

Abstract
Abnormal eye movements are commonly observed in movement disorders. Ocular motility examination should include bedside evaluation and laboratory recording of ocular misalignment, involuntary eye movements, including nystagmus and saccadic intrusions/oscillations, triggered nystagmus, saccades, smooth pursuit (SP), and the vestibulo-ocular reflex. Patients with Parkinson’s disease (PD) mostly show hypometric saccades, especially for the self-paced saccades, and impaired SP. Early vertical saccadic palsy is characteristic of progressive supranuclear palsy-Richardson’s syndrome. Patients with cortico-basal syndrome typically show a delayed onset of saccades. Downbeat and gaze-evoked nystagmus and hypermetric saccades are characteristic ocular motor findings in ataxic disorders due to cerebellar dysfunction. In this review, we discuss various ocular motor findings in movement disorders, including PD and related disorders, ataxic syndromes, and hyperkinetic movement disorders. Systemic evaluation of the ocular motor functions may provide valuable information for early detection and monitoring of movement disorders, despite an overlap in the abnormal eye movements among different movement disorders.
Keywords: Eye movements, parkinsonism, ataxia, saccades
Abnormal eye movements are common and occasionally a prominent finding in movement disorders [1,2]. These abnormal eye movements may follow or precede the motor symptoms of movement disorders. Accordingly, careful evaluation of the ocular motor functions may provide valuable information for early detection and monitoring of the disorders [3]. Patterns of abnormal eye movements may provide a key for differential diagnosis among the disorders presenting similar clinical features. Furthermore, an accurate scale of the abnormal ocular motor function helps to elucidate the mechanisms of abnormal eye movements and the pathophysiology of the underlying disorders [1,2].
Ocular motility examination should include bedside evaluation and laboratory recording of ocular misalignment, range of eye motion, involuntary eye movements, triggered nystagmus, saccades, smooth pursuit (SP), the vestibulo-ocular reflex (VOR), optokinetic nystagmus (OKN), and vergence eye movements [3,4]. Presence of any ocular misalignment should be confirmed with a cover test in the eccentric, as well as primary gaze, during both near and distant fixation. A moderate restriction of upward gaze is common in elderly individuals with or without parkinsonism [5]. Involuntary eye movements include nystagmus or inappropriate saccades. Nystagmus should be distinguished from saccadic intrusions and oscillations. In nystagmus, the eyes drift away from the fixation target by slow eye motion, while the eyes become off-target by saccades in saccadic intrusion oscillations. Nystagmus is called “jerky” when the returning eye movements are saccades, and “pendular” when they are slow. Saccadic intrusions and oscillations may be further divided according to the presence [e.g., square wave jerks (SWJs) and macrosaccadic oscillations] and absence (ocular flutter and opsoclonus) of the intersaccadic interval. For proper evaluation of spontaneous nystagmus, one should observe the direction and effects of gaze on the intensity and direction of the nystagmus. Nystagmus may be triggered or modulated by various maneuvers, such as eccentric gazes (gaze-evoked nystagmus, GEN), horizontal headshaking, and positional changes [6].
Saccades, both horizontal and vertical, should be assessed for latency, velocity, accuracy, and conjugacy [7]. SP may be evaluated by asking the patient to track a small target moving slowly in the horizontal or vertical direction with the head still. If the pursuit movement does not match the target velocity, corrective catch-up or back-up saccades would occur. Since many neural structures are involved in generation of SP, and various factors, including age, drugs, and alertness, can influence pursuit eye movements, impaired pursuit generally does not have a localizing value [7]. Induction of OKN may be useful in some patients who have difficulty initiating voluntary saccades. The VOR may be evaluated bedside using head impulse tests (HIT), visual enhancement of the VOR, and visual cancellation of the VOR [6]. Vergence is tested by having the patients look back and forth between a distant and a near target.
Although bedside evaluation is mostly sufficient to detect abnormal eye movements in movement disorders, laboratory recording of eye motion, mostly with video-oculography currently, may be utilized to detect subtle abnormalities or to quantify the impairments.
In this review, we will discuss various ocular motor findings in movement disorders including Parkinson’s disease (PD) and related disorders, ataxic syndromes, and hyperkinetic movement disorders.
This study followed the tenets of the Declaration of Helsinki and was approved by Institutional Review Board of Seoul National University Bundang Hospital (IRB number: N-1811-502-601).
PARKINSON’S DISEASE AND RELATED DISORDERS
Parkinson’s disease
In general, ocular motor deficits in PD are not as prominent as in progressive supranuclear palsy syndrome (PSPS) or Huntington’s disease (HD) and frequently require laboratory testing to bring out abnormalities [1]. In PD, steady fixation may be disrupted by saccadic intrusions, such as SWJs, that are characterized by involuntary saccadic movements from and back to the fixation point with an intersaccadic interval of approximately 200 ms and an amplitude of 0.5°–5° [8]. SWJs may be observed in normal elderly subjects [9], but the frequent and large SWJs in PD have been ascribed to compensatory increased activity in the frontal eye field [5,10].
Saccades are typically hypometric, especially vertically [5]. Downward gaze paresis is not seen in PD, and if it is present, one should consider the diagnosis of progressive supranuclear palsy-Richardson’s syndrome (PSP-RS) (Table 1) [2,11]. Most patients with PD have difficulty making self-paced saccades between two continuously visible targets. When patients are verbally instructed to look between two widely spaced targets, PD patients typically make near-accurate saccades. However, when they are instructed to maintain this activity on their own, their saccades invariably become hypometric [12]. Hypometric voluntary saccades are supposed to be caused by increased inhibition of the superior colliculus (SC) and reduced preoculomotor drive due to dysfunction of frontal-basal ganglia-SC circuits [5]. However, reflexive saccades can be generated by direct projections from the parietal cortex onto the saccade-related neurons in the intermediate layer of the SC [13]. The latency and velocity of reflexive saccades are usually normal in PD. Since patients with PD have impaired inhibition of reflexive saccades to visual stimuli, testing of antisaccades may reveal abnormal executive function, even in the early stage of PD [14]. In one study, errors in antisaccades were related to freezing of gait, and increased latency was associated with impaired postural control [15]. Impaired inhibition of unwanted saccades may be associated with dopaminergic depletion in the prefrontal cortex, resulting in lack of suppression of the SC by the basal ganglia.
Table 1.
Characteristics of ocular motor abnormalities in PD-related disorders
| Diagnosis | SI | Horizontal saccades | Vertical saccades | Smooth pursuit | Blepharospasm or eyelid apraxia |
|---|---|---|---|---|---|
| PD | + | Hypometric | Hypometric | Mild | Very rare |
| PSPS | ++ | Slowed, late | Slowed, early | Severe | Common |
| CBS | + | Delayed | Impaired, late | Mild | Common |
| MSA | + | Hypometric | Hypometric | Moderate | Rare |
PD: Parkinson’s disease, SI: saccadic intrusions, PSPS: progressive supranuclear palsy syndrome, CBS: corticobasal syndrome, MSA: multiple systemic atrophy.
SP is usually impaired in PD [5]. Combined eye-head tracking is abnormal to a similar degree as SP with the head stationary in most patients with PD [16]. Even though low-frequency rotational and caloric vestibular responses may be reduced in patients with PD [17], the VOR gain is close to 1.0 at higher frequencies of head rotation, corresponding to natural activities, especially visual fixation [18]. Patients also show abnormal eyelid movements, including reduced blinking, lid retraction and lid lag [19,20]. Unlike patients with PSP-RS, patients with PD habituate their blink responses when a flashlight is repetitively shone into the eyes [19].
Levodopa treatment may improve saccadic accuracy, SP, and convergence insufficiency [21-23]. Electrical stimulation of the pallidum or subthalamic nuclei was reported to improve performance on memory-guided or antisaccade tasks [24,25].
Progressive supranuclear palsy syndrome
PSPS, also known as the PSP-RS, is a clinical syndrome comprising vertical gaze palsy and balance difficulties with backward falls and mild dementia [26]. The disturbance of eye movements is usually present early in the course of the disease but occasionally develops later [27]. Patients with PSP-RS may present with visual complaints, such as blurred vision or photophobia [26].
Steady fixation is frequently disrupted by saccadic intrusions [28-30]. The SWJs are more common and larger in PSP-RS than in other parkinsonian disorders [27,28]. The prominent saccadic intrusions might be related to involvement of the SC and the adjacent central midbrain reticular formation [31].
Horizontal saccades are initially hypometric but normal in speed. As the disease progresses, they also become slow. Vertical saccades are slower than horizontal ones of similar size (Figure 1, Supplementary Video 1 in the online-only Data Supplement) [18,32]. The limitation of vertical saccades is more or less asymmetric [27]. Early on, vertical saccades may take a curved or oblique trajectory to the target (“round the houses” sign) [33]. During large-field, vertical optokinetic stimulation, PSP-RS patients often show tonic deviation of the eyes in the direction of stripe motion, with small or absent resetting quick phases [34]. Vertical saccades are generated by “burst neurons” in the midbrain but horizontal saccades are generated by burst neurons in the pons. Thus, the selective involvement of vertical saccades in the early stage of PSP-RS indicates that the initial pathology involves the midbrain burst neurons, or their local circuitry (SC and the adjacent central mesencephalic reticular formation) [32]. The latency of horizontal saccades may be prolonged in some patients with PSP-RS, but others retain the ability to make short-latency or “express” saccades [35]. Patients with PSP-RS also make errors in antisaccade tasks [35]. Both the presence of express saccades and errors in the antisaccade tasks suggest frontal lobe dysfunction [36].
Figure 1.
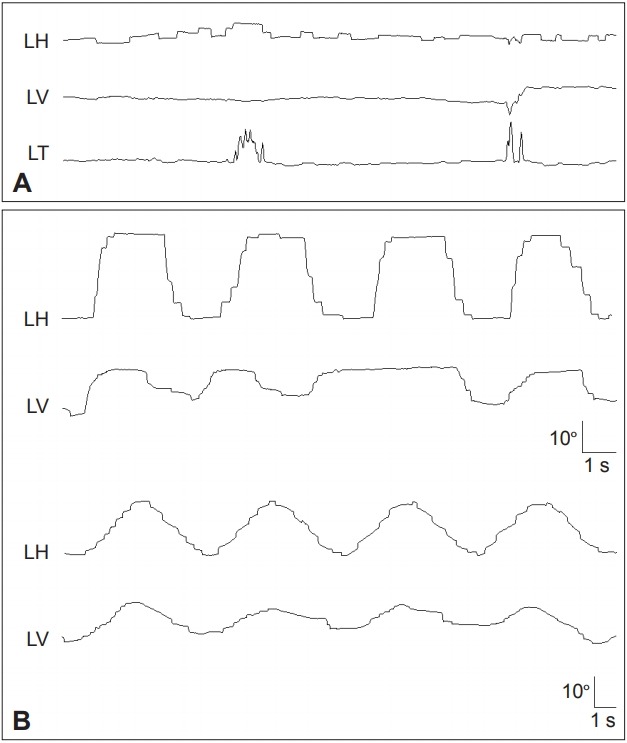
Frequent square wave jerks (A), slowed and hypometric saccades (B), and impaired SP are characteristic findings of progressive supranuclear palsy syndrome. Saccades and SP are more severely impaired in the vertical direction (B). LH: horizontal position of the left eye, LV: vertical position of the left eye, LT: torsional position of the left eye, SP: smooth pursuit.
Vertical SP or combined eye-head tracking may be relatively spared. Eventually, SP and saccades are both lost, constituting voluntary gaze palsy. The angular VOR is relatively preserved until the late stages of PSP-RS, however, the linear VOR is markedly impaired [37,38]. Bell’s phenomenon is usually absent. Convergence eye movements are commonly impaired [39]. Late in the disease, the ocular motor deficit progresses into a complete ophthalmoplegia.
Patients with PSP-RS may show several eyelid abnormalities, including blepharospasm, lid-opening or eye-closing apraxia, lid retraction, and lid lag [40,41]. Quite consistently, patients cannot inhibit a blink when a penlight is shone into the eyes (visual glabelar or Myerson’s sign) [40].
Other disorders
Patients with cortico-basal syndrome (CBS) frequently present with the clinical features of PSPS and may be misdiagnosed as having PSPS due to vertical supranuclear gaze palsy and falls within the first 2 years [18,42]. However, patients with CBS had a delayed onset of vertical supranuclear gaze palsy, i.e., more than 3 years after symptom onset, and had predominant downgaze abnormalities. Furthermore, patients with CBS show a significantly increased latency of horizontal saccades while the patients with PSPS mostly manifest with decreased saccadic velocity [18]. In the early stage of CBS, SP may be slow, but the range of movements is generally full. As the illness progresses, patients with CBS gradually lose the ability to make saccades to verbal commands, with retained spontaneous saccades and OKN [43]. Blepharospasm and eyelid opening apraxia may be observed [44].
Multiple systemic atrophy (MSA) is subdivided into cerebellar (MSA-C) and parkinsonian MSA (MSA-P) [45]. Patients with MSA-C may show saccadic intrusions, downbeat and gazeevoked nystagmus, perverted downbeat head-shaking nystagmus and positional downbeat nystagmus along with impaired smooth ocular and eye-head pursuit and saccadic dysmetria [46,47].
Dementia with Lewy bodies may be associated with vertical gaze palsy [48,49], but systematic measurements of vertical saccades are not available to date.
ATAXIC DISORDERS
Substantial advances have been made in defining and understanding the molecular biology of the hereditary ataxias [50]. With the identification of genes responsible for the hereditary ataxias, attempts have been made to identify distinctive syndromes of abnormal eye movements and to link those phenotypes with genotypes [51-54]. Even though findings are quite distinctive for some gene mutations causing ataxia, there is an overlap in the abnormal eye movements among different genotypes, and substantial phenotypic variation even in those with the same mutation [55].
There are three principal cerebellar syndromes; 1) the syndrome of the flocculus and paraflocculus, 2) the syndrome of the nodulus and ventral uvula, and 3) the syndrome of the dorsal vermis (lobules VI and VII) and underlying caudal fastigial nuclei. In addition, the cerebellar hemispheres contribute to the control of eye movements [56]. Understanding the characteristic eye movement abnormalities in each syndrome seems vital for interpretation of underlying cerebellar dysfunction in various ataxic disorders.
Lesions involving the flocculus and paraflocculus give rise to downbeat, and gaze-evoked and rebound nystagmus [57]. Inactivation of the flocculus severely impairs smooth tracking with the eyes alone, but active eye-head tracking is spared. Saccades are generally spared, but postsaccadic drift may be observed due to a pulse-step mismatch. Unilateral lesions produce ipsilateral deficits in pursuit and gaze holding. In patients with cerebellar disease, defects in SP, combined eye-head tracking, and gazeholding frequently occur together [58]. The VOR may by enhanced during low frequency and velocity stimulation, but may be impaired during HIT with high velocity and acceleration [59]. Adaptation of the VOR is also impaired in floccular lesions [60].
Lesions of the nodulus and ventral uvula generate positional nystagmus, especially downbeat and apogeotropic, periodic alternating nystagmus (PAN), perverted downbeat head-shaking nystagmus, increase in the duration of vestibular responses, and a failure of tilt-suppression of postrotatory nystagmus [61-67].
Experimental lesions of the dorsal vermis and fastigial nuclei cause saccadic dysmetria, typically hypometria if the vermis alone is involved, and hypermetria if the deep nuclei are involved [68]. Lesions involving the dorsal vermis also produce deficits of SP, especially at onset [69]. Patients with dorsal vermis lesions show moderate, ipsilateral deficits of sustained pursuit. Similarly, inactivation of one fastigial nucleus impairs the onset of contralateral pursuit but enhances acceleration of ipsilateral pursuit [69].
Spinocerebellar ataxia
Spinocerebellar Ataxia (SCA) is a group of autosomal dominant neurodegenerative disorders that are genetically and clinically heterogenous [50]. Previous reports have documented various abnormalities of eye movements in patients with SCA, providing clues for the differential diagnosis of SCA based on ocular motor phenotypes (Table 2) [51-54,70]. Due to a significant overlap in the phenotypes, however, caution is necessary in trying to assign a patient to one group on the basis of eye movement findings alone. Nonetheless, the presence of very slow saccades is suggestive of SCA2 (Figure 2), in which pontine saccadic burst neurons are involved. However, saccades may also be slow in patients with SCA1, SCA7, and SCA28. Impaired VOR is a common feature in SCA3. The presence of prominent downbeat, gaze-evoked, and rebound nystagmus with normal saccade velocity is typical of SCA6 and episodic ataxia type 2 (EA2).
Table 2.
Characteristic ocular motor abnormalities in ataxic disorders
| Diagnosis | Nystagmus or SI | Saccades abnormality | SP/OKN impairment | VOR gain |
|---|---|---|---|---|
| SCA1 | GEN | Moderately slow | Moderate | Normal |
| SCA2 | Severely slow | Mild | Normal | |
| SCA3 | Nystagmus, SWJ | Mildly slow | Moderate | Moderate |
| SCA6 | Downbeat, GEN, SWJ | Normal velocity | Severe | Normal |
| SCA7 | Nystagmus, SI | Severely slow in the late stage and hypo or hypermetria | Moderate | |
| EA2 | Downbeat GEN, positional nystagmus | Normal | Severe | Normal |
| FA | SWJ or ocular flutter | Normal velocity and prolonged latency | Impaired | Impaired |
SCA: spinocerebellar ataxia, EA2: episodic ataxia type 2, FA: Friedreich ataxia, SI: saccadic intrusions, GEN: gaze-evoked nystagmus, SP: smooth pursuit, SWJ: square wave jerks, OKN: optokinetic nystagmus, VOR: vestibulo-ocular reflex.
Figure 2.
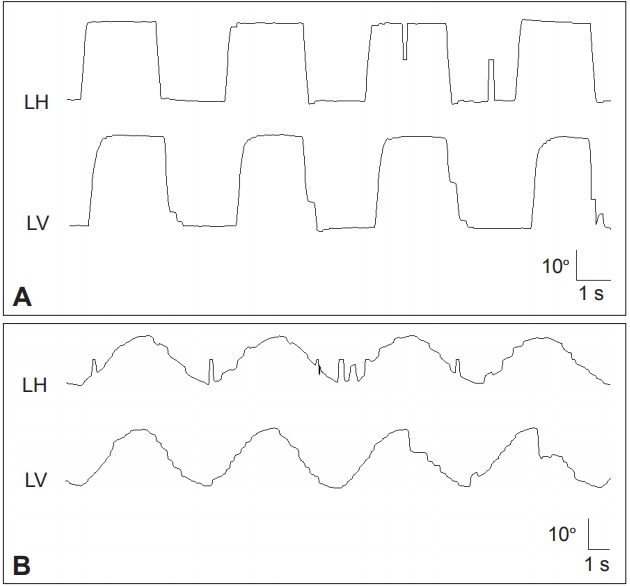
Patient with spinocerebellar ataxia type 2 shows slowed saccades especially in the vertical plane (A) and impaired smooth pursuit in both horizontal and vertical planes (B). LH: horizontal position of the left eye, LV: vertical position of the left eye.
In SCA1, GEN and pursuit abnormalities are frequently observed, and the prevalence of ophthalmoplegia is higher than that of other SCAs. The presence of ophthalmoplegia is correlated with higher ataxia score and poorer functional stage [70]. This indicates involvement of ocular motor neurons in later stages of SCA1. Since the characteristic pathological finding of SCA2 is the loss of burst neurons in the brainstem, saccadic slowing as well as increased saccadic latency is common in this disorder [71]. During follow-up, annual progression rates are significantly higher in patients with SCA2 [72]. SCA3, also named Machado-Joseph disease, is the most common form of autosomal dominant ataxia. Nystagmus is present in 88% of patients with SCA3 and the symptomatic patients with nystagmus tend to have a higher length of the CAG tract in the expanded allele [73]. Saccadic intrusions such as SWJs are frequent in SCA3. SCA6 is regarded a pure cerebellar form of SCA [74,75]. Downbeat and gaze-evoked nystagmus are frequent in SCA6 [76]. Perverted downbeat head shaking nystagmus may be a distinct feature of SCA6 [76]. The VOR gain during horizontal HIT shows a negative correlation with the ataxia score while the VOR gain in response to relatively low frequency stimuli remains normal or increased regardless of disease severity (Figure 3) [77]. SCA7 is characterized by retinal degeneration and ophthalmoplegia [78]. Oculomotor findings of SCA7 include nystagmus, saccadic intrusions, saccadic hypoand hypermetria, slow saccades, impaired SP and VOR cancellation, and late in the illness, ophthalmoparesis and ophthalmoplegia [79]. SCA31 is also a pure cerebellar form of ataxia, making it difficult to distinguish SCA31 from SCA6 based on clinical findings only. The onset age is slightly higher, and the frequency of positional downbeat nystagmus is lower, in SCA31 than in SCA6 [80].
Figure 3.

Results of the vestibular function tests in a representative patient with spinocerebellar ataxia type 6. A. Caloric tests. The sum of the peak SPV of the nystagmus in response to each caloric stimulus is increased at 174°/s (normal range=45–157°/s). B. Rotatory chair test. The gains of the VOR are normal during sinusoidal horizontal accelerations. Gray areas represent the normal ranges (mean ± 2SD). C. Head impulse tests. Head impulses in the plane of each SCC revealed decreased gain of the VOR for all six semicircular canals with overt catch-up saccades during stimulation of the horizontal and posterior SCCs. The blue lines indicate head velocity and the red lines represent eye velocity. D. The head impulse VOR gain for the HC is negatively correlated with the ataxia score as measured using the ICARS. SPV: slow-phase velocity, VOR: vestibulo-ocular reflex, SCC: semicircular canal, LAC: left anterior SCC, LHC: left horizontal SCC, LPC: left posterior SCC, RAC: right anterior SCC, RHC: right horizontal SCC, RPC: right posterior SCC, ICARS: International Cooperative Ataxia Rating Scale, HIT-HC: head impulse testing in the horizontal semicircular canal plane.
Friedreich ataxia
Friedreich ataxia (FA) is the most common cause of autosomal recessive ataxias with an onset usually before age 20 years. FA is characterized by ataxia, hyporeflexia, extensor plantar reflexes, neuropathy, cardiomyopathy, and diabetes. FA is mostly due to an unstable GAA repeat expansion within intron 1 of frataxin [81]. Abnormal ocular motor findings of FA include fixation instability manifesting as SWJs and ocular flutter [82,83]. While saccadic velocity is essentially normal, saccadic latency is prolonged. The latency correlates with clinical measures of disease severity. Saccades may be both hypo- and hypermetric. SP and the VOR may be impaired [84-86]. Caloric tests are abnormal in the majority of FA patients [85,87]. Thus, severe vestibulopathy with essentially normal saccadic velocity are hallmarks of FA and differentiate it from a number of dominant SCA [82].
Episodic ataxia
Episodic ataxia (EA) is a genetic disorder characterized by recurrent episodes of truncal ataxia and incoordination lasting minutes to hours [88]. EA is a heterogeneous group of disorders mostly with an autosomal dominant inheritance [89,90]. Several subtypes have been defined according to clinical and genetic characteristics [91]. Among the subtypes, EA2 is most common. Patients with EA2 mostly suffer from recurrent ataxia and slurred speech for several hours [88]. Even between the attacks, downbeat, gazeevoked, rebound and positional nystagmus are typically observed (Figure 4) [92]. The onset is mostly before the age of 20 years, but may be delayed [93]. The common triggers of the attacks are exercise, and physical or emotional stress [94]. Acetazolamide and aminopyridine have been found to reduce the frequency of attacks and improve the quality of life in patients with EA2 [88].
Figure 4.
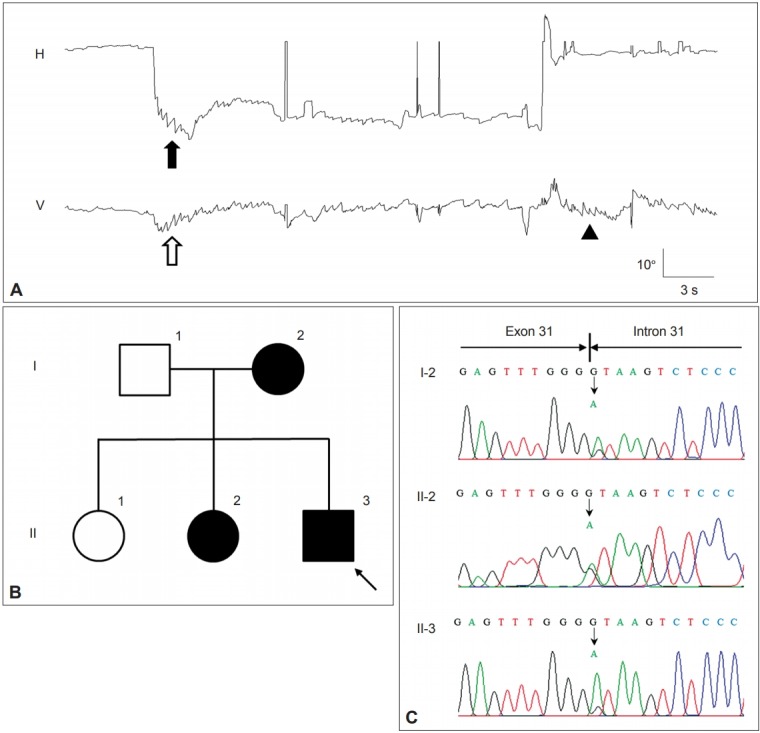
Findings in a patient with episodic ataxia type 2. A: The patient shows gaze-evoked (filled arrow) and downbeat nystagmus (open arrow) during leftward gaze that decrease gradually. On resuming the neutral position (arrow head), upbeat nystagmus develops without rebound nystagmus in the horizontal plane. Upward deflection indicates rightward and upward eye motion. H: horizontal eye position, V: vertical eye position. B: Pedigree of the patient. Squares males, circles females, open symbols unaffected, solid symbols affected. C. The chromatograms of a part of exon 31 and intron 31 of CACNA1A show a heterozygous point mutation at the splice donor site (c.4953+1G>A).
Ataxia telangiectasia
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized clinically by progressive ataxia, dystonia, parkinsonism, choreoathetosis, myoclonus, tremor, and ocular and cutaneous telangiectasia [95-97]. Patients with AT also have immunodeficiency, endocrine abnormalities, radiosensitivity, and a predisposition to neoplasia [98]. Eye movement abnormalities in AT include 1) spontaneous, gaze-evoked and periodic alternating nystagmus, microsaccadic oscillations, and SWJs [99], 2) impaired generation of volitional and reflexive saccades (characterized by prolonged latency and hypometria) in association with head “thrusts” during attempted shifts of gaze, and 3) impairment of pursuit eye movements, VOR and convergence [100,101]. These abnormal eye movements most likely result from dysfunction of the cerebellar flocculus and paraflocculus, and from abnormal supranuclear control of the SC due to dysfunction of the cerebellar vermis or the basal ganglia.
Ataxia-oculomotor apraxia
Ataxia-oculomotor apraxia (AOA) is an autosomal recessive disorder characterized by early onset ataxia, choreoathetosis, and ocular motor apraxia mimicking AT but without extraneurological features of this disease [102]. AOA type 1 (AOA1) is caused by mutations in the aprataxin gene (APTX) on chromosome 9p13 [103]. Aprataxin is a nuclear protein that repairs single-strand DNA breaks [104,105]. Patients with AOA1 also may show hypercholesterolemia, hypoalbuminaemia, and deficiency of muscle coenzyme Q10 [106,107]. Most patients with AOA1 show saccade initiation difficulties. When the head is restrained, patients make a series of small saccades (horizontal or vertical) at normal latency to shift their gaze. Although small, the saccades are normal in speed for their size. When the head is free, patients make large head movements that overshoot the target, with their eyes in contraversion, and then rotating their head back to bring their eyes back to the central position in the orbit. Other findings include SWJs, gaze-evoked nystagmus, and impaired SP [108].
AOA type 2 (AOA2) is caused by mutations of the senataxin gene on chromosome 9q34, which also causes a failure to repair single-strand DNA breaks [109-112]. The clinical features of AOA2 are similar to those of AOA1 with prominent hypometria of horizontal and vertical saccades, requiring a staircase of saccades to shift the gaze, but with normal saccadic latency and speed [113]. Other ocular motor findings include SWJs, gaze-evoked, rebound, and downbeat nystagmus, impaired SP, vergence dysfunction with exophoria, and normal or increased vestibulo-ocular responses. However, head movements are not prominent in AOA2 and are not used to shift gaze by overshooting the target [113]. Patients also show impaired antisaccades and memoryguided saccades [113].
Paraneoplastic cerebellar degeneration
Patients with paraneoplastic cerebellar degeneration (PCD) usually present with rapidly progressive dizziness and imbalance. In addition to gait and appendicular ataxia, examination almost always shows abnormal eye movements due to cerebellar dysfunction, which include downbeat, gaze-evoked, seesaw and positional nystagmus, ocular flutter, opsoclonus, saccadic dysmetria, and impaired SP (Figure 5) [63,114-117].
Figure 5.

Findings in a patient with paraneoplastic cerebellar degeneration. A and B: Recording of eye movements using 3-dimensional video-oculography shows spontaneous downbeat nystagmus (DBN) with decreasing slow phases, which increases without fixation (A). DBN increases during lateral gazes with horizontal gaze-evoked (hollow arrows), centripetal (arrows), and rebound nystagmus (arrow heads) (B). C and D: Whole body 2-deoxy-2-[F18]fluoro-D-glucose-positron emission tomography (FDG-PET) (C) and chest CT (D) show a hypermetabolic mass in the right lower lobe of the lung (arrows). E: Brain FDG-PET reveals increased metabolism, especially in the nodulus. F: The hypermetabolism observed on the initial PET disappeared after right lung lobectomy and chemotherapy.
HUNTINGTON’S DISEASE
Patients with HD often show impaired initiation of saccades with prolonged latencies, especially when the saccade is made to command or in anticipation of a predictable target [118]. Saccadic latency may be used as a marker of disease progression given its direct correlation with the severity of HD [119,120]. Patients frequently use an obligatory blink or head turn to start eye movements [118,121]. Paradoxically, patients often cannot suppress saccades during fixation, with increased visual distractibility [122]. Saccades to visual stimuli are made at normal latency, while those made to command are delayed [120]. Patients with HD also make increased errors during the antisaccade task. In preclinical stages of HD, increase in saccadic latency and error rates may predict the age of symptom onset [123]. In some patients, saccades may be slow in both horizontal and vertical planes. SP may also be impaired, but gaze holding and the VOR are well-preserved [121]. Fixation may be abnormal due to saccadic intrusions (Figure 6) [121].
Figure 6.
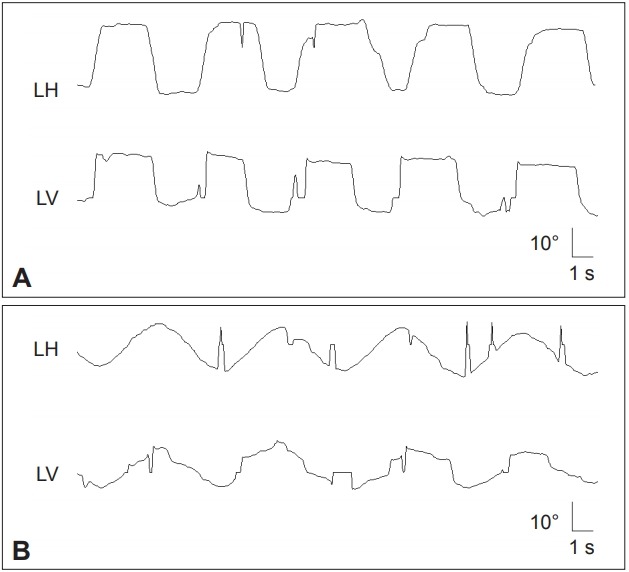
In a patient with Huntington’s disease, the horizontal saccades are markedly slowed (A). In the vertical plane, slowing is more prominent during downward saccades. In contrast, vertical SP is severely impaired while the horizontal SP is relatively preserved (B). LH: horizontal position of the left eye, LV: vertical position of the left eye, SP: smooth pursuit.
OTHER DISORDERS
Essential tremor
Compared with normal controls, patients with essential tremor more frequently show perverted downbeat head-shaking nystagmus, positional downbeat nystagmus, reduced gain of pursuit initiation, and impaired suppression of postrotational nystagmus with head tilt, all suggestive of underlying cerebellar dysfunction [124,125]. Another study reported higher prevalence of slowed SWJs and delayed saccades, which were independent of disease duration, tremor severity, and medication [126].
Wilson’s disease
Detection of Kayser-Fleischer rings is diagnostic of Wilson’s disease. Ocular motor abnormalities in Wilson’s disease include distractibility of gaze, slow vertical saccades, and lid-opening apraxia (Figure 7) [127,128].
Figure 7.
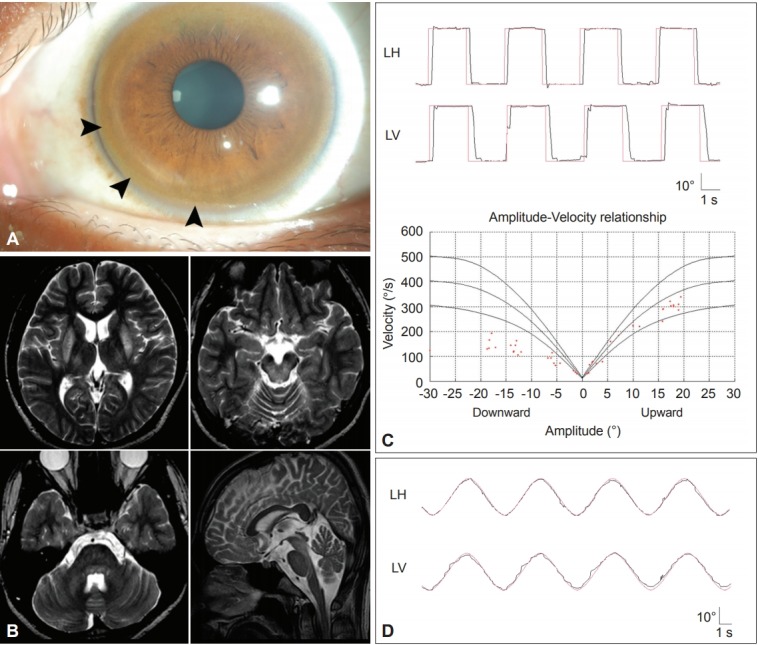
Findings in a patient with Wilson’s disease. A: Kayser-Fleischer ring. Color external photograph shows Kayser-Fleischer ring (arrows), a golden-brown discoloration of the cornea due to deposition of sulfur-copper complexes within the Descemet’s membrane. B: T2-weighted MRIs reveal symmetrical high signal intensity lesions in the putamina, thalami, midbrain and pontine tegmentum. C: Video-oculographic recording (SMI®, Teltow, Germany) of saccades shows selective slowing of downward saccades while the velocities of horizontal and upward saccades are normal. D: Smooth pursuit is impaired in the vertical direction. LH: horizontal position of the left eye, LV: vertical position of the left eye.
Whipple’s disease
Whipple’s disease may mimic parkinsonism or PSPS by involving the basal ganglia and rostral mesencephalon [129]. Initially, vertical saccades may be slow and curved while horizontal saccades may be relatively preserved. It is almost always accompanied by a supranuclear vertical gaze palsy. Eventually, all eye movements may be lost. Oculomasticatory myorhythmia is virtually pathognomonic of this disease, and consists of rhythmic movements of the masticatory and occasionally other skeletal muscles, synchronized with pendular vergence oscillations of the eyes [130]. However, many patients with Whipple’s disease have no ocular oscillations.
CONCLUSIONS
Patients with movement disorders show nearly all kinds of eye movement abnormalities. In general, increased SI, increased saccadic latency and saccadic hypometria, and impaired SP are quite common in movement disorders and have little differential diagnostic value among the disorders. However, saccadic slowing indicates damage to the burst neurons in the pons (horizontal) and mesodiencephalic junction (vertical) and their outflow tracts and thus characterizes the disorders involving the brainstem. This phenomenon is also observed in PSPS and MSA. Decreased VOR also suggests damage to the brainstem and peripheral vestibular organs, In contrast, downbeat, gaze-evoked, rebound and positional nystagmus, hypermetric saccades, and hyperactive VOR are ocular motor findings specific for cerebellar dysfunction. Thus, despite the wide overlap in ocular motor abnormalities among movement disorders, scrutinized evaluation of all subclasses of eye movements would provide valuable information on the underlying pathology, differential diagnosis and monitoring of disease progression.
Footnotes
Conflicts of Interest
The authors have no financial conflicts of interest.
Supplementary Materials
The online-only Data Supplement is available with this article at http://dx.doi.org/10.14802/jmd.18034.
Video 1.
Saccades are severely impaired in the vertical direction in progressive supranuclear palsy syndrome.
REFERENCES
- 1.Pinkhardt EH, Kassubek J. Ocular motor abnormalities in Parkinsonian syndromes. Parkinsonism Relat Disord. 2011;17:223–230. doi: 10.1016/j.parkreldis.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Antoniades CA, Kennard C. Ocular motor abnormalities in neurodegenerative disorders. Eye (Lond) 2015;29:200–207. doi: 10.1038/eye.2014.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leigh JR, Zee DS. The neurology of eye movements. 5th ed. London: Oxford University Press; 2015. [Google Scholar]
- 4.Jung I, Kim JS. Approach to dizziness in the emergency department. Clin Exp Emerg Med. 2015;2:75–88. doi: 10.15441/ceem.15.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White OB, Saint-Cyr JA, Tomlinson RD, Sharpe JA. Ocular motor deficits in Parkinson’s disease: II. control of the saccadic and smooth pursuit systems. Brain. 1983;106(Pt 3):571–587. doi: 10.1093/brain/106.3.571. [DOI] [PubMed] [Google Scholar]
- 6.Huh YE, Kim JS. Bedside evaluation of dizzy patients. J Clin Neurol. 2013;9:203–213. doi: 10.3988/jcn.2013.9.4.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaymard B, Pierrot-Deseilligny C. Neurology of saccades and smooth pursuit. Curr Opin Neurol. 1999;12:13–19. doi: 10.1097/00019052-199902000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Herishanu YO, Sharpe JA. Normal square wave jerks. Invest Ophthalmol Vis Sci. 1981;20:268–272. [PubMed] [Google Scholar]
- 9.Shallo-Hoffmann J, Sendler B, Muhlendyck H. Normal square wave jerks in differing age groups. Invest Ophthalmol Vis Sci. 1990;31:1649–1652. [PubMed] [Google Scholar]
- 10.Sharpe JA, Herishanu YO, White OB. Cerebral square wave jerks. Neurology. 1982;32:57–62. doi: 10.1212/wnl.32.1.57. [DOI] [PubMed] [Google Scholar]
- 11.Anderson T, Luxon L, Quinn N, Daniel S, David Marsden C, Bronstein A. Oculomotor function in multiple system atrophy: clinical and laboratory features in 30 patients. Mov Disord. 2008;23:977–984. doi: 10.1002/mds.21999. [DOI] [PubMed] [Google Scholar]
- 12.Terao Y, Fukuda H, Ugawa Y, Hikosaka O. New perspectives on the pathophysiology of Parkinson’s disease as assessed by saccade performance: a clinical review. Clin Neurophysiol. 2013;124:1491–1506. doi: 10.1016/j.clinph.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.May PJ. The mammalian superior colliculus: laminar structure and connections. Prog Brain Res. 2006;151:321–378. doi: 10.1016/S0079-6123(05)51011-2. [DOI] [PubMed] [Google Scholar]
- 14.Antoniades CA, Demeyere N, Kennard C, Humphreys GW, Hu MT. Antisaccades and executive dysfunction in early drug-naive Parkinson’s disease: the discovery study. Mov Disord. 2015;30:843–847. doi: 10.1002/mds.26134. [DOI] [PubMed] [Google Scholar]
- 15.Walton CC, O’Callaghan C, Hall JM, Gilat M, Mowszowski L, Naismith SL, et al. Antisaccade errors reveal cognitive control deficits in Parkinson’s disease with freezing of gait. J Neurol. 2015;262:2745–2754. doi: 10.1007/s00415-015-7910-5. [DOI] [PubMed] [Google Scholar]
- 16.White OB, Saint-Cyr JA, Tomlinson RD, Sharpe JA. Ocular motor deficits in Parkinson’s disease. III. Coordination of eye and head movements. Brain. 1988;111(Pt 1):115–129. doi: 10.1093/brain/111.1.115. [DOI] [PubMed] [Google Scholar]
- 17.White OB, Saint-Cyr JA, Sharpe JA. Ocular motor deficits in Parkinson’s disease. I. The horizontal vestibulo-ocular reflex and its regulation. Brain. 1983;106(Pt 3):555–570. doi: 10.1093/brain/106.3.555. [DOI] [PubMed] [Google Scholar]
- 18.Rottach KG, Riley DE, DiScenna AO, Zivotofsky AZ, Leigh RJ. Dynamic properties of horizontal and vertical eye movements in parkinsonian syndromes. Ann Neurol. 1996;39:368–377. doi: 10.1002/ana.410390314. [DOI] [PubMed] [Google Scholar]
- 19.Litvan I. Atypical pakinsonian disorders-clinical and research aspects. New York: Humana Press; 2005. [Google Scholar]
- 20.Corin MS, Elizan TS, Bender MB. Oculomotor function in patients with Parkinson’s disease. J Neurol Sci. 1972;15:251–265. doi: 10.1016/0022-510x(72)90068-8. [DOI] [PubMed] [Google Scholar]
- 21.Sharpe JA, Fletcher WA, Lang AE, Zackon DH. Smooth pursuit during dose-related on-off fluctuations in Parkinson’s disease. Neurology. 1987;37:1389–1392. doi: 10.1212/wnl.37.8.1389. [DOI] [PubMed] [Google Scholar]
- 22.Gibson JM, Pimlott R, Kennard C. Ocular motor and manual tracking in Parkinson’s disease and the effect of treatment. J Neurol Neurosurg Psychiatry. 1987;50:853–860. doi: 10.1136/jnnp.50.7.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Racette BA, Gokden MS, Tychsen LS, Perlmutter JS. Convergence insufficiency in idiopathic Parkinson’s disease responsive to levodopa. Strabismus. 1999;7:169–174. doi: 10.1076/stra.7.3.169.636. [DOI] [PubMed] [Google Scholar]
- 24.Antoniades CA, Rebelo P, Kennard C, Aziz TZ, Green AL, FitzGerald JJ. Pallidal deep brain stimulation improves higher control of the oculomotor system in Parkinson’s disease. J Neurosci. 2015;35:13043–13052. doi: 10.1523/JNEUROSCI.2317-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antoniades CA, Carpenter RH, Temel Y. Deep brain stimulation of the subthalamic nucleus in Parkinson’s disease: similar improvements in saccadic and manual responses. Neuroreport. 2012;23:179–183. doi: 10.1097/WNR.0b013e32834f6daa. [DOI] [PubMed] [Google Scholar]
- 26.Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8:270–279. doi: 10.1016/S1474-4422(09)70042-0. [DOI] [PubMed] [Google Scholar]
- 27.Chen AL, Riley DE, King SA, Joshi AC, Serra A, Liao K, et al. The disturbance of gaze in progressive supranuclear palsy: implications for pathogenesis. Front Neurol. 2010;1:147. doi: 10.3389/fneur.2010.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otero-Millan J, Schneider R, Leigh RJ, Macknik SL, Martinez-Conde S. Saccades during attempted fixation in parkinsonian disorders and recessive ataxia: from microsaccades to square-wave jerks. PLoS One. 2013;8:e58535. doi: 10.1371/journal.pone.0058535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rascol O, Sabatini U, Simonetta-Moreau M, Montastruc JL, Rascol A, Clanet M. Square wave jerks in parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 1991;54:599–602. doi: 10.1136/jnnp.54.7.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee ES, Choi JY, Kim JS. Teaching video neuroimages: alternating horizontal single saccadic pulses in progressive supranuclear palsy. Neurology. 2017;88:e32–e33. doi: 10.1212/WNL.0000000000003520. [DOI] [PubMed] [Google Scholar]
- 31.Otero-Millan J, Serra A, Leigh RJ, Troncoso XG, Macknik SL, Martinez-Conde S. Distinctive features of saccadic intrusions and microsaccades in progressive supranuclear palsy. J Neurosci. 2011;31:4379–4387. doi: 10.1523/JNEUROSCI.2600-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhidayasiri R, Riley DE, Somers JT, Lerner AJ, Buttner-Ennever JA, Leigh RJ. Pathophysiology of slow vertical saccades in progressive supranuclear palsy. Neurology. 2001;57:2070–2077. doi: 10.1212/wnl.57.11.2070. [DOI] [PubMed] [Google Scholar]
- 33.Quinn N. The “round the houses” sign in progressive supranuclear palsy. Ann Neurol. 1996;40:951. doi: 10.1002/ana.410400630. [DOI] [PubMed] [Google Scholar]
- 34.Garbutt S, Riley DE, Kumar AN, Han Y, Harwood MR, Leigh RJ. Abnormalities of optokinetic nystagmus in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2004;75:1386–1394. doi: 10.1136/jnnp.2003.027367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pierrot-Deseilligny C, Rivaud S, Pillon B, Fournier E, Agid Y. Lateral visually-guided saccades in progressive supranuclear palsy. Brain. 1989;112(Pt 2):471–487. doi: 10.1093/brain/112.2.471. [DOI] [PubMed] [Google Scholar]
- 36.Goffinet AM, De Volder AG, Gillain C, Rectem D, Bol A, Michel C, et al. Positron tomography demonstrates frontal lobe hypometabolism in progressive supranuclear palsy. Ann Neurol. 1989;25:131–139. doi: 10.1002/ana.410250205. [DOI] [PubMed] [Google Scholar]
- 37.Das VE, Leigh RJ. Visual-vestibular interaction in progressive supranuclear palsy. Vision Res. 2000;40:2077–2081. doi: 10.1016/s0042-6989(00)00046-8. [DOI] [PubMed] [Google Scholar]
- 38.Liao K, Wagner J, Joshi A, Estrovich I, Walker MF, Strupp M, et al. Why do patients with PSP fall? Evidence for abnormal otolith responses. Neurology. 2008;70:802–809. doi: 10.1212/01.wnl.0000304134.33380.1e. [DOI] [PubMed] [Google Scholar]
- 39.Kitthaweesin K, Riley DE, Leigh RJ. Vergence disorders in progressive supranuclear palsy. Ann N Y Acad Sci. 2002;956:504–507. doi: 10.1111/j.1749-6632.2002.tb02867.x. [DOI] [PubMed] [Google Scholar]
- 40.Golbe LI, Davis PH, Lepore FE. Eyelid movement abnormalities in progressive supranuclear palsy. Mov Disord. 1989;4:297–302. doi: 10.1002/mds.870040402. [DOI] [PubMed] [Google Scholar]
- 41.Dehaene I. Apraxia of eyelid opening in progressive supranuclear palsy. Ann Neurol. 1984;15:115–116. doi: 10.1002/ana.410150129. [DOI] [PubMed] [Google Scholar]
- 42.Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496–503. doi: 10.1212/WNL.0b013e31827f0fd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rinne JO, Lee MS, Thompson PD, Marsden CD. Corticobasal degeneration. A clinical study of 36 cases. Brain. 1994;117(Pt 5):1183–1196. doi: 10.1093/brain/117.5.1183. [DOI] [PubMed] [Google Scholar]
- 44.Riley DE, Lang AE, Lewis A, Resch L, Ashby P, Hornykiewicz O, et al. Cortical-basal ganglionic degeneration. Neurology. 1990;40:1203–1212. doi: 10.1212/wnl.40.8.1203. [DOI] [PubMed] [Google Scholar]
- 45.Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372:249–263. doi: 10.1056/NEJMra1311488. [DOI] [PubMed] [Google Scholar]
- 46.Bertholon P, Bronstein AM, Davies RA, Rudge P, Thilo KV. Positional down beating nystagmus in 50 patients: cerebellar disorders and possible anterior semicircular canalithiasis. J Neurol Neurosurg Psychiatry. 2002;72:366–372. doi: 10.1136/jnnp.72.3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JY, Lee WW, Kim JS, Kim HJ, Kim JK, Jeon BS. Perverted headshaking and positional downbeat nystagmus in patients with multiple system atrophy. Mov Disord. 2009;24:1290–1295. doi: 10.1002/mds.22559. [DOI] [PubMed] [Google Scholar]
- 48.Brett FM, Henson C, Staunton H. Familial diffuse Lewy body disease, eye movement abnormalities, and distribution of pathology. Arch Neurol. 2002;59:464–467. doi: 10.1001/archneur.59.3.464. [DOI] [PubMed] [Google Scholar]
- 49.de Bruin VM, Lees AJ, Daniel SE. Diffuse Lewy body disease presenting with supranuclear gaze palsy, parkinsonism, and dementia: a case report. Mov Disord. 1992;7:355–358. doi: 10.1002/mds.870070410. [DOI] [PubMed] [Google Scholar]
- 50.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. doi: 10.1016/S1474-4422(10)70183-6. [DOI] [PubMed] [Google Scholar]
- 51.Buttner N, Geschwind D, Jen JC, Perlman S, Pulst SM, Baloh RW. Oculomotor phenotypes in autosomal dominant ataxias. Arch Neurol. 1998;55:1353–1357. doi: 10.1001/archneur.55.10.1353. [DOI] [PubMed] [Google Scholar]
- 52.Pula JH, Gomez CM, Kattah JC. Ophthalmologic features of the common spinocerebellar ataxias. Curr Opin Ophthalmol. 2010;21:447–453. doi: 10.1097/ICU.0b013e32833eaf71. [DOI] [PubMed] [Google Scholar]
- 53.Zee DS, Yee RD, Cogan DG, Robinson DA, Engel WK. Ocular motor abnormalities in hereditary cerebellar ataxia. Brain. 1976;99:207–234. doi: 10.1093/brain/99.2.207. [DOI] [PubMed] [Google Scholar]
- 54.Moschner C, Perlman S, Baloh RW. Comparison of oculomotor findings in the progressive ataxia syndromes. Brain. 1994;117(Pt 1):15–25. doi: 10.1093/brain/117.1.15. [DOI] [PubMed] [Google Scholar]
- 55.Durr A, Chneiweiss H, Khati C, Stevanin G, Cancel G, Feingold J, et al. Phenotypic variability in autosomal dominant cerebellar ataxia type I is unrelated to genetic heterogeneity. Brain. 1993;116(Pt 6):1497–1508. doi: 10.1093/brain/116.6.1497. [DOI] [PubMed] [Google Scholar]
- 56.Lewis RF, Zee DS. Ocular motor disorders associated with cerebellar lesions: pathophysiology and topical localization. Rev Neurol (Paris) 1993;149:665–677. [PubMed] [Google Scholar]
- 57.Zee DS, Yamazaki A, Butler PH, Gücer G. Effects of ablation of flocculus and paraflocculus of eye movements in primate. J Neurophysiol. 1981;46:878–899. doi: 10.1152/jn.1981.46.4.878. [DOI] [PubMed] [Google Scholar]
- 58.Zee DS, Leigh RJ, Mathieu-Millaire F. Cerebellar control of ocular gaze stability. Ann Neurol. 1980;7:37–40. doi: 10.1002/ana.410070108. [DOI] [PubMed] [Google Scholar]
- 59.Park HK, Kim JS, Strupp M, Zee DS. Isolated floccular infarction: impaired vestibular responses to horizontal head impulse. J Neurol. 2013;260:1576–1582. doi: 10.1007/s00415-013-6837-y. [DOI] [PubMed] [Google Scholar]
- 60.Lisberger SG, Miles FA, Zee DS. Signals used to compute errors in monkey vestibuloocular reflex: possible role of flocculus. J Neurophysiol. 1984;52:1140–1153. doi: 10.1152/jn.1984.52.6.1140. [DOI] [PubMed] [Google Scholar]
- 61.Huh YE, Kim JS. Patterns of spontaneous and head-shaking nystagmus in cerebellar infarction: imaging correlations. Brain. 2011;134(Pt 12):3662–3671. doi: 10.1093/brain/awr269. [DOI] [PubMed] [Google Scholar]
- 62.Lee SU, Choi JY, Kim HJ, Park JJ, Zee DS, Kim JS. Impaired tilt suppression of post-rotatory nystagmus and cross-coupled head-shaking nystagmus in cerebellar lesions: image mapping study. Cerebellum. 2017;16:95–102. doi: 10.1007/s12311-016-0772-2. [DOI] [PubMed] [Google Scholar]
- 63.Choi SY, Park SH, Kim HJ, Kim JS. Paraneoplastic downbeat nystagmus associated with cerebellar hypermetabolism especially in the nodulus. J Neurol Sci. 2014;343:187–191. doi: 10.1016/j.jns.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 64.Jeong HS, Oh JY, Kim JS, Kim J, Lee AY, Oh SY. Periodic alternating nystagmus in isolated nodular infarction. Neurology. 2007;68:956–957. doi: 10.1212/01.wnl.0000257111.24769.d2. [DOI] [PubMed] [Google Scholar]
- 65.Moon IS, Kim JS, Choi KD, Kim MJ, Oh SY, Lee H, et al. Isolated nodular infarction. Stroke. 2009;40:487–491. doi: 10.1161/STROKEAHA.108.527762. [DOI] [PubMed] [Google Scholar]
- 66.Nam J, Kim S, Huh Y, Kim JS. Ageotropic central positional nystagmus in nodular infarction. Neurology. 2009;73:1163. doi: 10.1212/WNL.0b013e3181bacfde. [DOI] [PubMed] [Google Scholar]
- 67.Oh YM, Choi KD, Oh SY, Kim JS. Periodic alternating nystagmus with circumscribed nodular lesion. Neurology. 2006;67:399. doi: 10.1212/01.wnl.0000219818.35451.10. [DOI] [PubMed] [Google Scholar]
- 68.Takagi M, Tamargo R, Zee DS. Effects of lesions of the cerebellar oculomotor vermis on eye movements in primate: binocular control. Prog Brain Res. 2003;142:19–33. doi: 10.1016/S0079-6123(03)42004-9. [DOI] [PubMed] [Google Scholar]
- 69.Takagi M, Zee DS, Tamargo RJ. Effects of lesions of the oculomotor cerebellar vermis on eye movements in primate: smooth pursuit. J Neurophysiol. 2000;83:2047–2062. doi: 10.1152/jn.2000.83.4.2047. [DOI] [PubMed] [Google Scholar]
- 70.Moscovich M, Okun MS, Favilla C, Figueroa KP, Pulst SM, Perlman S, et al. Clinical evaluation of eye movements in spinocerebellar ataxias: a prospective multicenter study. J Neuroophthalmol. 2015;35:16–21. doi: 10.1097/WNO.0000000000000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geiner S, Horn AK, Wadia NH, Sakai H, Buttner-Ennever JA. The neuroanatomical basis of slow saccades in spinocerebellar ataxia type 2 (Wadia-subtype) Prog Brain Res. 2008;171:575–581. doi: 10.1016/S0079-6123(08)00683-3. [DOI] [PubMed] [Google Scholar]
- 72.Rodriguez-Labrada R, Velazquez-Perez L, Auburger G, Ziemann U, Canales-Ochoa N, Medrano-Montero J, et al. Spinocerebellar ataxia type 2: measures of saccade changes improve power for clinical trials. Mov Disord. 2016;31:570–578. doi: 10.1002/mds.26532. [DOI] [PubMed] [Google Scholar]
- 73.Raposo M, Vasconcelos J, Bettencourt C, Kay T, Coutinho P, Lima M. Nystagmus as an early ocular alteration in Machado-Joseph disease (MJD/SCA3) BMC Neurol. 2014;14:17. doi: 10.1186/1471-2377-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stevanin G, Durr A, David G, Didierjean O, Cancel G, Rivaud S, et al. Clinical and molecular features of spinocerebellar ataxia type 6. Neurology. 1997;49:1243–1246. doi: 10.1212/wnl.49.5.1243. [DOI] [PubMed] [Google Scholar]
- 75.Stefanescu MR, Dohnalek M, Maderwald S, Thürling M, Minnerop M, Beck A, et al. Structural and functional MRI abnormalities of cerebellar cortex and nuclei in SCA3, SCA6 and Friedreich’s ataxia. Brain. 2015;138(Pt 5):1182–1197. doi: 10.1093/brain/awv064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim JS, Kim JS, Youn J, Seo DW, Jeong Y, Kang JH, et al. Ocular motor characteristics of different subtypes of spinocerebellar ataxia: distinguishing features. Mov Disord. 2013;28:1271–1277. doi: 10.1002/mds.25464. [DOI] [PubMed] [Google Scholar]
- 77.Huh YE, Kim JS, Kim HJ, Park SH, Jeon BS, Kim JM, et al. Vestibular performance during high-acceleration stimuli correlates with clinical decline in SCA6. Cerebellum. 2015;14:284–291. doi: 10.1007/s12311-015-0650-3. [DOI] [PubMed] [Google Scholar]
- 78.Enevoldson TP, Sanders MD, Harding AE. Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A clinical and genetic study of eight families. Brain. 1994;117(Pt 3):445–460. doi: 10.1093/brain/117.3.445. [DOI] [PubMed] [Google Scholar]
- 79.Horton LC, Frosch MP, Vangel MG, Weigel-DiFranco C, Berson EL, Schmahmann JD. Spinocerebellar ataxia type 7: clinical course, phenotype-genotype correlations, and neuropathology. Cerebellum. 2013;12:176–193. doi: 10.1007/s12311-012-0412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yabe I, Matsushima M, Yoshida K, Ishikawa K, Shirai S, Takahashi I, et al. Rare frequency of downbeat positioning nystagmus in spinocerebellar ataxia type 31. J Neurol Sci. 2015;350:90–92. doi: 10.1016/j.jns.2014.12.042. [DOI] [PubMed] [Google Scholar]
- 81.Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- 82.Fahey MC, Cremer PD, Aw ST, Millist L, Todd MJ, White OB, et al. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain. 2008;131(Pt 4):1035–1045. doi: 10.1093/brain/awm323. [DOI] [PubMed] [Google Scholar]
- 83.Spieker S, Schulz JB, Petersen D, Fetter M, Klockgether T, Dichgans J. Fixation instability and oculomotor abnormalities in Friedreich’s ataxia. J Neurol. 1995;242:517–521. doi: 10.1007/BF00867423. [DOI] [PubMed] [Google Scholar]
- 84.Kirkham TH, Guitton D, Katsarkas A, Kline LB, Andermann E. Oculomotor abnormalities in Friedreich’s ataxia. Can J Neurol Sci. 1979;6:167–172. doi: 10.1017/s0317167100119584. [DOI] [PubMed] [Google Scholar]
- 85.Furman JM, Perlman S, Baloh RW. Eye movements in Friedreich’s ataxia. Arch Neurol. 1983;40:343–346. doi: 10.1001/archneur.1983.04050060043006. [DOI] [PubMed] [Google Scholar]
- 86.Monday LA, Lemieux B, St Vincent H, Barbeau A. Clinical and electronystagmographic findings in Friedreich’s ataxia. Can J Neurol Sci. 1978;5:71–73. [PubMed] [Google Scholar]
- 87.Ell J, Prasher D, Rudge P. Neuro-otological abnormalities in Friedreich’s ataxia. J Neurol Neurosurg Psychiatry. 1984;47:26–32. doi: 10.1136/jnnp.47.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW; CINCH investigators. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130(Pt 10):2484–2493. doi: 10.1093/brain/awm126. [DOI] [PubMed] [Google Scholar]
- 89.Jen JC, Baloh RW. Genetics of episodic ataxia. Adv Neurol. 2002;89:459–461. [PubMed] [Google Scholar]
- 90.Choi KD, Kim JS, Kim HJ, Jung I, Jeong SH, Lee SH, et al. Genetic variants associated with episodic ataxia in Korea. Sci Rep. 2017;7:13855. doi: 10.1038/s41598-017-14254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jen JC, Wan J. Episodic ataxias. Handb Clin Neurol. 2018;148:521–529. doi: 10.1016/B978-0-444-64076-5.00033-8. [DOI] [PubMed] [Google Scholar]
- 92.Kim HJ, Kim JS, Choi JH, Shin JH, Choi KD, Zee DS. Rebound upbeat nystagmus after lateral gaze in episodic ataxia type 2. Cerebellum. 2014;13:411–413. doi: 10.1007/s12311-014-0547-6. [DOI] [PubMed] [Google Scholar]
- 93.Choi KD, Jen JC, Choi SY, Shin JH, Kim HS, Kim HJ, et al. Late-onset episodic ataxia associated with SLC1A3 mutation. J Hum Genet. 2017;62:443–446. doi: 10.1038/jhg.2016.137. [DOI] [PubMed] [Google Scholar]
- 94.Choi JH, Seo JD, Choi YR, Kim MJ, Shin JH, Kim JS, et al. Exercise-induced downbeat nystagmus in a Korean family with a nonsense mutation in CACNA1A. Neurol Sci. 2015;36:1393–1396. doi: 10.1007/s10072-015-2157-6. [DOI] [PubMed] [Google Scholar]
- 95.Levy A, Lang AE. Ataxia-telangiectasia: a review of movement disorders, clinical features, and genotype correlations. Mov Disord. 2018;33:1372. doi: 10.1002/mds.27319. [DOI] [PubMed] [Google Scholar]
- 96.Lohmann E, Kruger S, Hauser AK, Hanagasi H, Guven G, Erginel-Unaltuna N, et al. Clinical variability in ataxia-telangiectasia. J Neurol. 2015;262:1724–1727. doi: 10.1007/s00415-015-7762-z. [DOI] [PubMed] [Google Scholar]
- 97.Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 1958;21:526–554. [PubMed] [Google Scholar]
- 98.Teive HAG, Camargo CHF, Munhoz RP. More than ataxia - Movement disorders in ataxia-telangiectasia. Parkinsonism Relat Disord. 2018;46:3–8. doi: 10.1016/j.parkreldis.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 99.Shaikh AG, Marti S, Tarnutzer AA, Palla A, Crawford TO, Straumann D, et al. Gaze fixation deficits and their implication in ataxia-telangiectasia. J Neurol Neurosurg Psychiatry. 2009;80:858–864. doi: 10.1136/jnnp.2008.170522. [DOI] [PubMed] [Google Scholar]
- 100.Lewis RF, Lederman HM, Crawford TO. Ocular motor abnormalities in ataxia telangiectasia. Ann Neurol. 1999;46:287–295. doi: 10.1002/1531-8249(199909)46:3<287::aid-ana3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 101.Baloh RW, Yee RD, Boder E. Eye movements in ataxia-telangiectasia. Neurology. 1978;28:1099–1104. doi: 10.1212/wnl.28.11.1099. [DOI] [PubMed] [Google Scholar]
- 102.Aicardi J, Barbosa C, Andermann E, Andermann F, Morcos R, Ghanem Q, et al. Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol. 1988;24:497–502. doi: 10.1002/ana.410240404. [DOI] [PubMed] [Google Scholar]
- 103.Le Ber I, Moreira MC, Rivaud-Pechoux S, Chamayou C, Ochsner F, Kuntzer T, et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003;126:2761–2772. doi: 10.1093/brain/awg283. [DOI] [PubMed] [Google Scholar]
- 104.McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–112. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reynolds JJ, Stewart GS. A single strand that links multiple neuropathologies in human disease. Brain. 2013;136(Pt 1):14–27. doi: 10.1093/brain/aws310. [DOI] [PubMed] [Google Scholar]
- 106.Quinzii CM, Kattah AG, Naini A, Akman HO, Mootha VK, DiMauro S, et al. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology. 2005;64:539–541. doi: 10.1212/01.WNL.0000150588.75281.58. [DOI] [PubMed] [Google Scholar]
- 107.Renaud M, Moreira MC, Ben Monga B, Rodriguez D, Debs R, Charles P, et al. Clinical, biomarker, and molecular delineations and genotype-phenotype correlations of ataxia with oculomotor apraxia type 1. JAMA Neurol. 2018;75:495–502. doi: 10.1001/jamaneurol.2017.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee M, Kim NY, Huh JY, Kim YE, Kim YJ. Ataxia with oculomotor apraxia type 1 without oculomotor apraxia: a case report. J Clin Neurol. 2016;12:126–128. doi: 10.3988/jcn.2016.12.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132(Pt 10):2688–2698. doi: 10.1093/brain/awp211. [DOI] [PubMed] [Google Scholar]
- 110.Le Ber I, Bouslam N, Rivaud-Péchoux S, Guimarães J, Benomar A, Chamayou C, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127:759–767. doi: 10.1093/brain/awh080. [DOI] [PubMed] [Google Scholar]
- 111.Moreira MC, Klur S, Watanabe M, Németh AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxiaocular apraxia 2. Nat Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- 112.Tazir M, Ali-Pacha L, M’Zahem A, Delaunoy JP, Fritsch M, Nouioua S, et al. Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients. J Neurol Sci. 2009;278:77–81. doi: 10.1016/j.jns.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 113.Panouillères M, Frismand S, Sillan O, Urquizar C, Vighetto A, Pélisson D, et al. Saccades and eye-head coordination in ataxia with oculomotor apraxia type 2. Cerebellum. 2013;12:557–567. doi: 10.1007/s12311-013-0463-1. [DOI] [PubMed] [Google Scholar]
- 114.Wray SH, Martinez-Hernandez E, Dalmau J, Maheshwari A, Chen A, King S, et al. Paraneoplastic upbeat nystagmus. Neurology. 2011;77:691–693. doi: 10.1212/WNL.0b013e318229e6a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gordon LK. Paraneoplastic syndromes in neuro-ophthalmology. J Neuroophthalmol. 2015;35:306–314. doi: 10.1097/WNO.0000000000000280. [DOI] [PubMed] [Google Scholar]
- 116.Ko MW, Dalmau J, Galetta SL. Neuro-ophthalmologic manifestations of paraneoplastic syndromes. J Neuroophthalmol. 2008;28:58–68. doi: 10.1097/WNO.0b013e3181677fcc. [DOI] [PubMed] [Google Scholar]
- 117.Rizvi MT, Cameron L, Kilbane C, Shaikh AG. Paraneoplastic seesaw nystagmus and opsoclonus provides evidence for hyperexcitable reciprocally innervating mesencephalic network. J Neurol Sci. 2018;390:239–245. doi: 10.1016/j.jns.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 118.Lasker AG, Zee DS. Ocular motor abnormalities in Huntington’s disease. Vision Res. 1997;37:3639–3645. doi: 10.1016/S0042-6989(96)00169-1. [DOI] [PubMed] [Google Scholar]
- 119.Antoniades CA, Xu Z, Mason SL, Carpenter RH, Barker RA. Huntington’s disease: changes in saccades and hand-tapping over 3 years. J Neurol. 2010;257:1890–1898. doi: 10.1007/s00415-010-5632-2. [DOI] [PubMed] [Google Scholar]
- 120.Patel SS, Jankovic J, Hood AJ, Jeter CB, Sereno AB. Reflexive and volitional saccades: biomarkers of Huntington disease severity and progression. J Neurol Sci. 2012;313:35–41. doi: 10.1016/j.jns.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Leigh RJ, Newman SA, Folstein SE, Lasker AG, Jensen BA. Abnormal ocular motor control in Huntington’s disease. Neurology. 1983;33:1268–1275. doi: 10.1212/wnl.33.10.1268. [DOI] [PubMed] [Google Scholar]
- 122.Lasker AG, Zee DS, Hain TC, Folstein SE, Singer HS. Saccades in Huntington’s disease: initiation defects and distractibility. Neurology. 1987;37:364–370. doi: 10.1212/wnl.37.3.364. [DOI] [PubMed] [Google Scholar]
- 123.Hicks SL, Robert MP, Golding CV, Tabrizi SJ, Kennard C. Oculomotor deficits indicate the progression of Huntington’s disease. Prog Brain Res. 2008;171:555–558. doi: 10.1016/S0079-6123(08)00678-X. [DOI] [PubMed] [Google Scholar]
- 124.Helmchen C, Hagenow A, Miesner J, Sprenger A, Rambold H, Wenzelburger R, et al. Eye movement abnormalities in essential tremor may indicate cerebellar dysfunction. Brain. 2003;126:1319–1332. doi: 10.1093/brain/awg132. [DOI] [PubMed] [Google Scholar]
- 125.Kim YE, Kim JS, Yang HJ, Yun JY, Kim HJ, Ehm G, et al. Perverted head-shaking and positional downbeat nystagmus in essential tremor. Cerebellum. 2016;15:152–158. doi: 10.1007/s12311-015-0683-7. [DOI] [PubMed] [Google Scholar]
- 126.Gitchel GT, Wetzel PA, Baron MS. Slowed saccades and increased square wave jerks in essential tremor. Tremor Other Hyperkinet Mov (N Y) 2013;3 doi: 10.7916/D8251GXN. tre-03-178-4116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ingster-Moati I, Bui Quoc E, Pless M, Djomby R, Orssaud C, Guichard JP, et al. Ocular motility and Wilson’s disease: a study on 34 patients. J Neurol Neurosurg Psychiatry. 2007;78:1199–1201. doi: 10.1136/jnnp.2006.108415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jung HK, Choi SY, Kim JM, Kim JS. Selective slowing of downward saccades in Wilson’s disease. Parkinsonism Relat Disord. 2013;19:134–135. doi: 10.1016/j.parkreldis.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 129.Averbuch-Heller L, Paulson GW, Daroff RB, Leigh RJ. Whipple’s disease mimicking progressive supranuclear palsy: the diagnostic value of eye movement recording. J Neurol Neurosurg Psychiatry. 1999;66:532–535. doi: 10.1136/jnnp.66.4.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schwartz MA, Selhorst JB, Ochs AL, Beck RW, Campbell WW, Harris JK, et al. Oculomasticatory myorhythmia: a unique movement disorder occurring in Whipple’s disease. Ann Neurol. 1986;20:677–683. doi: 10.1002/ana.410200605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1.
Saccades are severely impaired in the vertical direction in progressive supranuclear palsy syndrome.