

Abstract
Inhibition of the proteasome leads to proteotoxic stress, which is characterized by the buildup of ubiquitinated proteins that cannot be degraded properly. The transcription factor Nrf1 (also called NFE2L1) counteracts proteotoxic stress by inducing transcription of proteasome subunit genes, resulting in the restoration of proteasome activity. Further understanding of the Nrf1 pathway is therefore of interest in both neurodegeneration, where proteasome activity could be enhanced, and cancer, where suppression of this pathway could potentiate the cell-killing effect mediated by proteasome inhibitor drugs. Here, to identify novel regulators of Nrf1, we performed an RNAi screen in an engineered cell line, reporting on Nrf1 transcriptional activity. In addition to validating known regulators, we discovered that the AAA+ ATPase RUVBL1 is necessary for Nrf1's transcriptional activity. Given that RUVBL1 is part of different multisubunit complexes that play key roles in transcription, we dissected this phenomenon further and found that the TIP60 chromatin-regulatory complex is essential for Nrf1-dependent transcription of proteasome genes. Consistent with these observations, Nrf1, RUVBL1, and TIP60 proteins were co-recruited to the promoter regions of proteasome genes after proteasome inhibitor treatments. More importantly, depletion of RUVBL1 or TIP60 in various cancer cells sensitized them to cell death induced by proteasome inhibition. Overall, our study provides a framework for manipulating the TIP60–Nrf1 axis to alter proteasome function in various human diseases, including cancer.
Keywords: proteasome, transcription coactivator, transcription regulation, cancer, proteostasis, NFE2L1, Nrf1, RUVBL1, TIP60
Introduction
Protein homeostasis or proteostasis refers to the sum total of highly regulated and often interconnected cellular processes that include synthesis, folding, and destruction of proteins (1). The ubiquitin–proteasome system plays a pivotal role in proteostasis by orchestrating protein degradation. A central component of the ubiquitin–proteasome system is the 26S proteasome, a multisubunit molecular machine with proteolytic activity that is directly responsible for degrading its protein substrates, which are often tagged with polyubiquitin signals. The 26S proteasome, in turn, is composed of a 20S core that serves as the main proteolytic chamber and is capped on one or both ends by the 19S regulatory particle (2). Cellular proteasome activity is regulated at multiple levels, including transcription, translation, assembly, and posttranslational modifications of subunits (3).
We and others have demonstrated previously that the transcription factor nuclear factor erythroid-derived 2–related factor 1 (Nrf1, also called NFE2L1) functions as a master regulator of proteasome subunit genes (4–7). Nrf1 belongs to the cap 'n' collar basic region leucine zipper family of transcription factors, which also includes p45 Nfe2, Nrf2, and Nrf3 (8, 9). Although Nrf2, the most studied of the cap 'n' collar basic region leucine zipper factors, responds to oxidative stress (10), Nrf1 takes center stage when cells experience proteotoxic stress (4, 6). In response to proteasome inhibition, the Nrf1 pathway is activated, leading to de novo synthesis of proteasome subunit genes, resulting in “bounce back” or recovery of proteasome activity (4). Apart from responding to proteotoxic stress, Nrf1 is also responsible for basal transcription of proteasome genes in a tissue-specific manner. Nrf1-deficient mouse neuronal cells display accumulation of ubiquitinated proteins and decreased proteasome activity concomitant with a reduction in proteasome gene expression (11). A similar phenomenon is also evident in hepatocytes from mice that have a liver-specific knockout of Nrf1 (12).
From a molecular perspective, Nrf1 is co-translationally inserted into the endoplasmic reticulum (ER)2 membrane in such a way that only a small portion of its N terminus protrudes into the cytosol, whereas the bulk of its polypeptide, including the transcriptional activation and DNA-binding domains, is embedded in the ER lumen. Thus, Nrf1 depends on the action of the ATPase p97 to be retrotranslocated to the cytosolic side either to be constitutively degraded in the absence of proteotoxic stress or activated and mobilized to the nucleus when cells are subjected to proteasome inhibition (13). Activation of Nrf1 also requires its deglycosylation, which is mediated by a p97-associated N-glycanase enzyme, NGLY1 (14), and a proteolytic cleavage step that serves to trim the N terminus of Nrf1, which harbors the transmembrane domain. The protease involved in this process was recently discovered to be DDI2, a member of the aspartic family of proteases (15, 16).
Proteasome inhibitors are currently being used in the clinic against multiple myeloma and mantle cell lymphoma (17). One way to make this therapy more effective would be to devise strategies to block the Nrf1 pathway, thus impairing the ability of the cancer cells to recover their proteasome activity in response to inhibition of the proteasome. In support of this notion, a recent study demonstrated that attenuation of Nrf1 activation via p97 inhibition increased the efficacy of proteasome inhibitor drugs in multiple myeloma models (18). On the flip side, in certain proteinopathies, especially neurodegenerative diseases where the neurons display ubiquitinated protein aggregates, indicative of proteasome dysfunction, there could be interest in enhancing the Nrf1 pathway to directly increase proteasomal capacity (3). Thus, further understanding the Nrf1–proteasome axis could help pave the way for devising novel therapeutic agents targeted at different human diseases. In this study, we identified a requirement for the TIP60 chromatin-modifying complex in enabling Nrf1-mediated proteasomal gene expression in response to inhibition of the proteasome. This expands our view of the inner workings of Nrf1-mediated transcription and could offer strategies to manipulate this pathway in human diseases.
Results
RUVBL1 is necessary for Nrf1-mediated transcriptional response during proteotoxic stress
To identify factors that regulate Nrf1 activity under conditions of proteasome inhibition, we constructed a cell-based screening system in WT NIH-3T3 mouse fibroblasts. This cell line was engineered to stably express firefly luciferase under the control of 8x antioxidant response element (ARE; the sequence to which Nrf1 is known to bind (8)) repeats coupled to a minimal promoter. In addition, as a control, these cells also expressed Renilla luciferase driven by the human phosphoglycerate kinase (hPGK) promoter (Fig. 1A). The resultant screening system is referred to as WT 8xARE-Luc, whereas a similar system in an NIH-3T3 Nrf1-deficient background (19) is referred to as Nrf1−/− 8xARE-Luc. We then confirmed the status of Nrf1 in these two cell lines. Treatment with the proteasome inhibitor carfilzomib (CFZ) resulted in the accumulation of Nrf1 p120 (precursor) and p110 (processed form; transcriptionally active) in the WT 8xARE-Luc but not in Nrf1−/− 8xARE-Luc cell line, as expected (Fig. 1B). More importantly, although the WT 8xARE-Luc cells showed a dose-dependent increase in normalized luciferase activity (ratio of firefly to Renilla luciferase values) in response to CFZ, the Nrf1−/− 8xARE-Luc cell line showed no such increase (Fig. 1C), implying that this effect is Nrf1-dependent. In light of our previous study, which demonstrated a strict reliance of Nrf1 function on p97 ATPase activity (13), we used NMS-873, an inhibitor of p97 (20), to further test our screening system. NMS-873 was able to effectively attenuate CFZ-induced increased luciferase activity in the WT 8xARE-Luc cell line (Fig. 1D), additionally validating our screening system.
Figure 1.

Identification of RUVBL1 as a factor required for Nrf1 transcriptional activity. A, schematic of the reporter construct used in the cell-based screening system. This lentiviral reporter construct expressed firefly luciferase under the control of 8xARE upstream of a minimal promoter (Pmin), along with Renilla luciferase driven by the hPGK promoter. B, NIH-3T3 cells with stable incorporation of the reporter system described above and that were either wild-type (WT 8xARE-Luc) or Nrf1-deficient (Nrf1−/− 8xARE-Luc) were treated with DMSO or 200 nm CFZ overnight and analyzed by immunoblotting using antibodies specific for Nrf1, ubiquitin, and β-actin. The experiments were performed three independent times, and a representative blot is shown. C, WT 8xARE-Luc and Nrf1−/− 8xARE-Luc cells were treated for 16 h with increasing concentrations of CFZ (0, 20, 50, 100, 150, and 200 nm) and then subjected to Dual-Luciferase assays to measure the firefly and Renilla luciferase activity values. Normalized luciferase activity is shown. Error bars denote S.D. (n = 3). D, WT 8xARE-Luc cells were treated with 200 nm CFZ alone or in combination with 10 μm NMS-873 and compared with the DMSO-treated control for 16 h. The cell lysates were then used for luciferase assays. Normalized luciferase activity is shown. Error bars denote S.D. (n = 3). E, WT 8xARE-Luc cells were transfected with a focused library of siRNAs targeting several epigenetic factors and other candidate genes. Forty-eight hours after transfection, the cells were further treated with 200 nm CFZ overnight and assayed for luciferase activity. Error bars denote S.D. (n = 3). F, WT NIH-3T3 cells were either control (Ctrl)-transfected or transfected with siRNAs targeting RUVBL1 and further treated with 200 nm CFZ, as indicated, for 8 h. RNA extracted from these cells was then subjected to quantitative RT-PCR with primers specific for representative proteasome subunit genes as shown. The transcript levels of 18S rRNA were used for normalization. Error bars denote S.D. (n = 3). G, NIH-3T3 cells treated as described in F were used for immunoblotting with antibodies against Nrf1, RUVBL1, RUVBL2, ubiquitin, and β-actin as indicated. The experiments were performed three independent times, and a representative blot is shown.
Next, we used our WT 8xARE-Luc cell system in an RNAi screen where we used a focused library of siRNAs that target various epigenetic regulators and possible Nrf1 pathway influencers gleaned from public databases and the literature (8, 9, 21, 22). As positive controls, we used Nrf1-specific siRNAs and also siRNAs targeting known Nrf1 regulators such as p97 and the protease DDI2. In the screen, we found that depletion of any of the controls (Nrf1, p97, or DDI2) strongly attenuated the CFZ-induced increase in luciferase activity (Fig. 1E). Of the other test genes in the library, we observed that knockdown of RUVBL1 elicited an effect that was quite similar to the ones produced by the three positive control siRNAs (Fig. 1E). The results from the entire screen are listed in Table S1.
RUVBL1 (also called Pontin/TIP49) and the related protein RUVBL2 (also called Reptin/TIP48) are AAA+ ATPases (ATPases associated with diverse cellular activities) that most often occur together and participate in various cellular functions, including transcriptional regulation (23). Given that we identified RUVBL1 as a “hit” in our screen, it most likely is involved in Nrf1-mediated transcriptional activation of its target genes. To further confirm the involvement of RUVBL1 in the Nrf1 pathway, we examined the changes in proteasome gene transcription in response to CFZ treatment in control and siRUVBL1-treated NIH3T3 cells. Although the control cells displayed a functional Nrf1 pathway by up-regulating representative proteasome subunit genes as expected, cells with RUVBL1 depletion were profoundly defective in this response (Fig. 1F). Under these circumstances, we did not see a significant difference in Nrf1 protein levels or its ability to be processed into the p110 form after RUVBL1 knockdown (Fig. 1G). Taken together, these results reinforce a model where RUVBL1 could regulate the transcriptional activity of Nrf1, as seen for certain other transcription factors such as c-Myc, β-catenin, E2F1, NF-κB, and hypoxia-inducible factor 1α (HIF1α) (23). Interestingly, in the control immunoblots, to verify knockdown efficiency in this experiment, we noticed that, apart from RUVBL1, the protein levels of RUVBL2 were also attenuated in lysates derived from siRUVBL1-treated samples (Fig. 1G). This is consistent with previous observations where depletion of either factor results in a concomitant decrease in the other, in line with the notion that RUVBL1 and RUVBL2 together need to be incorporated into a hexameric complex to attain stability (24–26).
To rule out any cell type-specific bias, we tested other cell lines of various origin: HCT116 colon cancer, MDA-MB-231 breast cancer, and MIA-PaCa2 pancreatic cancer cells. Regardless of the cell type, we observed that, although proteasome inhibition up-regulated the transcript levels of the tested proteasome genes, depletion of RUVBL1 (Fig. S1) severely compromised this effect (Fig. 2A). Similar to our observation with NIH-3T3 cells, in all of these cancer cell lines, depletion of RUVBL1 resulted in decreases in the protein levels of both RUVBL1 and RUVBL2 (Fig. 2B).
Figure 2.

Depletion of RUVBL1 impairs the transcriptional function of Nrf1 in different cancer cell lines. A, the cell lines HCT116, MDA-MB-231, and MIA-PaCa2 were either control (Ctrl)-transfected or transfected with siRNAs targeting RUVBL1. Forty-eight hours after transfection, the cells were treated with 200 nm CFZ for 8 h and then analyzed by quantitative RT-PCR to measure representative proteasome subunit gene mRNA levels. The mRNA levels of 18S rRNA were used for normalization. Error bars denote S.D. (n = 3). B, the cell lines above were treated similarly as described in A and subjected to immunoblotting with antibodies specific for RUVBL1, RUVBL2, Nrf1, ubiquitin, and β-actin. The experiments were performed three independent times, and a representative blot is shown.
The TIP60 complex is required for Nrf1-dependent transcription
Although, in some instances, RUVBL1 and RUVBL2 act as a heterohexamer to regulate gene transcription (27), in most cases these proteins are part of multisubunit complexes with roles in transcription (23). The most prominent RUVBL1/2-containing complexes are INO80, SRCAP, TIP60, and R2TP (Fig. 3A). To test whether one or more of these complexes could be involved in mediating Nrf1-dependent transcription under proteotoxic stress, we proceeded to deplete a key component in each complex (the INO80 subunit in the INO80 complex, the SRCAP subunit in the SRCAP complex, the TIP60 subunit in the TIP60 complex, and the PIH1 subunit in the R2TP complex, all of which are indicated by arrows in Fig. 3A) in the WT 8xARE-Luc cell line. We observed that depletion of TIP60, but not any of the other subunits tested (Fig. S2), recapitulated the effect of RUVBL1 knockdown in blocking the proteasome inhibitor–mediated increase in luciferase activity (Fig. 3B). Likewise, when we measured transcriptional up-regulation of proteasome genes in response to proteasome inhibition, depletion of TIP60 attenuated this response, similar to what was observed with RUVBL1 depletion (Fig. 3C). Taken together, our results attribute a critical role of the TIP60 complex in Nrf1-dependent transcriptional response to proteotoxic stress. Interestingly, in our control immunoblots, we observed that depletion of RUVBL1 also decreased TIP60 and, to a certain extent, INO80 and PIH1 protein levels (Fig. 3D). This is consistent with an earlier study that indicated that the RUVBL1/2 complex is essential for assembly of a functional TIP60 complex (28).
Figure 3.

The TIP60 complex is required for Nrf1 transcriptional activity. A, some of the multisubunit complexes that contain RUVBL1. The subunits indicated by arrows are targets of siRNAs in the panels below. B, WT 8xARE-Luc cells were transfected with siRNAs targeting RUVBL1, TIP60, INO80, SRCAP, and PIH1, as indicated. Forty-eight hours after transfection, the cells were further treated with 200 nm CFZ overnight, and luciferase assays were performed. Normalized luciferase activity is shown. Error bars denote S.D. (n = 3). Ctrl, control. C, WT NIH-3T3 cells were transfected, further treated as described in B, and subjected to quantitative RT-PCR to measure the mRNA levels of select proteasome subunit genes. The mRNA levels of 18S rRNA were used for normalization. Error bars denote S.D. (n = 3). D, WT NIH-3T3 cells were treated as described in C, and the lysates were used for immunoblotting to measure the levels of different proteins as indicated. β-Actin was used as a loading control. The experiments were performed three independent times, and a representative blot is shown.
Interaction between Nrf1 and the TIP60 complex
Based on the known biology of TIP60 as a transcriptional co-activator (29–33), it is possible that this complex interacts with Nrf1 on the chromatin to aid in its transcription function. To verify this hypothesis, we compared the ARE-containing promoter regions of the Nrf1 target genes PSMA7, PSMB7, and PSMD12 in WT and Nrf1−/− NIH3T3 cells using ChIP assays. Under the condition of proteasome inhibition with CFZ, we could observe recruitment of Nrf1 in the promoter regions of proteasome genes in WT but not Nrf1−/− cells, as expected (Fig. 4A). Importantly, we observed a similar trend for RUVBL1 and TIP60 subunits, implying Nrf1-dependent recruitment of these factors to the proteasome gene promoters. Consistent with this notion, we were also able to detect interaction between Nrf1 and RUVBL1/2 using co-immunoprecipitation assays (Fig. 4B).
Figure 4.
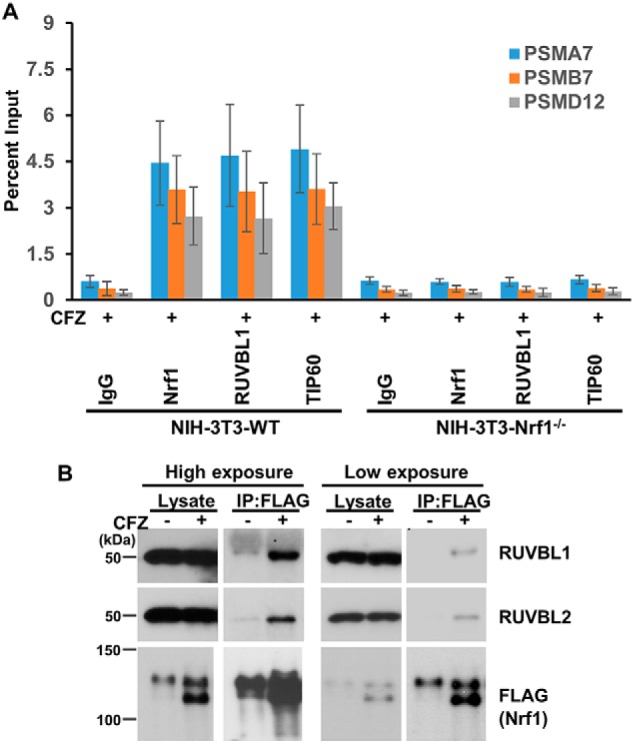
Nrf1 interacts with the TIP60 complex. A, WT and Nrf1−/− NIH-3T3 cell lines were treated with 200 nm CFZ for 8 h. The cells were then subjected to ChIP with IgG, Nrf1, RUVBL1, or TIP60 antibodies. These samples were then analyzed by quantitative PCR with primers specific for ARE-containing promoter regions of the proteasome genes PSMA7, PSMB7, and PSMD12. Error bars denote S.D. (n = 3). B, HEK293 cells stably expressing tagged Nrf1 (Nrf13xFLAG) were treated with 200 nm CFZ for 8 h or left untreated. The cell lysates were then subjected to immunoprecipitation with anti-FLAG beads and analyzed by immunoblotting with antibodies specific for FLAG, RUVBL1, and RUVBL2. The lysate lanes were loaded with 5% of the input that was used for immunoprecipitation. The experiments were performed three independent times, and a representative blot is shown.
Functional consequences of depletion of the TIP60 complex during proteotoxic stress
Our previous work conclusively demonstrated a cellular requirement for Nrf1 in recovering from inhibition of the proteasome (4). To verify whether this effect is recapitulated in cells deficient in RUVBL1 or TIP60, we performed proteasome recovery assays (Fig. 5A). Here we exploited the differences in the nature of binding of proteasome inhibitors to the active site of the proteasome. Although MG132 is a completely reversible proteasome inhibitor, CFZ binds to the proteasome active site in a covalent/irreversible manner. When cells are treated with MG132 briefly and then washed off, recovery of the proteasome activity is achieved by simple dissociation of the drug from the proteasome active site as well as via the action of the Nrf1 pathway during the time when the proteasome was inhibited. In the case of CFZ, given its covalent binding to the proteasome, the only way for the cells to recover after drug washout is by invoking the Nrf1 pathway to synthesize new proteasomes. Thus, impairing Nrf1 function in this context will severely undermine the ability of the cells to recover proteasome activity (4). To test these predictions in our study, we compared WT NIH-3T3 cells with siRUVBL1- or siTIP60-transfected cells for their relative ability to recover proteasome activity after pulse treatment with MG132 or CFZ at appropriate concentrations to achieve about 90% inhibition of proteasome activity in 1 h. Following drug washout, we found that cells depleted of RUVBL1 or TIP60 were able to recover their proteasome activity after MG132 pulse treatment but were partially impaired in their ability to do so when CFZ was employed (Fig. 5B). These results underscore the importance of RUVBL1 and TIP60 in mediating the recovery of proteasome activity after inhibition of the proteasome.
Figure 5.

Depletion of RUVBL1 or TIP60 impairs proteasome recovery and potentiates carfilzomib-mediated apoptosis. A, schematic of the proteasome recovery assay. B, NIH-3T3 cells were control-transfected or with siRNAs specific for RUVBL1 or TIP60. The cells were then treated for an hour with either 500 nm MG132 or 50 nm CFZ. The drugs were then washed out, and proteasome activity in the lysates was measured at the indicated time points. The results were normalized to the DMSO-treated control. Error bars denote S.D. (n = 3). C, the cell lines HCT116, MDA-MB-231, and MIA-PaCa2 were either control-transfected or transfected with siRNAs targeting RUVBL1 and TIP60 as indicated. Forty-eight hours after transfection, the cells were treated with increasing concentrations of CFZ for a further 24 h. The cells were then subjected to a viability assays. Error bars denote S.D. (n = 3). D, the different cancer cell lines were transfected as described in A and then further treated with 250 nm CFZ for 24 h. The cell lysates were then used for immunoblotting to measure the protein levels of cleaved caspase-3 along with the levels of RUVBL1, TIP60, ubiquitin, and β-actin as controls (Ctrl). The experiments were performed three independent times, and a representative blot is shown.
Irresolvable proteotoxic stress could lead to cell death, particularly in cancer cells, which are thought to be over-reliant on proteasome function (34, 35). Our previous studies have demonstrated potentiation of apoptosis in Nrf1-impaired cancer cells that were treated with proteasome inhibitors (4, 14). To evaluate the contribution of the TIP60 complex in this context in cancer cells, we first exposed control and siRUVBL1- or siTIP60-treated cells (HCT116, MDA-MB-231, and MIA-PaCa2) to different concentrations of CFZ. Although CFZ by itself caused a dose-dependent decrease in cell viability, this effect was exacerbated in siRUVBL1- and siTIP60-treated cells (Fig. 5C). Consistent with these results, immunoblotting experiments revealed that, compared with the control, the levels of cleaved caspase-3 (a marker for apoptosis) were markedly elevated in RUVBL1- or TIP60-depleted cells that were further treated with CFZ (Fig. 5D). Taken together, our results indicate that modulating the activity of the TIP60 complex could enable manipulation of the functional output from the Nrf1 pathway in cells experiencing proteotoxic stress.
Discussion
Nrf1 has emerged as a critical transcription factor in the cellular arsenal to combat proteotoxic stress or proteasome insufficiency. In response to inhibition of the proteasome, Nrf1 is mobilized to enable increased transcription of proteasome subunit genes, culminating in the formation of new proteasomes (4, 6, 13, 19). Several previous studies, including ours, have dissected the regulation of Nrf1 function that is achieved via multiple mechanisms in the cell: regulation of its abundance by the ubiquitin ligases HRD1, FBXW7, and β-TRCP; regulation of its extraction from the ER by p97; regulation of its proteolytic processing by DDI2; regulation of its deglycosylation by NGLY1; and regulation of its phosphorylation and, thereby, its transcriptional activity by casein kinase 2, to name a few (9, 14, 15). Our current study adds the TIP60 chromatin–modifying complex to the growing list of factors that impact Nrf1 activity. To our knowledge, this is the first example of an epigenetic regulator that appears to modulate Nrf1 function.
Although we have demonstrated that RUVBL1 and TIP60, both of which are subunits of the TIP60 complex, are necessary for Nrf1-mediated transcription after proteasome inhibition, the exact nature of their requirement remains to be uncovered. Based on the well-established role of RUVBL1/2 in chromatin remodeling, it could be that these factors serve to open up the chromatin at Nrf1 target gene promoters, as seen previously in the case of the E2F1 transcription factor (36). Also, TIP60, which harbors acetyl transferase activity, is known to acetylate histones H4 and H2A, thereby relaxing the chromatin in promoter regions and providing access to the transcriptional machinery (37). This is exemplified in the case of the hypoxia response, where HIF1α recruits TIP60, which enables acetylation of histones and subsequent recruitment and activation of RNA polymerase II at HIF1α target gene promoters (30). In our case, it could be that TIP60 provides such a function in Nrf1-dependent transcription. Given that TIP60 can also directly acetylate some transcription factors, such as c-myc and the androgen receptor, and modulate their function (38, 39), it could also be that Nrf1 is subject to such modification. It is important to note that all of these mechanisms may also occur together and are not necessarily mutually exclusive.
Mechanism aside, our findings could have important therapeutic implications, especially in cancer. RUVBL1 and RUVBL2 have been shown to be overexpressed in a number of cancer types, including colorectal, liver, breast, lung, gastric, esophageal, pancreatic, and kidney cancer and leukemia (23). More importantly, depletion of RUVBL1/2 seemed to delay or even shrink the tumors in preclinical models of some of the cancer types mentioned above. Accordingly, there is intense interest in developing small-molecule inhibitors of the ATPase activity of RUVBL1/2 (40, 41). These early-stage inhibitors already show promise in cell culture and xenograft studies in mice. Likewise, TIP60 has also been regarded as a viable target in cancer, and there have been efforts directed to identify inhibitors of its acetyl transferase activity (42). Given that in our study, depletion of RUVBL1 or TIP60 enhances cell killing by carfilzomib, this provides a framework for testing RUVBL1 or TIP60 inhibitors in combination with proteasome inhibitors in various cancer types. This combination would also make sense from the point of view of proteasome inhibitors, which, apart from their current use in the clinic against multiple myeloma and mantle cell lymphoma, are now being evaluated in combination with other chemotherapeutic agents in several types of solid tumors (43). Our findings, which demonstrates a strong reliance of the Nrf1-mediated proteasome recovery pathway on the functional TIP60 complex, prompt further consideration of combination trials with proteasome and TIP60 complex inhibitors.
Experimental procedures
Screening system
The lentiviral construct PLKO-luc-mcherry-puro-Renilla (Addgene plasmid 29783) was a gift from Dr. Carl Novina and has been described previously (44). This construct was digested with AgeI and EcoRI to generate PLKO-luc-mcherry-puro. The Renilla luciferase–neomycin cassette hRluc-neo was PCR-amplified from pF9A cytomegalovirus hRluc-neo Flexi (C9361, Promega) using primers containing XmaI and EcoRI. The XmaI/EcoRI-digested PCR product was ligated into PLKO-luc-mcherry-puro to generate PLKO-luc-mcherry-puro-Renilla-neo. This construct was further digested with SacII and BamHI to release the hPGK promoter driving the luc-mcherry-puro cassette and ligated with the 8xARE minimal promoter containing oligos harboring overhangs of SacII and BamHI to construct PLKO-8xARE-Pmin-luc-mcherry-puro-hPGK-Renilla-neo (referred to as 8xARE-Luc). Although this construct expressed firefly and Renilla luciferase products, as measured by luciferase assays, mcherry expression was undetectable by fluorescence microscopy. Using 8xARE-Luc together with the helper plasmids pCAGG-HIVgpco (gagpol), pCAG4-RTR2 (rev-tat), and pHDM.G (vesicular stomatitis virus glycoprotein G), lentiviral particles were produced and used to infect WT and Nrf1−/− NIH-3T3 cells. These cells were selected in Geneticin, and the resultant cell lines are referred to as WT 8xARE-Luc and Nrf1−/− 8xARE-Luc in the other sections of this paper.
Cell culture and siRNA transfections
All cell lines were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Atlanta Biologicals) and penicillin and streptomycin (Invitrogen) at 37 °C in a humidified incubator with 5% CO2. All siRNAs (sequences are shown in Tables S2 and S3) were purchased from Dharmacon and were transfected into the cell lines, typically at 40 nm concentration, using DharmaFECT-1 reagent (Dharmacon) according to the manufacturer's recommendations. Forty-eight hours after transfection, the cells were used for downstream experiments.
Quantitative RT-PCR
TRIzol reagent (Life Technologies) was added directly to the cell culture plates, and total RNA was isolated according to the manufacturer's instructions. Complementary DNA was then prepared from 1000 ng of total RNA using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was carried out with iTaq universal SYBR Green Supermix (Bio-Rad) using the C1000 Touch Thermal cycler (Bio-Rad). Data were analyzed using CFX Manager 3.1 (Bio-Rad). The levels of 18S rRNA were used for normalization. The primers used for the assays are listed in Table S4.
Immunoblot analysis
Cells were washed twice with ice-cold PBS, and pellets were collected under cold conditions. To obtain lysates, cell pellets were resuspended in radioimmune precipitation assay lysis buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 0.1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease and phosphatase inhibitor mixture (Thermo Fisher Pierce) and incubated on ice for 30 min, followed by high-speed centrifugation at 13,200 rpm for 30 min. Total protein was quantified by Bradford reagent. Typically, 30 μg of protein was used for SDS-PAGE, followed by electrotransfer onto polyvinylidene difluoride membranes. The membranes were then blocked for 1 h with 5% nonfat dry milk powder in TBS with Tween and then incubated with the appropriate primary antibodies overnight at 4 °C, followed by a secondary antibody at room temperature for an hour. The antibodies used were specific for Nrf1 (1:5000), RUVBL1 (1:2500), RUVBL2 (1:2500), ubiquitin (1:3000), and cleaved caspase-3 (1:3000) (all from Cell Signaling Technology); INO80 (1:500, a gift from Dr. Landry (45)); PIH1 (1:1000, Proteintech); and TIP60 (1:1500, Abcam) and β-actin (1:10,000, Sigma-Aldrich). The secondary antibodies used were rabbit IgG horseradish peroxidase and mouse IgG horseradish peroxidase (1:10,000, both from Bio-Rad).
Chromatin immunoprecipitation
Ten million cells were fixed with freshly prepared formaldehyde solution (1% final volume) and incubated at room temperature for 10 min, followed by addition of glycine to quench the formaldehyde. Plates were then washed twice with ice-cold PBS and collected in PBS supplemented with a protease inhibitor mixture. Pellets were collected after centrifugation at 800 × g at 4 °C. The EZ-Magna ChIP A/G (Millipore) kit was used for further steps according to the manufacturer's protocol. Briefly, cells were lysed in cell lysis buffer, and the nuclei were pelleted. These pellets were suspended in nuclear lysis buffer and used for shearing chromatin with Covaris M220. Shearing was done at 10% duty factor (df) for 16 min. Sheared chromatin was centrifuged at 10,000 × g for 10 min, and the supernatant was collected. 20 μl of protein A/G magnetic beads was used for preclearing the sheared chromatin for 1 h at 4 °C on a rotor. 50 μl of precleared chromatin was used for each immunoprecipitation with 5 μg of specific antibody overnight at 4 °C. Beads were then washed with low salt buffer followed by high salt buffer, LiCl wash buffer, and TE buffer (20 mm Tris (pH 8.0) and 5 mm EDTA). Elution was done with elution buffer at 65 °C for 4 h, followed by column purification (Qiagen) to obtain purified chromatin. Quantitative PCR was used to analyze the chromatin. Primers used for analysis are shown in Table S4.
Cell viability assays
Cells were typically grown in 96-well plates and treated with drugs as necessary. For quantifying cell viability, the Cell-Titer Glo kit (Promega) was used. This kit measures the level of ATP, which, in turn, is proportional to the number of viable cells. Qualitative measure of cell viability was assessed using immunoblots for cleaved caspase-3.
Proteasome activity recovery assays
Cells grown in 96-well plates were treated for an hour with 500 nm MG132 or 50 nm carfilzomib, doses that were determined to inhibit proteasome activity by 90%. The cells were then washed with PBS three times and allowed to recover in fresh medium. The cells were frozen in TE buffer at different time points. At the time of the assays, the cells were thawed and used for measuring proteasome activity as described previously (4). Briefly, the cell lysates obtained by freeze–thaw lysis were mixed with the fluorogenic substrate succinyl-Leu-Leu-Val-Tyr-amino-4-methylcoumarin (specific for measuring the chymotrypsin-like activity of the proteasome), and the resulting fluorescence was measured at 360/460 nm excitation/emission. The fluorescence values were then normalized by cell number, as determined using the Cell-Titer Glo kit (Promega), which measures cellular ATP levels.
Co-immunoprecipitation assays
Human embryonic kidney 293 cells stably expressing C-terminally tagged Nrf1 (Nrf13xFLAG) after appropriate treatments were pelleted and resuspended in IP buffer (50 mm Tris (pH 7.4), 0.5 m NaCl, 1 mm EDTA, 1% Triton X-100) supplemented with protease and phosphatase inhibitor mixture (Pierce) and incubated on ice for 30 min. These were then centrifuged at 13,000 rpm in a refrigerated tabletop centrifuge for 20 min. The lysate supernatants were then used for immunoprecipitation with anti-FLAG beads (Sigma-Aldrich) according to the manufacturer's recommendations. The beads (20 μl of the 50% slurry for each pulldown) were usually blocked with 5% BSA for 2 h before use. Typically, 500 μg of total protein (as determined by Bradford assay) was used for each immunoprecipitation that was allowed to proceed overnight. The beads were then washed three times with IP buffer, eluted in Laemmli buffer, and analyzed by SDS-PAGE, followed by immunoblotting.
Author contributions
J. R. V. and S. K. R. conceptualization; J. R. V. and S. K. R. formal analysis; J. R. V. and S. K. R. methodology; J. R. V. and S. K. R. writing-original draft; J. R. V. and S. K. R. writing-review and editing; S. K. R. supervision; S. K. R. funding acquisition.
Supplementary Material
Acknowledgments
We thank A. Feygin for technical assistance and Dr. Landry (Virginia Commonwealth University) for the anti-INO80 antibody.
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Tables S1–S4.
- ER
- endoplasmic reticulum
- ARE
- antioxidant response element
- hPGK
- human phosphoglycerate kinase
- Luc
- luciferase
- CFZ
- carfilzomib
- IP
- immunoprecipitation.
References
- 1. Lindquist S. L., and Kelly J. W. (2011) Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis. Cold Spring Harb. Perspect. Biol. 3, a004507 10.1101/cshperspect.a004507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rousseau A., and Bertolotti A. (2018) Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 19, 697–712 10.1038/s41580-018-0040-z [DOI] [PubMed] [Google Scholar]
- 3. Schmidt M., and Finley D. (2014) Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 1843, 13–25 10.1016/j.bbamcr.2013.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Radhakrishnan S. K., Lee C. S., Young P., Beskow A., Chan J. Y., and Deshaies R. J. (2010) Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 38, 17–28 10.1016/j.molcel.2010.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sekine H., Okazaki K., Kato K., Alam M. M., Shima H., Katsuoka F., Tsujita T., Suzuki N., Kobayashi A., Igarashi K., Yamamoto M., and Motohashi H. (2018) O-GlcNAcylation signal mediates proteasome inhibitor resistance in cancer cells by stabilizing NRF1. Mol. Cell. Biol. 38, e00252–18 10.1128/mcb.00252-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steffen J., Seeger M., Koch A., and Krüger E. (2010) Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 40, 147–158 10.1016/j.molcel.2010.09.012 [DOI] [PubMed] [Google Scholar]
- 7. Tsuchiya Y., Morita T., Kim M., Iemura S., Natsume T., Yamamoto M., and Kobayashi A. (2011) Dual regulation of the transcriptional activity of Nrf1 by β-TrCP- and Hrd1-dependent degradation mechanisms. Mol. Cell. Biol. 31, 4500–4512 10.1128/MCB.05663-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biswas M., and Chan J. Y. (2009) Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 244, 16–20 10.1016/j.taap.2009.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim H. M., Han J. W., and Chan J. Y. (2016) Nuclear factor erythroid-2 like 1 (NFE2L1): structure, function and regulation. Gene 584, 17–25 10.1016/j.gene.2016.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rojo de la Vega M., Chapman E., and Zhang D. D. (2018) NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43 10.1016/j.ccell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee C. S., Lee C., Hu T., Nguyen J. M., Zhang J., Martin M. V., Vawter M. P., Huang E. J., and Chan J. Y. (2011) Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 8408–8413 10.1073/pnas.1019209108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee C. S., Ho D. V., and Chan J. Y. (2013) Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J. 280, 3609–3620 10.1111/febs.12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Radhakrishnan S. K., den Besten W., and Deshaies R. J. (2014) p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. eLife 3, e01856 10.7554/eLife.01856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomlin F. M., Gerling-Driessen U. I. M., Liu Y.-C., Flynn R. A., Vangala J. R., Lentz C. S., Clauder-Muenster S., Jakob P., Mueller W. F., Ordoñez-Rueda D., Paulsen M., Matsui N., Foley D., Rafalko A., Suzuki T., et al. (2017) Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Central Science 3, 1143–1155 10.1021/acscentsci.7b00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koizumi S., Irie T., Hirayama S., Sakurai Y., Yashiroda H., Naguro I., Ichijo H., Hamazaki J., and Murata S. (2016) The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 5, e18357 10.7554/elife.18357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lehrbach N. J., and Ruvkun G. (2016) Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 5, e17721 10.1101/052936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orlowski R. Z., and Kuhn D. J. (2008) Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 14, 1649–1657 10.1158/1078-0432.CCR-07-2218 [DOI] [PubMed] [Google Scholar]
- 18. Le Moigne R., Aftab B. T., Djakovic S., Dhimolea E., Valle E., Murnane M., King E. M., Soriano F., Menon M. K., Wu Z. Y., Wong S. T., Lee G. J., Yao B., Wiita A. P., Lam C., et al. (2017) The p97 inhibitor CB-5083 is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol. Cancer Ther. 16, 2375–2386 10.1158/1535-7163.MCT-17-0233 [DOI] [PubMed] [Google Scholar]
- 19. Vangala J. R., Sotzny F., Krüger E., Deshaies R. J., and Radhakrishnan S. K. (2016) Nrf1 can be processed and activated in a proteasome-independent manner. Curr. Biol. 26, R834–R835 10.1016/j.cub.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Magnaghi P., D'Alessio R., Valsasina B., Avanzi N., Rizzi S., Asa D., Gasparri F., Cozzi L., Cucchi U., Orrenius C., Polucci P., Ballinari D., Perrera C., Leone A., Cervi G., et al. (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 9, 548–556 10.1038/nchembio.1313 [DOI] [PubMed] [Google Scholar]
- 21. Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N. K., O'Donnell L., Oster S., Theesfeld C., Sellam A., Stark C., Breitkreutz B. J., Dolinski K., and Tyers M. (2017) The BioGRID interaction database: 2017 update. Nucleic Acids Res. 45, D369–D379 10.1093/nar/gkw1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ru B., Sun J., Tong Y., Wong C. N., Chandra A., Tang A. T. S., Chow L. K. Y., Wun W. L., Levitskaya Z., and Zhang J. (2018) CR2Cancer: a database for chromatin regulators in human cancer. Nucleic Acids Res. 46, D918–D924 10.1093/nar/gkx877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mao Y. Q., and Houry W. A. (2017) The Role of Pontin and Reptin in cellular physiology and cancer etiology. Front. Mol. Biosci. 4, 58 10.3389/fmolb.2017.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venteicher A. S., Meng Z., Mason P. J., Veenstra T. D., and Artandi S. E. (2008) Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132, 945–957 10.1016/j.cell.2008.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Izumi N., Yamashita A., Iwamatsu A., Kurata R., Nakamura H., Saari B., Hirano H., Anderson P., and Ohno S. (2010) AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci. Signal. 3, ra27 10.1126/scisignal.2000468 [DOI] [PubMed] [Google Scholar]
- 26. Rajendra E., Garaycoechea J. I., Patel K. J., and Passmore L. A. (2014) Abundance of the Fanconi anaemia core complex is regulated by the RuvBL1 and RuvBL2 AAA+ ATPases. Nucleic Acids Res. 42, 13736–13748 10.1093/nar/gku1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gnatovskiy L., Mita P., and Levy D. E. (2013) The human RVB complex is required for efficient transcription of type I interferon-stimulated genes. Mol. Cell. Biol. 33, 3817–3825 10.1128/MCB.01562-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jha S., Gupta A., Dar A., and Dutta A. (2013) RVBs are required for assembling a functional TIP60 complex. Mol. Cell. Biol. 33, 1164–1174 10.1128/MCB.01567-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim J. H., Kim B., Cai L., Choi H. J., Ohgi K. A., Tran C., Chen C., Chung C. H., Huber O., Rose D. W., Sawyers C. L., Rosenfeld M. G., and Baek S. H. (2005) Transcriptional regulation of a metastasis suppressor gene by Tip60 and β-catenin complexes. Nature 434, 921–926 10.1038/nature03452 [DOI] [PubMed] [Google Scholar]
- 30. Perez-Perri J. I., Dengler V. L., Audetat K. A., Pandey A., Bonner E. A., Urh M., Mendez J., Daniels D. L., Wappner P., Galbraith M. D., and Espinosa J. M. (2016) The TIP60 complex is a conserved coactivator of HIF1A. Cell Rep. 16, 37–47 10.1016/j.celrep.2016.05.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rowe A., Weiske J., Kramer T. S., Huber O., and Jackson P. (2008) Phorbol ester enhances KAI1 transcription by recruiting Tip60/Pontin complexes. Neoplasia 10, 1421–1432 10.1593/neo.08850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taubert S., Gorrini C., Frank S. R., Parisi T., Fuchs M., Chan H. M., Livingston D. M., and Amati B. (2004) E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol. Cell. Biol. 24, 4546–4556 10.1128/MCB.24.10.4546-4556.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao L. J., Loewenstein P. M., and Green M. (2016) Ad E1A 243R oncoprotein promotes association of proto-oncogene product MYC with the NuA4/Tip60 complex via the E1A N-terminal repression domain. Virology 499, 178–184 10.1016/j.virol.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frankland-Searby S., and Bhaumik S. R. (2012) The 26S proteasome complex: an attractive target for cancer therapy. Biochim. Biophys. Acta 1825, 64–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Petrocca F., Altschuler G., Tan S. M., Mendillo M. L., Yan H., Jerry D. J., Kung A. L., Hide W., Ince T. A., and Lieberman J. (2013) A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 24, 182–196 10.1016/j.ccr.2013.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tarangelo A., Lo N., Teng R., Kim E., Le L., Watson D., Furth E. E., Raman P., Ehmer U., and Viatour P. (2015) Recruitment of Pontin/Reptin by E2f1 amplifies E2f transcriptional response during cancer progression. Nat. Commun. 6, 10028 10.1038/ncomms10028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sapountzi V., Logan I. R., and Robson C. N. (2006) Cellular functions of TIP60. Int. J. Biochem. Cell Biol. 38, 1496–1509 10.1016/j.biocel.2006.03.003 [DOI] [PubMed] [Google Scholar]
- 38. Gaughan L., Logan I. R., Cook S., Neal D. E., and Robson C. N. (2002) Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 277, 25904–25913 10.1074/jbc.M203423200 [DOI] [PubMed] [Google Scholar]
- 39. Patel J. H., Du Y., Ard P. G., Phillips C., Carella B., Chen C. J., Rakowski C., Chatterjee C., Lieberman P. M., Lane W. S., Blobel G. A., and McMahon S. B. (2004) The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 24, 10826–10834 10.1128/MCB.24.24.10826-10834.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elkaim J., Castroviejo M., Bennani D., Taouji S., Allain N., Laguerre M., Rosenbaum J., Dessolin J., and Lestienne P. (2012) First identification of small-molecule inhibitors of Pontin by combining virtual screening and enzymatic assay. Biochem. J. 443, 549–559 10.1042/BJ20111779 [DOI] [PubMed] [Google Scholar]
- 41. Elkaim J., Lamblin M., Laguerre M., Rosenbaum J., Lestienne P., Eloy L., Cresteil T., Felpin F. X., and Dessolin J. (2014) Design, synthesis and biological evaluation of Pontin ATPase inhibitors through a molecular docking approach. Bioorg. Med. Chem. Lett. 24, 2512–2516 10.1016/j.bmcl.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 42. Gao C., Bourke E., Scobie M., Famme M. A., Koolmeister T., Helleday T., Eriksson L. A., Lowndes N. F., and Brown J. A. (2014) Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci. Rep. 4, 5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roeten M. S. F., Cloos J., and Jansen G. (2018) Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 81, 227–243 10.1007/s00280-017-3489-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Levy C., Khaled M., Iliopoulos D., Janas M. M., Schubert S., Pinner S., Chen P. H., Li S., Fletcher A. L., Yokoyama S., Scott K. L., Garraway L. A., Song J. S., Granter S. R., Turley S. J., et al. (2010) Intronic miR-211 assumes the tumor suppressive function of its host gene in melanoma. Mol. Cell 40, 841–849 10.1016/j.molcel.2010.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qiu Z., Elsayed Z., Peterkin V., Alkatib S., Bennett D., and Landry J. W. (2016) Ino80 is essential for proximal-distal axis asymmetry in part by regulating Bmp4 expression. BMC Biol. 14, 18 10.1186/s12915-016-0238-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.