

Abstract

Flavin-dependent halogenases are known to regioselectively introduce halide substituents into aromatic moieties, for example, the indole ring of tryptophan. The process requires halide salts and oxygen instead of molecular halogen in the chemical halogenation. However, the reduced cofactor flavin adenine dinucleotide (FADH2) has to be regenerated using a flavin reductase. Consequently, coupled biocatalytic steps are usually applied for cofactor regeneration. Nicotinamide adenine dinucleotide (NADH) mimics can be employed stoichiometrically to replace enzymatic cofactor regeneration in biocatalytic halogenation. Chlorination of l-tryptophan is successfully performed using such NADH mimics. The efficiency of this approach has been compared to the previously established enzymatic regeneration system using the two auxiliary enzymes flavin reductase (PrnF) and alcohol dehydrogenase (ADH). The reaction rates of some of the tested mimics were found to exceed that of the enzymatic system. Continuous enzymatic halogenation reaction for reaction scale-up is also possible.
Keywords: regioselective chlorination, flavin-dependent halogenases, hydride transfer, NADH mimics, enzymatic cofactor regeneration, FADH2
Halogenated natural products are of high biological and biochemical relevance. Many halogenated metabolites or their derivatives found their way into pharmaceutical and agricultural applications.1−4 Although different chemical halogenation methods are known, most of them suffer from low regioselectivity and/or environmental burden.5,6 In view of the biosynthesis of halogenated metabolites, FAD-dependent halogenases have become promising alternatives for the regioselective halogenation of natural and synthetic molecules using only halide salts and molecular oxygen under mild reaction conditions.7−9 However, this enzyme class still faces several drawbacks that compromise a wider application in biocatalytic reactions: low activity, combined with a substrate scope that is mainly limited to certain core structures,10,11 as well as their dependence on the cofactor FADH2.12 Hence, the enzymatic halogenation reaction requires a complex cofactor regeneration system that maintains a steady FADH2 supply. The regeneration systems for this group of enzymes usually includes two auxiliary enzymes in addition to the halogenase in an enzyme cascade system (Figure 1).12−14
Figure 1.
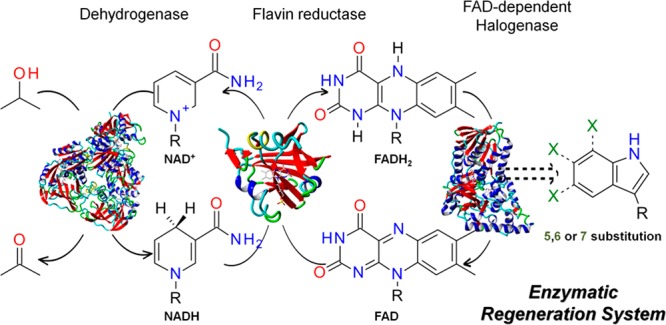
Enzymatic tryptophan halogenation with cofactor regeneration comprising a flavin reductase and a dehydrogenase.
Enzymatic cofactor regeneration systems for biocatalytic halogenation rely on the combination of a flavin reductase like PrnF from Pseudomonas fluorescens,8,12 RebF from Lechevalieria aerocolonigenes,15,16 or SsuE from Thermus thermophilus(12) with either an alcohol dehydrogenase (ADH) or a glucose dehydrogenase (CDX-901). The ADH (e.g., from Rhodococcus ruber) regenerates nicotinamide adenine dinucleotide (NADH) from NAD+ by catalyzing the oxidation of isopropanol to acetone.8 A glucose dehydrogenase regenerates NAD(P)H by catalyzing the oxidation of d-glucose to d-glucono-1,5-lactone, followed by a nonenzymatic hydrolysis to gluconic acid.15−17 The complexity of these enzymatic cascades limits the usability of halogenases in further applications and combination with other synthetic reactions like in chemoenzymatic cascades.
As cofactor regeneration is a common prerequisite for many biocatalytic reactions, alternative nonenzymatic regeneration systems have been proposed and investigated. The direct regeneration of FADH2 for tryptophan 7-halogenase PrnA was first reported using the organometallic complex pentamethylcyclopentadienyl rhodium bipyridine ([Cp*Rh(bpy)(H2O)]2+) as catalyst and formate as electron donor without requiring flavin reductase and NADH (additional regeneration systems are summarized in Table S3, Supporting Information).18 The use of a synthetic mimic of the natural cofactor system is a promising approach. NADH mimics have recently attracted attention for their implementation as a regeneration system for cofactor-dependent enzymes.19,20 They are simpler compared to enzymatic cofactor regeneration system; their application in the reactions is easy and does not require special apparatus or preparations, making them more robust and easier to control. Moreover, the reaction is scalable and proceeds in an aqueous medium under mild conditions without affecting the enzymatic activity in the biocatalysis. For these reasons we became interested in using NADH mimics as a regeneration system for flavin-dependent halogenases.
NADH mimics have been applied in organic reductions as model systems for understanding the mechanism of the natural biochemical reactions or for their use in organic synthesis, for instance, the stereoselective reduction of pyruvate under mild conditions using NADH mimics and perchlorate.21 Moreover, the reduction of thiobenzophenone to benzhydryl mercaptan has been described using 1-benzyl-1,4-dihydronicotinamide (BNAH).22 Medicinal applications have also been reported, where the reduced nicotinamide derivative 1-carbamoylmethyl-3-carbamoyl-1,4-dihydropyridine was used as a cosubstrate for activating the NAD(P)H quinone oxidoreductase 2 (NQO2), which further activates the antitumor prodrug 5-(aziridin-1-yl)-2,4-dinitrobenzamide with higher efficiency and stability than the natural reduced nicotinamide riboside.23 Other reports used the NADH mimics to explain the mechanism of artemisinin’s antimalaria activity in vitro, where the hydride transfer from BNAH reduced the drug to give a product that can access the malaria parasite.24 Recently, the application of NADH mimics was further investigated in biocatalysis.25
The use of NADH mimics was first studied with respect to the functional role of the pyridine ring in NADH,26 followed by mechanistic investigation with BNAH. The application of NADH mimics on NADH-dependent enzymes was first reported for the horse liver ADH,27,28 and further expanded to other enzymes including HbpA monooxygenase.29 Recently, several NADH mimics were synthesized to replace the natural cofactor required by ene reductases from the Old Yellow Enzyme family for biocatalytic reductions.19,25,30,31
As Paul et al. described previously, different synthetic NADH mimics can be easily obtained from inexpensive starting materials in two synthetic steps. A pyridine derivative is N-alkylated under reflux for 15 h to form a pyridinium salt and subsequently reduced under inert atmosphere with Na2S2O4 forming the NADH mimicking dihydropyridine (Figure 2).19,32 The synthesis is straightforward and does not require any harsh conditions, toxic reagents, or further purification after the synthesis.
Figure 2.

Synthesis of NADH mimics for FADH2 regeneration.18
The application of NADH mimics for the regeneration of FADH2 required by FAD-dependent tryptophan halogenases leads to a much simpler enzymatic halogenation system as both auxiliary enzymes would become redundant (Figure 3). To explore the applicability of NADH mimics as an alternative cofactor regeneration system for tryptophan halogenases, three flavin-dependent halogenases with different activities and regioselectivities were investigated in detail.
Figure 3.
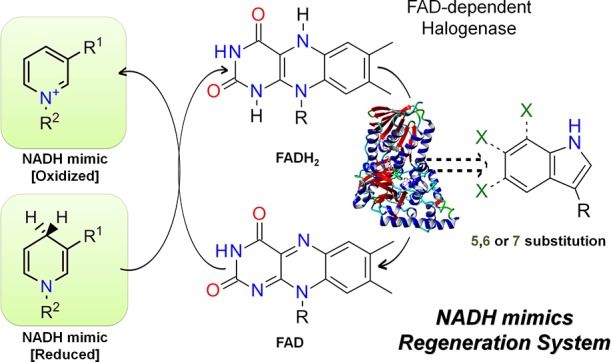
Enzymatic tryptophan halogenation with cofactor regeneration by NADH mimics.
The tryptophan 5-halogenase PyrH from Streptomyces rugosporus,33 the tryptophan 6-halogenase Thal from Streptomyces albogriseolus,34 and the tryptophan 7-halogenase RebH from Lechevalieria aerocolonigenes(16) were used as model systems. The purified enzymes RebH and PyrH were first tested using the standard protocol for the flavin-dependent halogenation (see Supporting Information p S9), while flavin reductase and alcohol dehydrogenase usually required for enzymatic cofactor regeneration were omitted. The halogenation reactions were performed employing 10 equiv of the NADH mimic BNAH (3a, 10 mM). Intriguingly, nearly 50% of tryptophan was halogenated within 2 h and the reaction proceeded to full conversion after 24 h (Figure 4, Figure S2).
Figure 4.

FADH2 regeneration with BNAH (3a) in the enzymatic halogenation with RebH. (A) Reverse-phase high-performance liquid chromatography chromatogram. (B) Liquid chromatography-mass spectrometry (LC-MS) confirms the correct mass of the product. Reduced 3a was extracted with organic solvent before the analysis. Reaction conditions: 10 mM Na2HPO4 buffer, pH 7.4, 30 mM NaCl, 650 U mL–1 catalase, 10 mM NADH mimic, 1 mM tryptophan, 100 μM FAD, 0.6 mol % (6 μM) RebH, final volume 400 μL, 600 rpm at 25 °C.
To identify the NADH mimic with the highest FADH2 regeneration efficiency, several literature-known NADH mimics 3 (BNAH 3a, mBU 3b, mAC 3c, and mCN 3d) were synthesized and tested with FAD-dependent halogenases (Table 1; RebH: Figure 5; Thal: Figure 6A; PyrH: Figure S3B).
Table 1. Initial Reaction Rates (v0) for Different NADH Mimics and ERS with Different Flavin-Dependent Halogenases RebH, Thal, and PyrH.
| initial reaction rate (v0) [×10–3 μmol min–1]a |
||||
|---|---|---|---|---|
| enzyme | NADH mimic 3a | NADH mimic 3b | NADH mimic 3c | ERS |
| RebH | 6.01 ± 0.003 | 4.95 ± 0.012 | 4.83 ± 0.016 | 1.26 ± 0.002 |
| Thal | 7.62 ± 0.001 | 6.88 ± 0.005 | 6.81 ± 0.008 | 1.87 ± 0.001 |
| PyrH | 4.07 ± 0.001 | 4.29 ± 0.003 | 2.23 ± 0.01 | 4.51 ± 0.002 |
Reaction conditions: 10 mM Na2HPO4 buffer, pH 7.4, 30 mM NaCl, 650 U mL–1 catalase, 10 mM NADH mimic, 1 mM tryptophan, 100 μM FAD, 1.2 mol % tryptophan halogenase (RebH, PyrH, or Thal), final volume 200 μL, shaking at 600 rpm at 25 °C. In the case of the ERS, 2.5 U mL–1 flavin reductase PrnF, 1 U mL–1 alcohol dehydrogenase, 5% (v/v) isopropanol, and 100 μM NAD+ were used instead of the NADH mimic.
Figure 5.

Screening of different NADH mimics to regenerate FADH2 for the halogenase RebH in comparison to the conventional enzymatic regeneration system (■ 3a, ● 3b, ▲ 3c, ▼ 3d, ◆ ERS). The conversion of tryptophan was monitored with LC-MS. Reactions were done in triplicate using 1 mM tryptophan, 100 μM FAD, 1.2 mol % RebH, 650 U mL–1 catalase, and 10 mM NADH mimic or PrnF, ADH at pH 7.4 and 25 °C. In the case of the ERS, 2.5 U mL–1 flavin reductase PrnF, 1 U mL–1 alcohol dehydrogenase, 5% (v/v) isopropanol and 100 μM NAD+ were used instead of the NADH mimic.
Figure 6.

Application of different Thal protein concentrations ((A) 12 μM (1.2 mol %), (B) 2.4 μM (0.24 mol %) using NADH mimics for flavin cofactor regeneration (■ 3a, ● 3b, ▲ 3c, ▼ 3d; 3b activity was not detected in (B)). Error bars represent the mean values ± standard deviation from three independent reactions. Reaction conditions: 10 mM Na2HPO4 buffer, pH 7.4, 30 mM NaCl, 650 U mL–1 catalase, 10 mM NADH mimic, 1 mM tryptophan, 100 μM FAD, final volume 200 μM, shaking at 600 rpm at 25 °C.
Interestingly, RebH (Figure 5) and Thal (Figure 6) showed the highest initial reaction rate and fastest conversion with 3a and 3c, even outperforming the enzymatic cofactor regeneration system (ERS). The reaction proceeded 3.5–4.7 times faster than that with the enzymatic cofactor regeneration system (Table 1, Figure S5). The highest FADH2 regeneration efficiency was achieved with 3a, of which the structure most closely resembles NADH. With RebH, 100% conversion of 1 mM tryptophan was achieved after only 2 h using 3a, whereas 5 h was needed to achieve full conversion under optimal reaction conditions using ERS.8 This is in agreement with previous reports on the NADH mimic 3a, where it was found to effect 40 times faster hydride transfer rate than NADH.29 With 3c the reaction rate is comparable to 3a and 3b with a butyl group at the pyridine nitrogen initially shows high efficiency, exceeding that of 3c. However, the reaction with 3b seems to stop at approximately 47% conversion, most likely because of the instability and rapid oxidation of this cofactor. Mimic 3d with a nitrile group at the pyridine ring displays the lowest reaction rate and activity, a common trend for this mimic, which has a higher redox potential (ca. −220 mV) compared to the others.35
Hence, the FADH2 regeneration efficiency of the NADH mimics is strongly affected by the R1 substituent in the first place with preference for the carboxamide moiety (mimicking the natural NADH) followed by the acetyl group. On the other hand, the substituent at the pyridine nitrogen also affects the activity of the mimic as observed for 3b (Table 1).
The stability of the mimic would also play an important role in the regeneration of FADH2, in particular in the case of 3b and 3d. This might be attributed to a more rapid consumption of these NADH mimics in the reduction of FAD to FADH2. Besides reaction of enzyme-bound FADH2 with O2 eventually leading to halogenation, FADH2 also reacts with molecular oxygen freely in solution. This uncoupling reaction forms hydrogen peroxide and consumes NADH mimic in a nonproductive manner. In addition, the accumulation of H2O2 may compromise the stability of the mimic and halogenase activity.20 Catalase was added to the reactions to prevent H2O2 accumulation.36
Notably, PyrH displays a lower reaction rate and activity compared to RebH and Thal with the NADH mimics but a higher reaction rate with the ERS. However, both regeneration reactions are assumed to proceed via reduction of FAD in solution independent of the halogenase. Subsequently, FADH2 is rapidly bound to the enzyme.37 Recently, the direct regeneration of enzyme-bound FAD in PyrH by photochemical regeneration using a reducing agent (ethylenediaminetetraacetic acid) and light has been reported.38 To test whether 3a might also directly reduce FAD inside the halogenase, PyrH was reconstituted with FAD and washed thoroughly to remove free FAD from the sample.
The spectrum of the sample shows a distinct fine structure in the band at 450 nm (Figure 7) and a shift of the band at 373–350 nm (Figure S6), which is characteristic of protein-bound FAD. Bleaching of the band at 450 nm is observed upon addition of 3a to the oxygen-free solution, giving proof of FADH2 formation.39 Within 23 min, the conversion is complete. After admission of oxygen to the cuvette, FADH2 is consumed, forming FAD within the protein as evidenced by the same fine structure as that prior to addition of mimic 3a (Figure 7).
Figure 7.
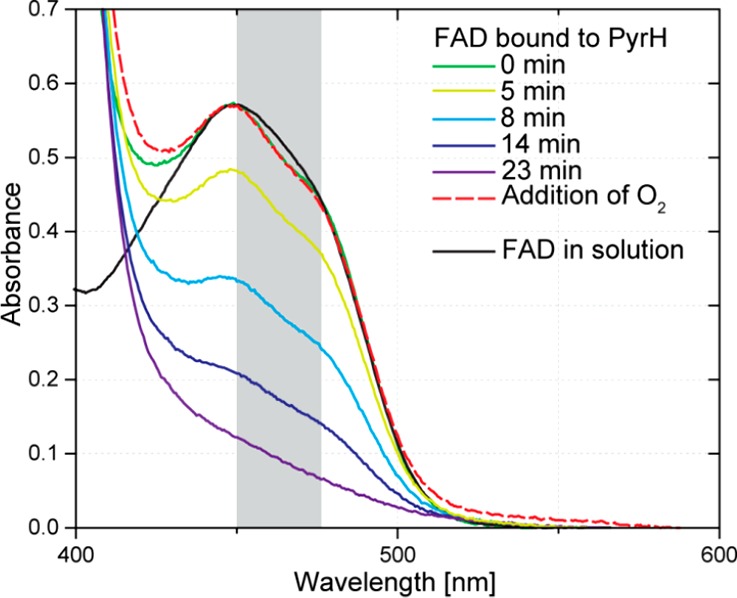
UV/vis spectra monitoring the reduction of FAD bound to PyrH after addition of 3a. Binding of FAD to PyrH is characterized by a fine structure between 450 and 470 nm (highlighted in gray). Reaction conditions: 40 μM PyrH, 10 mM BNAH (3a), FAD:protein ratio 1:2, 50 mM phosphate buffer at pH 8.3 and 20 °C, anaerobic.
These results indicate that FAD might be slowly reduced by the mimic directly inside the enzyme PyrH, which would explain the observed differences between the two regeneration approaches of the halogenases (Table 1) by their different affinities to FAD. It should be noted that we cannot exclude from these experiments a reduction via catalytic amounts of free FAD in solution which is involved in a rapid binding equilibrium with the enzyme.
As the activity of PyrH is much lower than RebH and Thal when combined with the NADH mimics, halogenation in the presence of NADH mimics as the regeneration system might depend not only on the affinity of FAD to the protein and the concentration of the flavin cofactor but also on the enzymatic activity. Different concentrations of Thal were used for testing the different NADH mimics (Figure 6). Halogenation of l-tryptophan was faster with 12 μM Thal (1.2 mol %) compared to halogenation at a 5 times lower halogenase concentration (2.4 μM Thal; 0.24 mol %).
Full conversion could not be obtained in the latter case with the tested NADH mimics. This indicates that the halogenation reaction using NADH mimics as a FAD cofactor regeneration system is halogenase-dependent, where the halogenase is rate-limiting. The FADH2 formed reacts with oxygen to the flavin hydroperoxide, which either generates hypohalous acid for halogenation or H2O2. On the basis of the substrate conversion and the used amounts of mimic with RebH and PyrH, the coupling efficiency was calculated to be less than 1%. Therefore, increasing the halogenase load or engineering the existing halogenase for higher efficiency would increase the halogenation rate by directing the excess FADH2 toward halogenation instead of H2O2 formation.31 The same reactions were performed with higher PyrH concentrations (12 μM, 1.2 mol % and 24 μM, 2.4 mol %, Figure S3C). The reaction rate and conversion using the NADH mimics increased upon doubling enzyme concentration, but still remained lower than those in the reactions catalyzed by 1.2 mol % RebH and Thal.
Since 3a had been identified as the best NADH mimic for enzymatic halogenation with RebH and Thal, reaction conditions and NADH mimic concentration were further optimized for RebH and PyrH because RebH and Thal have nearly similar activities and reaction rates. Tris-HCl buffer (50 mM), pH 7.4 at 25 °C and 20 mM 3a were found as optimal conditions for both RebH and PyrH. With use of tryptophan 7-halogenase RebH after a prolonged reaction period, a dichlorinated tryptophan was formed as side product. This side reaction can be avoided by monitoring the progress of the reaction and through the enzyme load applied to the reaction (Figure S7, Table S1).
To investigate FAD reduction by 3a independently from the halogenation, the reaction was performed at fixed concentration of either FAD or 3a, while altering the other one and the oxidation of 3a was measured via absorption at 360 nm. As expected, the reaction rate was almost proportional to the FAD concentration (Figure 8A). Continuous increase in FAD concentration showed an increased rate of 3a oxidation. However, FAD concentrations exceeding that of 3a resulted in no further improvement. The same was observed with increasing concentration of 3a at a fixed FAD concentration (data not shown). A steady FADH2 supply is crucial for the biohalogenation reaction; however, its accumulation results in H2O2 formation. As the coupling efficiency of the halogenation reaction is rather low, a certain ratio of FAD:3a is required to ensure a continuous biohalogenation without shifting the equilibrium toward H2O2 formation. Therefore, different FAD concentrations were tested in the biohalogenation reaction with 3a. As expected, lower FAD concentrations (20, 50 μM) showed higher conversion of tryptophan, while using 1:1 molar ratio of FAD:3a did not result in any product formation (Figure 8B). This phenomenon was also observed previously with styrene monooxygenase, where the higher FAD concentration results in aerobic reoxidation of FADH2 and consequently H2O2 formation instead of the product chlorotryptophan.31
Figure 8.

(A) Hydride transfer reaction between 3a and FAD. The reaction was monitored by measuring the oxidation of 3a at 360 nm. Reaction conditions: 50 mM Tris-HCl buffer, pH 7.4, 30 mM NaCl, 150 μM 3a, 5–300 μM FAD in 1 mL volume. (B) Biohalogenation reaction using 3a at variable FAD concentrations. Reaction conditions: 50 mM Tris-HCl buffer, pH 7.4, 30 mM NaCl, 10 mM 3a, 1–5000 μM FAD, 1.2 mol % RebH, 650 U mL–1 catalase, 25 °C for 1.5 h in 200 μL volume.
To prove that the NADH mimic regeneration system is also suitable for enzymatic halogenation using immobilized halogenases, we employed this system for the chlorination of l-tryptophan. RebH and PyrH were immobilized as cross-linked enzyme aggregates (CLEAs) as described previously, but without the addition of the auxiliary enzymes.40,41 At a 20 mM concentration of 3a, full conversion of 0.5 mM tryptophan was achieved within 5 h, a relatively shorter time compared to that for the enzymatic cofactor regeneration system (Figure S8, Table S2). Notably, the initial reaction rates of each RebH and PyrH under the same reaction conditions and enzyme concentrations are 1.3–1.9 times faster using 3a compared to that with the ERS, which has also been found when using the purified enzymes (Figure S9). This proves the utility of the NADH mimics as a valuable alternative regeneration system for FAD-dependent halogenases.19,29
With most of the synthetic mimics, the overall reaction proceeds faster than the enzymatic cofactor regeneration system (Figure 5, Figure 6A, Figure S9). However, after an initial phase, rapid consumption of the NADH mimics and a significant decline in the halogenation reaction rates were detected using immobilized enzymes.
Continuous addition of the mimics is one approach to overcome this problem. Incremental addition of the substrate and the NADH mimic 3a was selected after several attempts for scaling up the reaction using the immobilized enzymes. It was possible to run the reactions for up to 24 days using RebH CLEAs and 15 days using PyrH CLEAs without the necessity of exchanging the reaction buffer or adding more halogenase or cofactor.
A semipreparative scale reaction using the cross-linked enzyme aggregates of RebH with continuous addition of 3a as the sole regeneration factor was performed. Five equivalents of 3a was initially applied for biocatalytic chlorination of 10 mg of l-tryptophan (0.05 mmol) by RebH CLEAs in 500 mL of reaction buffer. On the basis of the consumption of the reactants, l-tryptophan and 3a were added incrementally in the course of the reaction (Figure S11, Scheme S3). After 8 days, 102 mg of l-tryptophan (0.5 mmol) were chlorinated to 7-chlorotryptophan by using 17.8 mol equiv of 3a and RebH CLEAs obtained from 6 L of Escherichia coli culture. For the product purification, Boc-protection was conducted, followed by extraction with dichloromethane. Finally, a yield of 74.9% of Boc-7-chlorotryptophan was obtained (Figure S12). Currently, the low coupling efficiency of tryptophan halogenases represents a major drawback for the applicability of the preparative scale reaction. Because of the competing H2O2 formation, a large percentage of NADH mimic is being consumed.
This result, in comparison to the previous report on the upscaling of the enzymatic halogenation reaction,41 has revealed the usability of the NADH mimic regeneration system for the scale-up reaction comparable to the reported results using ERS. FAD-dependent halogenases such as RebH can further function for prolonged periods (>20 days) using the NADH mimics regeneration system; most likely, the instability of the auxiliary enzymes impedes longer reaction times using ERS. Halogenated products or byproducts of the auxiliary enzyme (ADH) may have a detrimental effect on the catalytic activity of the enzymes (product/byproduct inhibition). This provides another attractive advantage of the NADH mimic over the ERS.
In conclusion, NADH mimics have successfully been exploited for FADH2 regeneration as an alternative to the ERS in the enzymatic halogenation of tryptophan. Their synthesis is simple and they are obtained from inexpensive starting material. FAD reduction using NADH mimics circumvents the implementation of two auxiliary enzymes for cofactor regeneration, facilitating the reaction setup. Mimics 3a and 3b led to faster halogenation reactions using purified halogenase than did a commonly used ERS. In addition, the mimics were used in a semipreparative tryptophan chlorination reaction catalyzed by immobilized tryptophan 7-halogenase RebH to produce 126.7 mg (74.9% yield) over the course of 8 days. Because of the simplicity of the NADH mimic regeneration system, it could be employed in the course of halogenases engineering via directed evolution. In particular, the mimics might show potential when improving halogenase thermostability because the auxiliary enzymes lack the required stability at elevated temperature. Halogenase engineering might improve the coupling efficiency that eventually would improve the performance of NADH mimics applied together with FAD-dependent halogenases. This will facilitate and improve the applicability of FAD-dependent halogenases for chemical biotransformation.
Acknowledgments
C.E.P. acknowledges a NWO Veni Grant No. 722.015.011. M.I. acknowledges funding from the German Academic Exchange Service (DAAD) and Bielefelder Nachwuchsfonds. T.K. acknowledges a Heisenberg fellowship by the DFG (KO3580/4-2).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b04500.
Details for the synthesis of the NADH mimics and enzymatic reactions using mimics and ERS and semipreparative scale reaction (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hernandes M.; Cavalcanti S. M.; Moreira D. R.; de Azevedo W. F. Jr.; Leite A. C. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11, 303–314. 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- Chen X.; van Pée K.-H. Catalytic Mechanisms, Basic Roles, and Biotechnological and Environmental Significance of Halogenating Enzymes. Acta Biochim. Biophys. Sin. 2008, 40, 183–193. 10.1111/j.1745-7270.2008.00390.x. [DOI] [PubMed] [Google Scholar]
- Smith D. R. M.; Grüschow S.; Goss R. J. M. Scope and Potential of Halogenases in Biosynthetic Applications. Curr. Opin. Chem. Biol. 2013, 17, 276–283. 10.1016/j.cbpa.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Vaillancourt F. H.; Yeh E.; Vosburg D. A.; Garneau-Tsodikova S.; Walsh C. T. Nature’s Inventory of Halogenation Catalysts: Oxidative Strategies Predominate. Chem. Rev. 2006, 106, 3364–3378. 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- Smith K.; El-Hiti G. A. Regioselective Control of Electrophilic Aromatic Substitution Reactions. Curr. Org. Synth. 2004, 1, 253–274. 10.2174/1570179043366747. [DOI] [Google Scholar]
- Alonso F.; Beletskaya I. P.; Yus M. Metal-Mediated Reductive Hydrodehalogenation of Organic Halides. Chem. Rev. 2002, 102, 4009–4091. 10.1021/cr0102967. [DOI] [PubMed] [Google Scholar]
- Dong C.; Flecks S.; Unversucht S.; Haupt C.; van Pée K.-H.; Naismith J. H. Tryptophan 7-Halogenase (PrnA) Structure Suggests a Mechanism for Regioselective Chlorination. Science 2005, 309, 2216–2219. 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese M.; Guzowska P. H.; Voß H.; Sewald N. Regioselective Enzymatic Halogenation of Substituted Tryptophan Derivatives Using the FAD-Dependent Halogenase RebH. ChemCatChem 2014, 6, 1270–1276. 10.1002/cctc.201301090. [DOI] [Google Scholar]
- Schnepel C.; Sewald N. Enzymatic Halogenation: A Timely Strategy for Regioselective C–H Activation. Chem. - Eur. J. 2017, 23, 12064–12086. 10.1002/chem.201701209. [DOI] [PubMed] [Google Scholar]
- Andorfer M. C.; Grob J. E.; Hajdin C. E.; Chael J. R.; Siuti P.; Lilly J.; Tan K. L.; Lewis J. C. Understanding Flavin-Dependent Halogenase Reactivity via Substrate Activity Profiling. ACS Catal. 2017, 7, 1897–1904. 10.1021/acscatal.6b02707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd S. A.; Karthikeyan C.; Latham J.; Struck A. W.; Thompson M. L.; Menon B. R. K.; Styles M. Q.; Levy C.; Leys D.; Micklefield J. Extending the Biocatalytic Scope of Regiocomplementary Flavin-Dependent Halogenase Enzymes. Chem. Sci. 2015, 6, 3454–3460. 10.1039/C5SC00913H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S.; Wage T.; Hohaus K.; Hölzer M.; Eichhorn E.; van Pée K.-H. Purification and Partial Characterization of Tryptophan 7- Halogenase (PrnA) from Pseudomonas fluorescens. Angew. Chem., Int. Ed. 2000, 39, 2300–2302. . [DOI] [PubMed] [Google Scholar]
- Zhao H.; van der Donk W. A. Regeneration of Cofactors for Use in Biocatalysis. Curr. Opin. Biotechnol. 2003, 14, 583–589. 10.1016/j.copbio.2003.09.007. [DOI] [PubMed] [Google Scholar]
- van der Donk W. A.; Zhao H. Recent Developments in Pyridine Nucleotide Regeneration. Curr. Opin. Biotechnol. 2003, 14, 421–426. 10.1016/S0958-1669(03)00094-6. [DOI] [PubMed] [Google Scholar]
- Payne J. T.; Andorfer M. C.; Lewis J. C. Regioselective Arene Halogenation Using the FAD-Dependent Halogenase RebH. Angew. Chem., Int. Ed. 2013, 52, 5271–5274. 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E.; Garneau S.; Walsh C. T. Robust in Vitro Activity of RebF and RebH, a Two-Component Reductase/Halogenase, Generating 7-Chlorotryptophan during Rebeccamycin Biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3960–3965. 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J. T.; Poor C. B.; Lewis J. C. Directed Evolution of RebH for Site-Selective Halogenation of Large Biologically Active Molecules. Angew. Chem., Int. Ed. 2015, 54, 4226–4230. 10.1002/anie.201411901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unversucht S.; Hollmann F.; Schmid A.; van Pée K.-H. FADH2-Dependence of Tryptophan 7-Halogenase. Adv. Synth. Catal. 2005, 347, 1163–1167. 10.1002/adsc.200505029. [DOI] [Google Scholar]
- Paul C. E.; Gargiulo S.; Opperman D. J.; Lavandera I.; Gotor-Fernández V.; Gotor V.; Taglieber A.; Arends I. W. C. E.; Hollmann F. Mimicking Nature: Synthetic Nicotinamide Cofactors for C=C Bioreduction Using Enoate Reductases. Org. Lett. 2013, 15, 180–183. 10.1021/ol303240a. [DOI] [PubMed] [Google Scholar]
- Paul C. E.; Churakova E.; Maurits E.; Girhard M.; Urlacher V. B.; Hollmann F. In Situ Formation of H2O2 for P450 Peroxygenases. Bioorg. Med. Chem. 2014, 22, 5692–5696. 10.1016/j.bmc.2014.05.074. [DOI] [PubMed] [Google Scholar]
- Ohnishi Y.; Kagami M.; Ohno A. Reduction by a Model of NAD(P)H. Effect of Metal Ion and Stereochemistry on the Reduction of α-Keto Esters by 1,4-Dihydronicotinamide Derivatives. J. Am. Chem. Soc. 1975, 97, 4766–4768. 10.1021/ja00849a055. [DOI] [PubMed] [Google Scholar]
- Abeles R. H.; Hutton R. F.; Westheimer F. H. The Reduction of Thioketones by a Model for a Coenzyme. J. Am. Chem. Soc. 1957, 79, 712–716. 10.1021/ja01560a058. [DOI] [Google Scholar]
- Knox R. J.; Jenkins T. C.; Hobbs S. M.; Chen S.; Melton R. G.; Burke P. J. Bioactivation of 5-(Aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) by Human NAD(P)H Quinone Oxidoreductase 2: A Novel Co-substrate-mediated Antitumor Prodrug Therapy. Cancer Res. 2000, 60, 4179–4186. [PubMed] [Google Scholar]
- Haynes R. K.; Chan W. C.; Wong H. N.; Li K. Y.; Wu W. K.; Fan K. M.; Sung H. H. Y.; Williams I. D.; Prosperi D.; Melato S.; Coghi P.; Monti D. Facile Oxidation of Leucomethylene Blue and Dihydroflavins by Artemisinins: Relationship with Flavoenzyme Function and Antimalarial Mechanism of Action. ChemMedChem 2010, 5, 1282–1299. 10.1002/cmdc.201000225. [DOI] [PubMed] [Google Scholar]
- Knaus T.; Paul C. E.; Levy C. W.; De Vries S.; Mutti F. G.; Hollmann F.; Scrutton N. S. Better than Nature: Nicotinamide Biomimetics That Outperform Natural Coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. 10.1021/jacs.5b12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrer P.; Warburg O. Iodomethylate of Nicotinic Amide. Biochem. Z. 1936, 285, 297–298. [Google Scholar]
- Taylor K. E.; Jones J. B. Nicotinamide Coenzyme Regeneration by Dihydropyridine and Pyridinium Compounds. J. Am. Chem. Soc. 1976, 98, 5689–5694. 10.1021/ja00434a047. [DOI] [PubMed] [Google Scholar]
- Lo H. C.; Fish R. H. Biomimetic NAD+ Models for Tandem Cofactor Regeneration, Horse Liver Alcohol Dehydrogenase Recognition of 1,4-NADH Derivatives, and Chiral Synthesis. Angew. Chem., Int. Ed. 2002, 41, 478–481. . [DOI] [PubMed] [Google Scholar]
- Lutz J.; Hollmann F.; Ho T. V.; Schnyder A.; Fish R. H.; Schmid A. Bioorganometallic Chemistry: Biocatalytic Oxidation Reactions with Biomimetic NAD+/NADH Co-Factors and [Cp*Rh(Bpy)H]+ for Selective Organic Synthesis. J. Organomet. Chem. 2004, 689, 4783–4790. 10.1016/j.jorganchem.2004.09.044. [DOI] [Google Scholar]
- Paul C. E.; Arends I. W. C. E.; Hollmann F. Is Simpler Better? Synthetic Nicotinamide Cofactor Analogues for Redox Chemistry. ACS Catal. 2014, 4, 788–797. 10.1021/cs4011056. [DOI] [Google Scholar]
- Paul C. E.; Tischler D.; Riedel A.; Heine T.; Itoh N.; Hollmann F. Nonenzymatic Regeneration of Styrene Monooxygenase for Catalysis. ACS Catal. 2015, 5, 2961–2965. 10.1021/acscatal.5b00041. [DOI] [Google Scholar]
- Mauzerall D.; Westheimer F. H. 1-Benzyldihydronicotinamide—A Model for Reduced DPN. J. Am. Chem. Soc. 1955, 77, 2261–2264. 10.1021/ja01613a070. [DOI] [Google Scholar]
- Zehner S.; Kotzsch A.; Bister B.; Süssmuth R. D.; Méndez C.; Salas J. A.; van Pée K.-H. A Regioselective Tryptophan 5-Halogenase is Involved in Pyrroindomycin Biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 2005, 12, 445–452. 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Seibold C.; Schnerr H.; Rumpf J.; Kunzendorf A.; Hatscher C.; Wage T.; Ernyei A. J.; Dong C.; Naismith J. H.; van Pée K.-H. A Flavin-Dependent Tryptophan 6-Halogenase and Its Use in Modification of Pyrrolnitrin Biosynthesis. Biocatal. Biotransform. 2006, 24, 401–408. 10.1080/10242420601033738. [DOI] [Google Scholar]
- Ostović D.; Lee I. S. H.; Roberts R. M. G.; Kreevoy M. M. Hydride Transfer and Oxyanion Addition Equilibria of NAD+ Analogues. J. Org. Chem. 1985, 50, 4206–4211. 10.1021/jo00222a006. [DOI] [Google Scholar]
- Massey V. Activation of Molecular Oxygen by Flavins and Flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462. [PubMed] [Google Scholar]
- Yeh E.; Cole L. J.; Barr E. W.; Bollinger J. M.; Ballou D. P.; Walsh C. T. Flavin Redox Chemistry Precedes Substrate Chlorination during the Reaction of the Flavin-Dependent Halogenase RebH. Biochemistry 2006, 45, 7904–7912. 10.1021/bi060607d. [DOI] [PubMed] [Google Scholar]
- Schroeder L.; Frese M.; Müller C.; Sewald N.; Kottke T. Photochemically Driven Biocatalysis of Halogenases for the Green Production of Chlorinated Compounds. ChemCatChem 2018, 10, 3336–3341. 10.1002/cctc.201800280. [DOI] [Google Scholar]
- Massey V. The Chemical and Biological Versatility of Riboflavin. Biochem. Soc. Trans. 2000, 28, 283. 10.1042/bst0280283. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A. Characteristic Features and Biotechnological Applications of Cross-Linked Enzyme Aggregates (CLEAs). Appl. Microbiol. Biotechnol. 2011, 92, 467–477. 10.1007/s00253-011-3554-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese M.; Sewald N. Enzymatic Halogenation of Tryptophan on a Gram Scale. Angew. Chem., Int. Ed. 2015, 54, 298–301. 10.1002/anie.201408561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.