

Abstract
Background
The pain-relief properties of tricyclic antidepressants can be attributed to several actions. Recent observations suggest that adenosine is involved in the antinociceptive effect of amitriptyline. The A3 adenosine receptor (A3AR) is the only adenosine subtype overexpressed in inflammatory and cancer cells. This study was performed to investigate the role of A3AR in the anti-nociceptive effect of amitriptyline.
Methods
Spinal nerve-ligated neuropathic pain was induced by ligating the L5 and L6 spinal nerves of male Sprague-Dawley rats. The neuropathic rats were randomly assigned to one of the following three groups (8 per group): a neuropathic pain with normal saline group, a neuropathic pain with amitriptyline group, and a neuropathic pain with amitriptyline and 3-ethyl-5-benzyl- 2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (MRS) group. Amitriptyline or saline was administered intraperitoneally and 3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (MRS-1191), an A3AR antagonist, was injected subcutaneously immediately before amitriptyline administration. The level of extracellular signal-regulated kinase P44/42 (ERK1/2), cyclic AMP response element-binding protein (CREB), and proinflammatory cytokines were assessed using immunoblotting or reverse-transciption polymerase chain reaction.
Results
Amitriptyline increased the mechanical withdrawal threshold of the neuropathic rats. The level of phospho-ERK1/2 and phospho-CREB proteins, and proinflammatory cytokines produced by spinal nerve ligation were significantly reduced by amitriptyline administration. However, the use of MRS-1191 before amitriptyline administration not only reduced the threshold of mechanical allodynia, but also increased the signaling protein and proinflammatory cytokine levels, which were reduced by amitriptyline.
Conclusions
The results of this study suggest that the anti-nociceptive effect of amitriptyline involves the suppression of ERK1/2 and CREB signaling proteins, and A3AR activation also affects the alleviation of the inflammatory response.
Keywords: Adenosine A3, Amitriptyline, Cyclic AMP response element-binding protein, Cytokine, Mitogen-activated protein kinase
Introduction
Neuropathic pain is a chronic pain condition that can be caused by many disorders such as diabetic polyneuropathy, post-spinal surgical pain, post-herpetic neuralgia, stroke, and spinal cord injury [1].
The inflammatory response of the nerves is one of the causes associated with the progress of neuropathic pain. Pro-inflammatory cytokines are involved in the demyelination and degeneration of peripheral nerves, increased sensory afferent excitability, and induction of neuropathic pain [2].
Recent studies have shown that the levels of extracellular signal-regulated kinase P44/42 (ERK1/2), which belongs to the mitogen-activated protein kinase (MAPK) family, and cyclic AMP response element-binding protein (CREB) are increased in rat neuropathic pain models [3,4]. ERK1/2 and CREB signaling proteins, which are induced by the activation of G-protein coupled receptors (GPCRs), cause plasticity changes and inflammatory responses in the spinal nerves. These changes are important factors in causing neuropathic pain [4].
Amitriptyline, a member of the tricyclic antidepressant (TCA) family, is currently the most widely prescribed antidepressant for neuropathic pain. Many studies have demonstrated that amitriptyline can profoundly change symptoms such as hyperalgesia and allodynia [5,6]. However, the mechanisms underlying the anti-hyperalgesic and anti-allodynic actions of amitriptyline have not yet been fully understood. Previous studies have shown that interactions with the endogenous adenosine system seem most important for the anti-nociceptive effect of amitriptyline [6]. Moreover, caffeine, a non-selective adenosine receptor (AR) antagonist, has been shown to reduce the antinociceptive effect of amitriptyline in both inflammatory and neuropathic pain models [7]. Evidences also suggest the involvement of an adenosine component in this action [5,6].
The A3 adenosine receptor (A3AR) is the only adenosine subtype overexpressed in inflammatory and cancer cells. Although the relationship between early proinflammatory cytokines and A1AR on the inflammatory responses of nerves in the development of neuropathic pain has been studied [8], the association with A3AR has not yet been explored.
Therefore, the aim of the present study was to investigate whether (1) amitriptyline was effective in relieving the symptoms of neuropathic pain in a rat model of spinal nerve ligation (SNL), and (2) whether the effect of amitriptyline was associated with endogenous adenosine, particularly A3AR.
Materials and Methods
All experiments conformed to the guidelines of and were approved by the Institutional Laboratory Animal Care and the Ethical Committee of Catholic University of Korea, Seoul, Korea, on March 26, 2015 (IRB 15-12). All animals were kept in separate cages with a certain photoperiodism (a 12-h light-dark cycle) and sufficient water and food. The rats were allowed to acclimatize for 7 days before the experiments. Twenty-four male Sprague-Dawley rats (weighing 250–300 g) were randomly assigned to one of three groups (8 per group): a neuropathic pain with normal saline group (NP + NS group, control group, n = 8), a neuropathic pain with amitriptyline group (NP + Ami group, n = 8), and a neuropathic pain with amitriptyline and MRS-1191 (CAS 185222-90-6, USA, 3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate) group (NP + Ami + MRS group, n = 8). In the NP + Ami + MRS group, amitriptyline was administered intraperitoneally immediately after the subcutaneous injection of MRS-1191.
Neuropathic pain models and behavioral tests
The Kim and Chung [9] method was used to create the neuropathic pain models by ligating the spinal nerves. Rats were anesthetized using pentobarbital sodium (50 mg/kg i.p.). A dorsal midline incision was made from L3 to S2. Under the microscopic guidance, the left L6 transverse process was partially resected to visualize the L5 and L6 spinal nerves. These nerves were tied up with 5-0 black silk. The wound was sutured again with a 4-0 soluble Vicryl suture, and the wound was closed using wound clips. Before the behavioral testing, the rats were housed in groups for 7 days. Among the animals, those exhibiting a foot withdrawal response to 8 von Frey filaments (0.6, 1.0, 2.0, 4.0, 6.0, 8.0, 10.0, and 15.0 g; Stoelting, USA) with an applied bending force of 4 g or less were considered neuropathic, and they were used in this study [10]. The rats were excluded from this experiment if they either showed the motor deficiency or did not exhibit subsequent mechanical allodynia. The mechanical withdrawal threshold test was performed for 1 week, daily after drug administration based on each group’s designation. Changes in the levels of pERK1/2, and pCREB signaling proteins and mRNA levels of proinflammatory cytokines determined using immunoblotting or reverse-transciption polymerase chain reaction (RT-PCR) techniques on tissues harvested from the spinal cord were compared among the three groups. All tests and analyses were performed by observers blinded to the assignment of animals in the treatment groups. All drug treatments were also blinded. The drug was administered intraperitoneally to neuropathic rats on the basis of their assigned treatment group of saline and amitriptyline (10 mg/kg). In the NP + Ami + MRS group, amitriptyline was administered immediately after the subcutaneous injection of MRS-1191 (1 mg/kg) 7 days after SNL [5,7,11]. The doses of amitriptyline and MRS-1191 were determined on the basis of pilot experiments conducted in our laboratory, in which the doses selected in the current study were found to have no effect on normal and pain behaviors. After an initial observation period, the rats were housed with their cage mates in an enriched environment during the experiment. Experiments for evaluating static tactile mechanical thresholds were carried out by examiners blinded to the assigned treatment groups.
Tissue preparation and immunoblot analysis
After assessing the static tactile mechanical thresholds for 7 days, by using oxygen and 5% sevoflurane, the rats were anesthetized. The third lumbar vertebra and the sacrum were cut almost simultaneously, and the spinal cord from the lumbar enlargement was harvested by rapidly injecting saline into the spinal canals and pushing the spinal cord toward the lumbar vertebra. The harvested spinal cord tissues were stored at −80°C after quick-freezing with liquid nitrogen. Spinal cord tissue was dissected on ice and immediately placed in ice-cold radioimmunoprecipitation assay buffer (150 mmol NaCl, 50 mmol Tris-HCl, 1 mmol EDTA, and 1% Triton-X100 [pH 7.4]) and homogenized for 10 s on ice. Tissue was super-centrifuged (32,500 g, at 4°C, and in a vacuum) for 1 h. For the immunoblotting described above, the supernatant was collected. Bovine serum albumin was used as a standard solution and analyzed using the Bradford assay (BioRad, USA), and the sample was stabilized using the Laemmli buffer. The protein bands, obtained by electrophoresing each protein in 10% sodium dodecyl sulfate polyacrylamide gel for 4 h at 80 V, were transferred to a polyvinylidene fluoride membrane. The membranes were washed with Tris-Buffered Saline Tween-20 (TBST) solution (0.1% Tween 20 in 1 × TBS) and coated with a blocking solution (5% dry milk in TBST). The membranes were incubated with primary phospho-ERK1/2 (pERK1/2) and phospho-CREB (pCREB) antibodies (Cell Signaling Technology, USA) for 16 h. After washing with TBST solution, the membranes were incubated with the secondary antibody (goat anti-rabbit or anti-mouse IgG, Santa Cruz Biotechnology, USA) for 1 h, and developed using electrochemiluminescence reagents (Amersham, USA) and a UVP Bio-imaging System (Upland, USA).
Semiquantitative RT-PCR for the mRNA of proinflammatory cytokines
The inflammation in the harvested spinal cord was also determined by measuring mRNA encoding markers of inflammation, including tumor necrosis factor α (TNF-α), intercellular adhesion molecule 1 (ICAM-1), macrophage inflammatory protein 2 (MIP-2), and monocyte chemoattractant protein 1 (MCP-1) [11]. Primers were designed on the basis of published Gen-Bank sequences (Table 1). A 1-ml aliquot of Trizol reagent (MRC, USA) was added to the spinal cord tissues of the rats, which were then homogenized and left for 5 min. A 0.2-ml aliquot of 1-bromo-3-chloropropane (Sigma, USA) was added, followed by centrifugation at 14,000 g for 15 min. The supernatant was then separated and centrifuged at 14,000 g for 10 min after adding the same amount of phenol:chloroform (5:1, pH 4.7) (Sigma, USA). After separating the supernatant, 100% ethanol was added and reacted at −20°C for 1 h, followed by centrifugation at 14,000 g for 10 min. The supernatant was discarded, and the RNA precipitate was washed with 75% ethanol. Diethylpyrocarbonate was added and reacted at 60°C for 5 min and stored in a refrigerator (−70°C). The RNA concentration was determined using a UVP Bio-imaging System (Upland, USA) at an absorbance of 260 nm. cDNA (50 μl) was constructed using the extracted RNA (0.5 μg) as a template and using avian marrow astrovirus reverse transcriptase (Promega, USA). The PCR was performed by generating 10 μl of the final reaction solution containing dNTPs (2.5 mM/L each) and 1 unit I Start Taq DNA polymerase (Intron, Korea), and each primer of 20 μM/L using composed cDNA of 2 μl. The PCR included 35 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension for 1 min at 72°C, and the reaction was terminated at 72°C for 10 min [12]. A 228-bp region of the β-actin gene was used as an internal control to determine whether appropriate mRNA was separated from the cells. After electrophoresis on 2.0% agarose gel and staining with ethidium bromide, the obtained PCR products were quantified using Gel DocTM XR (BioRad, USA).
Table 1.
RT-PCR Primer Sequences
| Primer | Accession number | Sequence (5' → 3') | Size (bp) | Cycle number | Annealing temperature (°C) |
|---|---|---|---|---|---|
| β-actin | NM031144 | F: AGCCATGTACGTAGCCATCC | 228 | 35 | 55 |
| R: CTCTCAGCTGTGGTGGTGAA | |||||
| TNF-α | X66539 | F: TACTGAACTTCGGGGTGATCGGTCC | 295 | 35 | 62 |
| S40199 | R: CAGCCTTGTCCCTTGAAGAGAACC | ||||
| ICAM-1 | S46779 | F: TTTCGATCTTCCGACTAGGG | 113 | 35 | 55 |
| R: AGCTTCAGAGGCAGGAAACA | |||||
| MIP-2 | AB060092 | F: CCACCAACCATCAGGGTACA | 214 | 35 | 56 |
| R: TGGACGATCCTCTGAACCAA | |||||
| MCP-1 | M57441 | F: CACCTGCTGCTACTCATTCACT | 349 | 35 | 62 |
| R: GTTCTCTGTCATACTGGTCACTTCT |
RT-PCR: reverse-transcription polymerase chain reaction, TNF-α: tumor necrosis factor α, ICAM-1: intercellular adhesion molecule 1, MIP-2: macrophage inflammatory protein 2, MCP-1: monocyte chemoattractant protein 1.
Statistical analysis
Data are expressed as mean ± SD. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software Inc., USA). Data were analyzed using one-way ANOVA, Dunnett’s test, and Kruskal-Wallis nonparametric test for comparing of the three groups. A P value < 0.050 was considered statistically significant.
Results
After 7 days of SNL, with a foot withdrawal threshold < 4 g, all rats used in the experiment showed mechanical allodynia and hyperalgesia. The withdrawal thresholds more significantly increased on the 4th, 5th, 6th, and 7th days after drug injection in the NP + Ami group than in the NP + NS and the NP + Ami + MRS groups. The use of MRS-1191 before amitriptyline administration reduced the threshold of mechanical allodynia (Fig. 1).
Fig. 1.
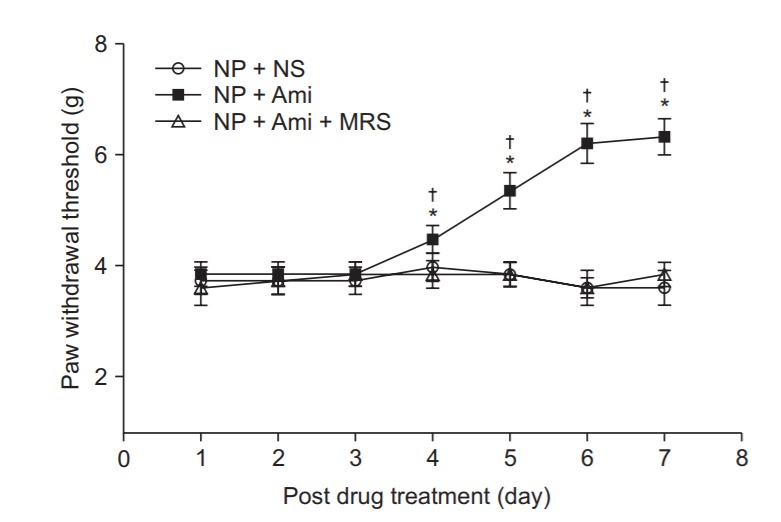
Time course of the paw withdrawal threshold to mechanical stimuli applied to the plantar surface of the affected left paw by using von Frey filaments in a neuropathic pain model of rats. Values are mean ± SD (n = 8 in each group). NP: neuropathic pain, NS: normal saline, Ami: amitriptyline, MRS: 3-ethyl-5benzyl-2-methyl-4-phenylehynyl-6-phenyl1,4(±)-dihydropyridine-3,5-dicarboxylate. *P < 0.05 versus the NP + NS group and †P < 0.05 versus the NP + Ami + MRS group by using analysis of variance (ANOVA) with Dunnett’s Post Hoc Test.
In the western blots of the spinal cord tissues, the expression of pERK1/2 and pCREB signaling proteins in the NP + Ami group was significantly decreased by 32% and 33%, respectively, in comparison with the NP + NS group (P = 0.022 and 0.003, respectively). However, the expression of pERK1/2 and pCREB signaling proteins in the NP + Ami + MRS group was significantly increased to levels similar to those in the NP + NS group. Moreover, no significant difference was observed in the changes of pERK1/2 and pCREB signaling protein levels between the NP + NS and NP + Ami + MRS groups (Fig. 2).
Fig. 2.
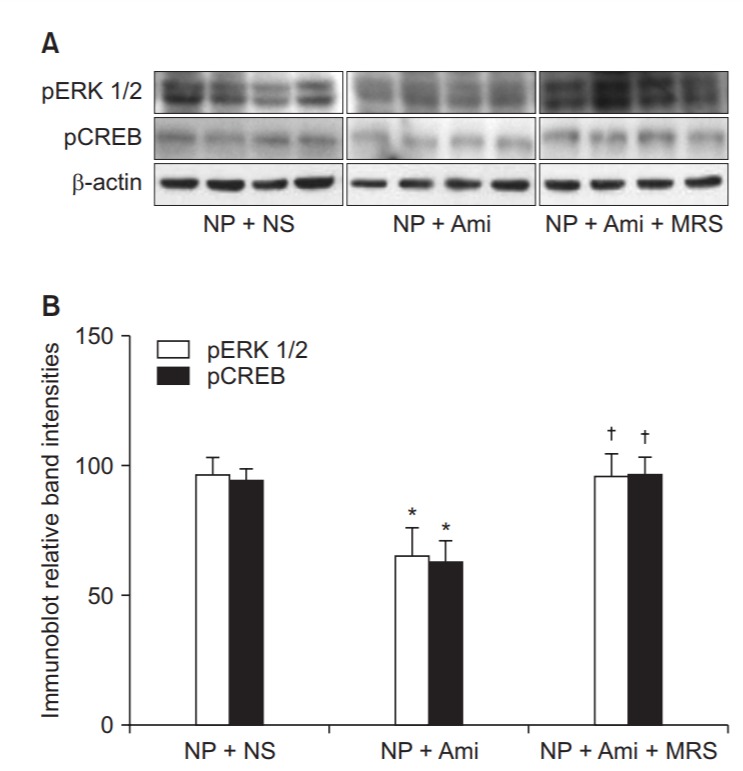
Comparison of the amount of signal proteins beween groups. (A) Representative immunoblots for phosphorylated extracellular signalregulated kinase 1/2 (pERK1/2) and phosphorylated cyclic AMP response element-binding protein (pCREB) from a spinal cord subjected to the NP + NS treatment (n = 8), NP + Ami treatment (n = 8), and NP + Ami + MRS treatment (n = 8). (B) Densitometric quantifications of band intensities for phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2) and phosphorylated cyclic AMP response element-binding protein (pCREB) from a spinal cord subjected to the NP + NS treatment (n = 8), NP + Ami treatment (n = 8), and NP + Ami + MRS treatment (n = 8). NP: neuropathic pain, NS: normal saline, Ami: amitriptyline, MRS: 3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate. *P < 0.05 versus the NP + NS group, and †P < 0.05 versus the NP + Ami group.
The mRNA levels of proinflammatory cytokines determined using the RT-PCR analysis of the spinal cord tissues were compared between the three groups. The NP + Ami group showed significantly decreased expression of TNF-α, ICAM-1, MIP-2, and MCP-1 (P = 0.016, 0.010, 0.024, and 0.018, respectively) than did the NP + NS group. Although, the expression of TNF-α, ICAM-1, MIP-2 and MCP-1 was significantly decreased in the NP + Ami group, it was significantly increased again in the NP + Ami + MRS group, similar to that in the NP + NS group. No significant difference was observed between the NP + NS and NP + Ami + MRS groups (Fig. 3).
Fig. 3.
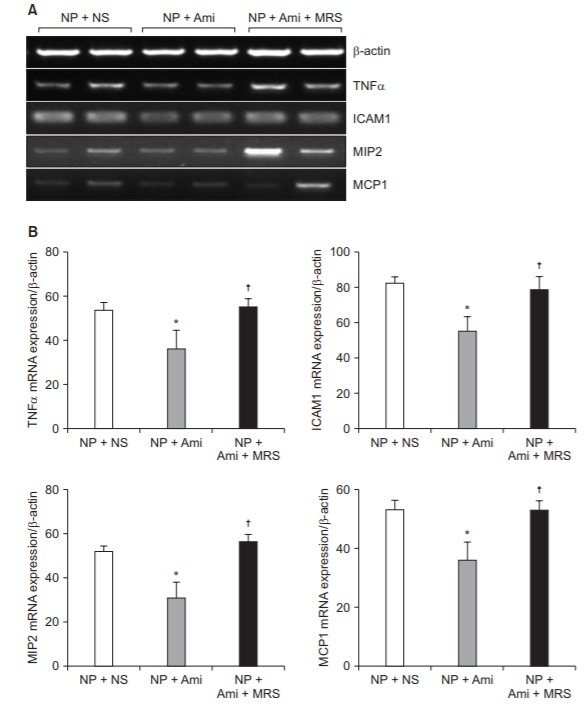
Comparison of the amount of proinflammatory cytokines between groups. (A) Representative RT-PCR images for TNF-α, ICAM-1, MIP-2, and MCP-1 from a spinal cord subjected to the NP+NS treatment (n = 8), NP + Ami treatment (n = 8), and NP + Ami + MRS treatment (n = 8). Representative of 3–5 experiments. β-actin is included to control for mRNA input. (B) Densitometric quantifications of band intensities for TNF-α, ICAM-1, MIP-2, and MCP1 from a spinal cord subjected to the NP + NS treatment (n = 8), NP + Ami treatment (n = 8), and NP + Ami + MRS treatment (n = 8). NP: neuropathic pain, NS: normal saline, Ami: amitriptyline, MRS: 3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate. RTPCR: reverse-transcriptase polymerase chain reaction, TNF-α: tumor necrosis factor α, ICAM-1: intercellular adhesion molecule 1, MIP-2: macrophage inflammatory protein 2, MCP-1: monocyte chemoattractant protein 1. *P < 0.05 versus the NP + NS group, †P < 0.05 versus the NP + Ami group
Discussion
The results of this study showed that the use of TCAs in neuropathic rats resulted in a reduction of pain and that the anti-inflammatory reaction of A3AR played a significant role in its action, in addition to the known actions of antidepressants. Neuropathic pain is one of the most debilitating chronic pain conditions, caused by traumatic nerve injury, various infections, autoimmune diseases, diabetes mellitus or nerve damage due to bone compression of cancer. Many studies have focused on neurons to understand the mechanism of neuropathic pain [13–15].
Neuropathic pain is a reflection of abnormal excitability of secondary sensory neurons in the spinal dorsal horn induced by peripheral sensory input and is the anatomic reconstruction of pain pathways in the peripheral and central nervous systems [14]. Persistent inflammatory responses in the spinal cord may also cause spontaneous pain and allodynia by generating ectopic discharges in the primary afferent nociceptors [15]. Faden and Simon [16] showed that excitatory amino acids (EEAs), acting at the N-methyl-D-aspartate receptor, contribute to secondary tissue damage following traumatic spinal cord injury. Furthermore, this study showed that elevated EEAs lead to the activation of MAPKs, including ERK1/2, JNK, and p38. Recent studies have shown an increase in the levels of ERK1/2, which belongs to the MAPK family, and CREB signaling protein in a rat neuropathic pain model [17,18]. Many studies on central sensitization after peripheral insults have also emphasized the role of ERK1/2 and CREB signaling proteins and inflammatory activities in nociceptive hyper-reactivity [3,17,18]. In a previous study, we demonstrated that the expression of ERK1/2 and CREB proteins was increased by 43% and 2.5 times, respectively, in the neuropathic pain group than in the control group [18].
TNF-α levels are elevated in inflammatory conditions, and its presence may indicate an ongoing inflammatory process. It also appears to play a key role in both peripheral and central neurodegenerative pathologies and in the development of neuropathic pain after following nerve injury [19]. Above study analyzed the proinflammatory cytokine mRNA markers KC, MIP-2, ICAM-1, MCP-1, TNF-α, interferon-induced protein 10 (IP-10), and regulated on activation normal T-cell expressed and secreted (RANTES). For each experiment, the researchers also performed semiquantitative RT-PCR under conditions yielding linear results for GAPDH to confirm equal RNA input between the groups [20]. We also demonstrated in previous study that neuropathic pain induced significant mRNA expression of TNF-α, ICAM-1, MIP-2, and MCP-1 compared to that in the control group [18].
ARs modulate inflammation and apoptosis in many organs, including the heart, kidney, lungs, and liver, and these organs are subject to multi-organ injury in sepsis [21]. Previous study showed that ARs play an important role in regulating the outcome after renal ischemia and reperfusion injury, as well as after cecal ligation and puncture-induced sepsis, partly by modulating inflammation [19–22]. Purinergic signals are involved in pain signal transduction, transmission, modulation, and sensitization in the nociceptive sensory nervous system. Furthermore, endogenous adenosine has been well established to induce anti-nociception in a variety of pain conditions [23]. Adenosine induces analgesia via the activation of A1 receptors expressed in the peripheral sensory nerve fibers. Previous studies have demonstrated that blocking A1 receptors abolishes the adenosine-induced anti-nociception in various inflammatory pain models [20–22].
Endogenous adenosine mediates antinociception by activating A2A, A2B, or A3 receptors expressed in nociceptive neurons, astrocytes, or immune cells [24–26]. A2AR agonists show some peripheral pronociceptive effects; moreover, they act on immune cells to suppress inflammation and on spinal glia to suppress pain signaling and may be useful in improving inflammatory and neuropathic pain [24]. Adenosine can also mediate antinociception by enhancing GABA inhibition through the activation of A3 receptors [25]. Selective A3AR agonists, like IB-MECA or its 2-chloro analog Cl-IB-MECA, block neuropathic pain caused by various chemotherapeutics such as paclitaxel, oxaliplatin, and bortezomib without interfering with their anticancer effects [26]. A3AR have now been demonstrated to produce antinociception in several preclinical neuropathic pain models, with mechanistic actions on glial cells, and they may be useful in improving neuropathic pain [25,26]. In this study, we also observed a significant increase in the mechanical allodynia threshold with the use of amitriptyline in rats with neuropathic pain, and had planned the use of the selective A3AR antagonist MRS-1191 to determine the role of A3 receptor because we were confident that adenosine was involved in its effect.
In particular, A3ARs appear to have a complex role in inflammation because of their demonstrated pro-inflammatory and anti-inflammatory effects in many cases [27]. In another study, a selective A3AR agonist (IB-MECA) reduced inflammation in mouse models of colitis and reduced LPS-induced mortality in mice [28]. However, Sullivan et al. [29] speculated that the protective effects of IB-MECA against endotoxemia may actually be mediated by A2aARs because high doses of IB-MECA may activate both A2aARs and A3ARs. Conversely, another study reported that sometimes A3AR activation before renal ischemia led to the worsening of renal function, and that the mice lacking A3ARs displayed improved renal function after ischemia-reperfusion injury [30]. Therefore, the role of A3ARs in protecting against sepsis and inflammation is not clear.
Treatment of neuropathic pain is still difficult because of the inadequate understanding of the mechanisms involved in the development and maintenance of neuropathic pain and the relative absence of clinically effective therapies. Amitriptyline is a commonly used antidepressant for the treatment of neuropathic pain with multiple actions, some of which are still incompletely understood. While this diversity of action may provide amitriptyline a unique analgesic effect on chronic pain, it may also be a major factor limiting the use of this drug because it can interact with multiple mechanisms acting simultaneously.
In this study, we found that amitriptyline significantly reduced the amount of pERK1/2 and pCREB signaling proteins already induced in rats with neuropathic pain. Furthermore, amitriptyline significantly reduced the expression of the mRNAs of proinflammatory cytokines, which are markers of inflammatory response. However, the positive protective effect of amitriptyline on neuropathic pain was completely reversed by the subcutaneous injection of the selective A3AR antagonist. On the basis of these experimental results, we could confirm that the therapeutic effect of amitriptyline on neuropathic pain was caused by the activation of A3ARs.
In conclusion, amitriptyline, currently used as a treatment for neuropathic pain, is an excellent drug that can be effectively used to alleviate the symptoms of neuropathic pain owing to its unique anti-inflammatory action in addition to its various known actions. Moreover, the previously known antinociceptive effect of amitriptyline on the neuropathic pain resulting from SNL is closely associated with the activation of A3ARs as well as A1ARs and A2ARs.
References
- 1.Farghaly HS, Abd-Ellatief RB, Moftah MZ, Mostafa MG, Khedr EM, Kotb HI. The effects of dexmedetomidine alone and in combination with tramadol or amitriptyline in a neuropathic pain model. Pain Physician. 2014;17:187–95. [PubMed] [Google Scholar]
- 2.Jancálek R, Dubový P, Svízenská I, Klusáková I. Bilateral changes of TNF-alpha and IL-10 protein in the lumbar and cervical dorsal root ganglia following a unilateral chronic constriction injury of the sciatic nerve. J Neuroinflammation. 2010;7:11. doi: 10.1186/1742-2094-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Hulsebosch CE. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp Neurol. 2006;199:397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Ji RR, Gereau RW 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–48. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esser MJ, Sawynok J. Caffeine blockade of the thermal antihyperalgesic effect of acute amitriptyline in a rat model of neuropathic pain. Eur J Pharmacol. 2000;399:131–9. doi: 10.1016/s0014-2999(00)00336-8. [DOI] [PubMed] [Google Scholar]
- 6.Ulugol A, Karadag HC, Tamer M, Firat Z, Aslantas A, Dokmeci I. Involvement of adenosine in the anti-allodynic effect of amitriptyline in streptozotocin-induced diabetic rats. Neurosci Lett. 2002;328:129–32. doi: 10.1016/s0304-3940(02)00491-3. [DOI] [PubMed] [Google Scholar]
- 7.Esser MJ, Chase T, Allen GV, Sawynok J. Chronic administration of amitriptyline and caffeine in a rat model of neuropathic pain: multiple interactions. Eur J Pharmacol. 2001;430:211–8. doi: 10.1016/s0014-2999(01)01276-6. [DOI] [PubMed] [Google Scholar]
- 8.Katz NK, Ryals JM, Wright DE. Central or peripheral delivery of an adenosine A1 receptor agonist improves mechanical allodynia in a mouse model of painful diabetic neuropathy. Neuroscience. 2015;285:312–23. doi: 10.1016/j.neuroscience.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 10.Dubé GR, Kohlhaas KL, Rueter LE, Surowy CS, Meyer MD, Briggs CA. Loss of functional neuronal nicotinic receptors in dorsal root ganglion neurons in a rat model of neuropathic pain. Neurosci Lett. 2005;376:29–34. doi: 10.1016/j.neulet.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 11.Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol. 2006;291:R959–69. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- 12.Joo JD, Kim M, Horst P, Kim J, D'Agati VD, Emala CW Sr, et al. Acute and delayed renal protection against renal ischemia and reperfusion injury with A1 adenosine receptors. Am J Physiol Renal Physiol. 2007;293:F1847–57. doi: 10.1152/ajprenal.00336.2007. [DOI] [PubMed] [Google Scholar]
- 13.Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9:807–19. doi: 10.1016/S1474-4422(10)70143-5. [DOI] [PubMed] [Google Scholar]
- 14.Bridges D, Thompson SW, Rice AS. Mechanisms of neuropathic pain. Br J Anaesth. 2001;87:12–26. doi: 10.1093/bja/87.1.12. [DOI] [PubMed] [Google Scholar]
- 15.Baron R. Peripheral neuropathic pain: from mechanisms to symptoms. Clin J Pain. 2000;16(2 Suppl):S12–20. doi: 10.1097/00002508-200006001-00004. [DOI] [PubMed] [Google Scholar]
- 16.Faden AI, Simon RP. A potential role for excitotoxins in the pathophysiology of spinal cord injury. Ann Neurol. 1988;23:623–6. doi: 10.1002/ana.410230618. [DOI] [PubMed] [Google Scholar]
- 17.Song XS, Cao JL, Xu YB, He JH, Zhang LC, Zeng YM. Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats. Acta Pharmacol Sin. 2005;26:789–98. doi: 10.1111/j.1745-7254.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- 18.Joo JD, Choi JW, In JH, Jung HS, Lee JA, Kim YS, et al. Lidocaine suppresses the increased extracellular signal-regulated kinase/cyclic AMP response element-binding protein pathway and pro-inflammatory cytokines in a neuropathic pain model of rats. Eur J Anaesthesiol. 2011;28:106–11. doi: 10.1097/eja.0b013e32834050fb. [DOI] [PubMed] [Google Scholar]
- 19.Kwon SY, Yeom JH, Joo JD. Ketamine reduces the induced spinal p38 MAPK and pro-inflammatory cytokines in a neuropathic rats. Korean J Anesthesiol. 2014;66:52–8. doi: 10.4097/kjae.2014.66.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am J Physiol Renal Physiol. 2005;289:F369–76. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- 21.Lee HT, Gallos G, Nasr SH, Emala CW. A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol. 2004;15:102–11. doi: 10.1097/01.asn.0000102474.68613.ae. [DOI] [PubMed] [Google Scholar]
- 22.Lee HT, Xu H, Nasr SH, Schnermann J, Emala CW. A1 adenosine receptor knockout mice exhibit increased renal injury following ischemia and reperfusion. Am J Physiol Renal Physiol. 2004;286:F298–306. doi: 10.1152/ajprenal.00185.2003. [DOI] [PubMed] [Google Scholar]
- 23.Sawynok J. Adenosine receptor targets for pain. Neuroscience. 2016;338:1–18. doi: 10.1016/j.neuroscience.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 24.Feoktistov I, Biaggioni I. Role of adenosine A(2B) receptors in inflammation. Adv Pharmacol. 2011;61:115–44. doi: 10.1016/B978-0-12-385526-8.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ford GK, Moriarty O, Okine BN, Tully E, Mulcahy A, Harhen B, et al. Involvement of the endocannabinoid system in attentional modulation of nociceptive behaviour in rats. Eur J Pain. 2015;19:1177–85. doi: 10.1002/ejp.646. [DOI] [PubMed] [Google Scholar]
- 26.Janes K, Esposito E, Doyle T, Cuzzocrea S, Tosh DK, Jacobson KA, et al. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain. 2014;155:2560–7. doi: 10.1016/j.pain.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem. 2000;275:4429–34. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 28.Mabley J, Soriano F, Pacher P, Haskó G, Marton A, Wallace R, et al. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5'-Nmethyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–9. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan GW, Fang G, Linden J, Scheld WM. A2A adenosine receptor activation improves survival in mouse models of endotoxemia and sepsis. J Infect Dis. 2004;189:1897–904. doi: 10.1086/386311. [DOI] [PubMed] [Google Scholar]
- 30.Lee HT, Ota-Setlik A, Xu H, D'Agati VD, Jacobson MA, Emala CW. A3 adenosine receptor knockout mice are protected against ischemiaand myoglobinuria-induced renal failure. Am J Physiol Renal Physiol. 2003;284:F267–73. doi: 10.1152/ajprenal.00271.2002. [DOI] [PubMed] [Google Scholar]