

Abstract
How contemporary plant genomes originated and evolved is a fascinating question. One approach uses reference genomes from extant species to reconstruct the sequence and structure of their common ancestors over deep timescales. A second approach focuses on the direct identification of genomic changes at a shorter timescale by sequencing ancient DNA preserved in subfossil remains. Merged within the nascent field of paleogenomics, these complementary approaches provide insights into the evolutionary forces that shaped the organization and regulation of modern genomes and open novel perspectives in fostering genetic gain in breeding programs and establishing tools to predict future population changes in response to anthropogenic pressure and global warming.
Introduction
Flowering plants, or angiosperms, have come to dominate terrestrial vegetation. They are an essential component of the carbon, oxygen and water cycles, and paramount to the stability of the climate and substrate of our planet. Through photosynthesis, angiosperms convert solar energy into the basal source of chemical energy that underlies the development of almost all terrestrial ecosystems. Flowering plants are also essential to human society as our principal source of food, animal fodder, medicines, and materials for building, clothing, and manufacturing, among many other uses. Molecular clock estimates [1] and paleontological data [2] suggest that angiosperms emerged some 120–170 million years ago (mya), during a period extending from the Cretaceous to the end of the Jurassic; whereas integrated timescale approaches suggest that they might have emerged even further in the past, some 200–250 mya [3]. Flowering plants rapidly diversified so that over 350,000 species are alive today [4–7]. These species are divided into two main groups, the monocots and eudicots, which account for 20% and 75%, respectively, of the diversity characterized to date [6]. Recent advances in high-throughput DNA sequencing and computational biology have helped researchers to develop the field of paleogenomics, making it possible to retrieve invaluable information about the evolutionary history that underlies the emergence and subsequent diversification of flowering plants.
This research field relies on two main complementary approaches that aim to track the evolutionary genomic changes at both the macro-evolutionary and micro-evolutionary temporal scales. The first, an indirect (or ‘synchronic’) approach, compares modern genomes to reconstruct ancestral genomes over deep timescales of several millions of years (macro-evolution). The second approach, a direct (or ‘allochronic’) strategy, relies on the direct sequencing of genomes from past plant subfossil materials that have been preserved over the past 10,000 years (micro-evolution). Here, we address the underlying methodologies for both paleogenomics approaches, as well as their major achievements and prospects in providing an understanding of the evolutionary trajectories that underpin the genetic makeup of modern plant species.
Reconstruction of an ancestral genome from modern genome sequences (synchronic reconstruction)
Background
The recent accumulation of plant genomic resources has provided an unprecedented opportunity to compare modern genomes with each other and to infer their evolutionary history from the reconstructed genomes of their most recent common ancestors (MRCA). Such ancestral genome reconstruction was initially used to investigate 105 million years of eutherian (placental) mammal evolution. The inferred ancestral karyotypes for the eutherians (2n = 44), boreoeutherians (2n = 46), and great apes (2n = 48) were used to increase our understanding of the mechanisms driving speciation and adaptation [8–11]. In particular, eutherian genomes have been found to be surprisingly stable, and affected by only a limited number of large-scale rearrangements during evolution. Higher rates of such chromosomal shuffling have been reported for the branch extending from the great ape ancestor to the ancestor of humans and chimpanzees, which diverged after the Cretaceous–Paleogene (K–Pg) boundary, at a time when the dinosaurs became extinct. Computational reconstructions of mammalian ancestral genomes were instrumental in suggesting that environmental changes may have driven genome plasticity through chromosome rearrangements. These changes may also have led to new variation in gene content and gene expression that gave rise to key adaptive biological functions, such as olfactory receptors [11–13]. Ancestral genome reconstruction has also shed light on plant evolution.
State-of-the-art methodology
The ancestral genome is a ‘median’ or ‘intermediate’ genome consisting of a clean reference gene order that is common to all of the investigated extant species (Fig. 1). The ancestral genomes that are inferred in silico are actually minimal shared ancestral genomes, which lack components of the ‘real’ (unknown) ancestral genomes that were either lost from all of the investigated descendants and/or retained by only one modern species. Such inferred ancestral (minimal) genomes are reconstructed following a four-step strategy [14]. First, sequence comparison across genomes is used to characterize conserved or duplicated gene pairs on the basis of alignment parameters and/or phylogenetic inferences that define genes that are conserved in pairs of species (i.e., putative protogenes (pPGs)). The pPGs that are conserved in all of the investigated species (i.e., core protogenes (core-pPGs)) are used for the definition of synteny blocks (SBs), with the filtering out of groups of fewer than five (pPGs) genes. SBs are then merged on the basis of chromosome-to-chromosome orthologous relationships between the compared genomes, delivering the ancestral protochromosomes (also referred to as contiguous ancestral regions (CARs)). These CARs correspond to independent sets of genomic blocks that display paralogous and/or orthologous relationships in modern species. Finally, the ordering of protogenes (including non-core-pPGs, i.e., genes that are conserved in only a subset of the investigated species) onto the previously defined protochromosomes yields an exhaustive set of ordered protogenes (oPGs).
Fig. 1.
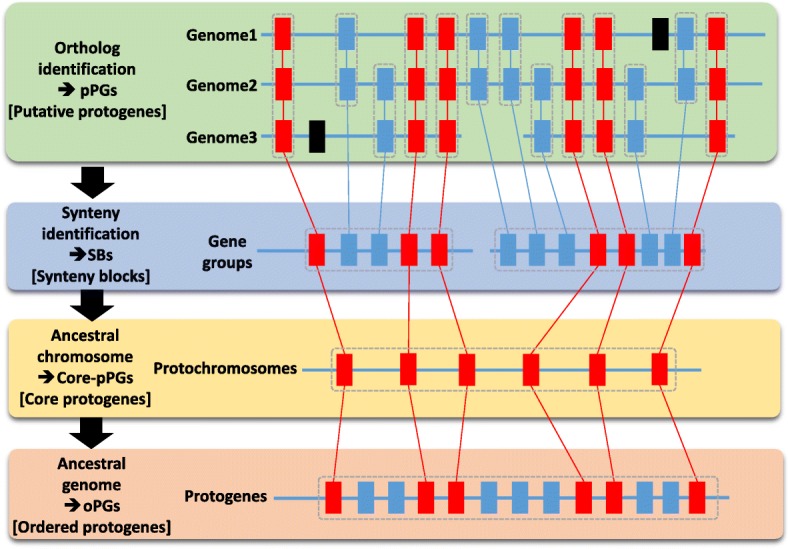
Methodological principles for ancestral genome reconstruction. The four-step strategy is as follows. Step 1: identification of conserved or duplicated genes (putative protogenes (pPGs)); here, genes are illustrated as rectangles and connected with red lines when conserved between all the investigated species or blue lines when conserved in a subset of these species. Species-specific genes, which are not present in the inferred ancestor, are shown as black rectangles. Step 2: identification of synteny in groups of conserved adjacent genes (synteny blocks (SBs)), which are highlighted by grey dashed rectangles. Step 3: reconstruction of contiguous ancestral regions (CARs; highlighted by grey dashed rectangles) containing genes that are conserved in all of the investigated species (referenced as core-pPGs). Step 4: ancestral genome reconstruction delivering protochromosomes (highlighted by grey dashed rectangles) and reordered protogenes (oPGs) [23]
Putative orthologous (or ancestral) genes that have either been transposed outside of CARs so that they are not conserved in synteny in the course of evolution, or that are only retained in one of the investigated species, or that are lost from all of the investigated species are not identified in SBs and therefore are missing from the inferred ancestral genomes. Several tools such as DRIMM-synteny [15], ADHoRe [16], DiagHunter [17], DAGchainer [18], SyMAP [19], and MCScanX [20] are publicly available for clustering or chaining collinear gene pairs, whereas ANGES [21], MRGA [22], and inferCARs [10] are used for reconstructing ancestral genomes. Finally, the reconstructed ancestral karyotypes can be used to infer a parsimonious evolutionary model that assumes minimal numbers of genomic rearrangements (including inversions, deletions, fusions, fissions, and translocations). Such a model fosters new investigations of the evolutionary fate of ancestral genes/genomes, through precise identification of the changes involved (chromosome fusion, fission, translocation, gains, and losses of genes) and their assignment to specific species or botanical families.
Major achievements
The ancestral angiosperm karyotype (AAK) has recently been reconstructed with a repertoire of 22,899 ancestral genes that are conserved in present-day crops and that date back 190–238 mya. The angiosperms have also been proposed to emerge some 250 mya using evolutionary timescale approaches [3]. This time period largely overlaps with the late Triassic era and predates the earliest recorded plant fossil [23]. The AAK then diverged, giving rise to the ancestral monocot karyotype (AMK), with five protochromosomes and 6707 ordered protogenes (or seven protochromosomes according to Ming et al. [24]), and the ancestral eudicot karyotype (AEK), with seven protochromosomes and 6284 ordered protogenes [23]. It is possible to reconstruct any investigated modern monocot or eudicot genome using these inferred ancestors (AAK and AMK or AEK), such that modern karyotypes can be seen as a mosaic of reconstructed ancestral protochromosomal segments (Fig. 2). The availability of the AAK, AMK, and AEK helps us to track the evolutionary plasticity acting at the gene, chromosome, genome, and species levels over more than 200 million years of plant evolution [23].
Fig. 2.

Plant genome evolution from reconstructed ancestors. The present-day monocot (right side, with grasses on a green background) and eudicot (left panel, on a pink background) genomes (bottom) are represented with color codes to illustrate the evolution of genomic segments from their founder ancestors over the time scale shown on the left (in mya). AGK and AEK paintings (bottom) represent, respectively, the modern monocot and eudicot genomes based on the ancestral grass karyotype (AGK) and the ancestral eudicot karyotype (AEK) of seven protochromosomes (color code). AAK painting (top) represents AGK, AEK, and the ancestral monocot karyotype (AMK), as well as oil palm and pineapple genomes, based on the ancestral angiosperm karyotype (AAK) of 15 CARs (color code). The polyploidization events that have shaped the structure of modern plant genomes during their evolution from inferred ancestors are indicated by red dots (duplication) and blue dots (triplication). Major angiosperm families are indicated on the tree branches as detailed in Table 1
At the gene level, the comparison of the AAK gene repertoire to those of outgroup species, such as gymnosperms, mosses, and single-cell green algae, uncovered genes that are specific to flowering plants. These genes were preferentially assigned to Gene Ontology (GO) terms such as ‘pollen–pistil interaction’, ‘response to endogenous stimuli’, ‘flower development’, and ‘pollination’, corresponding to the key biological processes that drove the transition between gymnosperms and angiosperms [23].
At the genome level, the genomic plasticity inherited through polyploidization events can be assessed, with ~ 60 % of AAK protogenes being present as singletons today in modern species despite recurrent polyploidization events (Fig. 2). This general phenomenon of gene repertoire contraction following polyploidy is also observed at the chromosome level, with a general decrease in chromosome number after whole-genome duplication (WGD) resulting from massive ancestral chromosome fusions through two mechanisms, centromeric chromosome fusion (CCF) and telomeric chromosome fusion (TCF). CCF, which is mainly observed in grasses, involves the insertion of an entire chromosome into a break in the centromeric region of another chromosome. TCF involves the ‘end-to-end’ joining of two chromosomes via their telomeres [25]. The observed general pattern of chromosome number reduction involves unequal reciprocal translocations and the loss of several centromeres, such that only a subset of the ancestral pool of telomeres or centromeres are re-used as functional telomeres or centromeres in modern species [25, 26]. Despite multiple rounds of WGD in the course of plant evolution, the number of genes and chromosomes has been kept constant by massive diploidization and fusion events, at the gene and genome levels, respectively. Diploidization did not occur at random in the genome, particularly where retained ancestral genes were partitioned between paralogous blocks so as to form ‘most fractionated’ (MF, also known as S for sensitive) and ‘least fractionated’ (LF, also known as D for dominant) chromosomal compartments [14]. ‘RNA binding’, ‘nucleic acid binding’, ‘receptor activity’, ‘signal transducer activity’, ‘receptor binding’, and ‘transcription factor activity’ are frequent GO terms associated with molecular functions that are enriched in extant genomes relative to the AAK. They correspond to adaptive or specialized biological functions for which multiple copies of genes were conserved after WGD and have survived the general diploidization phenomenon [23].
As has been proposed for mammalian evolution, paleopolyploidy events in angiosperms are usually considered rare, are likely to lead to an evolutionary dead-end, and may have served as the basis for species diversification and survival during episodes of mass species extinction [27–29]. Although still debated, the ancient paleopolyploidization as well as ancestral speciation events in angiosperms may have been associated with known periods of species extinction, such as the Cretaceous/Paleogene (called K-Pg, ~ 65 mya) transition [27] or the Triassic/Jurassic (called Tr-J, ~ 200 mya) transition [30]. More recent paleopolyploidization events that are specific to plant lineages (or even species) may be associated with more recent plant diversification periods during the Paleogene and Neogene (~ 20–30 mya), as observed from historical changes in dry forest communities and biomasses [31, 32]. Thus, polyploidy appears to have played a major role in (re-)shaping structural and functional genomic diversification during angiosperm evolution, with contrasting rates of changes between species, subgenomes, genes, and functions. It may also have delivered biological novelties that have enhanced tolerance of environmental changes, including those occurring during mass extinction events.
Grasses as a case study
Besides the recovery of extinct AMK, AEK, and AAK founder karyotypes, the synchronic approach has also enabled the computational reconstruction of the ancestral genomes of major angiosperm lineages. In eudicots, ancestral genomes have been proposed for the Rosaceae [33], Brassicaceae [34], and Cucurbitaceae [35] subfamilies, consisting of nine, eight (or seven), 12 (using the melon genome as pivot) protochromosomes, respectively, as well as for the legumes [36]. In grasses, the ancestral grass karyotype (AGK), which takes into account gene conservation between rice, wheat, barley, Brachypodium, sorghum, setaria, and maize, was structured into seven protochromosomes containing 8581 protogenes (9430 in Wang et al. [37]) and with a minimal gene space physical size of 30 Mb [23, 38, 39]. This ancestral genome went through a paleotetraploidization event (involving seven duplicated blocks shared by modern monocots) more than ~ 95 mya [37, 38, 40]. Two subsequent symmetric reciprocal translocations, one of which was centromeric (CCF) and the other telomeric (TCF), and two asymmetric reciprocal translocations resulted in a total of 12 chromosomes [23, 39] bearing 16,464 protogenes (18,860 according to Wang et al. [37]). All investigated modern grass genomes can then be reconstructed from this post-polyploidy ancestral karyotype of 12 protochromosomes, taking into account CCF, TCF, translocation, and inversion events (Fig. 2). Rice has retained the n = 12 structure of the AGK and has been proposed to be the slowest evolving species among the grasses [23, 37], whereas the other species underwent numerous chromosome rearrangements to reach their present-day karyotypes [23, 38, 39]. Rice can, therefore, be considered as a reference genome (also known as a ‘pivot’) for comparative genomics studies in grasses.
The grasses appear to constitute a key botanical family in which to investigate the role of polyploidizations in promoting species speciation and adaptation. Grasses experienced an ancestral paleotetraploidization event as well as species-specific polyploidization events, with a tetraploidization event in maize and tetraploidization or hexaploidization events in wheat. After a polyploidization event, homoeologous chromosome differentiation is necessary to stabilize meiosis by preventing incorrect pairing between homoeologs. This is achieved through massive partitioning of the organization and regulation of the subgenomes, involving the fusion, fission, inversion, and translocation of chromosomes, loss of genes or DNA, and neo- or sub-functionalization of gene pairs. Ultimately, such post-polyploidy genomic plasticity led to novel phenotypes that underlie the evolutionary success of polyploid plants and, ultimately, was selected for by humans during domestication (reviewed in [14, 29, 41, 42]).
Promising scientific avenues from inferred ancestral genomes
Inferred ancestral genomes are not only crucial for understanding how plant genomes have evolved at the chromosome and gene scales, but also offer the possibility to address, in novel ways, issues regarding translational research and post-polyploidy plasticity that are relevant to plant breeding.
Translational research
Ancestral genomes and related comparative genomics data are delivered through public web servers such as PlantSyntenyViewer (https://urgi.versailles.inra.fr/synteny [30, 34, 39]), Genomicus (http://www.genomicus.biologie.ens.fr/genomicus-plants [43]), COGE (https://genomevolution.org/coge/ [44]) and PLAZA (http://bioinformatics.psb.ugent.be/plaza/ [45]). The ancestral genomes (AAK, AEK, AMK, and AGK, as well as ancestral genomes for the Rosaceae, Brassicaceae, and Cucurbitaceae; Table 1) provide a list of accurate orthologs between species that can be used to improve the structural and functional annotation of genomes. The plant genomes shown in Fig. 2 have been sequenced, assembled, and finally annotated by different methods and groups, potentially resulting in some inconsistencies. With the use of reconstructed ancestral genomes, structural (intron and exon structure) and functional (GO) annotations of genes can be improved by comparing orthologous and paralogous gene sets that may share similar (ancestral) genomic features. Reconstructed ancestors can also be used as a useful resource for translational research on key agronomical traits, particularly from model species (such as Arabidopsis thaliana) to crops [46]. Modern monocot and eudicot crops can now be connected via the 22,899 protogenes that define the AAK [23], offering the opportunity to exploit the knowledge gained on genes underlying traits of interest in models based on orthologs or paralogs in crops delivered in the proposed evolutionary scenario and associated paleogenomic data (Fig. 2). Such translational-based dissection of traits has been performed successfully in several botanical families, including legumes (for example, between Medicato truncatula and pea, as described by Bordat et al. [47]) and grasses (for example, between Brachypodium distachyon and wheat, as described by Dobrovolskaya et al. [48]).
Table 1.
Ancestral plant genomes
| Family | Dating | Name | Chromosome number | Gene number | Reference |
|---|---|---|---|---|---|
| Angiosperms | 190–238 | AAK (post-ε/ζ) | 15 | 22,899 | [23] |
| Eudicots | 87–109 | AEK (pre-γ) | 7 | 6284 | [23] |
| Eudicots | 87–109 | AEK (post-γ) | 21 | 9022 | [23] |
| Monocots | 100–150 | AMK (pre-τ) | 5 | 6707 | [23] |
| Monocots | 100–150 | AMK (post-τ) | 10 | 13,916 | [23] |
| Grasses | 65–81 | AGK (pre-ρ) | 7 | 8581 | [39] |
| Grasses | 70–96 | AGK (pre-ρ) | 7 | 9430 | [37] |
| Grasses | 65–81 | AGK (post-ρ) | 12 | 16,464 | [39] |
| Grasses | 70–96 | AGK (post-ρ) | 12 | 18,860 | [37] |
| Brassicaceae | 27–40 | ABK (post-α/β) | 8 | 20,037 | [34] |
| Brassicaceae | 23–27 | ACaK (post-α/β) | 8 | 22,085 | [34] |
| Brassicaceae | 23–27 | PCK (post-α/β) | 7 | 21,227 | [34] |
| Rosaceae | 70–90 | ARK (post-WGD) | 9 | 8861 | [33] |
| Cucurbitaceae | 25–50 | ACuK (post-WGD) | 12 (Melon as pivot) | 18,534 | [35] |
| Legumes | 56–59 | ALK (post-WGD) | – | 28,900 | [36] |
Summary of reconstructed ancestral angiosperm genomes listing the targeted botanical family, dating (in mya) of the whole-genome duplication defining the delivered post- and pre-polyploidization ancestors, ancestral genome name, number of chromosomes, number of genes and associated references in the literature
Abbreviations: AAK ancestral angiosperm karyotype, ABK ancestral Brassicaceae karyotype, ACaK ancestral Camelineae karyotype, ACuK ancestral Cucurbitaceae karyotype, AEK ancestral eudicot karyotype, AGK ancestral grass karyotype, ALK ancestral legume karyotype, AMK ancestral monocot karyotype, ARK ancestral Rosaceae karyotype, PCK proto-Calepineae karyotype, WGD whole-genome duplication
Polyploidization
Polyploidization events have been proposed as a major source of genetic novelty during evolution. Such post-polyploidy genomic plasticity takes place in paleopolyploids that are subject to diploidization (evolution toward a reduction of duplicate redundancy) through (not exclusively): (i) differences in ancestral gene retention yielding contrasted plasticity between MF (or S) and LF (or D) compartments; (ii) bias in GO for the retention of multiple copies of genes displaying an enrichment in functional categories such as transcriptional regulation, ribosomes, response to abiotic or biotic stimuli, response to hormonal stimuli, cell organization, and transporter functions; (iii) partitioned gene expression with differences in transcript abundance or neo- and sub- functionalization patterns between retained pairs; (iv) contrasted single nucleotide polymorphisms (SNPs) at the population level between paralogous genomic fragments; and (v) contrast in small regulation as well as differences in epigenetic (CG methylation) marks between duplicated blocks/genes [49]. Such subgenome dominance phenomena, which partition the organization and regulation of diploidized paleopolyploids, have been particularly exemplified in Brassicaceae and maize [50–53] but are reportedly so far undetectable in soybean, banana, and poplar [54].
The evolutionary plasticity gained from recurrent polyploidization and diploidization (also known as post-polyploidization diploidization (PPD) [55]) processes has provided the basis for functional and phenotypic novelty in angiosperms. This plasticity may underlie a plant's ability to survive in or invade a novel environment, ultimately driving the observed evolutionary success of important plant families [27]. Nevertheless, the continuum and interplay between the reported structural and functional reprogramming after PPD processes remain poorly understood. The access to ancient DNA (aDNA) sequences from extinct diploid and polyploid ancestors contemporary to past polyploidization events will further expand our understanding of this major phenomenon driving plant evolutionary dynamics, making it possible to characterize the driving molecular mechanisms that have potential for use in breeding. In that regard, nascent polyploids (particularly in wheat and Brassicaceae) provide opportunities for testing the hypothesis that polyploidization accelerates evolutionary adaptation to environmental changes [56].
Evolutionary processes inferred from ancient DNA (allochronic reconstruction)
Background
Ancient DNA sequencing offers a unique opportunity to retrieve genetic information from past individuals. It has been applied successfully to ancient hominins and human individuals (see review in Marciniak and Perry [57]), and to a handful of mammal species, including woolly mammoths [58], aurochs [59], horses [60], and dogs [61], at both the genomic and the population scale. Ancient genomes have helped to unveil the complex population dynamics and processes that underlie evolution, involving admixture, migration, and adaptation [62–64]. Signatures of adaptation in response to natural or human-driven selection are embedded within the genomes of modern populations and species [65], making inferences about past selective processes possible. Such indirect approaches have clearly demonstrated the power of natural and artificial selection in shaping local adaptation, but have also shown limitations because evolutionary inferences are based on theoretical models with simplifying assumptions. The possibility of adding a temporal dimension to such analyses, overlapping key evolutionary transitions such as demographic or environmental changes, could provide enhanced statistical power for detecting and quantifying the genomic changes underlying adaptive [66, 67] and non-adaptive histories [68, 69]. Such studies are still embryonic in plants but will eventually help us to (i) chart the complex patterns of adaptation through space and time, and (ii) measure the evolutionary responses of plants to key evolutionary and/or environmental transitions.
State-of-the-art methodology
In addition to working under rigorous clean laboratory conditions, appropriate sequencing and computational methods are required to authenticate and analyze aDNA sequences correctly (Fig. 3). Over the past decade, aDNA research has moved from the characterization of short pieces of DNA, mostly mitochondrial, to complete genome sequencing (for a review, see Orlando et al. [70]). This impressive progress has been made possible due to the advent of high-throughput DNA sequencing platforms, which can sequence up to several billions of short nucleotide reads in no more than a few days [71, 72]. Such sequencing capacities have opened access to even the most minute fraction of DNA molecules preserved in fossil specimens, even though these molecules are often outnumbered by DNA fragments from environmental microbes [73].
Fig. 3.

Methodological principles for ancient DNA investigation. Documentation of tissue type (i.e., leaves, seeds, stems, wood), conservation (i.e., dessicated, waterlogged, charred), and age is followed by sample decontamination, DNA extraction (depending on DNA degradation, DNA size, quantity, and absence of inhibitors), and NGS library (single-stranded) preparation (depending on input quantity, aDNA fragment size, and the number of amplification cycles) performed in standardized clean conditions using laboratory procedures optimized for aDNA. Sequencing reads can be mapped against a reference genome (defining endogenous aDNA and unique mapped reads) and authenticated based on typical post-mortem damage patterns using bioinformatic pipelines tailored for aDNA. The exogenous DNA content can be evaluated using metagenomic analysis tools. Finally, the authenticated endogenous DNA can be compared to modern reference samples to unveil genomic footprints of species’ origin, migration, anthropogenic translocation, extinction, and hybridization events. Examples of bioinformatic tools used in investigating aDNA sequences are mentioned at the bottom of each panel (see main text for further details)
Current aDNA methodologies do not rely on brute-force sequencing of DNA extracts but rather leverage the specific biochemical features of aDNA molecules in subfossils. First, a number of paleontological and archaeological remains, such as hair [74], petrous bones [75], and tooth cementum [76], generally provide micro-environments with DNA preservation conditions that are generally better than those provided by other types of calcified remains, such as shells [77]. Second, DNA extraction methods tailored to the retrieval of the most fragmented DNA templates, which also represent the most abundant fraction of ancient DNA molecules, have been developed [78, 79]. As the information present in 25–35 bp fragments is generally compatible with accurate sequence alignment, the recovery of such ultra-short templates has greatly improved the sensitivity of aDNA analyses. Third, some extraction procedures, including pre-digestion [80], the washing steps prior to full digestion [81], and other techniques [82, 83], have proved useful for removing at least a fraction of environmental contamination.
In addition, a range of DNA library construction methods have also been developed, and important biases have been mitigated, including those that occur during adapter ligation [84] and PCR amplification [85]. The development of DNA library construction methods that exploit molecular features of aDNA, in particular the presence of damage in the form of single-strand breaks [86] and/or deaminated cytosines [87], has also enhanced our ability to access aDNA templates. Finally, target enrichment approaches, aimed at the characterization of organellar DNA [88] or of a limited number of mitochondrial and nuclear loci [89], or up to hundreds of millions of SNPs scattered throughout the nuclear genome [90] and even of the entire nuclear genome [91, 92], now contribute to the retrieval of the genome-scale information required to address major biological questions in both a cost- and time-effective manner. It is worth noting that a number of computational approaches have also helped to quantify DNA damage [93, 94], to reduce its impact on downstream analyses [94, 95], and to improve the sensitivity and accuracy of aDNA read alignments [96–99]. Unlike material from human or vertebrate taxa, plant material contains polyphenols, polysaccharides, and other molecules that can interfere with standard molecular tools and/or reagents. Therefore, the development of procedures that are tailor-made for the recovery, purification, and manipulation of DNA from botanical remains is necessary (Table 2).
Table 2.
Ancient plant DNA
| Species | Datinga | Site | Sample | Characterizationb | Extractionc | Reference |
|---|---|---|---|---|---|---|
| Oak | 500–9800 BP | Europe | Waterlogged wood | NGS | TrisHCL SDS CaCl2 EDTA DTT PK/Phchlo/Column | [142] |
| Japanese cedar | 3600 BP | Japan | Buried tree | PCR/sequencing | Column | [133] |
| Maize | 1100–6000 BP | New World | Desiccated cob | PCR/sequencing | CTAB TrisHCL NaCl EDTA/Chlo | [111] [112] |
| Maize | 360–1320 BP | New World | Desiccated cob | PCR/sequencing | Column | [114] |
| Maize | 670–5280 BP | New World | Desiccated cob | Capture, NGS | SDS DTT PK/Phchlo/Column | [115] |
| Maize | 650–4300 BP | New World | – | PCR/sequencing | SDS DTT PK/Phchlo/EDTA PTB/Column | [113] |
| Maize | 4700 BP | Chile, Peru | Charred and non-charred grain | PCR/sequencing | SDS DTT PK/Phchlo | [110] |
| Maize | 5310 BP | Mexico | Desiccated grain | NGS | TrisHCL SDS CaCl2 EDTA DTT PK/Phchlo/Column | [116] |
| Maize | 5300 BP | Mexico | Desiccated cob | NGS | – | [117] |
| Sunflower | 3100 BP | USA | Desiccated disk fragment, pericarp, kernel | NGS | TrisHCl NaCl SDS CaCl2 EDTA DTT PK/Phchlo/Column | [119] |
| Radish | 350–550 AD | Egypt | Desiccated seed | Chemical analysis/PCR/sequencing | CTAB TrisHCL NaCl EDTA/Chlo | [124] |
| Sorghum | 2800 BP | Egypt | Desiccated seed | PCR/sequencing | – | [125] |
| Rice | 1200–2400 BC | China | Desiccated seed and chaff | PCR/sequencing | Magnetic beads | [127] |
| Grape | 1600–2500 BP | Europe | Waterlogged and charred pip | PCR/sequencing | DTAB /Chlo/CTAB | [120] |
| Grape | 7th–15th century AD | Italy | Waterlogged pip | PCR | SDS DTT PK/Phchlo/Column | [121] |
| Grape, maize, olive, dogwood, cotton | 400–2400 BP | New World, Europe | Non-carbonized remain | PCR/sequencing | SDS DTT PK/Phchl | [122] |
| Barley | 6200–5800 BP | Israel | Desiccated seed | NGS | CTAB TrisHCL PVP βME/Phchlo/Column | [104] |
| Barley | 3000 BP | Egypt | Desiccated grain | PCR/sequencing | SDS DTT PK EDTA PTB/Column | [103] |
| Barley, wheat | 150–5250 BC | Spain | Charred, partially charred, waterlogged seed | PCR/sequencing | TrisHCL SDS EDTA PK/Phchlo or Column | [105] |
| Wheat | 700 AD–8400 BP | Anatolia | Charred grain | PCR/sequencing | CTAB TrisHCL NaCl EDTA/Chlo | [109] |
| Wheat | 340–3500 BP | Spain | Charred and desiccated seed | PCR/sequencing | Tris EDTA CTAB βME/Column | [108] |
| Cotton | 750–3750 BP | Brazil, Peru, Egypt | Desiccated seed | NGS | CTAB/Column | [132] |
| Arabidopsis | 300 BP | USA | Herbarium | NGS | CTAB or PTB DTT/Column | [131] |
Summary of ancient plant nuclear DNA recovery listing species, dating, location site, sample type, characterization method, extraction protocol, and associated references. aDatings are referenced as in the publication concerned using BC, AD, or BP. bPCR/sequencing polymerase chain reaction and sequence capture, NGS next-generation sequencing. cDDT dithiothreitol proteinase K, Phchlo phenol-chloroform, Ph phenol, Chlo chloroform, SDS sodium dodecyl sulphate, EDTA ethylene-diamine-tetraacetic acid, CTAB cetyltrimethylammonium-bromide, PTB phenacylthiazolium bromide, DTAB dodecyltrimethylammonium bromide, βME β-mercaptoethanol, Tris tris(hydroxymethyl) aminomethane, PK proteinase K
Major achievements
Plant seeds can provide a favorable environment for the preservation of nucleic acids over millennia, perhaps as a result of the active desiccation mechanisms involved in dormancy [100, 101]. While recent work has shown encouraging results, studies leveraging the information in the DNA (and/or RNA) fragments present in plant subfossils are still scarce (for review, see Gutaker and Burbano [102]). The number of the species studied spans a large taxonomic range and includes barley [103, 104], wheat [105–109], maize [110–118], sunflower [119], grape [120–122], bottle gourd [123], radish [124], sorghum [125], papyri [126], rice [127], olive [128], orchid [129], Prunus [130], Arabidopsis [131], cotton [132], and trees [133–136]. Similarly, the primary material used for DNA extraction includes a whole variety of tissues, such as fruits, seeds, leaves, and woods, preserved in a wide range of conditions, including charred, waterlogged, desiccated, or mineralized remains. Ancient DNA from organelles, which have sequences that are highly conserved among plant species and which is generally better preserved than the nuclear genome, have been widely used in paleogenomics studies on plants over the past decade [137]. Such organellar DNAs include ribosomal (rDNA) and chloroplast (cpDNA) markers such as the rbcL gene (which encodes the large subunit of ribulose-1,5-bisphosphate carboxylase, an important enzyme in photosynthesis), trn introns and spacers (which offer more variable non-coding information), and matK (the maturase K) gene. The internal transcribed spacer 1 of the ribosomal DNA gene (ITS1) has classically been used in characterizing plant aDNA in papyri [126], Prunus [130], bottle gourd [123], orchid [129], olive [128], wheat [105, 106], and trees [134–136]. Importantly, in contrast to studies on animals, mitochondrial DNA (mtDNA) has been overlooked in plant aDNA research, probably because of its more-than-100-times-slower mutation rate [138–140].
At the nuclear level, a handful of genetic markers, mostly carrying functional variants that are associated with flowering time and starch storage, have already been characterized, originally by PCR, and more recently by target-enrichment approaches, which helped track the genetic variation of major maize genes over the past 6000 years [116]. The presence of lipids (fatty acids and sterols) and nucleic acids in desiccated radish seeds from a 6th century storage vessel recovered from Qasr Ibrîm in Egypt has been reported [124]. Further DNA investigation of plant remains from archaeological sites in Egypt have been also reported for sorghum [125]. In rice, genomic sequences from remains found at Tianluoshan, a site of the local Hemudu Neolithic culture in the low Yangtze, were compared to current domesticated and wild rice populations in order to investigate the genetic changes underlying the domestication syndrome [127]. Microsatellite loci have also been used to investigate the origins of grape seeds preserved by waterlogging and charring at several European Celtic, Greek, and Roman sites [120, 122]. Cotton aDNA was used to investigate changes in transposon composition that have occurred over the past 1600 years of domestication [132]. Finally, moving towards more recent times, herbarium specimens have been found to generally yield excellent DNA preservation, compatible with the whole-genome sequencing of plants such as potato [141], Arabidopsis [131], or orchid [129], and even of some of the pathogens responsible for historical famines such as that caused by potato blight [141]. More recently, technological advances in next-generation sequencing (NGS; Table 2) as well as target enrichment approaches (such as sequence capture) have delivered highly relevant retrieval and authentication of aDNA in barley [104], maize [115–117], oak [142], and sunflower [119], which can now be used as standards for plant aDNA studies. In addition, the recent recovery of aDNA from waterlogged wood (oak) remains has opened up new avenues for investigating the recent evolution of forest cover in the face of climate and/or anthropogenic changes [142].
The most thorough plant aDNA studies have been probably carried out in maize and barley. In maize, the analysis of transposable elements (Mu) in pre-Columbian kernels [111], and of the alcohol dehydrogenase gene (adh) and microsatellite loci in desiccated maize cobs excavated in caves, provided the first insights into maize origin and domestication [112, 114]. Then, target enrichment through gene capture helped to decipher the early diffusion of maize into the American Southwest and to track genomic selection signals during different phases of domestication, in particular in loci relevant to drought tolerance and sugar content [115]. In barley, the genome (exome) sequence of 6000-year-old barley grains from (pre-)historic caves in the Judean Desert revealed close affinities with extant landraces from the Southern Levant and Egypt. These findings were consistent with a proposed origin of domesticated barley in the Upper Jordan Valley, as well as with gene flow between cultivated and wild populations during the early domestication phase [104]. In addition to aDNA, plant subfossils can also provide ancient RNA (including small RNAs) and epigenetic (i.e., DNA methylation) signatures [143–146]. Barley is the ancient crop that has garnered most of the attention at the DNA, RNA, and epigenetic levels. A series of investigations have addressed the complex process of local adaptation and domestication, or have identified the presence of barley stripe mosaic virus, one of the major diseases affecting this major crop [101, 144–146].
Wheat as a case study
Plant aDNA provides a catalog of ancient sequence polymorphisms that are extremely valuable in disentangling the temporal and geographical locus of domestication as well as the patterns of migration (diffusion). This catalog also helps to detect hybridization events between cultivated and wild relatives, including those that may have been advantageous. More than 2500 plant species are thought to have been domesticated over the past 12,000 years of evolution, since the last glacial period [147]. However, the domestication history of most crops is still contentious and several evolutionary models have been proposed. Ancient DNA has provided the data necessary to test a number of competing scenarios, mostly in cereals and especially in wheat, pertaining to the migration, translocation, extinction, hybridization, and demographic dynamics underpinning modern cultivars (Fig. 4).
Fig. 4.

Scientific insights gained from plant aDNA, as exemplified by wheat. Top: The geological periods are indicated at the top (orange arrow) above the associated temperature profiles (blue curve showing the variation of temperature at constant elevation (y-axis) over thousands of years before present (x-axis), modified from Cuffey and Clow [168]) and key climatic changes. Center: The known historical routes of wheat migration from the site of origin (Fertile Crescent) are indicated by green arrows: westwards via an inland route (through the Balkans to Central Europe) or along a coastal path (via Anatolia to the Mahgreb and Iberian peninsula); or eastwards via routes to the north and along the Inner Asian Mountain Corridor. Major migration phases are shown in different colors on the time scale. Diploid, tetraploid, and hexaploid wheats are depicted as colored circles (green, red, and blue colors) with mixed colors reflecting hybridization events. Bottom: Illustration of the hexaploid bread wheat paleohistory from progenitors (with some possibly extinct) A (green circles), B (red circles), and D (blue circles) shown at the left, along the time scale expressed in million years (left) at the bottom. Subgenomes (A, B, and D) are illustrated with circles so that hybridization events are highlighted with mixed color within circles (similar to the central panel). Modern (diploid, tetraploid, and hexaploid) species are illustrated at the right of the figure. Wheat aDNA offers the opportunity to investigate the impact of migration, translocation, extinction, and hybridization events in shaping the modern genetic diversity and in driving adaptation to environmental constraints (temperature variation) over 10,000 years of domestication and cultivation
It is currently thought that tetraploid wheats, which are used for pasta production, emerged some 0.5 mya from the hybridization of a wild Triticum urartu Tumanian ex Gandivan (AA) and an undiscovered species of the Aegilops speltoides Tausch lineage (BB). It is widely accepted that the domestication of this tetraploid wheat (wild emmer AABB) in southeastern Turkey—within the so-called ‘Fertile Crescent’ (a region extending from western Iran, Iraq, Jordan, Israel, Lebanon, and Syria to south-east Turkey)—followed by a north-eastern migration, led to its hybridization with A. tauschii (DD) and the emergence of bread wheat (Triticum aestivum, AABBDD). This hybridization event most probably occurred within a corridor spanning from Armenia to the south-western coast of the Caspian Sea [148, 149]. After the last glacial maximum, human sedentarism associated with the development of agriculture emerged in several sub-regions of the Fertile Crescent [150], where several major crops, including wheat and barley, and farm animals such as sheep, goats, cows, and pigs were domesticated.
Plant fossil remains displaying the characteristics of domesticated cereal crops have been discovered at multiple archaeological sites dating from 8000–10,000 years ago, a key historic period marking the human transition from a foraging lifestyle to early sedentary agricultural societies. The domestication of wheat involved selection for traits that are related to seed dormancy and dispersal, such as brittle rachis, tenacious glume, and non-free-threshing traits [151]. The spread outside of the original domestication center followed four major historical routes of human migration, including a westwards expansion through inland (via Anatolia and the Balkans to Central Europe) and coastal (via Egypt to the Mahgreb and Iberian peninsula) paths, and an eastwards expansion through the north and along the Inner Asian Mountain Corridor [152]. Following domestication, modern breeding activities starting after 1850 CE (Common Era) further reduced the genetic diversity in genomic regions harboring genes involved in agricultural performance or adaptation (such as photoperiodism, vernalization, flowering, accumulation of seed storage protein, plant architecture, and so on) [153].
Access to aDNA would enable the estimation of the loss of genetic diversity associated with the various processes operating during 10,000 years of domestication, hybridization, migration, and adaptation. Among the evolutionary processes underpinning the origins of wheat, hybridization (often referred to as reticulated evolution) remains controversial because of the lack of a modern representative of some diploid progenitors (especially for the B subgenome; Fig. 4). Future aDNA work, notably for the so-called naked wheat that was common in the Neolithic [154–156] or the ‘new glume wheat’ found at Neolithic and Bronze Age sites [157] and proposed to be an extinct wheat cultivar [158], may shed light on such controversies. Wheat aDNA recovered from waterlogged, desiccated, and (semi-)charred remains (grains, rachides, and/or spikes), provides a seminal resource for attempts to address wheat origin and evolution during the past 10,000 years of domestication [106–109, 159], as well as the impact of polyloidization (comparing diploid, tetraploid, and hexaploid wheats) in adaptation. Nevertheless, the extraction of aDNA from wheat remains is still challenging because of the diversity of the tissues considered and their conservation over time. The two different methods, classically used for plant aDNA extraction (proteinase K or cetyltrimethylammonium-bromide (CTAB); Table 2), still need to be refined for the retrieval of DNA from all types of plant remains, especially charred grains. Although charred seeds retain their morphological characteristics and are therefore suitable for botanical classification, the preservation of DNA within such materials remains so far unlikely [160].
Promising scientific avenues from sequenced ancient DNA
Beside solving phylogenetic questions regarding evolution, admixture, hybridization events, relationships between species or populations, domestication, and improvement processes, aDNA can provide new possibilities for addressing a number of issues related to plant adaptation and diversification.
Adaptation
Ongoing climate change, the steady growth of the human population worldwide, and increasing demand from emerging economies place food security at threat all over the world [161]. In addition, the demand for wood is growing continuously at a time when forest trees are exposed to rapidly increasing biotic and abiotic threats [162, 163]. These issues are of utmost importance in the context of ongoing climatic changes [164] and the rise in food and wood demand from the expanding world population [165]. The development of high-yielding, durably stress-resistant crops and trees is thus paramount for the sustenance of future human societies. This challenge can be addressed in part through the identification, conservation, and exploitation, through genome-informed conservation and breeding strategies, of key genetic polymorphisms that enhance plant resilience in the face of environmental pressures [166]. Plants have faced temperature and water constraints in the past, including some similar to the +0.3°C to +4.8°C average increment by 2100 (depending on the model considered) predicted by the Intergovernmental Panel on Climate Change (IPCC; http://www.ipcc.ch/) [167]. In particular, the extended Holocene period has included multiple periods of short- and long-term climate change, including a particularly steep temperature increase at the beginning of the Holocene (about 11,700 years ago) and a climate optimum reached during the mid-Holocene (Fig. 4) [168]. The Holocene also witnessed the domestication of crops in the early Neolithic of the Fertile Crescent 10,000 to 8000 years ago and in other farming centers in Asia, Africa, and the Americas.
There is much to learn from the aDNA of early crops and their propagation and local adaptation outside of their native domestication area [169]. In particular, the possibility to obtain reliable estimates of the allelic trajectory at virtually any genomic locus during the major climate transitions that have occurred in the past opens an avenue towards the identification of the genetic variants that underlie adaptation to novel environmental conditions. They could represent priority targets for breeders and/or top-candidates for reintroduction into modern germplasms.
Diversification
Domestication and recent breeding have reduced the genetic diversity of modern cultivated germplasm. Looking for novel sources of diversity (currently absent not only from the elite pool but also from extant wild species and landraces) is a major concern for the sustained improvement of commercial lines. aDNA can deliver the genetic diversity that has been lost at several key time points during plant domestication, with the potential of reintroducing extinct loci (gene alleles) for key traits. Mutant screening technologies offer the opportunity to validate such variants functionally prior to their reintroduction. If these variants still segregate amongst wild relatives, they could be reintroduced through conventional marker-assisted breeding/selection programs; otherwise, they could be introduced through genome editing. Ultimately, aDNA investigations would allow us to uncover the content of the ‘lost’ diversity to be resurrected in modern germplasm (a process referenced to as ‘de-extinction’), which might be of utmost value for current breeding and conservation initiatives.
Novel scientific insights in paleogenomics that merge synchronic and allochronic approaches
The evolution of modern species can be investigated in detail through the analysis of ancient genomes. The indirect (synchronic) approach, derived from the computational reconstruction of ancestral genomes of several million years old (macro-evolution), is built on the comparison of the genomes of modern species. The direct (allochronic) approach derives from the recovery of ancient DNA from remains that are up to several tens of thousands of years old (micro-evolution). Embracing both synchronic and allochronic approaches into the growing field of paleogenomics is paramount to understanding how past macro- and micro-evolutionary processes shaped modern plant diversity (Fig. 5). Future advances can be expected in addressing whether recurrent genomic rearrangements (including polyploidization and diploidization events) have affected recent adaptation to environmental constraints and how the partitioning of genomic plasticity following polyploidization (producing stable and plastic genomic compartments) may have influenced the selective response to novel natural and/or anthropogenic pressures. In particular, the evolutionary frameworks presented above holds the potential to unveil the extent to which post-polyploidy subgenome plasticity (comparing sensitive (MF or S) and dominant (LF or D) genomic compartments) can be considered as a driving evolutionary force that provides a reservoir of novel mutations to be selected during selection or domestication in particular genomic regions. Such research questions pertaining to the role of polyploidy and partitioned genomic plasticity in the adaptive response to selection or domestication and climate change are highly novel and may provide the basis for technological innovations aimed at further developing the breeding capacity of the crop industry.
Fig. 5.
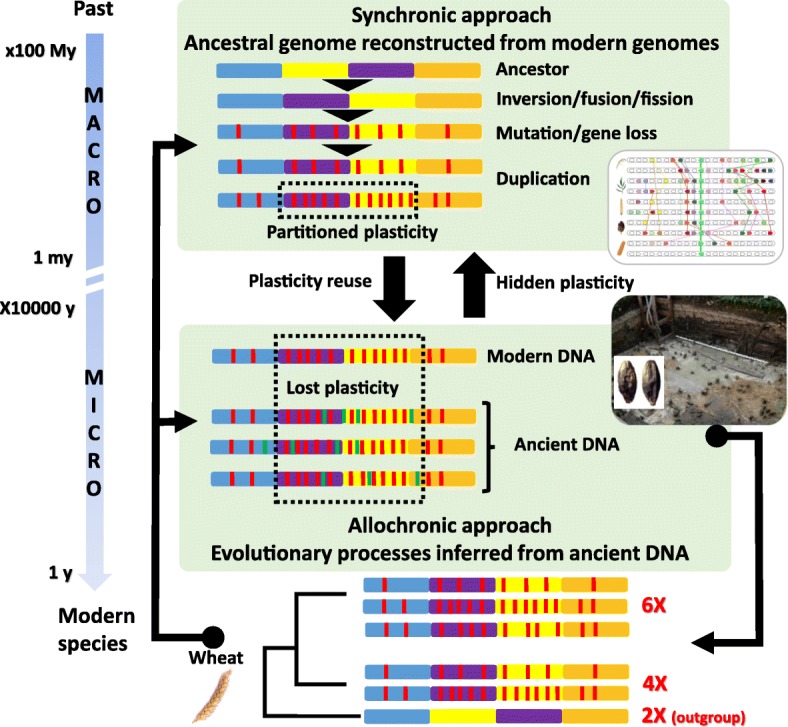
Paleogenomics from ancestral genome reconstruction and aDNA recovery. Paleogenomics encompasses a synchronic approach (top), involving the reconstruction of ancestral genomes of several million years old (macro-evolution) from comparisons of modern genome sequences, and an allochronic approach (bottom), involving the recovery and analysis of ancient DNA from archaeobotanical remains of several hundreds or thousands of years old (micro-evolution). Both approaches are complementary in unveiling the impact of past evolutionary processes (million-years-old genomic rearrangements such as duplications and inversions, illustrated with colored blocks) on the diversity of the modern germplasm (thousand-years-old mutations illustrated with red and green vertical bars, the latter representing mutations that have been lost during domestication and/or the adaptation of modern species). Comparison of DNA from modern and ancient diploid (2X), tetraploid (4X), and hexaploid (6X) species offers the opportunity to investigate the genomic drivers (duplication, inversion, deletion, fusion, fission, mutation…) of plant evolution and adaptation to environmental constraints, as exemplified by wheat (bottom). Excavation and photo A.-M. et P. Pétrequin (CRAVA, CNRS) from Clairvaux-les-Lacs (Jura), CL VII, IVth Millennium BC
Funding
Completion of this article was supported by the ‘Région Auvergne-Rhône-Alpes’ and FEDER 'Fonds Européen de Développement Régional' (#23000816 SRESRI 2015), Institut Carnot Plant2Pro (Project 2017 SyntenyViewer), AgreenSkills fellowship (Applicant ID #4146), ERC Advanced grant project TREEPEACE (#339728), and ANR projects H2oak (ANR-14-CE02-0013), PAGE (ANR-11-BSV6-0008) and GENOAK (ANR-11-BSV6-0009).
Abbreviations
- AAK
Ancestral angiosperm karyotype
- aDNA
Ancient DNA
- AEK
Ancestral eudicot karyotype
- AGK
Ancestral grass karyotype
- AMK
Ancestral monocot karyotype
- CAR
Contiguous ancestral region
- CCF
Centromeric chromosome fusion
- GO
Gene Ontology
- K–Pg boundary
Cretaceous–Paleogene boundary
- mya
Million years ago
- oPGs
Ordered protogenes
- PPD
Post-polyploidization diploidization
- pPGs
Putative protogenes
- SB
Synteny block
- SNP
Single nucleotide polymorphism
- TCF
Telomeric chromosome fusion
- WGD
Whole-genome duplication
Authors’ contributions
CP, SW, AK, LO, CP, and JS jointly contributed to the work. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bell CD, Soltis DE, Soltis PS. The age of the angiosperms: a molecular timescale without a clock. Evolution. 2005;59:1245–1258. doi: 10.1111/j.0014-3820.2005.tb01775.x. [DOI] [PubMed] [Google Scholar]
- 2.Magallon S. Using fossils to break long branches in molecular dating: a comparison of relaxed clocks applied to the origin of angiosperms. Syst Biol. 2010;59:384–399. doi: 10.1093/sysbio/syq027. [DOI] [PubMed] [Google Scholar]
- 3.Barba-Montoya J, Dos Reis M, Schneider H, Donoghue PCJ, Yang Z. Constraining uncertainty in the timescale of angiosperm evolution and the veracity of a Cretaceous Terrestrial Revolution. New Phytol. 2018;218:819–834. doi: 10.1111/nph.15011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friis EM, Pedersen KR, Crane PR. Cretaceous angiosperm flowers: innovation and evolution in plant reproduction. Palaeogeogr Palaeoclimatol Palaeoecol. 2006;232:251–293. doi: 10.1016/j.palaeo.2005.07.006. [DOI] [Google Scholar]
- 5.Friis EM, Pedersen KR, Crane PR. Diversity in obscurity: fossil flowers and the early history of angiosperms. Philos Trans R Soc Lond B Biol Sci. 2010;365:369–382. doi: 10.1098/rstb.2009.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soltis DE, Bell CD, Kim S, Soltis PS. Origin and early evolution of angiosperms. Ann N Y Acad Sci. 2008;1133:3–25. doi: 10.1196/annals.1438.005. [DOI] [PubMed] [Google Scholar]
- 7.Doyle JA. Molecular and fossil evidence on the origin of angiosperms. Annu Rev Earth Planet Sci. 2012;40:301–26.
- 8.Ferguson-Smith MA, Trifonov V. Mammalian karyotype evolution. Nat Rev Genet. 2007;8:950–962. doi: 10.1038/nrg2199. [DOI] [PubMed] [Google Scholar]
- 9.Nakatani Y, Takeda H, Kohara Y, Morishita S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 2007;17:1254–1265. doi: 10.1101/gr.6316407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma J, Zhang L, Suh BB, Raney BJ, Burhans RC, Kent WJ, et al. Reconstructing contiguous regions of an ancestral genome. Genome Res. 2006;16:1557–1565. doi: 10.1101/gr.5383506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Farré M, Auvil L, Capitanu B, Larkin DM, Ma J, Lewin HA. Reconstruction and evolutionary history of eutherian chromosomes. Proc Natl Acad Sci U S A. 2017;114:E5379–E5388. doi: 10.1073/pnas.1702012114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy WJ, Agarwala R, Schäffer AA, Stephens R, Smith C, Jr, Crumpler NJ, et al. A rhesus macaque radiation hybrid map and comparative analysis with the human genome. Genomics. 2005;86:383–395. doi: 10.1016/j.ygeno.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Hayden S, Bekaert M, Crider TA, Mariani S, Murphy WJ, Teeling EC. Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res. 2010;20:1–9. doi: 10.1101/gr.099416.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salse J. Ancestors of modern plant crops. Curr Opin Plant Biol. 2016;30:134–142. doi: 10.1016/j.pbi.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Pham SK, Pevzner PA. DRIMM-Synteny: decomposing genomes into evolutionary conserved segments. Bioinformatics. 2010;26:2509–2516. doi: 10.1093/bioinformatics/btq465. [DOI] [PubMed] [Google Scholar]
- 16.Proost S, Fostier J, De Witte D, Dhoedt B, Demeester P, Van de Peer Y, Vandepoele K. i-ADHoRe 3.0—fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 2012;40:e11. doi: 10.1093/nar/gkr955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cannon SB, Kozik A, Chan B, Michelmore R, Young ND. DiagHunter and GenoPix2D: programs for genomic comparisons, large-scale homology discovery and visualization. Genome Biol. 2003;4:R68. doi: 10.1186/gb-2003-4-10-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haas BJ, Delcher AL, Wortman JR, Salzberg SL. DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics. 2004;20:3643–3646. doi: 10.1093/bioinformatics/bth397. [DOI] [PubMed] [Google Scholar]
- 19.Soderlund C, Bomhoff M, Nelson WM. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 2011;39:e68. doi: 10.1093/nar/gkr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones BR, Rajaraman A, Tannier E, Chauve C. ANGES: reconstructing ANcestral GEnomeS maps. Bioinformatics. 2012;28:2388–2390. doi: 10.1093/bioinformatics/bts457. [DOI] [PubMed] [Google Scholar]
- 22.Lin CH, Zhao H, Lowcay SH, Shahab A, Bourque G. webMGR: an online tool for the multiple genome rearrangement problem. Bioinformatics. 2010;26:408–410. doi: 10.1093/bioinformatics/btp689. [DOI] [PubMed] [Google Scholar]
- 23.Murat F, Armero A, Pont C, Klopp C, Salse J. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat Genet. 2017;49:490–496. doi: 10.1038/ng.3813. [DOI] [PubMed] [Google Scholar]
- 24.Ming R, VanBuren R, Wai CM, Tang H, Schatz MC, Bowers JE, et al. The pineapple genome and the evolution of CAM photosynthesis. Nat Genet. 2015;47:1435–1442. doi: 10.1038/ng.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schubert I, Lysak MA. Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 2011;27:207–216. doi: 10.1016/j.tig.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Jin D, Wang Z, Guo H, Zhang L, Wang L, et al. Telomere-centric genome repatterning determines recurring chromosome number reductions during the evolution of eukaryotes. New Phytol. 2015;205:378–389. doi: 10.1111/nph.12985. [DOI] [PubMed] [Google Scholar]
- 27.Lohaus R, Van de Peer Y. Of dups and dinos: evolution at the K/Pg boundary. Curr Opin Plant Biol. 2016;30:62–69. doi: 10.1016/j.pbi.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Van de Peer Y, Maere S, Meyer A. The evolutionary significance of ancient genome duplications. Nat Rev Genet. 2009;10:725–732. doi: 10.1038/nrg2600. [DOI] [PubMed] [Google Scholar]
- 29.Van de Peer Y, Mizrachi E, Marchal K. The evolutionary significance of polyploidy. Nat Rev Genet. 2017;18:411–424. doi: 10.1038/nrg.2017.26. [DOI] [PubMed] [Google Scholar]
- 30.Murat F, Zhang R, Guizard S, Gavranović H, Flores R, Steinbach D, et al. Karyotype and gene order evolution from reconstructed extinct ancestors highlight contrasts in genome plasticity of modern rosid crops. Genome Biol Evol. 2015;7:735–749. doi: 10.1093/gbe/evv014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Becerra JX. Timing the origin and expansion of the Mexican tropical dry forest. Proc Natl Acad Sci U S A. 2005;12:10919–10923. doi: 10.1073/pnas.0409127102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Couvreur TL, Franzke A, Al-Shehbaz IA, Bakker FT, Koch MA, Mummenhoff K. Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae) Mol Biol Evol. 2010;27:55–71. doi: 10.1093/molbev/msp202. [DOI] [PubMed] [Google Scholar]
- 33.Raymond O, Gouzy J, Just J, Badouin H, Verdenaud M, Lemainque A, et al. The Rosa genome provides new insights into the domestication of modern roses. Nat Genet. 2018;50:772–777. doi: 10.1038/s41588-018-0110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murat F, Louis A, Maumus F, Armero A, Cooke R, Quesneville H, et al. Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biol. 2015;16:262. doi: 10.1186/s13059-015-0814-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S, Shamimuzzaman M, Sun H, Salse J, Sui X, Wilder A, et al. The bottle gourd genome provides insights into Cucurbitaceae evolution and facilitates mapping of a Papaya ring-spot virus resistance locus. Plant J. 2017;92:963–975. doi: 10.1111/tpj.13722. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Sun P, Li Y, Liu Y, Yu J, Ma X, et al. Hierarchically aligning 10 legume genomes establishes a family-level genomics platform. Plant Physiol. 2017;174:284–300. doi: 10.1104/pp.16.01981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Wang J, Jin D, Guo H, Lee TH, Liu T, Paterson AH. Genome alignment spanning major Poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Mol Plant. 2015;8:885–898. doi: 10.1016/j.molp.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Murat F, Xu JH, Tannier E, Abrouk M, Guilhot N, Pont C. Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution. Genome Res. 2010;20:1545–1557. doi: 10.1101/gr.109744.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murat F, Zhang R, Guizard S, Flores R, Armero A, Pont C, et al. Shared subgenome dominance following polyploidization explains grass genome evolutionary plasticity from a seven protochromosome ancestor with 16K protogenes. Genome Biol Evol. 2014;6:12–33. doi: 10.1093/gbe/evt200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salse J, Bolot S, Throude M, Jouffe V, Piegu B, Quraishi UM, et al. Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell. 2008;20:11–24. doi: 10.1105/tpc.107.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soltis PS, Marchant DB, Van de Peer Y, Soltis DE. Polyploidy and genome evolution in plants. Curr Opin Genet Dev. 2015;35:119–125. doi: 10.1016/j.gde.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Hollister JD. Polyploidy: adaptation to the genomic environment. New Phytol. 2015;205:1034–1039. doi: 10.1111/nph.12939. [DOI] [PubMed] [Google Scholar]
- 43.Louis A, Murat F, Salse J, Crollius HR. GenomicusPlants: a web resource to study genome evolution in flowering plants. Plant Cell Physiol. 2015;56:e4. doi: 10.1093/pcp/pcu177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 2008;148:1772–1781. doi: 10.1104/pp.108.124867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Proost S, Van Bel M, Vaneechoutte D, Van de Peer Y, Inzé D, Mueller-Roeber B, Vandepoele K. PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res. 2015;42(Database issue):D974–D981. doi: 10.1093/nar/gku986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valluru R, Reynolds MP, Salse J. Genetic and molecular bases of yield-associated traits: a translational biology approach between rice and wheat. Theor Appl Genet. 2014;127:1463–1489. doi: 10.1007/s00122-014-2332-9. [DOI] [PubMed] [Google Scholar]
- 47.Bordat A, Savois V, Nicolas M, Salse J, Chauveau A, Bourgeois M, et al. Translational genomics in legumes allowed placing in silico 5460 unigenes on the pea functional map and identified candidate genes in Pisum sativum L. G3 (Bethesda) 2011;1:93–103. doi: 10.1534/g3.111.000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobrovolskaya O, Pont C, Sibout R, Martinek P, Badaeva E, Murat F, et al. FRIZZY PANICLE drives supernumerary spikelets in bread wheat. Plant Physiol. 2015;167:189–199. doi: 10.1104/pp.114.250043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang Z, Schnable JC. Functional divergence between subgenomes and gene pairs after whole genome duplications. Mol Plant. 2018;11:388–397. doi: 10.1016/j.molp.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 50.Cheng F, Wu J, Fang L, Sun S, Liu B, Lin K, et al. Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS One. 2012;7:e36442. doi: 10.1371/journal.pone.0036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng F, Sun C, Wu J, Schnable J, Woodhouse MR, Liang J, et al. Epigenetic regulation of subgenome dominance following whole genome triplication in Brassica rapa. New Phytol. 2016;211:288–299. doi: 10.1111/nph.13884. [DOI] [PubMed] [Google Scholar]
- 52.Cheng F, Sun R, Hou X, Zheng H, Zhang F, Zhang Y, et al. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat Genet. 2016;48:1218–1224. doi: 10.1038/ng.3634. [DOI] [PubMed] [Google Scholar]
- 53.Pophaly SD, Tellier A. Population level purifying selection and gene expression shape subgenome evolution in mMaize. Mol Biol Evol. 2015;32:3226–3235. doi: 10.1093/molbev/msv191. [DOI] [PubMed] [Google Scholar]
- 54.Garsmeur O, Schnable JC, Almeida A, Jourda C, D'Hont A, Freeling M. Two evolutionarily distinct classes of paleopolyploidy. Mol Biol Evol. 2014;31:448–454. doi: 10.1093/molbev/mst230. [DOI] [PubMed] [Google Scholar]
- 55.Mandáková T, Lysak MA. Post-polyploid diploidization and diversification through dysploid changes. Curr Opin Plant Biol. 2018;42:55–65. doi: 10.1016/j.pbi.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Gou X, Bian Y, Zhang A, Zhang H, Wang B, Lv R, et al. Transgenerationally precipitated meiotic chromosome instability fuels rapid karyotypic evolution and phenotypic diversity in an artificially constructed allotetraploid wheat (AADD) Mol Biol Evol. 2018;35:1078–1091. doi: 10.1093/molbev/msy009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marciniak S, Perry GH. Harnessing ancient genomes to study the history of human adaptation. Nat Rev Genet. 2017;18:659–674. doi: 10.1038/nrg.2017.65. [DOI] [PubMed] [Google Scholar]
- 58.Lynch VJ, Bedoya-Reina OC, Ratan A, Sulak M, Drautz-Moses DI, Perry GH, et al. Elephantid genomes reveal the molecular bases of woolly mammoth adaptations to the Arctic. Cell Rep. 2015;12:217–228. doi: 10.1016/j.celrep.2015.06.027. [DOI] [PubMed] [Google Scholar]
- 59.Park SD, Magee DA, McGettigan PA, Teasdale MD, Edwards CJ, Lohan AJ, et al. Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 2015;16:234. doi: 10.1186/s13059-015-0790-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaunitz C, Fages A, Hanghøj K, Albrechtsen A, Khan N, Schubert M, et al. Ancient genomes revisit the ancestry of domestic and Przewalski’s horses. Science. 2018;360:111–114. doi: 10.1126/science.aao3297. [DOI] [PubMed] [Google Scholar]
- 61.Frantz LA, Mullin VE, Pionnier-Capitan M, Lebrasseur O, Ollivier M, Perri A, et al. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science. 2016;352:1228–1231. doi: 10.1126/science.aaf3161. [DOI] [PubMed] [Google Scholar]
- 62.Rasmussen M, Guo X, Wang Y, Lohmueller KE, Rasmussen S, Albrechtsen A, et al. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–98. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 2015;16:359–371. doi: 10.1038/nrg3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leonardi M, Librado P, Der Sarkissian C, Schubert M, Alfarhan AH, Alquraishi SA, et al. Evolutionary patterns and processes: lessons from ancient DNA. Syst Biol. 2017;66:e1–29. doi: 10.1093/sysbio/syw059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malaspinas AS, Lao O, Schroeder H, Rasmussen M, Raghavan M, Moltke I, et al. Two ancient human genomes reveal Polynesian ancestry among the indigenous Botocudos of Brazil. Curr Biol. 2014;24:R1035–R1037. doi: 10.1016/j.cub.2014.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schraiber JG, Evans SN, Slatkin M. Bayesian inference of natural selection from allele frequency time series. Genetics. 2016;203:493–511. doi: 10.1534/genetics.116.187278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramakrishnan U, Hadly EA. Using phylochronology to reveal cryptic population histories: review and synthesis of 29 ancient DNA studies. Mol Ecol. 2009;18:1310–1330. doi: 10.1111/j.1365-294X.2009.04092.x. [DOI] [PubMed] [Google Scholar]
- 69.Mourier T, Ho SY, Gilbert MT, Willerslev E, Orlando L. Statistical guidelines for detecting past population shifts using ancient DNA. Mol Biol Evol. 2012;29:2241–2251. doi: 10.1093/molbev/mss094. [DOI] [PubMed] [Google Scholar]
- 70.Orlando L, Gilbert MT, Willerslev E. Reconstructing ancient genomes and epigenomes. Nat Rev Genet. 2015;16:395–408. doi: 10.1038/nrg3935. [DOI] [PubMed] [Google Scholar]
- 71.Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, Waterston RH. DNA sequencing at 40: past, present and future. Nat Rev. 2017;550:345–353. doi: 10.1038/nature24286. [DOI] [PubMed] [Google Scholar]
- 72.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 73.Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, et al. Analysis of one million base pairs of Neanderthal DNA. Nature. 2006;444:330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- 74.Gilbert MT, Drautz DI, Lesk AM, Ho SY, Qi J, Ratan A, et al. Intraspecific phylogenetic analysis of Siberian woolly mammoths using complete mitochondrial genomes. Proc Natl Acad Sci U S A. 2008;105:8327–8332. doi: 10.1073/pnas.0802315105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gamba C, Jones ER, Teasdale MD, McLaughlin RL, Gonzalez-Fortes G, Mattiangeli V, et al. Genome flux and stasis in a five millennium transect of European prehistory. Nat Commun. 2014;5:5257. doi: 10.1038/ncomms6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Damgaard PB, Margaryan A, Schroeder H, Orlando L, Willerslev E, Allentoft ME. Improving access to endogenous DNA in ancient bones and teeth. Sci Rep. 2015;5:11184. doi: 10.1038/srep11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Der Sarkissian C, Pichereau V, Dupont C, Ilsøe PC, Perrigault M, Butler P, et al. Ancient DNA analysis identifies marine mollusc shells as new metagenomic archives of the past. Mol Ecol Resour. 2017;17:835–853. doi: 10.1111/1755-0998.12679. [DOI] [PubMed] [Google Scholar]
- 78.Dabney J, Meyer M, Pääbo S. Ancient DNA damage. Cold Spring Harb Perspect Biol. 2013;5(7). [DOI] [PMC free article] [PubMed]
- 79.Gamba C, Hanghøj K, Gaunitz C, Alfarhan AH, Alquraishi SA, Al-Rasheid KA, et al. Comparing the performance of three ancient DNA extraction methods for high-throughput sequencing. Mol Ecol Resour. 2016;16:459–469. doi: 10.1111/1755-0998.12470. [DOI] [PubMed] [Google Scholar]
- 80.Der Sarkissian C, Ermini L, Jónsson H, Alekseev AN, Crubezy E, Shapiro B, Orlando L. Shotgun microbial profiling of fossil remains. Mol Ecol. 2014;23:1780–1798. doi: 10.1111/mec.12690. [DOI] [PubMed] [Google Scholar]
- 81.Korlević P, Gerber T, Gansauge MT, Hajdinjak M, Nagel S, Aximu-Petri A, Meyer M. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques. 2015;59:87–93. doi: 10.2144/000114320. [DOI] [PubMed] [Google Scholar]
- 82.Boessenkool S, Hanghøj K, Nistelberger HM, Der Sarkissian C, Gondek AT, Orlando L, et al. Combining bleach and mild predigestion improves ancient DNA recovery from bones. Mol Ecol Resour. 2017;17:742–751. doi: 10.1111/1755-0998.12623. [DOI] [PubMed] [Google Scholar]
- 83.Slon V, Hopfe C, Weiß CL, Mafessoni F, de la Rasilla M, Lalueza-Fox C, et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science. 2017;356:605–608. doi: 10.1126/science.aam9695. [DOI] [PubMed] [Google Scholar]
- 84.Seguin-Orlando A, Schubert M, Clary J, Stagegaard J, Alberdi MT, Prado JL, et al. Ligation bias in Illumina next-generation DNA libraries: implications for sequencing ancient genomes. PLoS One. 2013;8:e78575. doi: 10.1371/journal.pone.0078575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dabney J, Meyer M. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques. 2012;52:87–94. doi: 10.2144/000113809. [DOI] [PubMed] [Google Scholar]
- 86.Gansauge MT, Meyer M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat Protoc. 2013;8:737–748. doi: 10.1038/nprot.2013.038. [DOI] [PubMed] [Google Scholar]
- 87.Gansauge MT, Meyer M. Selective enrichment of damaged DNA molecules for ancient genome sequencing. Genome Res. 2014;24:1543–1549. doi: 10.1101/gr.174201.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One. 2011;5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fabre PH, Vilstrup JT, Raghavan M, Sarkissian CD, Willerslev E, Douzery EJ, Orlando L. Rodents of the Caribbean: origin and diversification of hutias unravelled by next- generation museomics. Biol Lett. 2014;10. [DOI] [PMC free article] [PubMed]
- 90.Lipson M, Skoglund P, Spriggs M, Valentin F, Bedford S, Shing R, et al. Population turnover in remote Oceania shortly after initial settlement. Curr Biol. 2018;28:1157–1165. doi: 10.1016/j.cub.2018.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carpenter ML, Buenrostro JD, Valdiosera C, Schroeder H, Allentoft ME, Sikora M, et al. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am J Hum Genet. 2013;93:852–864. doi: 10.1016/j.ajhg.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Enk JM, Devault AM, Kuch M, Murgha YE, Rouillard JM, Poinar HN. Ancient whole genome enrichment using baits built from modern DNA. Mol Biol Evol. 2014;31:1292–1294. doi: 10.1093/molbev/msu074. [DOI] [PubMed] [Google Scholar]
- 93.Ginolhac A, Rasmussen M, Gilbert MT, Willerslev E, Orlando L. mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics. 2011;27:2153–2155. doi: 10.1093/bioinformatics/btr347. [DOI] [PubMed] [Google Scholar]
- 94.Jónsson H, Ginolhac A, Schubert M, Johnson PL, Orlando L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013;29:1682–1684. doi: 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Skoglund P, Sjödin P, Skoglund T, Lascoux M, Jakobsson M. Investigating population history using temporal genetic differentiation. Mol Biol Evol. 2014;31:2516–2527. doi: 10.1093/molbev/msu192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schubert M, Ginolhac A, Lindgreen S, Thompson JF, Al-Rasheid KA, Willerslev E, et al. Improving ancient DNA read mapping against modern reference genomes. BMC Genomics. 2012;13:178. doi: 10.1186/1471-2164-13-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schubert M, Jónsson H, Chang D, Der Sarkissian C, Ermini L, Ginolhac A, et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc Natl Acad Sci U S A. 2014;111:E5661–E5669. doi: 10.1073/pnas.1416991111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kerpedjiev P, Frellsen J, Lindgreen S, Krogh A. Adaptable probabilistic mapping of short reads using position specific scoring matrices. BMC Bioinformatics. 2014;15:100. doi: 10.1186/1471-2105-15-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Taron UH, Lell M, Barlow A, Paijmans JLA. Testing of alignment parameters for ancient samples: evaluating and optimizing mapping parameters for ancient samples using the TAPAS tool. Genes (Basel). 2018;9(3). [DOI] [PMC free article] [PubMed]
- 100.Fordyce SL, Ávila-Arcos MC, Rasmussen M, Cappellini E, Romero-Navarro JA, Wales N, et al. Deep sequencing of RNA from ancient maize kernels. PLoS One. 2013;8:e50961. doi: 10.1371/journal.pone.0050961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Willerslev E, Cooper A. Ancient DNA. Proc Biol Sci. 2005 ;272(1558):3-16. [DOI] [PMC free article] [PubMed]
- 102.Gutaker RM, Burbano HA. Reinforcing plant evolutionary genomics using ancient DNA. Curr Opin Plant Biol. 2017;36:38–45. doi: 10.1016/j.pbi.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 103.Palmer SA, Moore JD, Clapham AJ, Rose P, Allaby RG. Archaeogenetic evidence of ancient nubian barley evolution from six- to two-row indicates local adaptation. PLoS One. 2009;4:e6301. doi: 10.1371/journal.pone.0006301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mascher M, Schuenemann VJ, Davidovich U, Marom N, Himmelbach A, Hübner S, et al. Genomic analysis of 6,000-year-old cultivated grain illuminates the domestication history of barley. Nat Genet. 2016;48:1089–1093. doi: 10.1038/ng.3611. [DOI] [PubMed] [Google Scholar]
- 105.Fernández E, Thaw S, Brown TA, Arroyo-Pardo E, Buxó R, Serret MD, Araus JL. DNA analysis in charred grains of naked wheat from several archaeological sites in Spain. J Archaeol Sci. 2013;40:659–670. doi: 10.1016/j.jas.2012.07.014. [DOI] [Google Scholar]
- 106.Allaby RG, O'Donoghue K, Sallares R, Jones MK, Brown TA. Evidence for the survival of ancient DNA in charred wheat seeds from European archaeological sites. Anc Biomol. 1997;1:119–129. [Google Scholar]
- 107.Li C, Lister DL, Li H, Xu Y, Cui Y, Bower MA, et al. Ancient DNA analysis of desiccated wheat grains excavated from a Bronze Age cemetery in Xinjiang. J Archaeol Sci. 2011;38:115–119. doi: 10.1016/j.jas.2010.08.016. [DOI] [Google Scholar]
- 108.Oliveira HR, Civáň P, Morales J, Rodríguez-Rodríguez A, Lister D, Jones MR. Ancient DNA in archaeological wheat grains: preservation conditions and the study of pre-Hispanic agriculture on the island of Gran Canaria (Spain) J Archaeol Sci. 2012;39:828–835. doi: 10.1016/j.jas.2011.10.008. [DOI] [Google Scholar]
- 109.Bilgic H, Hakki EE, Pandey A, Khan MK, Akkaya MS. Ancient DNA from 8400 year-old Çatalhöyük wheat: implications for the origin of Neolithic agriculture. PLoS One. 2016;11:e0151974. doi: 10.1371/journal.pone.0151974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goloubinoff P, Pääbo S, Wilson AC. Evolution of maize inferred from sequence diversity of an Adh2 gene segment from archaeological specimens. Proc Natl Acad Sci U S A. 1993;90:1997–2001. doi: 10.1073/pnas.90.5.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rollo F, Asci W, Sassaroli S. Conservation of plant genes II: utilization of ancient and modern DNA. Missouri: Missouri Botanical Garden; 1994. pp. 27–35. [Google Scholar]
- 112.Freitas FO, Bendel G, Allaby RG, Brown TA. DNA from primitive maize landraces and archaeological remains: implications for the domestication of maize and its expansion into South America. J Archaeol Sci. 2003;30:901–908. doi: 10.1016/S0305-4403(02)00269-8. [DOI] [Google Scholar]
- 113.Jaenicke-Després V, Buckler ES, Smith BD, Gilbert MT, Cooper A, Doebley J, Pääbo S. Early allelic selection in maize as revealed by ancient DNA. Science. 2003;302:1206–1208. doi: 10.1126/science.1089056. [DOI] [PubMed] [Google Scholar]
- 114.Lia VV, Confalonieri VA, Ratto N, Hernández JA, Alzogaray AM, Poggio L, Brown TA. Microsatellite typing of ancient maize: insights into the history of agriculture in southern South America. Proc R Soc Lond B Biol Sci. 2007;274:545–554. doi: 10.1098/rspb.2006.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.da Fonseca RR, Smith BD, Wales N, Cappellini E, Skoglund P, Fumagalli M, et al. The origin and evolution of maize in the Southwestern United States. Nat Plants. 2015;1:14003. doi: 10.1038/nplants.2014.3. [DOI] [PubMed] [Google Scholar]
- 116.Ramos-Madrigal J, Smith BD, Moreno-Mayar JV, Gopalakrishnan S, Ross-Ibarra J, Gilbert MTP, Wales N. Genome Sequence of a 5,310-year-old maize cob provides insights into the early stages of maize domestication. Curr Biol. 2016;26:3195–3201. doi: 10.1016/j.cub.2016.09.036. [DOI] [PubMed] [Google Scholar]
- 117.Vallebueno-Estrada M, Rodríguez-Arévalo I, Rougon-Cardoso A, Martínez González J, García Cook A, Montiel R, Vielle-Calzada JP. The earliest maize from San Marcos Tehuacán is a partial domesticate with genomic evidence of inbreeding. Proc Natl Acad Sci U S A. 2016;113:14151–14156. doi: 10.1073/pnas.1609701113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pérez-Zamorano B, Vallebueno-Estrada M, Martínez González J, García Cook A, Montiel R, Vielle-Calzada JP, Delaye L. Organellar genomes from a ∼5,000-year-old archaeological maize sample are closely related to NB genotype. Genome Biol Evol. 2017;9:904–915. doi: 10.1093/gbe/evx048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wales N, Akman M, Watson RHB, Sánchez Barreiro F, Smith BD, Gremillion KJ, et al. Ancient DNA reveals the timing and persistence of organellar genetic bottlenecks over 3,000 years of sunflower domestication and improvement. Evolutionary Applications. 2018;12(1):38–53. [DOI] [PMC free article] [PubMed]
- 120.Manen JF, Bouby L, Dalnoki O, Marinval P, Turgay M, Schlumbaum A. Microsatellites from archaeological Vitis vinifera seeds allow a tentative assignment of the geographical origin of ancient cultivars. J Archaeol Sci. 2003;30:721–729. doi: 10.1016/S0305-4403(02)00244-3. [DOI] [Google Scholar]
- 121.Cappellini E, Gilbert MT, Geuna F, Fiorentino G, Hall A, Thomas-Oates J, et al. A multidisciplinary study of archaeological grape seeds. Naturwissenschaften. 2010;97:205–217. doi: 10.1007/s00114-009-0629-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wales N, Andersen K, Cappellini E, Avila-Arcos MC, Gilbert MT. Optimization of DNA recovery and amplification from non-carbonized archaeobotanical remains. PLoS One. 2014;9:e86827. doi: 10.1371/journal.pone.0086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kistler L, Montenegro A, Smith BD, Gifford JA, Green RE, Newsom LA, Shapiro B. Transoceanic drift and the domestication of African bottle gourds in the Americas. Proc Natl Acad Sci U S A. 2014;111:2937–2941. doi: 10.1073/pnas.1318678111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.O’Donoghue K, Clapham A, Evershed RP, Brown TA. Remarkable preservation of biomolecules in ancient radish seeds. Proc Biol Sci. 1996;263:541–547. doi: 10.1098/rspb.1996.0082. [DOI] [PubMed] [Google Scholar]
- 125.Deakin WJ, Rowley-Conwy PJ, Shaw CH. Amplification and sequencing of DNA from preserved sorghum of up to 2800 years antiquity found at Qasr Ibrim. Ancient Biomolecules. 1998;2:27–41. [Google Scholar]
- 126.Marota I, Basile C, Ubaldi M, Rollo F. DNA decay rate in papyri and human remains from Egyptian archaeological sites. Am J Phys Anthropol. 2002;117:310–318. doi: 10.1002/ajpa.10045. [DOI] [PubMed] [Google Scholar]
- 127.Fan LJ, Gui YJ, Zheng YF, Wang Y, Cai DG, You XL. Ancient DNA sequences of rice from the low Yangtze reveal significant genotypic divergence. Chin Sci Bull. 2011;56:3108. doi: 10.1007/s11434-011-4691-9. [DOI] [Google Scholar]
- 128.Elbaum R, Melamed-Bessudo C, Boaretto E, Galili E, Lev-Yadun S, Levy AA, Weiner S. Ancient olive DNA in pits: preservation, amplification and sequence analysis. J Archaeol Sci. 2006;33:77–88. doi: 10.1016/j.jas.2005.06.011. [DOI] [Google Scholar]
- 129.Mazo L, Gómez A, Quintanilla S, Bernal J, Ortiz P, Valdivieso SJ. Extraction and amplification of DNA from orchid exsiccates conserved for more than half a century in a herbarium in Bogotá. Colombia. Lankesteriana. 2012;12:121–129. [Google Scholar]
- 130.Pollmann B, Jacomet S, Schlumbaum A. Morphological and genetic studies of waterlogged Prunus species from the Roman vicus Tasgetium (Eschenz, Switzerland) J Archaeol Sci. 2005;32:1471–1480. doi: 10.1016/j.jas.2005.04.002. [DOI] [Google Scholar]
- 131.Gutaker RM, Reiter E, Furtwängler A, Schuenemann VJ, Burbano HA. Extraction of ultrashort DNA molecules from herbarium specimens. Biotechniques. 2017;62:76–79. doi: 10.2144/000114517. [DOI] [PubMed] [Google Scholar]
- 132.Palmer SA, Clapham AJ, Rose P, Freitas FO, Owen BD, Beresford-Jones D, et al. Archaeogenomic evidence of punctuated genome evolution in Gossypium. Mol Biol Evol. 2012;29:2031–2038. doi: 10.1093/molbev/mss070. [DOI] [PubMed] [Google Scholar]
- 133.Tani N, Tsumura Y, Sato H. Nuclear gene sequences and DNA variation of Cryptomeria japonica samples from the postglacial period. Mol Ecol. 2003;12:859–868. doi: 10.1046/j.1365-294X.2003.01779.x. [DOI] [PubMed] [Google Scholar]
- 134.Deguilloux MF, Bertel L, Celant A, Pemonge MH, Sadori L, Magri D, Pett RJ. Genetic analysis of archaeological wood remains: first results and prospects. J Archaeol Sci. 2006;33:1216–1227. doi: 10.1016/j.jas.2005.12.012. [DOI] [Google Scholar]
- 135.Liepelt S, Sperisen C, Deguilloux MF, Petit RJ, Kissling R, Spencer M, et al. Authenticated DNA from ancient wood remains. Ann Bot. 2006;98:1107–1111. doi: 10.1093/aob/mcl188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lendvay B, Hartmann M, Brodbeck S, Nievergelt D, Reinig F, Zoller S, et al. Improved recovery of ancient DNA from subfossil wood—application to the world's oldest Late Glacial pine forest. New Phytol. 2017;217:1737–1748. doi: 10.1111/nph.14935. [DOI] [PubMed] [Google Scholar]
- 137.Di Donato A, Filippone E, Ercolano MR, Frusciante L. Genome sequencing of ancient plant remains: findings, uses and potential applications for the study and improvement of modern crops. Front Plant Sci. 2018;9:441. doi: 10.3389/fpls.2018.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Parducci L, Jørgensen T, Tollefsrud MM, Elverland E, Alm T, Fontana SL, et al. Glacial survival of boreal trees in northern Scandinavia. Science. 2012;335:1083–1086. doi: 10.1126/science.1216043. [DOI] [PubMed] [Google Scholar]
- 139.Soltis PS, Soltis DE, Smiley CJ. An rbcL sequence from a Miocene Taxodium (bald cypress) Proc Natl Acad Sci U S A. 1992;89:449–451. doi: 10.1073/pnas.89.1.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wan QH, Wu H, Fujihara T, Fang SG. Which genetic marker for which conservation genetics issue? Electrophoresis. 2004;25:2165–2176. doi: 10.1002/elps.200305922. [DOI] [PubMed] [Google Scholar]
- 141.Weiß CL, Schuenemann VJ, Devos J, Shirsekar G, Reiter E, Gould BA, et al. Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens. R Soc Open Sci. 2016;3:160239. doi: 10.1098/rsos.160239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wagner S, Lagane F, Seguin-Orlando A, Schubert M, Leroy T, Guichoux E, et al. High-throughput DNA sequencing of ancient wood. Mol Ecol. 2018;27:1138–1154. doi: 10.1111/mec.14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rollo F. Characterisation by molecular hybridization of RNA fragments isolated from ancient (1400 B.C.) seeds. Theor Appl Genet. 1985;71:330–333. doi: 10.1007/BF00252076. [DOI] [PubMed] [Google Scholar]
- 144.Smith O, Clapham AJ, Rose P, Liu Y, Wang J, Allaby RG. Genomic methylation patterns in archaeological barley show de-methylation as a time-dependent diagenetic process. Sci Rep. 2014;4:5559. doi: 10.1038/srep05559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Smith O, Clapham A, Rose P, Liu Y, Wang J, Allaby RG. A complete ancient RNA genome: identification, reconstruction and evolutionary history of archaeological Barley Stripe Mosaic Virus. Sci Rep. 2014;4:4003. doi: 10.1038/srep04003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Smith O, Palmer SA, Clapham AJ, Rose P, Liu Y, Wang J, Allaby RG. Small RNA activity in archeological barley shows novel germination inhibition in response to environment. Mol Biol Evol. 2017;34:2555–2562. doi: 10.1093/molbev/msx175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Meyer RS, DuVal AE, Jensen HR. Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol. 2012;196:29–48. doi: 10.1111/j.1469-8137.2012.04253.x. [DOI] [PubMed] [Google Scholar]
- 148.Feldman M, Levy AA. Genome evolution due to allopolyploidization in wheat. Genetics. 2012;192:763–774. doi: 10.1534/genetics.112.146316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pont C, Salse J. Wheat paleohistory created asymmetrical genomic evolution. Curr Opin Plant Biol. 2017;36:29–37. doi: 10.1016/j.pbi.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 150.Asouti E, Fuller DQ. A contextual approach to the emergence of agriculture in Southwest Asia reconstructing Early Neolithic plant-food production. Curr Anthropol. 2013;54:299–345. doi: 10.1086/670679. [DOI] [Google Scholar]
- 151.Brown TA, Jones MK, Powell W, Allaby RG. The complex origins of domesticated crops in the Fertile Crescent. Trends Ecol Evol. 2009;24:103–109. doi: 10.1016/j.tree.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 152.Szécsényi-Nagy A, Brandt G, Haak W, Keerl V, Jakucs J, Möller-Rieker S, et al. Tracing the genetic origin of Europe's first farmers reveals insights into their social organization. Proc Biol Sci. 2015;282(1805). [DOI] [PMC free article] [PubMed]
- 153.Jordan KW, Wang S, Lun Y, Gardiner LJ, MacLachlan R, Hucl P, et al. A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol. 2015;16:48. doi: 10.1186/s13059-015-0606-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jacomet S, Schlichtherle H. Der kleine Pfahlbauweizen Oswald Heer's—Neue Untersuchungen zur Morphologie neolithischer Nacktweizen-Ähren. In: Van Zeist W, Casparie WA, editors. Plants and ancient man: studies in palaeoethnobotany. Rotterdam: A. A. Balkema; 1984. pp. 153–176. [Google Scholar]
- 155.Maier U. Morphological studies of free-threshing wheat ears from a Neolithic site in southwest Germany, and the history of the naked wheats. Veget Hist Archaeobot. 1996;5:39–55. doi: 10.1007/BF00189434. [DOI] [Google Scholar]
- 156.Zohary D, Hopf M. Domestication of plants in the Old World. New York: Oxford University Press; 2000. [Google Scholar]
- 157.Jones G, Valamoti S, Charles M. Early crop diversity: a ‘new’ glume wheat from northern Greece. Veget Hist Archaeobot. 2000;9:133–146. doi: 10.1007/BF01299798. [DOI] [Google Scholar]
- 158.Brown TA, Allaby RG, Sallares R, Jones G. Ancient DNA in charred wheats: taxonomic identification of mixed and single grains. Ancient Biomolecules. 1998;2:185–193. [Google Scholar]
- 159.Allaby RG, Banerjee M, Brown TA. Evolution of the high molecular weight glutenin loci of the A, B, D, and G genomes of wheat. Genome. 1999;42:296–307. doi: 10.1139/g98-114. [DOI] [PubMed] [Google Scholar]
- 160.Nistelberger HM, Smith O, Wales N, Star B, Boessenkool S. The efficacy of high-throughput sequencing and target enrichment on charred archaeobotanical remains. Sci Rep. 2016;6:37347. doi: 10.1038/srep37347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Baltos ULC, Hertel TW. Global food security in 2050: the role of agricultural productivity and climate change. Aust J Agric Resour Econ. 2014;58:554–570. doi: 10.1111/1467-8489.12048. [DOI] [Google Scholar]
- 162.Allen CD, Macalady AK, Chenchouni H, Bachele D, McDowell N, Vennetier M, et al. A global overview of drought and heat induced tree mortality reveals emerging climate change risks for forests. For Ecol Manage. 2010;259:660–684. doi: 10.1016/j.foreco.2009.09.001. [DOI] [Google Scholar]
- 163.Birdsey R, Pan Y. Trends in management of the world's forests and impacts on carbon stocks. For Ecol Manage. 2015;355:83–90. doi: 10.1016/j.foreco.2015.04.031. [DOI] [Google Scholar]
- 164.Habash DZ, Kehel Z, Nachit M. Genomic approaches for designing durum wheat ready for climate change with a focus on drought. J Exp Bot. 2009;60:2805–2815. doi: 10.1093/jxb/erp211. [DOI] [PubMed] [Google Scholar]
- 165.Boyer JS, Westgate ME. Grain yields with limited water. J Exp Bot. 2004;55:2385–2394. doi: 10.1093/jxb/erh219. [DOI] [PubMed] [Google Scholar]
- 166.Shafer AB, Wolf JB, Alves PC, Bergström L, Bruford MW, Brännström I, et al. Genomics and the challenging translation into conservation practice. Trends Ecol Evol. 2015;30:78–87. doi: 10.1016/j.tree.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 167.Marcott SA, Shakun JD, Clark PU, Mix AC. A reconstruction of regional and global temperature for the past 11,300 years. Science. 2013;339:1198–1201. doi: 10.1126/science.1228026. [DOI] [PubMed] [Google Scholar]
- 168.Cuffey KM, Clow GD. Temperature, accumulation, and ice sheet elevation in central Greenland through the last deglacial transition. Journal of Geophysical Research 1997;102:26383-26396.
- 169.Allaby RG, Gutaker R, Clarke AC, Pearson N, Ware R, Palmer SA, et al. Using archaeogenomic and computational approaches to unravel the history of local adaptation in crops. Philos Trans R Soc Lond B Biol Sci. 2015;370:20130377. [DOI] [PMC free article] [PubMed]