complex environment, evidence emerges that PROTACs can enhance selectivity and that kinase protein degradation can result in distinct pharmacological effects compared to classical inhibition. Kinases are particularly suited for a PROTAC approach because of the abundance of suitable starting points, namely the kinase inhibitors, many of them with crystallographic information and usually harboring a solvent exposed functional group that can be used for conjugation. In addition, kinase inhibitors often suffer from drug resistance, and PROTAC-mediated degradation is an exciting new strategy to modulate both phosphorylation activity and kinase scaffolding function.
2.3. PROTACs Targeting Receptors
Nuclear hormone receptors play key roles in obesity, diabetes and cancer, and they have been drug discovery targets for decades. Modulation of the activity of these receptors has been very successful, but pharmaceutical applications have been hampered by resistance through post-translational modifications, upregulation or mutations [109]. The first degraders of nuclear receptors made use of peptide-based molecules which required high concentrations of PROTACs for efficient degradation [23,24,55].
The first non-peptidic PROTAC hijacked MDM2 to degrade the androgen receptor (Table 3, entry 1). Treatment of Hela cells with 10 μM concentration of a bivalent molecule consisting of a SARM linked to nutlin, demonstrated decreased AR levels [26]. This study is somewhat atypical as AR is known to have endogenous interactions with MDM2 [110] and nutlin itself induces ubiquitination [111]. Hijacking the E3 ligase cIAP1 was shown to be effective for bivalent molecules called SNIPERs. The first example addressing cIAP1 consisted of a bestatin methyl ester linked to all-trans-retinoic acid, and degraded the cellular retinoic acid binding protein (CRABP-II) [28,29]. The same strategy was applied for three nuclear hormone receptors: RAR, ERα and AR. Degradation was observed for all three proteins with moderate cell potencies in the range of 30 μM [27].
Table 3.
E3 protein ligand, structure of the linker and POI ligand with a description of the properties for (nuclear) receptors.
| Table entry | E3 protein (ligand) | Linker | POI ligand | POI | Potency/efficacy | Reference |
|---|---|---|---|---|---|---|
| 1. | MDM2 (Nutlin) | 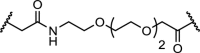 |
 |
AR | Upon treatment of HeLa cells with 10 μM compound for 7h, a decrease in androgen receptor levels was observed | Schneekloth [26] |
| 2. | cIAP1 (Bestatin) |  |
 |
CRABP-II | Degradation of CRABP-II in IMR-32 cells was observed at 10 μM PROTAC concentration. | Itoh [28] and Okuhira [29] |
| 3. | cIAP1 (Bestatin) |  |
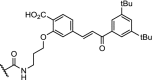 |
RAR | Maximal RAR degradation at 30 μM concentration in HT1080 cells | Itoh [27] |
| 4. | cIAP1 (Bestatin) | 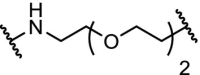 |
 |
ERα | Maximal ERα degradation at 30 μM concentration in human mammary tumor MCF7 cells | Itoh [27] |
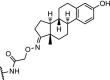
| 5. | cIAP1 (Bestatin) |
|
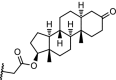 |
AR | Maximal AR degradation at 30 μM concentration in human mammary tumor MCF7 cells | Itoh [27] |
| 6. | VHL (9) | 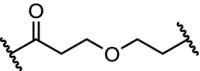 |
 |
ERRα | 50% ERRα degradation at 100 nM. Protein knockdown in tumor xenografts (mouse) | Bondeson [7] |
| 7. | HSP70 (adamantyl) | 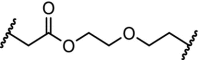 |
 |
AR | DC50: 1.1 μM, Dmax: 69%. Anti-proliferative activity remained in castration-resistant prostate cancer cells | Gustafson [113] |
| 8. | VHL (9) | 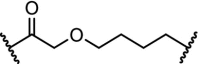 |
 |
AR | DC50: 5 nM, Dmax: 98%. ARCC-4 effectively degrades clinically relevant AR mutants. ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance | Salami [114] |
The structure of the CRBN ligands and the VHL ligand (9) are shown in Fig. 2.
High affinity and effective degradation was observed for the ERRα with a DC50 of 100 nM in MCF-7 breast cancer cells using a selective ERRα ligand linked to 9 (Table 3, entry 6). In addition to high potency in cells, the PROTAC showed in vivo depletion of ERRα in mice bearing xenograft tumors and subsequent significant reduction of tumor size [7].
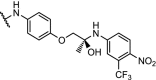
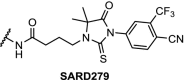
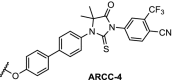
There are several antitumor agents targeting AR, including the antagonist enzalutamide. Recent reports have shown that the F876 L mutation in the AR ligand binding domain causes induced tolerance for enzalutamide [112]. In the hope of bypassing resistance through AR degradation, a hydrophobic tagging strategy was employed. An AR binder with an adamantyl tag bound to the protein will display this very lipophilic tag to the surface, mimicking a protein misfolded state. This exposes the protein to proteosomal degradation via the ubiquitination pathway. The adamantyl tag decreased the binding affinity of the AR ligand significantly, but nevertheless induction of AR degradation at sub-micromolar concentrations was observed [113]. Building on this result, highly effective AR PROTACs were prepared based on the structure of enzalutamide linked to 9. With a DC50 of 5 nM and Dmax of 98% for the wildtype AR, the PROTAC named ARCC-2 was also shown to degrade all clinically relevant AR mutants independent of elevated androgen levels [114].
Altogether, these results demonstrate the potential of PROTACs for the degradation of nuclear hormone receptor proteins. Global frontrunners in this area are two programs run at the company Arvinas: ARV-471 and ARV-110, targeting ERα and AR, respectively. The structures of these compounds have not yet been disclosed, and are therefore not included in this review. Based on the promising in vitro and in vivo data [45,114] Arvinas is preparing both programs for phase I clinical trials.
2.4. PROTACs Targeting Other Proteins
While the bulk of the reported PROTAC molecules target kinases, nuclear hormone receptors and epigenetic proteins, examples are appearing in other areas as well. The results described in Table 4 demonstrate the potential of PROTACs as research tools to investigate biochemical pathways, and also as therapeutic interventions. The cytosolic signalling protein FKBP12 plays a role in cardiac development as well as oncogenic signalling. Engineered mutants of human FKBP12 have been reported that are rapidly and constitutively degraded when expressed in mammalian cells, and this instability is conferred to proteins fused to these destabilizing domains [51,115]. This provides a new strategy to control protein function in living organisms, but is of course limited to non-endogenous fusion proteins. FKBP12 has also been subjected to degradation with more conventional PROTAC ligands by linking the FKBP12 ligand SLF to thalidomide, with two different spacers (Table 4, entry 1). Both molecules effectively reduce FKBP12 levels in MV4–11 leukemia cells [6].
Table 4.
E3 protein ligand, structure of the linker and POI ligand with a description of the properties research for FKBP12, E3 ligases, an enzyme, a Tau degrader and a recent non-natural fusion protein.
| Table entry | E3 protein (ligand) | Linker | POI ligand | POI | Potency/efficacy | Reference |
|---|---|---|---|---|---|---|
| 1. | CRBN (thalidomide) |
n-butyl and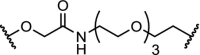
|
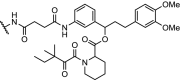 |
FKBP12 | 80% reduction of FKBP12 at 0.1 μM and 50% reduction at 0.01 μM in MV4-11 cells | Winter [6] |
| 2. | VHL (9) | 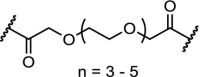 |
(9) | VHL | CM11 (n = 5) induced complete depletion of VHL after 4 h at 10 nM. Potent, long-lasting and selective degradation of VHL, with DC50 of < 100 nM | Maniaci [117] |
| 3. | VHL (9) |  |
 |
DHODH | IC50 for DHODH 93 nM. No degradation observed. Linker optimization needed to target the inner mitochondrial protein. | Madak [121] |
| 4. | Keap1 (Keap1 binding peptide)* | GSGS peptide | YQQYQDATADEQG | Tau | Poly-D-arginine was added for cell penetration. Strong in vitro binding with Keap1 and Tau. Keap1-dependent degradation by enhancing the ubiquitination of Tau. | Lu [124] |
| 5. | CRBN (thalidomide) | 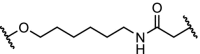 |
 |
FKBP12F36V Fusion proteins | Degradation of a panel of fusion chimeras with FKBP12F36V including: BRD4, HDAC1, EZH2, Myc, PLK1 and KRASG12V. Rapid degradation in vivo was shown | Nabet [33] |
| 6. | CRBN (pomalidomide) | 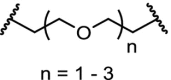 |
pomalidomide | CRBN | The homo-PROTAC with n = 2 was identified as the most potent degrader. Degradation observed at 10 nM after 16 h. Hook-effect observed at 100 μM | Steinebach [118] |
The structure of the CRBN ligands and the VHL ligand (9) are shown in Fig. 2. *Keap1 binding peptide: Ac-LDPETGEYL-OH.
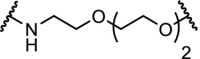
E3 ligases play an important role in normal cellular physiology and disease, and VHL itself is also an attractive drug target [49,116]. In an effort to achieve VHL degradation homo-PROTACs were designed. Two VHL ligands were linked via ethylene glycol linkers demonstrating very efficient depletion of a specific isoform of the VHL protein (pVHL30), allowing interrogations of biological functions of this VHL isoform [117]. Similarly, by linking two pomalidomide molecules, CRBN homo-PROTACs were made that were able to promote ubiquitination and degradation [118].
DHODH inhibitors are used in the treatment of autoimmune diseases such as rheumatoid arthritis, psoriasis and multiple sclerosis [119,120]. To study the therapeutic relevance of DHODH and to create an intracellular knockdown a PROTAC was developed based on the DHODH ligand brequinar (Table 4, entry 3). Although the bifunctional molecules inhibited DHODH with an IC50 of 93 nM, degradation of the target was not observed. The lack of degradation was explained by the lower expression of VHL in the mitochondrial ubiquitination system compared to the cytosol [121].
For many years, amyloid-β (Aβ) has been a key target for therapeutic intervention in Alzheimer's disease (AD), but clinical candidates have not demonstrated slowing of the disease progression. AD neuropathology is characterized by accumulation and aggregation of Aβ but also of Tau proteins. Therefore, tau pathology is an important area for the development of disease-modifying therapies [122,123]. A Keap1-Tau fused peptide PROTAC appended with a poly-D-Arg showed strong in vitro binding to Keap1 and Tau with decent cell permeability. Western blotting and flow cytometry confirmed time- and concentration-dependent degradation of Tau. The results suggested that Tau can be degraded via Keap1 dependent ubiquitination using PROTACs, and this approach holds promise as a strategy in the treatment of neurodegenerative diseases [124].
Temporal control of signal transduction pathways via chemical-genetic model systems provides insights into cellular processes. A series of dTAG tool molecules capable of recruiting the CRBN E3 ligase complex to several targets fused to FKBP12F36V including Brd4, HDAC1, EZH2, Myc, PLK-1 and KRASG12V has been described. Using a selective FKBP12F36V degrader (Table 4, entry 5), the downstream effects of degradation could be studied for these proteins [33].
3. Brd4: A Mechanistic Case Study
The bromodomain and extra-terminal domain (BET) proteins constitute a family of epigenetic readers that regulate gene expression by recruiting transcriptional complexes to acetylated chromatin domains [128,129]. The bromodomain subset of proteins, including the widely investigated Brd4, have a crucial function in the expression of oncogenes and are therefore attractive targets for cancer treatment [125,130,131]. Consequently, these epigenetic proteins were amongst the earliest candidates to be targeted by protein degradation. Specifically, Brd4-degraders have been used extensively in mechanistic chemical biology studies.
In 2015, three publications appeared almost simultaneously, all describing Brd4-targeting degraders with differentiating characteristics compared to normal small molecule inhibitors. Both the labs of Crews and Bradner reported degraders using Cereblon as ligase and JQ1 (ARV-825) and OTX015 (dBET1) as Brd-inhibitors (Fig. 3) [6,82]. Most interestingly, these papers demonstrate that the use of the small molecule inhibitors JQ1 and OTX015 led to significant accumulation of Brd4 protein, which likely limited the desired downstream effect on c-Myc levels and cell proliferation [82]. The degrader molecules, on the other hand, caused persistent suppression of c-Myc and yielded a significantly stronger antiproliferative effect in lymphoma and leukemia cells [6,82].
Fig. 3.

Structures of Brd4 inhibitors (+)-JQ1 and OTX015 [125]. Structures of the Brd4 PROTACs: ARV-825 [82], ARV-763 [126,127] dBET1 [6], and ARV-771 [83].
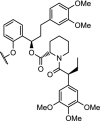
The group of Ciulli described a PROTAC of JQ1 as well, utilizing VHL as the ligase (MZ1, Fig. 4). Surprisingly, this approach resulted in significant selectivity for Brd4 over Brd2/3, a quality that the previously mentioned Cereblon-based degraders did not possess [38]. No small molecule inhibitors reported to date exhibit both substantial intra-BET selectivity and high potency [132]. This lack of selectivity is believed to cause side-effects in recent clinical trials and has limited elucidation of the specific functions of separate BET proteins [133]. Since the degrader molecules showed no binding preference for either protein, the observed selectivity was reasoned to be caused by direct interactions between the Brd and VHL proteins. This rationale gained credence when the same group published the first crystal structure of a ternary complex, in this case Brd4-MZ1-VHL (Fig. 4) [8]. The extensive interactions observed between Brd4 and VHL suggest a strong relative influence of the stability of the ternary complex, on both the effectiveness and the selectivity of the degrader. In follow-up work, the importance of ternary complex formation was further highlighted, by exchanging the original MZ1-based BET-protein ligand for a higher affinity inhibitor [134]. Despite possessing higher affinity for the BET-protein, the high-affinity PROTAC showed a negative cooperativity of ternary complex formation and was less effective as degrader. In addition, a change in selectivity profile from Brd4 to Brd3/Brd4 was observed.
Fig. 4.

Crystal structure of the Brd4–MZ1–VHL complex published by Gadd and co-workers (PDB entry 5T35) [8]. a) Ribbon representation of the X-ray co-crystal structure. b) Zoomed-in view of the MZ1 ligand at the interface of Brd4 and VHL. c) The chemical structure of MZ1.
Raina and co-workers reported ARV-771 (Fig. 3), a VHL-based PROTAC differing from MZ1 only in linker composition. The selectivity profile of ARV-771 appeared to be less pronounced, but clear in-vivo effects were demonstrated in mouse xenograft models of castration-resistant prostate cancer [83].
Structural and mechanistical insights into ternary complex formation may open the door to rational design of PROTAC degraders. This has recently been pioneered by the groups of Gray, Bradner and Fischer, using a combination of in silico protein-protein docking and X-ray crystallography [86]. Thus far, Cereblon-recruiting degraders were found to be unselective within the BET family [6,82]. However, this integrated approach revealed several low-energy Brd4-Cereblon binding modes, laying the foundation for the design and synthesis of ZXH-3-26 (Table 1, entry 10), a short-linker, pomalidomide-based, degrader with significant selectivity over Brd2/3. Notably, the authors argue that the short linker constrained the number of binding conformations, leading to increased selectivity. On the other hand, they state that the interprotein contacts between Brd4 and CRBN contribute relatively little to the overall binding affinity. The latter observation appears to, at least partially, differ from previous publications where pronounced cooperative effects were shown [8,10,134]. It is clear that the mechanistic understanding of induced protein degradation is still in its infancy, and there is need for more advanced, preferentially intra-cellular studies to understand the complicated processes involved [135].
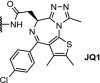
Recent work on the development of Brd4-degraders has produced interesting linker developments. For example, click chemistry has been applied to produce a set of ten triazole-containing linkers, based on JQ1 [47]. The DC50s were in a similar range as previously reported degraders, although certain linker lengths were found to result in inactive degraders, stressing the importance of linker lengths in PROTAC molecules. Another notable application of click chemistry was reported by researchers at Astex. Using their ‘CLIPTAC’ approach (click-formed proteolysis targeting chimera), the final degrader molecule is formed intracellularly by a click reaction from the two corresponding precursors [84]. Combining trans-cyclooctene-functionalized JQ1 with a tetrazine-functionalized Cereblon-ligand resulted in intracellular PROTAC formation, which led to near-complete downregulation of Brd4 after 24 h. This approach has the potential to circumvent the high molecular weight, high polar surface area, and poor permeability commonly associated with PROTAC molecules. The main challenge with CLIPTACs is to prevent PROTAC formation outside the cell, which amongst others entails the precursors to be administered separated in time. Also, it would be required to develop and obtain regulatory approval for two distinct chemical entities.
Two recent publications of the Wang group demonstrated the importance of linker optimization in the development of highly potent Brd4-degraders. Using a novel inhibitor, a CRBN-recruiting Brd4-PROTAC was designed through multiple optimization cycles [85]. The compound has an IC50 of 51 pM in RS4–11 acute leukemia cells and achieved >90% tumor regression in the corresponding xenograft model. Shortly after, the even more efficacious Brd4-degrader QCA570 was published (Table 1, entry 12) [87]. A rigidification tactic was applied by introducing an additional alkyne group in the linker. This ultimately resulted in QCA570, which possesses an IC50 as low as 8 pM in MV4–11 cells and achieves complete and long-lasting tumor regression in xenograft models.
In addition to the aforementioned in vivo models, extensive pre-clinical studies on primary multiple myeloma and mantle cell lymphoma cells have recently been reported with PROTACs ARV-763, ARV-771 and ARV-825 [127,136]. Detailed effects downstream from Brd4 were demonstrated, such as reduced levels of c-Myc and CDK4/6, increased p21 levels, and induction of apoptosis. Altogether, these results exemplified the potential therapeutic benefit of Brd4-PROTACs, with a profile that is differentiating from the clinical stage BET inhibitor OTX015 [137].
4. Conclusion and Outlook
The number of protein targets with therapeutic potential is steadily rising with advances in gene silencing techniques such as CRISPR/Cas9, antisense oligonucleotides and RNA interference. Many of those may prove difficult to tackle with conventional small-molecule inhibitors, but a plethora of new therapeutic modalities are being investigated to enlarge our drug discovery toolbox and success rate against these new targets. The “induction of protein degradation” represents a fundamentally different approach to modulate biological pathways and is rightfully receiving much increased attention.
Less than two decades ago, “a method of generating a compound for activating ubiquitination of a target protein which comprises covalently linking a target protein binding element able to bind specifically to said protein to a ubiquitination element” was first reported. Since then, the field of protein degradation has made tremendous strides. As described in this mini-review, there is now an abundance of examples of PROTACS that possess remarkable in vitro activity and, in a number of cases, in vivo activity in rodents.
In the coming years, we will learn if the distinct attributes ascribed to PROTACS such as prolonged efficacy (need for compensatory protein resynthesis), increased potency (potential for repeated, catalytic ligand action), higher/different selectivity profile (driven by ternary complex formation) and broader spectrum activity (thanks to whole protein degradation) will translate into clinical benefit for patients. Questions about bioavailability, systemic exposure and stability remain and all eyes are now directed towards the frontrunner PROTACs that are entering clinical testing.
In the meantime, as the potencies obtained in vitro and in animal models of various PROTAC constructs continue to improve there remains very significant interest in further defining the scope and limitations of the technology. Historically, target-degrading compounds have emerged from serendipity or target-specific campaigns in medicinal chemistry. The field soon evolved to employ a more rational approach based on bivalent ligands, actively targeting the proteome. These molecules can be considered inducers of protein-protein interaction, and improvements in our structural understanding of ternary complexes using x-ray crystallography will undoubtedly assist in the design of smaller molecules with better positioned linkers or even fused or merged dual binders that can be applied as “molecular glue” [138].
Another important area of research involves the identification of alternative E3 ligases and their corresponding ligase binding motifs, providing different degradation profiles, exploiting differences in ternary complex formation and subcellular localization. PROTAC technology seems to be particularly suited for the targeting of the undruggable proteome. Because ligands incorporated in PROTACs do not necessarily have to bind in the catalytic cavity of the proteins, label free biophysical tools will undoubtedly emerge within this area, allowing for the identification of both novel therapeutic targets and new ligases. Therefore, hits from HTS screens that were abandoned because they lacked functional activity or selectivity may gain renewed interest. Finally, it is important to further expand the arsenal of linkers with more differentiated physicochemical properties to optimize the bioavailability and cellular uptake of PROTAC constructs.
The field of induced protein degradation offers a lot of potential as a novel therapeutic paradigm, but it will take considerable effort and time for this field to become a recognized, validated complementary approach to the approaches that have dominated the pharmaceutical industry. If the promise of expanding druggable space in the vast proteome with PROTACS materializes, the impact on the treatment of human disease will be significant.
Conflicts of Interest
All authors are employees of Mercachem B.V.
Acknowledgements
We thank Bart DeCorte for critical reading of the manuscript.
References
- 1.Yin H., Kauffman K.J., Anderson D.G. Delivery technologies for genome editing. Nat Rev Drug Discov. 2017;16:387–399. doi: 10.1038/nrd.2016.280. [DOI] [PubMed] [Google Scholar]
- 2.Boettcher M., McManus M.T. Choosing the right Tool for the Job: RNAi, TALEN, or CRISPR. Mol Cell. 2015;58:575–585. doi: 10.1016/j.molcel.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lecker S.H., Goldberg A.L., Mitch W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J Am Soc Nephrol. 2006;17:1807–1819. doi: 10.1681/ASN.2006010083. [DOI] [PubMed] [Google Scholar]
- 5.Fisher S.L., Phillips A.J. Targeted protein degradation and the enzymology of degraders. Curr Opin Chem Biol. 2018;44:47–55. doi: 10.1016/j.cbpa.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Winter G.E., Buckley D.L., Paulk J., Roberts J.M., Souza A., Dhe-Paganon S. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bondeson D.P., Mares A., Smith I.E.D., Ko E., Campos S., Miah A.H. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gadd M.S., Testa A., Lucas X., Chan K.-H., Chen W., Lamont D.J. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gechijian L.N., Buckley D.L., Lawlor M.A., Reyes J.M., Paulk J., Ott C.J. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol. 2018;14:405–412. doi: 10.1038/s41589-018-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol. 2018;25:78–87. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An S., Fu L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553–562. doi: 10.1016/j.ebiom.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cromm P.M., Crews C.M. Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem Biol. 2017;24:1181–1190. doi: 10.1016/j.chembiol.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neklesa T.K., Winkler J.D., Crews C.M. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138–144. doi: 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- 14.Wang P., Zhou J. Proteolysis Targeting Chimera (PROTAC): a paradigm-shifting approach in small molecule drug discovery. Curr Top Med Chem. 2018;18:1354–1356. doi: 10.2174/1568026618666181010101922. [DOI] [PubMed] [Google Scholar]
- 15.Gu S., Cui D., Chen X., Xiong X., Zhao Y. PROTACs: an emerging targeting technique for protein degradation in drug discovery. Bioessays. 2018;40 doi: 10.1002/bies.201700247. [DOI] [PubMed] [Google Scholar]
- 16.Churcher I. Protac-induced protein degradation in drug discovery: breaking the rules or just making new ones? J Med Chem. 2018;61:444–452. doi: 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- 17.Kenten J.H., Roberts S.F. 2001. Controlling protein levels in eucayotic organisms. US 6,306,663 B1. [Google Scholar]
- 18.Rogers S., Wells R., Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 19.Glotzer M., Murray A.W., Kirschner M.W. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- 20.Belshaw P.J., Ho S.N., Crabtree G.R., Schreiber S.L. Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc Natl Acad Sci U S A. 1996;93:4604–4607. doi: 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spencer D.M., Wandless T.J., Schreiber S.L., Crabtree G.R. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto K.M., Kim K.B., Kumagai A., Mercurio F., Crews C.M., Deshaies R.J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneekloth J.S., Fonseca F.N., Koldobskiy M., Mandal A., Deshaies R., Sakamoto K. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 24.Bargagna-Mohan P., Baek S.-H., Lee H., Kim K., Mohan R. Use of PROTACS as molecular probes of angiogenesis. Bioorg Med Chem Lett. 2005;15:2724–2727. doi: 10.1016/j.bmcl.2005.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puppala D., Lee H., Kim K.B., Swanson H.I. Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: a potential tool for chemoprevention. Mol Pharmacol. 2008;73:1064–1071. doi: 10.1124/mol.107.040840. [DOI] [PubMed] [Google Scholar]
- 26.Schneekloth A.R., Pucheault M., Tae H.S., Crews C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh Y., Kitaguchi R., Ishikawa M., Naito M., Hashimoto Y. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg Med Chem. 2011;19:6768–6778. doi: 10.1016/j.bmc.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 28.Itoh Y., Ishikawa M., Naito M., Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 29.Okuhira K., Ohoka N., Sai K., Nishimaki-Mogami T., Itoh Y., Ishikawa M. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett. 2011;585:1147–1152. doi: 10.1016/j.febslet.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X., Crowley V.M., Wucherpfennig T.G., Dix M.M., Cravatt B.F. 2018. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Unpubl Results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward C.C., Kleinman J.I., Chung C.Y.S., Kim K., Petri Y., Lee P.S. 2018. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. Unpubl Results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spradlin J.N., Hu X., Ward C.C., Brittain S.M., Ou L., Bussiere D.E. 2018. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Unpubl Results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A. The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harling J.D., Smith I.E.D. 2016. IAP E3 Ligase Directed Proteolysis Targeting Chimeric Molecules. WO2016169989 (A1) [Google Scholar]
- 35.Galdeano C., Gadd M.S., Soares P., Scaffidi S., Van Molle I., Birced I. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J Med Chem. 2014;57:8657–8663. doi: 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salami J., Crews C.M. Waste disposal-An attractive strategy for cancer therapy. Science. 2017;355:1163–1167. doi: 10.1126/science.aam7340. [DOI] [PubMed] [Google Scholar]
- 37.Buhimschi A.D., Armstrong H.A., Toure M., Jaime-Figueroa S., Chen T.L., Lehman A.M. Targeting the C481S ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation. Biochemistry. 2018;57:3564–3575. doi: 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- 38.Zengerle M., Chan K.-H., Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang K., Song Y., Xie H., Wu H., Wu Y.-T., Leisten E.D. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg Med Chem Lett. 2018;28:2493–2497. doi: 10.1016/j.bmcl.2018.05.057. [DOI] [PubMed] [Google Scholar]
- 40.Yunus D., Muppala H., Hamer F., Clarke F. Three consecutive false negative pregnancy tests in a twin pregnancy: a case report. Int J Gynecol Obstet. 2006;6 [Google Scholar]
- 41.Zorba A., Nguyen C., Xu Y., Starr J., Borzilleri K., Smith J. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci. 2018;115:E7285–E7292. doi: 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ollivier J.F., Shahrezaei V., Swain P.S. Scalable rule-based modelling of allosteric proteins and biochemical networks. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roy R.D., Rosenmund C., Stefan M.I. Cooperative binding mitigates the high-dose hook effect. BMC Syst Biol. 2017;11 doi: 10.1186/s12918-017-0447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang C.H., Lee D.H., Lee C.O., Du Ha J., Park C.H., Hwang J.Y. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC) Biochem Biophys Res Commun. 2018;505:542–547. doi: 10.1016/j.bbrc.2018.09.169. [DOI] [PubMed] [Google Scholar]
- 45.Crew AP, Hornberger KR, Dong H, Wang J, Qian Y, Crews CM. Modulators of estrogen receptor proteolysis and associated methods of use. [US20180237418A1], 2018.
- 46.Crew AP, Dong H, Wang J, Ferraro C, Chen X, Qian Y. Compounds and methods for the targeted degradation of androgen receptor. US20170327469A1, 2017.
- 47.Wurz R.P., Dellamaggiore K., Dou H., Javier N., Lo M.-C., McCarter J.D. A “Click Chemistry Platform” for the rapid synthesis of bispecific molecules for inducing protein degradation. J Med Chem. 2018;61:453–461. doi: 10.1021/acs.jmedchem.6b01781. [DOI] [PubMed] [Google Scholar]
- 48.Lai A.C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew Chem Int Ed. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bulatov E., Ciulli A. Targeting Cullin–RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem J. 2015;467:365–386. doi: 10.1042/BJ20141450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jhaveri K., Taldone T., Modi S., Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banaszynski L.A., Sellmyer M.A., Contag C.H., Wandless T.J., Thorne S.H. Chemical control of protein stability and function in living mice. Nat Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buckley D.L., Crews C.M. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew Chem Int Ed Engl. 2014;53:2312–2330. doi: 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toure M., Crews C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew Chem Int Ed Engl. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 54.Collins I., Wang H., Caldwell J.J., Chopra R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem J. 2017;474:1127–1147. doi: 10.1042/BCJ20160762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-Gonzalez A., Cyrus K., Salcius M., Kim K., Crews C., Deshaies R. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cyrus K., Wehenkel M., Choi E.-Y., Swanson H., Kim K.-B. Two-headed PROTAC: an effective new tool for targeted protein degradation. Chembiochem Eur J Chem Biol. 2010;11:1531–1534. doi: 10.1002/cbic.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hines J., Gough J.D., Corson T.W., Crews C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc Natl Acad Sci U S A. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X., Feng S., Fan J., Li X., Wen Q., Luo N. New strategy for renal fibrosis: targeting Smad3 proteins for ubiquitination and degradation. Biochem Pharmacol. 2016;116:200–209. doi: 10.1016/j.bcp.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 59.Fontaine F., Overman J., François M. Pharmacological manipulation of transcription factor protein-protein interactions: opportunities and obstacles. Cell Regen Lond Engl. 2015:4:2. doi: 10.1186/s13619-015-0015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hagenbuchner J., Ausserlechner M.J. Targeting transcription factors by small compounds--current strategies and future implications. Biochem Pharmacol. 2016;107:1–13. doi: 10.1016/j.bcp.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 61.Roche J., Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur J Med Chem. 2016;121:451–483. doi: 10.1016/j.ejmech.2016.05.047. [DOI] [PubMed] [Google Scholar]
- 62.Duan Y., Guan Y., Qin W., Zhai X., Yu B., Liu H. Targeting Brd4 for cancer therapy: inhibitors and degraders. Med Chem Commun. 2018;9:1779–1802. doi: 10.1039/c8md00198g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pervaiz M., Mishra P., Günther S. Bromodomain drug discovery - the past, the present, and the future. Chem Rec N Y N. 2018;18:1808–1817. doi: 10.1002/tcr.201800074. [DOI] [PubMed] [Google Scholar]
- 64.Hohmann A.F., Martin L.J., Minder J.L., Roe J.-S., Shi J., Steurer S. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat Chem Biol. 2016;12:672–679. doi: 10.1038/nchembio.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark P.G.K., Vieira L.C.C., Tallant C., Fedorov O., Singleton D.C., Rogers C.M. LP99: Discovery and Synthesis of the first Selective BRD7/9 Bromodomain Inhibitor. Angew Chem Int Ed Engl. 2015;54:6217–6221. doi: 10.1002/anie.201501394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin L.J., Koegl M., Bader G., Cockcroft X.-L., Fedorov O., Fiegen D. Structure-based design of an in vivo active selective BRD9 inhibitor. J Med Chem. 2016;59:4462–4475. doi: 10.1021/acs.jmedchem.5b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Remillard D., Buckley D.L., Paulk J., Brien G.L., Sonnett M., Seo H.-S. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew Chem Int Ed. 2017;56:5738–5743. doi: 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.North B.J., Marshall B.L., Borra M.T., Denu J.M., Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+−dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 69.Donmez G., Outeiro T.F. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med. 2013;5:344–352. doi: 10.1002/emmm.201302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim H.-S., Vassilopoulos A., Wang R.-H., Lahusen T., Xiao Z., Xu X. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487–499. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schiedel M., Herp D., Hammelmann S., Swyter S., Lehotzky A., Robaa D. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals) J Med Chem. 2018;61:482–491. doi: 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- 72.Khetchoumian K., Teletin M., Tisserand J., Mark M., Herquel B., Ignat M. Loss of Trim24 (Tif1alpha) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat Genet. 2007;39:1500–1506. doi: 10.1038/ng.2007.15. [DOI] [PubMed] [Google Scholar]
- 73.Le Douarin B., Nielsen A.L., Garnier J.M., Ichinose H., Jeanmougin F., Losson R. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 1996;15:6701–6715. [PMC free article] [PubMed] [Google Scholar]
- 74.Zhan Y., Kost-Alimova M., Shi X., Leo E., Bardenhagen J.P., Shepard H.E. Development of novel cellular histone-binding and chromatin-displacement assays for bromodomain drug discovery. Epigenetics Chromatin. 2015;8 doi: 10.1186/s13072-015-0026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palmer W.S., Poncet-Montange G., Liu G., Petrocchi A., Reyna N., Subramanian G. Structure-guided design of IACS-9571, a selective high-affinity dual TRIM24-BRPF1 bromodomain inhibitor. J Med Chem. 2016;59:1440–1454. doi: 10.1021/acs.jmedchem.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Falkenberg K.J., Johnstone R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 77.Bazzaro M., Lin Z., Santillan A., Lee M.K., Wang M.-C., Chan K.C. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14:7340–7347. doi: 10.1158/1078-0432.CCR-08-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kanno K., Kanno S., Nitta H., Uesugi N., Sugai T., Masuda T. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncol Rep. 2012;28:867–873. doi: 10.3892/or.2012.1898. [DOI] [PubMed] [Google Scholar]
- 79.Bassi Z.I., Fillmore M.C., Miah A.H., Chapman T.D., Maller C., Roberts E.J. Modulating PCAF/GCN5 immune cell function through a PROTAC approach. ACS Chem Biol. 2018;13:2862–2867. doi: 10.1021/acschembio.8b00705. [DOI] [PubMed] [Google Scholar]
- 80.McCoull W., Abrams R.D., Anderson E., Blades K., Barton P., Box M. Discovery of pyrazolo[1,5-a]pyrimidine B-cell lymphoma 6 (BCL6) binders and optimization to high affinity macrocyclic inhibitors. J Med Chem. 2017;60:4386–4402. doi: 10.1021/acs.jmedchem.7b00359. [DOI] [PubMed] [Google Scholar]
- 81.McCoull W., Cheung T., Anderson E., Barton P., Burgess J., Byth K. Development of a novel B-Cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem Biol. 2018 doi: 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- 82.Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raina K., Lu J., Qian Y., Altieri M., Gordon D., Rossi A.M.K. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lebraud H., Wright D.J., Johnson C.N., Heightman T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent Sci. 2016;2:927–934. doi: 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou B., Hu J., Xu F., Chen Z., Bai L., Fernandez-Salas E. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J Med Chem. 2018;61:462–481. doi: 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nowak R.P., DeAngelo S.L., Buckley D., He Z., Donovan K.A., An J. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol. 2018;14:706–714. doi: 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qin C., Hu Y., Zhou B., Fernandez-Salas E., Yang C.-Y., Liu L. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J Med Chem. 2018;61:6685–6704. doi: 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hauser A.S., Attwood M.M., Rask-Andersen M., Schiöth H.B., Gloriam D.E. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang J., Yang P.L., Gray N.S. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lovly C.M., Shaw A.T. Molecular Pathways: Resistance to Kinase Inhibitors and Implications for Therapeutic strategies. Clin Cancer Res Off J Am Assoc Cancer Res. 2014;20:2249–2256. doi: 10.1158/1078-0432.CCR-13-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang H.-T., Dobrovolsky D., Paulk J., Yang G., Weisberg E.L., Doctor Z.M. A Chemoproteomic Approach to Query the Degradable Kinome using a Multi-kinase Degrader. Cell Chem Biol. 2018;25:88–99. doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jones L.H. Small-Molecule Kinase Downregulators. Cell Chem Biol. 2018;25:30–35. doi: 10.1016/j.chembiol.2017.10.011. [DOI] [PubMed] [Google Scholar]
- 93.Hrdinka M., Schlicher L., Dai B., Pinkas D.M., Bufton J.C., Picaud S. Small molecule inhibitors reveal an indispensable scaffolding role of RIPK2 in NOD2 signaling. EMBO J. 2018;37 doi: 10.15252/embj.201899372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buscà R., Pouysségur J., Lenormand P. ERK1 and ERK2 map kinases: specific roles or functional redundancy? Front Cell Dev Biol. 2016;4 doi: 10.3389/fcell.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Robb C.M., Contreras J.I., Kour S., Taylor M.A., Abid M., Sonawane Y.A. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC) Chem Commun (Camb) 2017;53:7577–7580. doi: 10.1039/c7cc03879h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Olson C.M., Jiang B., Erb M.A., Liang Y., Doctor Z.M., Zhang Z. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14:163–170. doi: 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bian J., Ren J., Li Y., Wang J., Xu X., Feng Y. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem. 2018;81:373–381. doi: 10.1016/j.bioorg.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 98.Burslem G.M., Smith B.E., Lai A.C., Jaime-Figueroa S., McQuaid D.C., Bondeson D.P. The advantages of targeted protein degradation over inhibition: An RTK Case Study. Cell Chem Biol. 2018;25:67–77. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Crew A.P., Raina K., Dong H., Qian Y., Wang J., Vigil D. Identification and characterization of Von Hippel-Lindau-recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-binding kinase 1. J Med Chem. 2018;61:583–598. doi: 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- 100.Li W., Gao C., Zhao L., Yuan Z., Chen Y., Jiang Y. Phthalimide conjugations for the degradation of oncogenic PI3K. Eur J Med Chem. 2018;151:237–247. doi: 10.1016/j.ejmech.2018.03.066. [DOI] [PubMed] [Google Scholar]
- 101.Zhang C., Han X.-R., Yang X., Jiang B., Liu J., Xiong Y. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK) Eur J Med Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Powell C.E., Gao Y., Tan L., Donovan K.A., Nowak R.P., Loehr A. Chemically induced degradation of anaplastic lymphoma kinase (ALK) J Med Chem. 2018;61:4249–4255. doi: 10.1021/acs.jmedchem.7b01655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hatcher J.M., Wang E.S., Johannessen L., Kwiatkowski N., Sim T., Gray N.S. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med Chem Lett. 2018;9:540–545. doi: 10.1021/acsmedchemlett.8b00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sun Y., Zhao X., Ding N., Gao H., Wu Y., Yang Y. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018;28:779–781. doi: 10.1038/s41422-018-0055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pinna L.A. Protein kinase CK2: a challenge to canons. J Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 106.Pagano M.A., Bain J., Kazimierczuk Z., Sarno S., Ruzzene M., Di Maira G. The selectivity of inhibitors of protein kinase CK2: an update. Biochem J. 2008;415:353–365. doi: 10.1042/BJ20080309. [DOI] [PubMed] [Google Scholar]
- 107.Chen H., Chen F., Liu N., Wang X., Gou S. Chemically induced degradation of CK2 by proteolysis targeting chimeras based on a ubiquitin-proteasome pathway. Bioorg Chem. 2018;81:536–544. doi: 10.1016/j.bioorg.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 108.Galkin A.V., Melnick J.S., Kim S., Hood T.L., Li N., Li L. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci. 2007;104:270–275. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gronemeyer H., Gustafsson J.-Å., Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 110.Lin H.-K., Wang L., Hu Y.-C., Altuwaijri S., Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002;21:4037–4048. doi: 10.1093/emboj/cdf406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Logan I.R., McNeill H.V., Cook S., Lu X., Lunec J., Robson C.N. Analysis of the MDM2 antagonist nutlin-3 in human prostate cancer cells. Prostate. 2007;67:900–906. doi: 10.1002/pros.20568. [DOI] [PubMed] [Google Scholar]
- 112.Liu N., Zhou W., Guo Y., Wang J., Fu W., Sun H. Molecular dynamics simulations revealed the regulation of ligands to the interactions between androgen receptor and its coactivator. J Chem Inf Model. 2018;58:1652–1661. doi: 10.1021/acs.jcim.8b00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gustafson J.L., Neklesa T.K., Cox C.S., Roth A.G., Buckley D.L., Tae H.S. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew Chem Int Ed. 2015;54:9659–9662. doi: 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Salami J., Alabi S., Willard R.R., Vitale N.J., Wang J., Dong H. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol. 2018;1 doi: 10.1038/s42003-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Banaszynski L.A., Chen L.-C., Maynard-Smith L.A., Ooi A.G.L., Wandless T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nalepa G., Rolfe M., Harper J.W. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 117.Maniaci C., Hughes S.J., Testa A., Chen W., Lamont D.J., Rocha S. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun. 2017;8 doi: 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Steinebach C., Lindner S., Udeshi N.D., Mani D.C., Kehm H., Köpff S. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem Biol. 2018;13:2771–2782. doi: 10.1021/acschembio.8b00693. [DOI] [PubMed] [Google Scholar]
- 119.Munier-Lehmann H., Vidalain P.-O., Tangy F., Janin Y.L. On dihydroorotate dehydrogenases and their inhibitors and uses. J Med Chem. 2013;56:3148–3167. doi: 10.1021/jm301848w. [DOI] [PubMed] [Google Scholar]
- 120.Norman P. Evaluation of WO2013076170: the use of a dihydroorotate dehydrogenase inhibitor for the treatment of psoriasis. Expert Opin Ther Pat. 2013;23:1391–1394. doi: 10.1517/13543776.2013.831075. [DOI] [PubMed] [Google Scholar]
- 121.Madak J.T., Cuthbertson C.R., Chen W., Showalter H.D., Neamati N. Design, synthesis, and characterization of brequinar conjugates as probes to study DHODH inhibition. Chem A Eur J. 2017;23:13875–13878. doi: 10.1002/chem.201702999. [DOI] [PubMed] [Google Scholar]
- 122.Medina M. An overview on the clinical development of Tau-based therapeutics. Int J Mol Sci. 2018;19:1160. doi: 10.3390/ijms19041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Takeda S. Progression of Alzheimer's disease, tau propagation, and its modifiable risk factors. Neurosci Res. 2018 doi: 10.1016/j.neures.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 124.Lu M., Liu T., Jiao Q., Ji J., Tao M., Liu Y. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251–259. doi: 10.1016/j.ejmech.2018.01.063. [DOI] [PubMed] [Google Scholar]
- 125.Zhang G., Smith S.G., Zhou M.-M. Discovery of chemical inhibitors of human bromodomains. Chem Rev. 2015;115:11625–11668. doi: 10.1021/acs.chemrev.5b00205. [DOI] [PubMed] [Google Scholar]
- 126.Qian Y, Dong H, Wang J, Berlin M, Crew AP, Crews CM. Compounds and methods for the targeted degradation of bromodomain-containing proteins. US20170065719A1, 2017.
- 127.Zhang X., Lee H.C., Shirazi F., Baladandayuthapani V., Lin H., Kuiatse I. Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia. 2018;32:2224–2239. doi: 10.1038/s41375-018-0044-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shi J., Vakoc C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gallenkamp D., Gelato K.A., Haendler B., Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem. 2014;9:438–464. doi: 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- 130.Belkina A.C., Denis G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Noguchi-Yachide T. BET Bromodomain as a Target of Epigenetic Therapy. Chem Pharm Bull(Tokyo) 2016;64:540–547. doi: 10.1248/cpb.c16-00225. [DOI] [PubMed] [Google Scholar]
- 132.Raux B., Voitovich Y., Derviaux C., Lugari A., Rebuffet E., Milhas S. Exploring selective inhibition of the first Bromodomain of the Human Bromodomain and Extra-terminal Domain (BET) proteins. J Med Chem. 2016;59:1634–1641. doi: 10.1021/acs.jmedchem.5b01708. [DOI] [PubMed] [Google Scholar]
- 133.Andrieu G., Belkina A.C., Denis G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol. 2016;19:45–50. doi: 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chan K.-H., Zengerle M., Testa A., Ciulli A. Impact of target warhead and linkage vector on inducing protein degradation: comparison of bromodomain and extra-terminal (BET) degraders derived from triazolodiazepine (JQ1) and tetrahydroquinoline (I-BET726) BET inhibitor scaffolds. J Med Chem. 2018;61:504–513. doi: 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Riching K.M., Mahan S., Corona C.R., McDougall M., Vasta J.D., Robers M.B. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem Biol. 2018;13:2758–2770. doi: 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- 136.Sun B., Fiskus W., Qian Y., Rajapakshe K., Raina K., Coleman K.G. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 2018;32:343–352. doi: 10.1038/leu.2017.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Berthon C., Raffoux E., Thomas X., Vey N., Gomez-Roca C., Yee K. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–e195. doi: 10.1016/S2352-3026(15)00247-1. [DOI] [PubMed] [Google Scholar]
- 138.Hughes S.J., Ciulli A. Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays Biochem. 2017;61:505–516. doi: 10.1042/EBC20170041. [DOI] [PMC free article] [PubMed] [Google Scholar]