

Abstract
Lipid transport is an essential process with manifest importance to human health and disease. Phospholipid flippases (P4-ATPases) transport lipids across the membrane bilayer and are involved in signal transduction, cell division, and vesicular transport. Mutations in flippase genes cause or contribute to a host of diseases, such as cholestasis, neurological deficits, immunological dysfunction, and metabolic disorders. Genome-wide association studies have shown that ATP10A and ATP10D variants are associated with an increased risk of diabetes, obesity, myocardial infarction, and atherosclerosis. Moreover, ATP10D SNPs are associated with elevated levels of glucosylceramide (GlcCer) in plasma from diverse European populations. Although sphingolipids strongly contribute to metabolic disease, little is known about how GlcCer is transported across cell membranes. Here, we identify a conserved clade of P4-ATPases from Saccharomyces cerevisiae (Dnf1, Dnf2), Schizosaccharomyces pombe (Dnf2), and Homo sapiens (ATP10A, ATP10D) that transport GlcCer bearing an sn2 acyl-linked fluorescent tag. Further, we establish structural determinants necessary for recognition of this sphingolipid substrate. Using enzyme chimeras and site-directed mutagenesis, we observed that residues in transmembrane (TM) segments 1, 4, and 6 contribute to GlcCer selection, with a conserved glutamine in the center of TM4 playing an essential role. Our molecular observations help refine models for substrate translocation by P4-ATPases, clarify the relationship between these flippases and human disease, and have fundamental implications for membrane organization and sphingolipid homeostasis.
Keywords: sphingolipid, glycolipid, lipid transport, membrane bilayer, enzyme mechanism, cerebroside, flippase, glucosylceramide, membrane asymmetry, P4-ATPase, membrane biology, glycosphingolipids
Introduction
Phospholipid flippases in the P4-ATPase family establish and repair the phospholipid asymmetry of cellular membranes and facilitate import of phospholipid from extracellular sources (1). These enzymes are integral membrane proteins that harness energy from ATP catalysis to translocate lipids from the exofacial leaflet to the cytofacial leaflet of cellular membranes. There are 14 distinct P4-ATPases in humans with important differences in tissue-specific expression, subcellular localization, and substrate specificity (2). These differences in P4-ATPase localization and enzymology are important for coordinating physiological processes, and their impairment results in diverse pathologies (3). For example, the P4-ATPase ATP8B1 transports phosphatidylcholine at the apical membrane of canalicular hepatocytes (4), and mutations in the ATP8B1 gene lead to membrane damage and hepatic cholestasis (3, 5). Conversely, ATP8A2 translocates phosphatidylserine within the central nervous system, and mutations in ATP8A2 cause cerebellar ataxia mental retardation and disequilibrium syndrome in humans and motor neuron degeneration in mice (6–8). Thus, defining the substrate specificity of the P4-ATPases is crucial to understanding their role in disease.
Genome-wide association (GWA)3 studies have found that SNPs in the human P4-ATPase genes ATP10A and ATP10D are strongly linked with metabolic disease. One GWA study of nondiabetic African American patients identified two SNPs in ATP10A that associated with insulin resistance (9), whereas two GWA studies of European and Japanese populations have revealed strong associations between commonly found ATP10D SNPs and both myocardial infarction and atherosclerosis (10, 11). Importantly, analyses of plasma sphingolipids from over 4000 individuals revealed a specific elevation in glucosylceramide (GlcCer) levels associated with ATP10D SNPs (10).
Mouse models with Atp10a and Atp10d mutations also demonstrate a link to metabolic disease. Two mouse strains with Atp10a mutations (formerly known as Atp10c) are susceptible to diet-induced insulin resistance and obesity (12, 13). Additionally, the C57BL/6 mouse strain, which carries a premature stop codon in an Atp10d exon (14), is specifically prone to develop obesity, hyperglycemia, and hyperinsulinemia under a high-fat diet (15), and these metabolic phenotypes can be attenuated by the transgenic complementation of Atp10d (16). Lipidomic examinations of Atp10d-deficient mice demonstrated that GlcCer was elevated in mouse plasma, similar to the human patients, and that complementation with the Atp10d transgene also mitigated this increase (16).
No substrates have been reported for the enzyme ATP10D. P4-ATPases were presumed to selectively translocate glycerophospholipids and not sphingolipids; therefore, the molecular basis for the links between ATP10D and GlcCer was mysterious. In addition, the ATP10D-related P4-ATPases in Saccharomyces cerevisiae (Dnf1 and Dnf2) are part of a regulatory circuit that controls sphingolipid synthesis through an as-yet-unclear mechanism (17). Sphingolipids are divided into four classes: (i) sphingoid bases, (ii) ceramides, (iii) phosphosphingolipids, and (iv) glycosphingolipids (18). GlcCer is the central metabolite between two of these major classes, the ceramides and glycosphingolipids. GlcCer is one commonly found sphingolipid in the circulation, although its transport to and from target tissues is poorly understood. Like ceramide, GlcCer is carried through the body by lipoproteins (19), although its amphipathic chemistry suggests that it requires active transport for absorption and distribution across the membranes of target tissues. Identifying and characterizing GlcCer transporters will be essential for testing the systemic and tissue-specific roles of this lipid in metabolic disease.
In previous studies, we and others have examined the molecular determinants of P4-ATPase substrate specificity (20–22). The yeast P4-ATPases in particular have been indispensable for understanding how phospholipid headgroups and backbones are coordinated/selected during transport. In this study, we have identified a conserved clade of yeast and human P4-ATPases that transport GlcCer. Further, a central glutamine in TM4 is conserved in all identified GlcCer-flippases and is necessary for GlcCer but not glycerophospholipid transport. Identifying and characterizing these transporters significantly advances our understanding of systemic lipid delivery in mammals and provides the enzymological basis for targeted pharmaceutical development. We propose that these enzymes function as critical sphingolipid importers within fungi and humans and that GlcCer transport likely evolved to support cellular lipid homeostasis.
Results
Fungal P4-ATPases transport glycosphingolipids
Dnf1 and Dnf2 are two related P4-ATPases that localize to the plasma membrane of Saccharomyces cerevisiae and transport phosphatidylcholine (PC) and phosphatidylethanolamine (PE). To probe how P4-ATPases distinguish glycerophospholipids from sphingolipids, we previously mutagenized the budding yeast P4-ATPase Dnf1 and selected variants capable of transporting sphingomyelin (SM). Gain-of-function mutations were identified (Dnf1N200S,L1202P) that changed the substrate preference of Dnf1 from PC/PE to SM (23). While testing the specificity of these SM-permissive Dnf1 mutants, we surprisingly discovered an existing capacity for WT Dnf1 to transport GlcCer and galactosylceramide (GalCer) (Fig. S1). Therefore, we broadly examined the requirement for Dnf1 and Dnf2 in sphingolipid transport relative to known substrates using WT; dnf1Δ; dnf2Δ; and dnf1,2Δ knockout (KO) strains. As previously reported, (7-nitro-2–1,3-benzoxadiazol-4-yl)-PC (NBD-PC) and NBD-PE transport progressively decreased in each KO strain (Fig. 1A and Fig. S2D) (24). WT S. cerevisiae surprisingly transported substantially more NBD-GlcCer and NBD-GalCer than NBD-PC, which was thought to be the preferred substrate for Dnf1 and Dnf2. Dnf2 is the primary NBD-GlcCer transporter at the plasma membrane because dnf2Δ caused the greatest reduction in transport relative to WT cells. Lem3 forms a heterodimer complex with both Dnf1 and Dnf2 that is necessary for their exit from the endoplasmic reticulum, and deletions of this gene are known to impair Dnf1,2-dependent lipid transport (25, 26). We tested the role of lem3 in glycosphingolipid transport and found that it recapitulated the transport defects of the dnf1,2Δ strain but did not enhance this defect in a dnf1,2Δ lem3Δ strain (Fig. S2C).
Figure 1.

S. cerevisiae NBD-monosaccharide glycosphingolipid uptake requires plasma membrane P4-ATPases, Dnf1 and Dnf2. A, NBD-lipid uptake was measured in WT (BY4741) and P4-ATPase knockout strains and presented as raw, arbitrary fluorescent units (A.F.U.) (n ≥ 9) ± S.D. (error bars). B, upon Dnf1,2-dependent uptake, NBD-GlcCer localized to mitochondria (mt-RFP). C and D, kinetics assessments of NBD-PC, NBD-GlcCer, and NBD-GalCer uptake in dnf1,2Δ cells expressing pRS313-DNF1 (C) or pRS313-DNF2 (D), normalized to empty vector controls (n = 6) ± S.D. E, velocities of substrate transport from linear regression fits of data in C and D, ± S.E. F, NBD-lipid uptake was measured in S. cerevisiae WT, S. pombe WT, and S. pombe dnf2Δ strains. Asterisks in F indicate differences in S. pombe KO strains from S. pombe WT. A one-way ANOVA was performed to assess variance in A and F, and comparisons were made with Tukey's post hoc analysis. A two-way repeated measures ANOVA was used to assess variance in the kinetic data, and comparisons with NBD-PC were made with Tukey's post hoc analysis: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
After uptake, NBD-GlcCer accumulated in the mitochondria (Fig. 1B) and was stable for up to 2 h after administration (Fig. S3). NBD-GlcCer was not found to accumulate in the ER or endosomes regardless of P4-ATPase expression (Fig. S4). None of the P4-ATPase KO strains displayed a decrease in NBD-SM, NBD-ceramide (Cer), or NBD-lactosylceramide (LacCer) transport (Fig. 1A), suggesting that these lipids are not Dnf1 or Dnf2 substrates. These experiments demonstrate that the Dnf1 and Dnf2 flippases are capable of transporting monosaccharide but not disaccharide glycosphingolipids.
The magnitude of NBD-GlcCer uptake suggested that it may be the preferred substrate of Dnf1 and Dnf2. Therefore, we measured the kinetics of NBD-PC, NBD-GlcCer, and NBD-GalCer transport (Fig. 1, C–E). Plasmid-borne DNF1 and DNF2 constructs complemented lipid transport in a dnf1,2Δ strain (Fig. S2A) and were used to normalize the genetic background to empty vector controls. Dnf1 transported NBD-PC and NBD-GlcCer at equivalent rates and NBD-GalCer to a lesser extent (Fig. 1, C and E). Comparatively, Dnf2 transported NBD-GlcCer at twice the rate of NBD-PC and NBD-GalCer (Fig. 1, D and E). Competition assays demonstrated that GlcCer lacking the N-acyl chain and NBD was capable of acutely inhibiting NBD-GlcCer uptake by Dnf2 (Fig. S2E), implying that the enzyme is capable of recognizing unmodified glycosphingolipid.
We then examined a distantly related fungal species, Schizosaccharomyces pombe, to test the conservation of P4-ATPase–mediated GlcCer transport. Measurements of NBD-lipid transport in S. pombe revealed robust uptake of GlcCer and GalCer, surpassing that of WT S. cerevisiae (Fig. 1F and Fig. S2, B and D). Deleting the S. pombe DNF2 ortholog elicited a substantial reduction in glycosphingolipid transport (Fig. 1F and Fig. S2 (B and D)). Surprisingly, S. pombe Dnf2 did not transport NBD-PC or NBD-PE, indicating that GlcCer/GalCer transport is the conserved function of this fungal Dnf2.
Human P4-ATPases specifically transport glucosylceramide
Eight of the 14 human P4-ATPases localize to the plasma membrane (27–29) and were tested for the ability to transport sphingolipids across the plasma membrane of HeLa cells (Fig. 2 (A–C) and Fig. S5). No substrate had previously been reported for ATP10D, yet this flippase was capable of transporting NBD-GlcCer (Fig. 2, A, C, and D) with exquisite specificity; NBD-GalCer was not transported (Fig. 2A). The ATP10DE215Q mutant lacks ATPase activity, and its inability to transport NBD-GlcCer demonstrates that translocation requires ATP catalysis (Fig. 2B). ATP10A preferred NBD-PC as previously reported (Fig. 2C) (30, 31), but measuring the kinetics of GlcCer uptake relative to parental (−) and ATP10DE215Q controls revealed that ATP10A was also capable of transporting NBD-GlcCer, although at half the rate of ATP10D (Fig. 2, D and E).
Figure 2.

H. sapiens ATP10A and -10D translocate NBD-GlcCer at the plasma membrane. A–C, parental HeLa cells (−) and cells stably expressing human P4-ATPases and mutants were incubated with the indicated NBD-lipids at 15 °C for 15 min. After extraction with fatty acid–free BSA, the residual fluorescence intensity associated with the cells was determined by flow cytometry. Graphs display averages from 3–4 independent experiments ± S.D. (error bars). -Fold increase of NBD-lipid uptake compared with parental cells (−) is shown. A one-way ANOVA was performed to assess variance in A–C, and comparisons with parental cells (−) were made with Tukey's post hoc analysis. D, HeLa cells were incubated with NBD-GlcCer at 15 °C for the indicated times (x axis). E, velocities of GlcCer transport from linear regression fits of data in D (AU, arbitrary units), ± S.D. Graphs display averages from three independent experiments ± S.D. A two-way repeated measures ANOVA was used to assess variance in the kinetic data, and comparisons with parental cells (−) were made with Tukey's post hoc analysis: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
TM1 and TM4 participate in GlcCer transport
P4-ATPase substrate selection and transport are coordinated by the first six transmembrane (TM) domains, and chimeric mapping of these segments has been a productive strategy for dissecting P4-ATPase enzymology (20, 32, 33). We used a series of chimeras between Dnf1 and Drs2 (a phosphatidylserine flippase (34)) to broadly map the transmembrane segments required for NBD-GlcCer transport (20). Dnf1 chimeras bearing Drs2 TM segments 1–4 displayed a significant reduction in NBD-GlcCer transport and 2-fold changes to the ratio of GlcCer to PC (a measure of substrate preference) (Fig. S6). A focused examination of these regions revealed one motif in TM1 (GA) and two in TM4 (WVAV and YQS) that were particularly important for GlcCer preference (Fig. S7). Two additional regions in the lumenal loop connecting TM1 and -2 (LL1–2) impaired both PC and GlcCer transport equivalently and thus were considered general loss of function (Fig. S7A).
Dnf2 is the primary PM-localized GlcCer transporter in S. cerevisiae (Fig. 1, A and D), and its transport activity provides a greater dynamic range of responses. All three GlcCer motifs are conserved in the Dnf1 paralog, Dnf2 (Fig. S7D); therefore, we performed a more thorough examination of these motifs in Dnf2. P4-ATPase homology models predict that the GA motif is positioned near the membrane/exoplasmic interface of TM1 (Fig. 4E). A sequence Logo and alignment illustrate the variability of this motif and the surrounding area (Fig. 3, A and B). Substitutions at the glycine and alanine positions reduced the GlcCer/PC transport ratio but did not impact PC or PE transport (Fig. 3, C and D). Double mutants in this region had the most profound impact on GlcCer preference and transport (Fig. 3, E and F). We previously linked the QQ motif at this same position to phosphatidylserine selection in Drs2, a Golgi-resident P4-ATPase (33). Additionally, the exoplasmic region of TM1 was also involved in substrate coordination and transport in a human phosphatidylserine-translocating P4-ATPase, ATP8A2 (35), although the QQ motif was not directly examined. These studies collectively indicate that this GA/QQ motif in TM1 is an important component of GlcCer/PS recognition but does not contribute to PE/PC selection.
Figure 4.

A Pro−4 glutamine in TM4 of Dnf2 is required for GlcCer transport. A, a sequence logo was created from an alignment of TM4 of P4-ATPases from different organisms, with letter size representing residue frequency and color denoting chemical characteristics. Hydrophilic residues are shown in green and purple, acidic residues in red, and hydrophobic residues in black. B, a focused alignment comparing a region of TM4 from S. cerevisiae, H. sapiens, and S. pombe, highlighting the YQS motif that was previously altered in S. cerevisiae Dnf1 (Fig. S7). C and D, the three YQS positions were tested for their influence on GlcCer preference (C) and selection (D), revealing that the central glutamine was the strongest determinant of GlcCer transport. E, homology model of Dnf1 with TM1–6 shown as pink cylinders and the rest of the protein colored green. GA, YQS, and WVAV motifs are represented by spheres and colored by element. PM boundaries are indicated. Variance was assessed with one-way ANOVAs, and comparisons with WT were made with Tukey's post hoc analysis. Although the YQS positions in C are presented in separate panels, their statistical variance was tested together. n ≥ 9, ± S.D. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Figure 3.
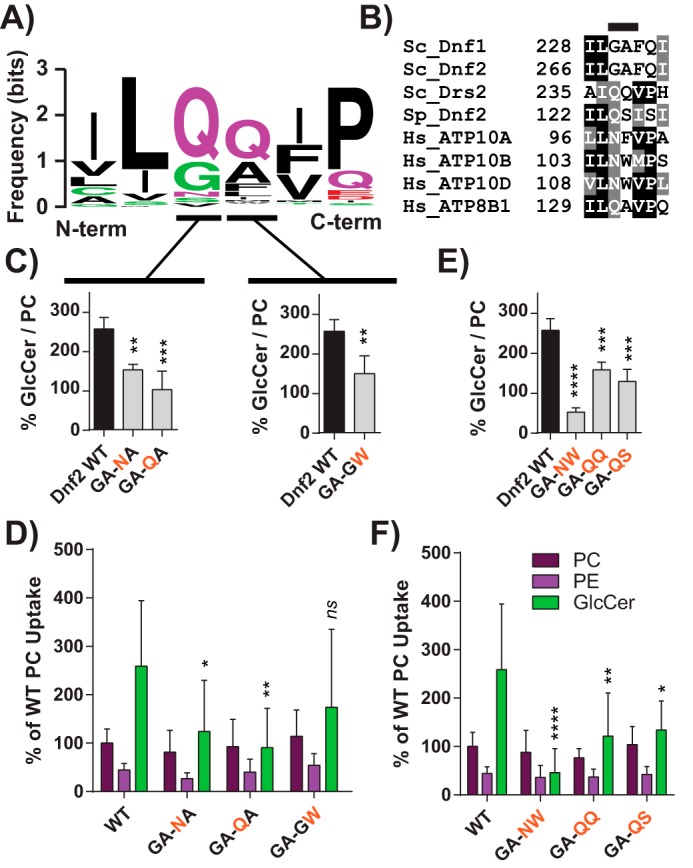
The exofacial TM1 “GA motif” facilitates Dnf2 selection and preference for GlcCer. A, a sequence logo was created from an alignment of TM1 of P4-ATPases from different organisms, with letter size representing residue frequency and color denoting chemical differences. Hydrophilic residues are shown in green and purple, acidic residues in red, and hydrophobic residues in black. B, a focused alignment comparing a region of TM1 from S. cerevisiae, H. sapiens, and S. pombe, highlighting the GA motif. C and D, dissecting the first and second positions of the GA motif reveals that substitutions in both positions can reduce GlcCer preference (C) but do not alter PC or PE recognition (D). E and F, double substitutions were created to examine S. pombe and H. sapiens sequences in the context of the S. cerevisiae Dnf2. These compound mutations reduced GlcCer preference (E) and selection (F) without altering the known glycerophospholipid substrates (F). Variance was assessed among data sets using one-way ANOVAs, and comparisons with WT were made with Tukey's post hoc analysis. Although first- and second-position GA analyses are presented in different panels (C), their statistical variance was tested together. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Error bars, S.D.
TM4 is predicted to be a long α-helix, broken in the center by a conserved proline found in all P-type ATPases; the WVAV and YQS motifs are exoplasmic relative to this central proline (Fig. 4E). Replacing WVAV in Dnf1 with LTFW from Drs2 reduced PC transport and eliminated GlcCer selection (Fig. S7B). However, single-position mutations in the WVAV motif of Dnf2 did not alter GlcCer preference or selection (Fig. S8, C and D), suggesting that the general integrity of this region may be more critical to lipid transport than individual amino acid side chains.
The YQS motif of TM4 is a functionally conserved island of polarity flanked by numerous hydrophobic residues (Fig. 4, A and B). Tyr654 mutations modestly influenced GlcCer selection, but changes to Ser656, including a Ser to Thr mutation, caused a more substantial and specific loss of GlcCer transport (Fig. 4, C and D). However, altering the central glutamine (Dnf2Q655A/N) elicited the strongest perturbation of GlcCer preference and transport (Fig. 4, C and D). The single hydrocarbon truncation of a glutamine to asparagine ablated GlcCer transport without substantially altering PC selection (Fig. 4, C and D). Gln655 is conserved in all five of the newly established GlcCer P4-ATPases (S. cerevisiae Dnf1 and Dnf2, S. pombe Dnf2, and Homo sapiens ATP10A and ATP10D; Figs. 1 and 2). These results demonstrate the importance of Gln655 in GlcCer recognition and suggest that it is a conserved determinant of GlcCer translocation. Dnf2Q655 precedes the conserved TM4 proline by four residues and thus will be called the Pro−4 position.
Conserved requirement for the Pro−4 glutamine and neighboring residues
Based on a Dnf1 homology model (23), we mutated residues predicted to surround the Pro−4 position in TM1, -2, -3, and -6 and tested their influence on GlcCer versus glycerophospholipid transport (Fig. 5, A–C). Two residues were mutated in TM1, one in TM2, two in TM3, and four in TM6 (Fig. 5, D and E). No change in GlcCer coordination was noted for the mutations in TM3 (Fig. 5, D and E). In contrast, all positions tested within TM1 and TM2 (mutations at Phe261, Leu264, and Leu285) decreased GlcCer preference (Fig. 5, D and E). Four residues were mutated in TM6, revealing three mutations that abrogated GlcCer preference. Ser1257 is predicted to be positioned in the lumenal loop between TM5 and TM6 (Fig. 5B), and Dnf2S1257Q reduced GlcCer and glycerophospholipid transport (Fig. 5E). Residues Thr1266 and Leu1270 may project directly toward Gln655 along a parallel segment of TM6, and mutating either position impaired GlcCer selection (Fig. 5, D and E). Thr1266 mutations in particular strongly affected the GlcCer transport without impairing PC transport (Fig. 5, D and E).
Figure 5.

Residues modeled in proximity to the Pro−4 glutamine influence GlcCer transport. A, homology model of Dnf1 (PDB code 3W5D) with TM1–6 shown as pink cylinders, the rest of the protein colored green, and the YQS motif represented by spheres and colored by element; PM boundaries are indicated. B, a 90° rotated and enhanced view of the peri-Gln655 region formed by TM2, -4, and -6. C, enhanced view of TM1, -2, -3, and -4, which surround Gln655. Residues were selected for mutagenesis by identifying positions that were predicated to be planar with the YQS motif and are shown in sticks and colored by element. D and E, GlcCer preference (D) and selection (E) were examined for all positions, revealing that TM1, TM2, and TM6 positions were capable of altering GlcCer transport. One position required chemical specificity to alter GlcCer transport (Leu264). Variance was assessed among data sets using one-way ANOVAs, and comparisons with WT were made with Tukey's post hoc analysis. n ≥ 9, ± S.D. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
The Thr1266 residue is not conserved in Dnf1 (Fig. S9, A and B); therefore, we generated a new homology model of Dnf2 to more closely evaluate this region of the enzyme. Leu1265 and Thr1266 are predicted to flank the Gln655 residue, potentially restricting the rotational flexibility of Gln655 (Fig. S9C). Dnf1 encodes two sequential methionines in the Dnf2 LT positions, and a Dnf2L1265M,T1266M construct recapitulated the Dnf1 preference for GlcCer (Fig. S9, F and G). Replacing the LT with the ATP8A2 sequence (IG; Dnf2L1265I,T1266G), an enzyme that is not predicted to transport GlcCer, reduced GlcCer transport without impacting PC coordination (Fig. S9, F and G). The lack of conservation in this region may suggest that these residues are not primary coordinators of GlcCer binding, but this position may help support a larger substrate-binding pocket or translocation pathway.
To test whether the mechanism of GlcCer recognition is conserved, we mutated the Pro−4 and Pro−3 positions in TM4 of ATP10A and ATP10D to assess their influence on PC and GlcCer transport (Fig. 6). The ATP10DQ381N,V382T mutant ablated GlcCer transport (Fig. 6B), similar to its impact in Dnf2. The necessity of the Pro−4 glutamine was confirmed by separating this TM4 motif into single mutants. Again, a simple change of a glutamine to asparagine impaired GlcCer transport (Fig. 6C); however, the single ATP10DV382T mutation elicited the unanticipated capacity to enhance GlcCer transport (Fig. 6C). Finally, the ATP10AQ370N,V371T mutant did not impact PC transport (Fig. 6D), thus reinforcing the specific and conserved role that the Pro−4 position plays in GlcCer but not PC translocation.
Figure 6.

Structure–function analysis of the H. sapiens ATP10D substrate pathway demonstrates primary structural conservation of TM4 Pro−4 position in GlcCer transport. A, sequence alignments of TM1, TM4, and TM6 of P4-ATPases are shown. Hydrophilic residues are indicated in green, red (negatively charged), and blue (positively charged), and hydrophobic residues are indicated in black. Three motifs, which were required for GlcCer preference in Dnf2, are underlined. The arrowheads indicate amino acids that were critical for ATP10D to transport GlcCer. B–H, NBD-lipid uptake was measured in HeLa cells stably expressing C-terminally HA-tagged ATP10A (WT), ATP10D (WT), each mutant (indicated), and parental cells (−); TM is numbered in parenthesis. B–D, the glutamine at TM4 Pro−4 was critical for ATP10D to transport GlcCer but was dispensable for ATP10A to transport PC. E and F, the motif in TM6 was critical for GlcCer transport of ATP10D but was dispensable for PC transport of ATP10A. The experiments of panels E were performed together with panels C, and thus graphs (−) and WT of E are equivalent to those of C. Graphs display averages from three independent experiments ± S.D. (error bars). -Fold increase of NBD-lipid uptake compared with parental cells (−) is shown. A one-way ANOVA was performed to assess variance in B–H, and comparisons with WT were made with Tukey's post hoc analysis. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. I, TM1, -2, -3, -4, -5, and -6 are indicated in red, orange, pink, magenta, purple, and blue, respectively, and others are indicated in gray. Critical residues for GlcCer transport are indicated by green sticks.
The TM1 and TM6 residues defined in yeast are not conserved in the mammalian enzymes, yet we tested the influence of these positions on ATP10D and ATP10A GlcCer and PC transport. ATP10DL1148,I1149 and ATP10AL1123,I1124 correspond with the TM6 LT motif of Dnf2, and an ATP10D homology model similarly predicted their proximity to the Pro−4 position (Fig. 6I). Both Leu1148 and Ile1149 in TM6 of ATP10D were critical for the transport of GlcCer (Fig. 6E), whereas the corresponding residues in ATP10A played a minor role in PC transport (Fig. 6F). ATP10DN110,W111 and ATP10AN98,F99 correspond to the GA motif in TM1 of Dnf2. ATP10DN110,W111 substitutions abolished GlcCer transport (Fig. 6G), whereas ATP10AN98,F99 mutations moderately affected PC transport (Fig. 6H).
Loss of lipid transport could be caused by enzyme misfolding, reduced expression, or improper trafficking. In addition, membrane proteins that misfold due to mutations are typically retained in the endoplasmic reticulum by the quality control machinery and degraded. Therefore, ATP10A and ATP10D expression and localization to the plasma membrane were determined by Western blotting, microscopy, and surface biotinylation (Fig. S5) All enzymes with mutations in TM1, TM4, and the ATPase domain were robustly expressed and localized to the plasma membrane (Fig. S5, A–C). Mutations within TM6 of ATP10D did show a slight reduction in overall expression (Fig. S5A), but plasma membrane localization was similar to WT (Fig. S5, A and B), suggesting general enzyme integrity and demonstrating proper trafficking. Moreover, the Dnf2 and ATP10A mutations described here abrogate GlcCer transport without substantially altering PC transport, indicating that the catalytic cycle is not perturbed by these mutations. Collectively, these experiments highlight conserved structural determinants of P4-ATPase–mediated GlcCer transport in TM1, TM4, and TM6. For TM4, a Pro−4 glutamine is critical for GlcCer transport, whereas positions in TM1 and TM6 are conserved even though the specific amino acids at these sites differ.
Discussion
Bioactive sphingolipids are linked to numerous cell and physiological processes, including cell death/survival, cell proliferation, senescence, autophagy, migration, differentiation, adhesion, and inflammatory responses (36). In addition, dysregulation of sphingolipid metabolism contributes to neurological disease, cancer, metabolic disorder, type 2 diabetes, hepatic steatosis, and cardiovascular disease (36–39). Whereas ceramides and sphingosines have received the most attention in this context, GlcCer is a central species in the metabolism of sphingolipids. GlcCer synthesis is a major consumer of ceramide in animal cells, and its breakdown can produce ceramide and sphingosine, yet GlcCer transport has been a particularly enigmatic topic. Although GlcCer is synthesized in the cytosolic leaflet of the Golgi, an unknown transporter carries it to the Golgi lumen, where it becomes the keystone substrate for a majority of more than 2000 different glycosphingolipids (40). Mechanisms for GlcCer transfer between serum lipoprotein particles (low-density lipoproteins and very low–density lipoproteins) and cells is also unclear, as is the distribution of GlcCer between leaflets of cellular membranes. Thus, defining the transport mechanisms of this key lipid species will be essential to understanding how sphingolipids contribute to pathogenesis and how they may be leveraged for the treatment of human disease.
We report here the identification and molecular characterization of a family of GlcCer transporters conserved from yeast to humans. Phylogenetic analyses of S. cerevisiae, S. pombe, and H. sapiens P4-ATPases suggests that S. cerevisiae Dnf1,2 and S. pombe Dnf2 cluster with the ATP10 family of human P4-ATPases (Fig. 7). Given the evolutionary distance between these three organisms, we propose that these five P4-ATPases represent a functional clade of glycosphingolipid flippases (Fig. 7). We establish that the yeast enzymes are capable of transporting both NBD-GlcCer and NBD-GalCer, whereas the human enzymes appear to be specific for NBD-GlcCer. The difference between these two substrates is simply the orientation of a distal hydroxyl, highlighting the exquisite specificity of these enzymes. We believe that the translocation of these NBD-lipids accurately reflects the transport of unlabeled lipids. Reconstituted lipid transport assays have shown that the NBD-lipids reflect endogenous substrates (41, 42), and our experiments here demonstrate that we are able to compete NBD-lipid transport with unlabeled lipids (Fig. S2). The linkage between human and mice ATP10D and increased plasma GlcCer levels also suggests GlcCer is a substrate of these enzymes. However, additional work is needed to quantitatively assess transport of unmodified GlcCer by these P4-ATPases.
Figure 7.
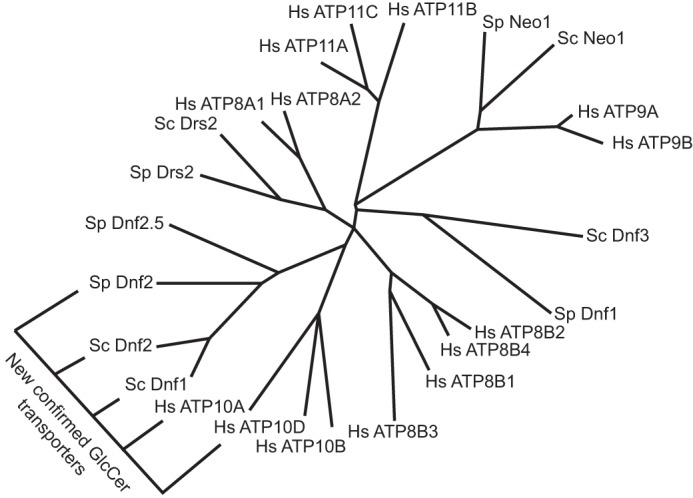
Yeast Dnf1,2 phylogenically clusters with the human ATP10D family members. An unrooted phylogenic tree of S. cerevisiae, S. pombe, and H. sapiens P4-ATPases with branch length indicating character change. A new clade of GlcCer P4-ATPases is indicated. Protein accession numbers and tools used for analysis are found under “Experimental procedures.”
We have shown that GlcCer transport by yeast and human P4-ATPases is strongly influenced by components of TM1, TM4, and TM6, at positions that are exoplasmic relative to the invariant TM4 proline. These residues do not affect PC transport (Fig. S10 and Tables S3 and S4), leading us to believe that we have not altered essential components required to appropriately fold, traffic, and complete the P4-ATPase catalytic cycle. This mutagenic dissection of yeast and human enzymes leads us to propose that GlcCer is coordinated by a cluster of residues that act cooperatively.
There are two prevailing hypotheses that describe the mechanism of P4-ATPase substrate transport: (i) a hydrophobic gate model and (ii) a two-gate model (1, 2, 43). The hydrophobic gate model proposes that substrate translocation is facilitated by the restriction and dilation of a cluster of hydrophobic residues centered around the middle of TM4 (21). The two-gate model suggests that “entry” and “exit” gates at the exoplasmic and cytoplasmic membrane interface will bind and transfer substrate along TM4 (33). These models are complementary as they each describe different aspects of the enzymatic process, and indeed, the sequential passage of a substrate through a hydrophobic barrier would facilitate the directionality of P4-ATPase translocation as it transitions from entry to exit gates.
The principal determinant of GlcCer transport is a conserved TM4 glutamine at the Pro−4 position. It was surprising that a Gln-to-Asn mutation was sufficient to ablate GlcCer transport in yeast and human enzymes. Glutamine and asparagine are chemically similar residues, only differing in the extension of their terminal amine (Fig. S11A). However, the difference in a hydrocarbon provides additional rotational flexibility to the glutamine side chain, relative to asparagine. Homology models of Dnf2 and ATP10D predict that the Pro−4 glutamine occupies a relatively open pocket within the TM domain of the enzyme (Fig. S11, B and C). The glutamine side chain was easily sculpted around the β-γ carbon bond within the Dnf2 and ATP10D models, and a 360° rotation around this bond was only restricted by Dnf2Y1262, Dnf2L1265, and Dnf2T1266 or ATP10DY1145, ATP10DL1148, and ATP10DI1149 (Fig. S11, B and C). Conversely, the rotation of the terminal amide of asparagine around the β-γ carbon bond was unimpeded (Fig. S11, D and E). Thus, the flexibility and rotational capacity of the Pro−4 amide may be an important component of GlcCer selection and translocation.
The Pro−4 position was previously proposed to modulate the hydration of a pocket that preceded the hydrophobic gate (21). Vestergaard et al. (21) defined a conserved isoleucine at the Pro+1 position as the primary component of a hydrophobic gate. The proximity of these two residues raises the possibility that the Pro−4 glutamine may anchor a prehydrophobic GlcCer-binding pocket, similar to that proposed for phosphatidylserine coordination in ATP8A2 (21). However, it is clear that not all P4-ATPase substrates require these residues for transport, and they possibly use an alternative substrate pathway. The separation of function mutations in yeast Dnf1 and Dnf2 demonstrates that the Pro−4 and its surrounding residues do not impact PC or PE transport, and mutagenesis data with human ATP10A and ATP10D support this separation of function. These results demonstrate that PC and GlcCer are coordinated differently and may be transported by independent pathways.
We previously proposed a substrate translocation pathway along a cleft bounded by TM1-3-4; this hypothesis was based on the position of residues we have previously mapped that are involved in PS selection by Drs2 and gain-of-function PS flipping variants of Dnf1 (20, 33, 44). The TM1-3-4 cleft includes the TM1 GA-QQ motif that is important for PS and GlcCer selection. In contrast, the Pro−4 Gln is modeled to project into a cleft bounded by TM2-4-6, and residues in TM6 also contribute to GlcCer selection. Moreover, prior mutagenesis work implicated this TM2-4-6 cleft in PS transport by ATP8A1. Thus, it is possible that different substrates can follow different translocation pathways along the protein–lipid interface. However, these respective models assume that the membrane domain is a relatively rigid body that would prevent penetration of the phospholipid between membrane segments. TM1 and -2 move substantially during the catalytic cycle of SERCA relative to the rest of the TM domain (45). It is possible that phospholipid substrate penetrates between TM1 and -2 and TM3, -4, and -6 as it flips from the extracellular leaflet to the cytosolic leaflet, allowing substrate to engage side chains from each lateral side of the membrane domain. The depth of this penetration could be substrate-dependent, and changing the chemical composition of residues in this region would facilitate the restriction or accessibility of different physically and/or chemically disparate substrates. Elucidating the precise mechanism of substrate translocation will be challenging, but work with other transporter families has yielded promising advances through the combination of crystallographic, computational, enzymatic, and spectroscopic approaches (46).
Fungi are heterotrophic eukaryotic organisms that will absorb materials from their environment to facilitate growth. Most yeast produce GlcCer, but both S. pombe and S. cerevisiae have independently lost the GlcCer synthase enzyme (47). Therefore, the GlcCer transport activity in these organisms may have evolved to scavenge this lipid from plant material that yeast grow upon (48, 49). This possibility would explain why Dnf1 and Dnf2 are intimately linked to sphingolipid homeostatic pathways (17), even though S. cerevisiae and S. pombe do not endogenously produce these glycolipids. The translocation of this substrate could be leveraged to change membrane properties, as GlcCer supplementation has been shown to increase alkaline resistance in yeast (50). Alternatively, imported GlcCer could be catabolized, and its backbone, N-linked acyl chain, and/or headgroup could be repurposed.
Membrane asymmetry is the difference in lipid composition between the two leaflets of the membrane and is a critical property of the plasma membrane. Simple alterations of this asymmetric structure can elicit substantial changes in biology, influencing micro- and macro-processes, such as vesicle budding (51–54), apoptosis (55–57), phagocytosis (58–60), and many others. The majority of research has thus far examined the cellular and physiologic impact of glycerophospholipid asymmetry, yet our discovery of two endogenous human GlcCer flippases raises important questions about the impact of sphingolipid asymmetry. For example, it is known that there are three human glucocerebrosidases, enzymes that cleave the glucose headgroup from the ceramide backbone. Glucocerebrosidase 1 is the best understood, and mutations in the GBA1 gene lead to Gaucher's disease and Parkinson's disease (61–63). Glucocerebrosidase 1 localizes to the lumen of the lysosome, yet glucocerebrosidase 2 and 3 localize to the cytosol (38). It is unclear where glucocerebrosidase 2 and 3 obtain their cytosolic GlcCer substrates and what role they perform in sphingolipid homeostasis. ATP10A and ATP10D are importers of GlcCer and therefore may be providing cytosolic substrates for metabolism or signaling. Thus, we hypothesize that the transport activity of ATP10A and ATP10D toward GlcCer may be an important component of sphingolipid homeostasis and lipid metabolism in mammals.
Experimental procedures
Reagents
All yeast culture reagents were purchased from Sigma-Aldrich, as well as NBD-hexanoic acid. All other lipids used in this study were purchased from Avanti Polar Lipids (Fig. S2). TLC materials and solvents used for lipid extraction were purchased from Fisher.
Strains and culture
S. pombe were cultured on YES medium, and S. cerevisiae were grown on synthetic defined (SD) medium unless examined in parallel with S. pombe. If examined in parallel with S. pombe, S. cerevisiae were grown and analyzed on YPD. All yeast and human cell lines used in the study are listed in Table S1, and plasmids are listed in Table S2.
Establishment of HeLa stable cell lines, antibodies, and immunofluorescence analysis
HeLa cells were cultured as described previously (29). HeLa cells expressing each C-terminally HA-tagged ATP8B4 and ATP10D (E215Q) were established as described previously (4, 30). Sources of antibodies used in the present study were as follows: monoclonal rabbit anti-ATP1A1 (EP1845Y), Abcam; monoclonal rat anti-HA (3F10), Roche Applied Science; Alexa Fluor–conjugated secondary antibodies, Molecular Probes; Cy3- and horseradish peroxidase-conjugated secondary antibodies, Jackson ImmunoResearch Laboratories. Immunostaining was performed as described previously (64) and visualized using an Axiovert 200MAT microscope (Carl Zeiss, Thornwood, NY)
Lipid administration and flow cytometry (yeast cells)
NBD-lipid administration and analysis were performed as described previously (23). Briefly, overnight yeast cultures were subcultured to 0.15 A600/ml and cultured to mid-log phase, and 0.5 ml of cells were collected per sample. The designated NBD-lipid was solubilized in 100% ethanol and added to the designated ice-cold growth medium at a final concentration of 2 μg/ml with final ethanol volumes ≤0.5%. Cells were suspended with the designated medium + NBD-lipids and incubated on ice for 30 min, unless otherwise stated (kinetics analyses and TLC examinations). After 30 min, the cells were washed twice with ice-cold SA (SD medium + 2% (w/v) sorbitol + 20 mm NaN3) supplemented with 4% (w/v) fatty acid–free BSA. All cells regardless of original medium growth were washed with ice-cold SD medium containing either dropout or complete amino acid supplements, respective to the strain/transformant requirements. Once washed with chilled 4% BSA-SA, cells were washed with ice-cold SA, resuspended in chilled SA containing 5 μm propidium iodide, and analyzed immediately. A gating example is provided (Fig. S12). The sequence of sample preparation and processing was varied in each experiment to mitigate potential positional experimental bias. Additional detail is provided in the supplemental Experimental procedures.
NBD-lipid administration and flow cytometry (human cells)
The incorporation of NBD-lipids was analyzed by flow cytometry as described (4) with some modifications. In brief, HeLa cells (in a 24-well plate) were washed and equilibrated at 15 °C for 15 min in 500 μl of Hanks' balanced salt solution (pH 7.4) containing 1 g/liter glucose (HBSS-glucose). The buffer was replaced with 1 μm NBD-lipid in HBSS-glucose, and cells were further incubated at 15 °C. After incubation for the indicated times, the buffer was replaced with ice-cold PBS(−) containing 2.5% (w/v) fatty acid–free BSA (Wako), 5 mm EDTA, and 0.5 μg/ml propidium iodide (Nacalai Tesque), and cells were incubated on ice for 30 min. The detached cells (more than 104 cells/sample) were analyzed with a FACSCalibur (BD Biosciences) to measure the fluorescence of NBD-lipids incorporated and translocated into the cytoplasmic leaflet of the plasma membrane. Graphs for NBD-lipid flippase activities are expressed as the averages of three independent experiments ± S.D. settings.
Protein homology modeling
All structural images were generated using PyMOL. The Dnf1 homology model was previously published (23). Dnf2 and ATP10D homology models used for sculpting were generated using the intensive Phyre2 modeling (65). The Phyre2 intensive modeling process represents an unbiased approach. First, homologous templates are surveyed, and crude backbone models are generated and ranked by coverage and confidence; then loop fragments are modeled in 2–15–residue chunks. A protein folding simulator uses a heuristic process to synthesize multiple templates and leverages ab initio strategies for regions without predictions. Finally, side chains are placed using a rotamer library, optimized to avoid steric clashes. Unlike the threaded modeling approach, the intensive modeling process facilitates subsequent model manipulation. The Dnf2 model was generated from the synthesis of PDB entries 1MHS (66), 4WIT (67), 3B9B (68), 3IXZ (69), 2ZXE (70), 3B8C (71), 3B8E (72), whereas the ATP10D intensive model used PDB entries 1MHS (66), 3B9B (68), 3IXZ (69), 2ZXE (70), 3B8C (71), and 3B8E (72). Residue sculpting within these models was manually performed via PyMOL, with residue shells and cushions set at 6 Å. The ATP10D model used in Fig. 6 was generated by threading the sequence over the structure of the Na/K pump (PDB entry 2ZXE) (70) using Phyre2 (65).
Data analysis
Yeast substrate uptake data analyses were performed as outlined previously (23). Briefly, three independent clonal transformants were selected for each yeast experiment and examined in parallel per sample. When KO strains were examined, three clonal strain cultures were assessed in parallel. When reported as arbitrary fluorescent units, raw fluorescence data are presented. When normalized to WT NBD-PC transport, substrate uptake in dnf1,2Δ vector controls are subtracted from the experimental group, and the data are normalized relative to WT PC uptake, which is set at 100%. Median fluorescence values were averaged from the respective experiments, and substrate preference was determined by taking ratios of clonal replicates.
Author contributions
B. P. R. and T. R. G. conceived of the project; B. P. R., T. N., C. A.-Y., R. J. Y., H.-W. S. and T. R. G. designed the experiments; B. P. R., T. N., J. T. B., R. J. Y., H. T., and C. A.-Y. performed the experiments; B. P. R., T. N., J. T. B., H.-W. S., and T. R. G. analyzed the data; and B. P. R., H.-W. S., and T. R. G. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank the laboratory of Kathleen L. Gould (Vanderbilt University) for the gift of the S. pombe strains. We thank Toshio Kitamura (University of Tokyo) and Hiroyuki Miyoshi (RIKEN BioResource Center) for providing plasmids for retroviral infection. Yeast flow cytometry experiments were performed with the Vanderbilt Medical Center Flow Cytometry Shared Resource, supported by National Institutes of Health Grants P30-CA68485 (to the Vanderbilt Ingram Cancer Center) and P30-DK058404 (to the Vanderbilt Digestive Disease Research Center).
This work was supported by National Institutes of Health Grants R01-GM107978 (to T. R. G.) and F32-GM116310 (to B. P. R.), Japan Society for the Promotion of Science (JSPS) KAKENHI Grants JP16H00764 and JP17H03655 and a grant from the Japan Foundation for Applied Enzymology (to H.-W. S), and Grant-in-Aid for JSPS Research Fellow JP16J05381 (to T. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains supporting Experimental procedures, Tables S1–S4, and Figs. S1–S12.
- GWA
- genome-wide association
- GlcCer
- glucosylceramide
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- SM
- sphingomyelin
- GalCer
- galactosylceramide
- KO
- knockout
- NBD
- 7-nitro-2–1,3-benzoxadiazol-4-yl
- TM
- transmembrane
- SD
- synthetic defined
- HA
- hemagglutinin
- HBSS
- Hanks' balanced salt solution
- PDB
- Protein Data Bank
- ANOVA
- analysis of variance
- PM
- plasma membrane.
References
- 1. Roland B. P., and Graham T. R. (2016) Decoding P4-ATPase substrate interactions. Crit. Rev. Biochem. Mol. Biol. 51, 513–527 10.1080/10409238.2016.1237934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andersen J. P., Vestergaard A. L., Mikkelsen S. A., Mogensen L. S., Chalat M., and Molday R. S. (2016) P4-ATPases as phospholipid flippases–structure, function, and enigmas. Front. Physiol. 7, 275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Folmer D. E., Elferink R. P., and Paulusma C. C. (2009) P4 ATPases–lipid flippases and their role in disease. Biochim. Biophys. Acta 1791, 628–635 10.1016/j.bbalip.2009.02.008 [DOI] [PubMed] [Google Scholar]
- 4. Takatsu H., Tanaka G., Segawa K., Suzuki J., Nagata S., Nakayama K., and Shin H. W. (2014) Phospholipid flippase activities and substrate specificities of human type IV P-type ATPases localized to the plasma membrane. J. Biol. Chem. 289, 33543–33556 10.1074/jbc.M114.593012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bull L. N., van Eijk M. J., Pawlikowska L., DeYoung J. A., Juijn J. A., Liao M., Klomp L. W., Lomri N., Berger R., Scharschmidt B. F., Knisely A. S., Houwen R. H., and Freimer N. B. (1998) A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 18, 219–224 10.1038/ng0398-219 [DOI] [PubMed] [Google Scholar]
- 6. Coleman J. A., Zhu X., Djajadi H. R., Molday L. L., Smith R. S., Libby R. T., John S. W., and Molday R. S. (2014) Phospholipid flippase ATP8A2 is required for normal visual and auditory function and photoreceptor and spiral ganglion cell survival. J. Cell Sci. 127, 1138–1149 10.1242/jcs.145052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Onat O. E., Gulsuner S., Bilguvar K., Nazli Basak A., Topaloglu H., Tan M., Tan U., Gunel M., and Ozcelik T. (2013) Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur. J. Hum. Genet. 21, 281–285 10.1038/ejhg.2012.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu X., Libby R. T., de Vries W. N., Smith R. S., Wright D. L., Bronson R. T., Seburn K. L., and John S. W. (2012) Mutations in a P-type ATPase gene cause axonal degeneration. PLoS Genet. 8, e1002853 10.1371/journal.pgen.1002853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Irvin M. R., Wineinger N. E., Rice T. K., Pajewski N. M., Kabagambe E. K., Gu C. C., Pankow J., North K. E., Wilk J. B., Freedman B. I., Franceschini N., Broeckel U., Tiwari H. K., and Arnett D. K. (2011) Genome-wide detection of allele specific copy number variation associated with insulin resistance in African Americans from the HyperGEN study. PLoS One 6, e24052 10.1371/journal.pone.0024052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hicks A. A., Pramstaller P. P., Johansson A., Vitart V., Rudan I., Ugocsai P., Aulchenko Y., Franklin C. S., Liebisch G., Erdmann J., Jonasson I., Zorkoltseva I. V., Pattaro C., Hayward C., Isaacs A., et al. (2009) Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genet. 5, e1000672 10.1371/journal.pgen.1000672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kengia J. T., Ko K. C., Ikeda S., Hiraishi A., Mieno-Naka M., Arai T., Sato N., Muramatsu M., and Sawabe M. (2013) A gene variant in the Atp10d gene associates with atherosclerotic indices in Japanese elderly population. Atherosclerosis 231, 158–162 10.1016/j.atherosclerosis.2013.08.034 [DOI] [PubMed] [Google Scholar]
- 12. Dhar M., Hauser L., and Johnson D. (2002) An aminophospholipid translocase associated with body fat and type 2 diabetes phenotypes. Obes. Res. 10, 695–702 10.1038/oby.2002.94 [DOI] [PubMed] [Google Scholar]
- 13. Dhar M. S., Sommardahl C. S., Kirkland T., Nelson S., Donnell R., Johnson D. K., and Castellani L. W. (2004) Mice heterozygous for Atp10c, a putative amphipath, represent a novel model of obesity and type 2 diabetes. J. Nutr. 134, 799–805 10.1093/jn/134.4.799 [DOI] [PubMed] [Google Scholar]
- 14. Flamant S., Pescher P., Lemercier B., Clément-Ziza M., Képès F., Fellous M., Milon G., Marchal G., and Besmond C. (2003) Characterization of a putative type IV aminophospholipid transporter P-type ATPase. Mamm. Genome 14, 21–30 10.1007/s00335-002-3032-3 [DOI] [PubMed] [Google Scholar]
- 15. Surwit R. S., Feinglos M. N., Rodin J., Sutherland A., Petro A. E., Opara E. C., Kuhn C. M., and Rebuffé-Scrive M. (1995) Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism 44, 645–651 10.1016/0026-0495(95)90123-X [DOI] [PubMed] [Google Scholar]
- 16. Sigruener A., Wolfrum C., Boettcher A., Kopf T., Liebisch G., Orsó E., and Schmitz G. (2017) Lipidomic and metabolic changes in the P4-type ATPase ATP10D deficient C57BL/6J wild type mice upon rescue of ATP10D function. PLoS One 12, e0178368 10.1371/journal.pone.0178368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roelants F. M., Baltz A. G., Trott A. E., Fereres S., and Thorner J. (2010) A protein kinase network regulates the function of aminophospholipid flippases. Proc. Natl. Acad. Sci. U.S.A. 107, 34–39 10.1073/pnas.0912497106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vance D. E., and Vance J. E. (2008) Biochemistry of Lipids, Lipoproteins and Membranes, 5th Ed., pp. 364–397, Elsevier, Amsterdam [Google Scholar]
- 19. Dawson G., Kruski A. W., and Scanu A. M. (1976) Distribution of glycosphingolipids in the serum lipoproteins of normal human subjects and patients with hypo- and hyperlipidemias. J. Lipid Res. 17, 125–131 [PubMed] [Google Scholar]
- 20. Baldridge R. D., and Graham T. R. (2012) Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc. Natl. Acad. Sci. U.S.A. 109, E290–E298 10.1073/pnas.1115725109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vestergaard A. L., Coleman J. A., Lemmin T., Mikkelsen S. A., Molday L. L., Vilsen B., Molday R. S., Dal Peraro M., and Andersen J. P. (2014) Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. U.S.A. 111, E1334–E1343 10.1073/pnas.1321165111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stone A., Chau C., Eaton C., Foran E., Kapur M., Prevatt E., Belkin N., Kerr D., Kohlin T., and Williamson P. (2012) Biochemical characterization of P4-ATPase mutations identified in patients with progressive familial intrahepatic cholestasis. J. Biol. Chem. 287, 41139–41151 10.1074/jbc.M112.413039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roland B. P., and Graham T. R. (2016) Directed evolution of a sphingomyelin flippase reveals mechanism of substrate backbone discrimination by a P4-ATPase. Proc. Natl. Acad. Sci. U.S.A. 113, E4460–E4466 10.1073/pnas.1525730113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pomorski T., Lombardi R., Riezman H., Devaux P. F., van Meer G., and Holthuis J. C. (2003) Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol. Biol. Cell 14, 1240–1254 10.1091/mbc.e02-08-0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saito K., Fujimura-Kamada K., Furuta N., Kato U., Umeda M., and Tanaka K. (2004) Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 3418–3432 10.1091/mbc.e03-11-0829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kato U., Emoto K., Fredriksson C., Nakamura H., Ohta A., Kobayashi T., Murakami-Murofushi K., Kobayashi K., and Umeda M. (2002) A novel membrane protein, Ros3p, is required for phospholipid translocation across the plasma membrane in Saccharomyces cerevisiae. J. Biol. Chem. 277, 37855–37862 10.1074/jbc.M205564200 [DOI] [PubMed] [Google Scholar]
- 27. Bryde S., Hennrich H., Verhulst P. M., Devaux P. F., Lenoir G., and Holthuis J. C. (2010) CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem. 285, 40562–40572 10.1074/jbc.M110.139543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van der Velden L. M., Wichers C. G., van Breevoort A. E., Coleman J. A., Molday R. S., Berger R., Klomp L. W., and van de Graaf S. F. (2010) Heteromeric interactions required for abundance and subcellular localization of human CDC50 proteins and class 1 P4-ATPases. J. Biol. Chem. 285, 40088–40096 10.1074/jbc.M110.139006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takatsu H., Baba K., Shima T., Umino H., Kato U., Umeda M., Nakayama K., and Shin H. W. (2011) ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J. Biol. Chem. 286, 38159–38167 10.1074/jbc.M111.281006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naito T., Takatsu H., Miyano R., Takada N., Nakayama K., and Shin H. W. (2015) Phospholipid flippase ATP10A translocates phosphatidylcholine and is involved in plasma membrane dynamics. J. Biol. Chem. 290, 15004–15017 10.1074/jbc.M115.655191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takada N., Naito T., Inoue T., Nakayama K., Takatsu H., and Shin H. W. (2018) Phospholipid-flipping activity of P4-ATPase drives membrane curvature. EMBO J. 37, e97705 10.15252/embj.201797705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jensen M. S., Costa S. R., Duelli A. S., Andersen P. A., Poulsen L. R., Stanchev L. D., Gourdon P., Palmgren M., Günther Pomorski T., and López-Marqués R. L. (2017) Phospholipid flipping involves a central cavity in P4 ATPases. Sci. Rep. 7, 17621 10.1038/s41598-017-17742-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baldridge R. D., and Graham T. R. (2013) Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc. Natl. Acad. Sci. U.S.A. 110, E358–E367 10.1073/pnas.1216948110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Natarajan P., Wang J., Hua Z., and Graham T. R. (2004) Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc. Natl. Acad. Sci. U.S.A. 101, 10614–10619 10.1073/pnas.0404146101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gantzel R. H., Mogensen L. S., Mikkelsen S. A., Vilsen B., Molday R. S., Vestergaard A. L., and Andersen J. P. (2017) Disease mutations reveal residues critical to the interaction of P4-ATPases with lipid substrates. Sci. Rep. 7, 10418 10.1038/s41598-017-10741-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hannun Y. A., and Obeid L. M. (2018) Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 19, 175–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sabourdy F., Astudillo L., Colacios C., Dubot P., Mrad M., Ségui B., Andrieu-Abadie N., and Levade T. (2015) Monogenic neurological disorders of sphingolipid metabolism. Biochim. Biophys. Acta 1851, 1040–1051 10.1016/j.bbalip.2015.01.010 [DOI] [PubMed] [Google Scholar]
- 38. Astudillo L., Therville N., Colacios C., Ségui B., Andrieu-Abadie N., and Levade T. (2016) Glucosylceramidases and malignancies in mammals. Biochimie 125, 267–280 10.1016/j.biochi.2015.11.009 [DOI] [PubMed] [Google Scholar]
- 39. Holland W. L., and Summers S. A. (2008) Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 29, 381–402 10.1210/er.2007-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fahy E., Subramaniam S., Murphy R. C., Nishijima M., Raetz C. R., Shimizu T., Spener F., van Meer G., Wakelam M. J., and Dennis E. A. (2009) Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 50, S9–S14 10.1194/jlr.R800095-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coleman J. A., Kwok M. C., and Molday R. S. (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J. Biol. Chem. 284, 32670–32679 10.1074/jbc.M109.047415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou X., and Graham T. R. (2009) Reconstitution of phospholipid translocase activity with purified Drs2p, a type-IV P-type ATPase from budding yeast. Proc. Natl. Acad. Sci. U.S.A. 106, 16586–16591 10.1073/pnas.0904293106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. López-Marqués R. L., Poulsen L. R., Bailly A., Geisler M., Pomorski T. G., and Palmgren M. G. (2015) Structure and mechanism of ATP-dependent phospholipid transporters. Biochim. Biophys. Acta 1850, 461–475 10.1016/j.bbagen.2014.04.008 [DOI] [PubMed] [Google Scholar]
- 44. Baldridge R. D., Xu P., and Graham T. R. (2013) Type IV P-type ATPases distinguish mono- versus diacyl phosphatidylserine using a cytofacial exit gate in the membrane domain. J. Biol. Chem. 288, 19516–19527 10.1074/jbc.M113.476911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Primeau J. O., Armanious G. P., Fisher M. E., and Young H. S. (2018) The sarcoendoplasmic reticulum calcium ATPase. Subcell. Biochem. 87, 229–258 10.1007/978-981-10-7757-9_8 [DOI] [PubMed] [Google Scholar]
- 46. Kazmier K., Claxton D. P., and Mchaourab H. S. (2017) Alternating access mechanisms of LeuT-fold transporters: trailblazing towards the promised energy landscapes. Curr. Opin. Struct. Biol. 45, 100–108 10.1016/j.sbi.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leipelt M., Warnecke D., Zähringer U., Ott C., Müller F., Hube B., and Heinz E. (2001) Glucosylceramide synthases, a gene family responsible for the biosynthesis of glucosphingolipids in animals, plants, and fungi. J. Biol. Chem. 276, 33621–33629 10.1074/jbc.M104952200 [DOI] [PubMed] [Google Scholar]
- 48. Jiang Y., Wang W., Xie Q., Liu N., Liu L., Wang D., Zhang X., Yang C., Chen X., Tang D., and Wang E. (2017) Plants transfer lipids to sustain colonization by mutualistic mycorrhizal and parasitic fungi. Science 356, 1172–1175 10.1126/science.aam9970 [DOI] [PubMed] [Google Scholar]
- 49. Luginbuehl L. H., Menard G. N., Kurup S., Van Erp H., Radhakrishnan G. V., Breakspear A., Oldroyd G. E. D., and Eastmond P. J. (2017) Fatty acids in arbuscular mycorrhizal fungi are synthesized by the host plant. Science 356, 1175–1178 10.1126/science.aan0081 [DOI] [PubMed] [Google Scholar]
- 50. Sawada K., Sato T., Hamajima H., Jayakody L. N., Hirata M., Yamashiro M., Tajima M., Mitsutake S., Nagao K., Tsuge K., Abe F., Hanada K., and Kitagaki H. (2015) Glucosylceramide contained in Koji mold-cultured cereal confers membrane and flavor modification and stress tolerance to Saccharomyces cerevisiae during coculture fermentation. Appl. Environ. Microbiol. 81, 3688–3698 10.1128/AEM.00454-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen C. Y., Ingram M. F., Rosal P. H., and Graham T. R. (1999) Role for Drs2p, a P-type ATPase and potential aminophospholipid translocase, in yeast late Golgi function. J. Cell Biol. 147, 1223–1236 10.1083/jcb.147.6.1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gall W. E., Geething N. C., Hua Z., Ingram M. F., Liu K., Chen S. I., and Graham T. R. (2002) Drs2p-dependent formation of exocytic clathrin-coated vesicles in vivo. Curr. Biol. 12, 1623–1627 10.1016/S0960-9822(02)01148-X [DOI] [PubMed] [Google Scholar]
- 53. Liu K., Surendhran K., Nothwehr S. F., and Graham T. R. (2008) P4-ATPase requirement for AP-1/clathrin function in protein transport from the trans-Golgi network and early endosomes. Mol. Biol. Cell 19, 3526–3535 10.1091/mbc.e08-01-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu P., Baldridge R. D., Chi R. J., Burd C. G., and Graham T. R. (2013) Phosphatidylserine flipping enhances membrane curvature and negative charge required for vesicular transport. J. Cell Biol. 202, 875–886 10.1083/jcb.201305094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bratton D. L., Fadok V. A., Richter D. A., Kailey J. M., Guthrie L. A., and Henson P. M. (1997) Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J. Biol. Chem. 272, 26159–26165 10.1074/jbc.272.42.26159 [DOI] [PubMed] [Google Scholar]
- 56. Fadok V. A., Bratton D. L., Frasch S. C., Warner M. L., and Henson P. M. (1998) The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 5, 551–562 10.1038/sj.cdd.4400404 [DOI] [PubMed] [Google Scholar]
- 57. Uchida K., Emoto K., Daleke D. L., Inoue K., and Umeda M. (1998) Induction of apoptosis by phosphatidylserine. J. Biochem. 123, 1073–1078 10.1093/oxfordjournals.jbchem.a022045 [DOI] [PubMed] [Google Scholar]
- 58. Yoshida H., Kawane K., Koike M., Mori Y., Uchiyama Y., and Nagata S. (2005) Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 437, 754–758 10.1038/nature03964 [DOI] [PubMed] [Google Scholar]
- 59. Fadok V. A., Bratton D. L., Rose D. M., Pearson A., Ezekewitz R. A., and Henson P. M. (2000) A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405, 85–90 10.1038/35011084 [DOI] [PubMed] [Google Scholar]
- 60. Hoffmann P. R., deCathelineau A. M., Ogden C. A., Leverrier Y., Bratton D. L., Daleke D. L., Ridley A. J., Fadok V. A., and Henson P. M. (2001) Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 155, 649–659 10.1083/jcb.200108080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Platt F. M. (2014) Sphingolipid lysosomal storage disorders. Nature 510, 68–75 10.1038/nature13476 [DOI] [PubMed] [Google Scholar]
- 62. Vitner E. B., and Futerman A. H. (2013) Neuronal forms of Gaucher disease. Handb. Exp. Pharmacol. 405–419 10.1007/978-3-7091-1511-4_20 [DOI] [PubMed] [Google Scholar]
- 63. Sidransky E., and Lopez G. (2012) The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998 10.1016/S1474-4422(12)70190-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shin H. W., Morinaga N., Noda M., and Nakayama K. (2004) BIG2, a guanine nucleotide exchange factor for ADP-ribosylation factors: its localization to recycling endosomes and implication in the endosome integrity. Mol. Biol. Cell 15, 5283–5294 10.1091/mbc.e04-05-0388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kühlbrandt W., Zeelen J., and Dietrich J. (2002) Structure, mechanism, and regulation of the Neurospora plasma membrane H+-ATPase. Science 297, 1692–1696 10.1126/science.1072574 [DOI] [PubMed] [Google Scholar]
- 67. Brunner J. D., Lim N. K., Schenck S., Duerst A., and Dutzler R. (2014) X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212 10.1038/nature13984 [DOI] [PubMed] [Google Scholar]
- 68. Olesen C., Picard M., Winther A. M., Gyrup C., Morth J. P., Oxvig C., Møller J. V., and Nissen P. (2007) The structural basis of calcium transport by the calcium pump. Nature 450, 1036–1042 10.1038/nature06418 [DOI] [PubMed] [Google Scholar]
- 69. Abe K., Tani K., Nishizawa T., and Fujiyoshi Y. (2009) Inter-subunit interaction of gastric H+,K+-ATPase prevents reverse reaction of the transport cycle. EMBO J. 28, 1637–1643 10.1038/emboj.2009.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shinoda T., Ogawa H., Cornelius F., and Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4 Å resolution. Nature 459, 446–450 10.1038/nature07939 [DOI] [PubMed] [Google Scholar]
- 71. Focht D., Croll T. I., Pedersen B. P., and Nissen P. (2017) Improved model of proton pump crystal structure obtained by interactive molecular dynamics flexible fitting expands the mechanistic model for proton translocation in P-Type ATPases. Front. Physiol. 8, 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., and Nissen P. (2007) Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049 10.1038/nature06419 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.