

Abstract
Lignocellulosic biomass is the only renewable source of carbon for the chemical industry. Alkylmethoxyphenols can be obtained in good yield from woody biomass by reductive fractionation, but these compounds are of limited value for large-scale applications. We present a method to convert lignocellulose-derived alkylmethoxyphenols to phenol that can be easily integrated in the petrochemical industry. The underlying chemistry combines demethoxylation catalyzed by a titania-supported gold nanoparticle catalyst and transalkylation of alkyl groups to a low-value benzene-rich stream promoted by HZSM-5 zeolite. In this way, phenol can be obtained in good yield, and benzene can be upgraded to more valuable propylbenzene, cumene, and toluene. We demonstrate that intimate contact between the two catalyst functions is crucial to transferring the methyl groups from the methoxy functionality to benzene instead of phenol. In a mixed-bed configuration, we achieved a yield of 60% phenol and 15% cresol from 4-propylguaiacol in a continuous one-step reaction at 350 °C at a weight hourly space velocity of ∼40 h–1.
Keywords: biomass, lignin, phenol, transalkylation, demethoxylation
Introduction
Phenol is one of the most important intermediates in the chemical industry. Nearly all phenol is obtained from petroleum-derived benzene in the cumene process, which covers about 20% of global benzene demand. Besides drawbacks of a high energy intensity and the coproduction of acetone, there is a growing interest in replacing petroleum as a feedstock by renewable biomass.1 Lignin is recognized as a promising source of bioaromatics, because it is already available as a byproduct of the pulp and paper industry and also increasingly of the processing of second-generation lignocellulosic biomass.2 Converting the polyphenolic network of lignin into useful aromatics such as BTX (benzene, toluene, and xylenes) and phenol remains a formidable challenge.3 A recent promising approach is reductive fractionation of woody biomass in which lignin fragments are released from lignocellulosic biomass (in planta) and depolymerized into alkylmethoxyphenols and a carbohydrate residue in a one-pot reaction (Scheme 1).4−11 Depending on the type of wood, aromatics yields higher than 50 wt % (based on lignin content) have been obtained. These alkylmethoxyphenols, whose structure derives from the phenylpropane units that are randomly linked through C–C and C–O–C bonds in lignin, are primarily phenols functionalized at the para position with an alkyl group (typically propyl) and methoxy groups at one or both of the ortho positions. Their overfunctionalized nature limits the number of applications. Accordingly, it is highly desirable to develop a chemical route to selectively convert these alkylmethoxyphenols into phenol, which requires selective demethoxylation and dealkylation (Scheme 1).
Scheme 1. Pathways for the Production of Phenol from Lignin Fraction of Lignocellulosic Biomass.

The black arrows indicate the main focus of the present study, the green arrows known technologies outside the scope of the present study.
The challenge of demethoxylating these compounds is to avoid hydrogenation of the aromatic ring and removal of the phenolic hydroxyl group.12,13 Early studies have shown that supported Au and Mo catalysts exhibit high selectivity to phenolics in the hydrodeoxygenation of guaiacol.14,15 Steam dealkylation of 4-propylphenol to phenol and propylene has also been achieved on HZSM-5.16,17 Dealkylation typically yields olefins that are prone to oligomerization on the used acid catalysts. A combination of demethoxylation and dealkyation for lignin-derived monomers has also been discussed.18 Here, we report an approach that readily converts methoxyalkylphenols into phenol based on a combination of selective demethoxylation with transalkylation of the alkyl groups to a conventional benzene feedstock producing toluene, cumene, and propylbenzene as coproducts (Scheme 1). Notably, toluene and cumene can respectively be further upgraded to more valuable xylenes using mature industrialized technologies. We will demonstrate that intimate contact between a TiO2-supported Au nanoparticle catalyst for demethoxylation and a HZSM-5 transalkylation catalyst can result in a phenol yield of nearly 60% from 4-propylguaiacol in a single step. Our approach builds on earlier examples in which upgrading of biomass is coupled to a low-value petroleum refinery stream in order to increase the economic competitivess.19−21
Experimental Section
Materials
4-Propylphenol, 4-propylguaiacol, guaiacol, catechol, phenol, and benzene were supplied by Sigma-Aldrich and used without further treatment. HZSM-5 zeolite (Si/Al 20) was obtained from Albemarle. HZSM-5 zeolites (Si/Al 15 and 30) were purchased from Conteka. All the mordenite catalysts were received from Zeolyst. HY zeolites (Si/Al 60, 120, and 160) were obtained by calcining parent ammonium forms of the Y zeolite (purchased from Alfa Aesar) at 550 °C for 6 h at a ramping rate of 1 °C/min.
The Au/TiO2 (P25) catalyst was prepared by deposition-precipitation with NaOH, an appropriate amount of HAuCl4 aqueous solution (10 mmol/L) was added to 200 mL of deionized water at room temperature. The pH was adjusted to 7 by dropwise addition of NaOH (1 M), then 2 g of TiO2 support (P25, Evonik-Degussa) was dispersed in the solution and the pH was readjusted to 7 with NaOH. The suspension was vigorously stirred for 2 h at room temperature before filtration. The catalyst was washed by using extensive deionized water until the filtrate was free of chloride monitored by AgNO3. The obtained residue was dried overnight at 110 °C. The dried material was calcined at 350 °C for 2 h after heating at a ramping rate of 2 °C/min. The obtained material was used without pretreatment. The same procedure was applied to prepare supported Au on anatase and rutile TiO2 catalysts.
Catalyst Characterization
The gold content of the supported gold catalysts was determined by inductively coupled plasma atomic emission spectrometry (ICP-AES) using a Spectro Ciros CCD ICP optical emission spectrometer with axial plasma viewing. All the samples were dissolved in aqua regia (HCl/HNO3 = 3:1).
XPS measurements were performed using a Kratos AXIS Ultra spectrometer, equipped with a monochromatic X-ray source and a delay-line detector (DLD). Spectra were obtained using an aluminum anode (Al Kα = 1486.6 eV) operating at 150 W. Data analysis was performed using CasaXPS software.
Catalytic Activity Measurements
Demethoxylation of 4-Propylguaiacol
A Parr batch autoclave with a volume of 100 mL was used for 4-propylguaiacol demethoxylation. Typically, 100 mg of catalyst and 3000 mg of feedstock were added to the reactor together with 30 mL of benzene solvent. After sealing, purging with H2, and checking for leaks, the autoclave was brought to 50 bar by introducing H2. Then the reactor was heated to 350 °C in ca. 1 h and maintained at this temperature for 2 h. A small amount of sample was taken via a sampling valve after 1 and 2 h of reaction time. An aliquot amount of 1 mL of sample was accurately taken from the reaction mixture. After adding an amount of 10 μL of dodecane internal standard, the sample was measured by GC-MS for product identification and quantification. After the reaction, the reactor was allowed to naturally cool down by removing the heating oven.
Dealkylation of 4-Propylphenol
A mini-autoclave with a volume of 10 mL was used for carrying out 4-propylphenol dealkylation reactions. Typically, 40 mg of catalyst and 400 mg of feedstock were added to the reactor together with 4 mL of solvent. After sealing, the autoclave was transferred to a GC oven, which was preheated to the desired reaction temperature and maintained at that temperature for 2 h. After the reaction, the autoclave was removed from the oven and quenched in an ice bath. After opening the reactor, an amount of 10 μL of dodecane was added to the reaction mixture as an internal standard. After separating the catalyst using membrane filtration or centrifugation, an aliquot of ca. 1 mL of liquid sample was taken from the reaction mixture and directly subjected to GC-MS analysis for product identification and quantification. The liquid phase product mixture was analyzed by a Shimadzu 2010 GC-MS system equipped with an RTX-1701 column (60 m × 0.25 mm × 0.25 μm) and a flame ionization detector (FID) together with a mass spectrometer detector. Identification of products was achieved based on a search of the MS spectra with the NIST11 and NIST11s MS libraries.
Demethoxylation and Transalkylation of 4-Propylguaiacol
A home-built down-flow fixed-bed reactor (stainless steel, 6 mm inner diameter, 150 mm length) was used for the dealkylation and demethoxylation reaction. In a typical experiment, 25–100 mg of Au/TiO2 (125–250 μm) and 100–200 mg of HZSM-5 catalyst powder (125–250 μm) were loaded in a tubular reactor either stacked (Au/TiO2 on top of HZSM-5) or homogeneously mixed. For the stacked catalyst bed, a small amount of quartz wool was placed between the two catalyst beds. The pressure was maintained at 100 bar by a back-pressure controller. H2 was introduced at a flow rate of 30 N mL/min. Typically, the temperature was maintained at 350 °C. After reaching the reaction temperature, a 4-propylguaiacol-benzene solution (5 mol % 4-propylguaiacol) was fed through a preheated (200 °C) feeding line using an HPLC pump at a flow rate in the 2.3–9.0 mL/h range. The liquid products were collected in a cold trap installed directly after the reactor. Samples were collected every 1 h. An aliquot of 1 mL was accurately taken from the reaction mixture. After adding 10 μL of dodecane internal standard, the sample was analyzed by GC-MS for product identification and quantification.
Kinetic Investigations
A Parr autoclave with a volume of 100 mL was used for investigating the reaction pathways using guaiacol, catechol, and phenol as reactants. When phenol was used as a reactant, a stoichiometric amount of methanol was used. Either 50 mg of Au/TiO2 or 100 mg of HZSM-5 catalyst and 10 mmol of substrate were added to the reactor together with 30 mL of benzene solvent. All the other procedures are the same as in the previous section.
Results and Discussion
We first explored the transalkylation of the propyl groups to benzene using 4-propylphenol as a model reactant. We screened a number of microporous zeolites using 10 mL batch autoclaves at 350 °C for 2 h. Among the explored 10-membered (HZSM-5) and 12-membered ring (HMOR and HY) zeolites, HZSM-5 with a Si/Al ratio of 15 stood out as a highly active and selective catalyst for the transalkylation of 4-propylphenol with benzene. The major products from benzene were n-propylbenzene (58 mol %) and cumene (28 mol %; Figure 1b). HZSM-5 zeolite with a higher Si/Al ratio did not significantly alter the product distribution, although a slight decrease of the activity was observed. Such a trend was also observed in an earlier dealkylation study, where HZSM-5 zeolites with different Si/Al ratios were compared for the dealkylation of 4-propylphenol into phenol and propylene.16 Although in the present study HZSM-5 zeolites with Si/Al ratios of 15 and 40 gave comparable results at 350 °C, the performance of the zeolite with a higher Si/Al ratio was substantially lower when the reaction temperature was below 325 °C. The pore size of the zeolites also affects the product distribution. The most striking observation is that a large-pore zeolite like HY results in more n-propylbenzene and less cumene than the medium-pore HZSM-5 zeolite. Characterization of the spent catalysts by thermogravimetric analysis shows that spent HY and HMOR zeolites contain more carbonaceous products (11–16% by weight) than HZSM-5 (6–7%; Figure S1). This difference is due to the shape selectivity of the 10-membered ring pores in HZSM-5, which limits the formation of larger products. These coking reactions likely also involve propylene molecules, which were not efficiently transferred to the benzene solvent. This is consistent with the lower rate of alkylbenzene formation for the large-pore zeolites. Thus, HZSM-5 is the preferred zeolite for transalkylation of propylphenol with benzene.
Figure 1.

Product distribution of (a) 4-propylphenol- and (b) benzene-derived products of the transalkylation of 4-propylphenol with benzene over different zeolites (conditions: 400 mg 4-propylphenol, 4 mL benzene, 40 mg catalyst, 350 °C, 2 h, the zeolite Si/Al ratio given in brackets, the selectivity and yield of alkylbenzenes are based on the 4-propylphenol feedstock).
Increasing the 4-propylphenol/benzene ratio resulted in a significant decrease of the activity and product selectivity (Figure S2). Without benzene, only 5 mol % phenol was obtained, and the main reaction products were polyalkyphenols. The nonclosure of the mass balance indicates that extensive polymerization occurred. We verified that similar transalkylation chemistry is also possible with toluene instead of benzene, albeit the phenol yield decreased slightly (Figure S3). The only side-products are propylphenol isomers. No methylphenol was formed, confirming that the reverse transalkylation reaction between phenols and solvent did not occur. On the other hand, the use of ethylbenzene and propylbenzene significantly decreased the transalkylation efficiency. With these solvents, a small amount of solvent-derived alkylphenols (e.g., ethylphenols and dipropylphenols) was formed, meaning that reverse transalkylation took place. We also found that the presence of water has a pronounced negative effect on the transalkylation activity. For all the water-containing experiments, hardly any phenol was obtained, and extensive polymerization occurred. From these experiments, we infer that the use of benzene and toluene as a solvent can prevent unwanted side-reactions involving reactive olefins and radical species obtained during the dealkylation of 4-propylphenol.
Experiments involving transalkylation of methoxy-containing phenols such as guaiacol and 4-propylguaiacol with benzene using a zeolite catalyst resulted in severe coke formation (Figure S4). A survey of the literature shows that polymerization starts with the homolysis of the O–CH3 group, which results in reactive methyl radicals and catechol.22 Accordingly, we speculated that removing the methoxy group prior to dealkylation is a necessary step for obtaining phenol. For the demethoxylation step, a set of supported Au catalysts were prepared through a deposition–precipitation method. The 1 wt % Au catalysts were prepared on different forms of TiO2 and other oxide supports (Figure S5). Au was selected as the hydrogenation component because of its low activity for aromatic ring hydrogenation. On the basis of this and the higher Ar–OH bond energy (414 kJ/mol) compared to the Ar–OCH3 bond energy (356 kJ/mol),23 we expected that the methoxy group could be removed without losing aromaticity and the phenolic hydroxyl moiety. The performance of these materials was evaluated in a 100 mL Parr autoclave at 350 °C for 2 h using propylguaiacol as the model reactant in benzene. The results demonstrated that 1 wt % Au/TiO2 (P25) exhibits the highest activity and selectivity. Full conversion of propylguaiacol was achieved. The main products were propylphenol (∼50 mol %), methylpropylphenols (∼25 mol %), and polyalkylphenols (∼10 mol %). Au supported on anatase is less effective compared to the P25 TiO2 support. All the other catalysts are nearly inactive for this reaction. We also confirmed that the P25 TiO2 support only displays negligible activity for demethoxylation (∼ 5 mol % phenol). These results suggest that the interface between Au and TiO2 support is involved in the selective demethoxylation reaction. XPS analysis of the calcined Au/TiO2(P25) catalyst shows that 80% of the Au (0.76 wt %) is in the metallic state with the remaining Au being in the +1 oxidation state (Figure S6).
Initial experiments involving a two-step approach in which 4-propylguaiacol in benzene was first demethoxylated over Au/TiO2 followed by dealkylation using HZSM-5 in a flow reactor showed promising phenol yields (Figure S7). In further experiments, we combined both catalysts in a flow reactor at 350 °C and a H2 pressure of 100 bar in order to achieve a one-step process. First, we investigated a stacked-bed configuration with Au/TiO2 on top of HZSM-5. The product selectivity was optimized by changing the amount of Au/TiO2 (25–200 mg) using a fixed amount of 100 mg of HZSM-5. When the amount of Au/TiO2 was small (25 mg), not all the methoxy groups were removed, and the conversion rapidly decreased from 92% to 50%, resulting in a very low phenol yield after 6 h of reaction (Figure 2a). Increasing the amount of Au/TiO2 to 50 mg significantly increased the phenol yield and lowered the rate of deactivation of HZSM-5 (Figure 2b). When 100 mg of Au/TiO2 was used, a phenol yield of ∼50 mol % could be maintained at a feeding rate in the 4.5–6.8 mL/h range. A higher feeding rate (9.0 mL/h) resulted in a lower phenol yield, because the transalkylation activity was insufficient as followed from a higher yield of methylpropylphenols and cresol (Figure 2c). This lower phenol yield could be offset by increasing the amount of HZSM-5 to 200 mg, resulting in good phenol yield at a feed rate of 9.0 mL/h (Figure 2d). We also varied the reaction temperature (300–400 °C) using 100 mg of Au/TiO2 and 100 mg of HZSM-5 at a feed rate of 4.5 mL/h (Figure S8). Efficient one-step demethoxylation and transalkylation can already be achieved at a reaction temperature of 325 °C. At 300 °C, the dealkylation rate is too slow as evidenced by the large amount of propylphenol formation. A too high reaction temperature (375–400 °C) results in lower phenol yield, likely due to more pronounced side-reactions.
Figure 2.

One-step process for demethoxylation and dealkylation over (a–d) stacked-bed and (e,f) mixed-bed Au/TiO2 and HZSM-5 (Si/Al = 15) catalysts in benzene using a fixed-bed down-flow reactor (conditions: 5 mol % 4-propylguaiacol in benzene, 350 °C, 100 bar, gas flow rate 30 N mL/min H2, weight hourly space velocity (WHSV): (a) 48 h–1, (b and e) 40 h–1, (c and f) 30 h–1, and (d) 20 h–1.
In order to gain better insight into the underlying chemistry, we employed model compounds such as guaiacol, catechol, and phenol dissolved in benzene over either Au/TiO2 or HZSM-5 in a batch reactor at 350 °C (Scheme 2). When phenol was used, an equimolar amount of methanol was added to the mixture. For guaiacol, the results between the Au/TiO2 and HZSM-5 catalysts are very different. Au/TiO2 can rapidly convert guaiacol into phenol (∼50.6%), coproducing cresol (19.6%) and polymethylphenols (13.4%). The methyl groups will transfer preferentially to the aromatic ring of phenolic products rather than being transferred to benzene. From the observations that catechol was absent as a product and Au/TiO2 is also active in the alkylation of phenol with methanol (Scheme 2c), we can infer that the methoxy group is removed from guaiacol as methanol followed by alkylation of the phenol product with methanol. However, given the fact that one of the hydroxyl groups in catechol can be rapidly removed by Au/TiO2 to form phenol (Scheme 2b), we cannot rule out the possibility that the methyl group is transferred directly to the aromatic ring involving methyl catechol as an intermediate compound. HZSM-5 is only slightly active in demethoxylating guaiacol, and instead of phenol, catechol is the main product. The formation of catechol might cause char formation, which leads to catalyst deactivation. This is confirmed by the significant nonclosure of the mass balance and the nearly identical results obtained after 1 and 2 h of reaction (Scheme 2b). Notably, the methyl group could be effectively transferred to benzene, evidenced by the formation of toluene (Scheme 2a). An alkylation experiment between phenol and methanol catalyzed by HZSM-5 confirms this as well (Scheme 2c).
Scheme 2. Conversion of (a) Guaiacol, (b) Catechol, and (c) Phenol over the Au/TiO2 and HZSM-5 Catalysts in Benzene Solvent Using a Batch Reactor (Conditions: 10 mmol Feedstock, 100 mg HZSM-5 or 50 mg Au/TiO2, 30 mL Benzene, 350 °C, 50 bar H2).

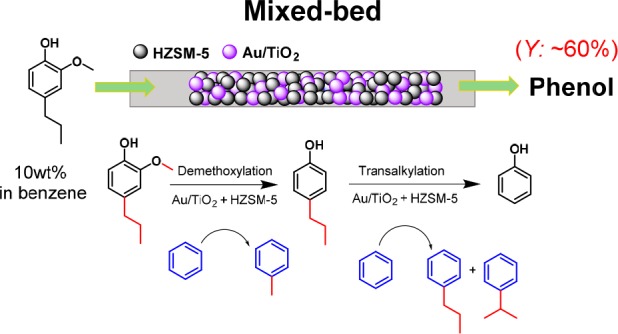
With these mechanistic insights in hand, we were able to further improve the process by using a bed configuration in which the two catalysts were mixed. When 50 mg or 100 mg of Au/TiO2 was physically mixed with 100 mg of HZSM-5, the yield of phenol could be significantly improved to nearly 60 mol %. Notably, much less methylpropylphenols and cresols were obtained compared to the experiments using a stacked-bed configuration. Scheme 3 summarizes the possible differences between these two cases. In the stacked-bed configuration, the phenol yield is limited by the formation of methylated phenols in the top Au/TiO2 bed. These methylated phenols cannot be removed by HZSM-5 and will lead to cresol and methylpropylphenols (Scheme 3a). The benefit of using a mixed-bed configuration is that the methyl group, which is removed from 4-propylguaiacol via HZSM-5 catalyzed demethylation or via Au/TiO2 catalyzed demethoxylation, can be efficiently transferred to the benzene solvent via HZSM-5 catalyzed transalkylation, minimizing the formation of methylated phenols (Scheme 3b). Overall, this leads to a 2.5-fold higher yield of toluene for the mixed-bed compared to the stacked-bed configuration, and a phenol yield close to 60 mol % yield was achieved with 15 mol % cresol and 20 mol % of methylpropylphenols as side-products.
Scheme 3. Proposed Reaction Network of the One-Step Process for Demethoxylation and Dealkylation over (a) Stacked-Bed and (b) Mixed-Bed of Au/TiO2 and HZSM-5 Catalysts in Benzene Using a Fixed-Bed Reactor (Conditions: 5 mol % 4-Propylguaiacol in Benzene, 350 °C, 100 bar, Gas Flow Rate 30 mL/min H2, WHSV 30 h–1).

Conclusions
This work presents a novel process to selectively produce phenol from lignocellulose-derived alkylmethoxyphenols via demethoxylation and transalkylation using a mixture of Au/TiO2 and HZSM-5 catalysts in benzene. The use of benzene is crucial for obtaining a high yield of phenol, minimizing coke formation related to reactive intermediates obtained from methoxy and propyl side chains. This is because benzene is able to effectively react with these alkyl groups by forming alkylbenzenes. In this way, benzene can be upgraded to more valuable propylbenzene, cumene, and toluene. We also demonstrated that intimate contact between the Au/TiO2 demethoxylation catalyst and the HZSM-5 transalkylation catalyst is crucial for obtaining a high yield of phenol. This ensures that the methyl group of 4-propylguaiacol effectively ends up in toluene rather than in methylated phenols. This work offers a promising next step toward obtaining bio-based phenol from lignocellulosic biomass.
Acknowledgments
This work was performed under the framework of Chemelot InSciTe and is supported by contributions from the European Interreg V Flanders, the European Regional Development Fund (ERDF), the Provinces of Brabant and Limburg and the Dutch Ministry of Economy. A patent application entitled “Catalytic conversion of biomass to biophenol” with filing number NL2020595 was filed on March 14, 2018 by the inventors X.H. and E.J.M.H.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b03430.
TG analysis of the spent zeolite catalysts, effect of feedstock to benzene ratio for transalkylation, effect of solvent for transalkylation, catalyst screening for demethoxylation, XPS analysis of the Au/TiO2 catalyst, two-step demethoxylation-transalkylation, effect of temperature on one-step demethoxylation-transalkylation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sato T.; Haryu E.; Adschiri T.; Arai K. Non-catalytic Recovery of Phenol Through Decomposition of 2-isopropylphenol in Supercritical Water. Chem. Eng. Sci. 2004, 59, 1247–1253. 10.1016/j.ces.2003.12.018. [DOI] [Google Scholar]
- Li C. Z.; Zhao X. C.; Wang A. Q.; Huber G. W.; Zhang T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–11624. 10.1021/acs.chemrev.5b00155. [DOI] [PubMed] [Google Scholar]
- Ragauskas A. J.; Beckham G. T.; Biddy M. J.; Chandra R.; Chen F.; Davis M. F.; Davison B. H.; Dixon R. A.; Gilna P.; Keller M.; Langan P.; Naskar A. K.; Saddler J. N.; Tschaplinski T. J.; Tuskan G. A.; Wyman C. E. Lignin Valorization: Improving Lignin Processing in The Biorefinery. Science 2014, 344, 1246843. 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- Van den Bosch S.; Schutyser W.; Vanholme R.; Driessen T.; Koelewijn S. F.; Renders T.; De Meester B.; Huijgen W. J. J.; Dehaen W.; Courtin C. M.; Lagrain B.; Boerjan W.; Sels B. F. Reductive Lignocellulose Fractionation into Soluble Lignin-derived Phenolic Monomers and Dimers and Processable Carbohydrate Pulps. Energy Environ. Sci. 2015, 8, 1748–1763. 10.1039/C5EE00204D. [DOI] [Google Scholar]
- Parsell T.; Yohe S.; Degenstein J.; Jarrell T.; Klein I.; Gencer E.; Hewetson B.; Hurt M.; Kim J. I.; Choudhari H.; Saha B.; Meilan R.; Mosier N.; Ribeiro F.; Delgass W. N.; Chapple C.; Kenttamaa H. I.; Agrawal R.; Abu-Omar M. M. A Synergistic Biorefinery Based on Catalytic Conversion of Lignin Prior to Cellulose Starting from Lignocellulosic Biomass. Green Chem. 2015, 17, 1492–1499. 10.1039/C4GC01911C. [DOI] [Google Scholar]
- Ferrini P.; Rezende C. A.; Rinaldi R. Catalytic Upstream Biorefining through Hydrogen Transfer Reactions: Understanding the Process from the Pulp Perspective. ChemSusChem 2016, 9, 3171–3180. 10.1002/cssc.201601121. [DOI] [PubMed] [Google Scholar]
- Song Q.; Wang F.; Cai J. Y.; Wang Y. H.; Zhang J. J.; Yu W. Q.; Xu J. Lignin Depolymerization (LDP) in Alcohol over Nickel-based Catalysts via a Fragmentation-hydrogenolysis Process. Energy Environ. Sci. 2013, 6, 994–1007. 10.1039/c2ee23741e. [DOI] [Google Scholar]
- Huang X.; Zhu J.; Koranyi T. I.; Boot M. D.; Hensen E. J. Effective Release of Lignin Fragments from Lignocellulose by Lewis Acid Metal Triflates in the Lignin-First Approach. ChemSusChem 2016, 9, 3262–3267. 10.1002/cssc.201601252. [DOI] [PubMed] [Google Scholar]
- Xiao L.; Wang S.; Li H.; Li Z.; Shi Z.; Xiao L.; Sun R.; Fang Y.; Song G. Catalytic Hydrogenolysis of Lignins into Phenolic Compounds over Carbon Nanotube Supported Molybdenum Oxide. ACS Catal. 2017, 7, 7535–7542. 10.1021/acscatal.7b02563. [DOI] [Google Scholar]
- Anderson E. M.; Stone M. L.; Katahira R.; Reed M.; Beckham G. T.; Roman-Leshkov Y. Flowthrough Reductive Catalytic Fractionation of Biomass. Joule 2017, 1, 613–622. 10.1016/j.joule.2017.10.004. [DOI] [Google Scholar]
- Xia Q.; Chen Z.; Shao Y.; Gong X.; Wang H.; Liu X.; Parker S. F.; Han X.; Yang S.; Wang Y. Direct Hydrodeoxygenation of Raw Woody Biomass into Liquid Alkanes. Nat. Commun. 2016, 7, 11162. 10.1038/ncomms11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H.; Feng B.; Kobayashi H.; Hara K.; Fukuoka A. Selective Hydrodeoxygenation of Lignin-related 4-Propylphenol into n-propylbenzene in Water by Pt-Re/ZrO2 Catalysts. Catal. Today 2014, 234, 139–144. 10.1016/j.cattod.2014.01.022. [DOI] [Google Scholar]
- Cao Z.; Engelhardt J.; Dierks M.; Clough M. T.; Wang G. H.; Heracleous E.; Lappas A.; Rinaldi R.; Schüth F. Catalysis Meets Nonthermal Separation for the Production of (Alkyl)phenols and Hydrocarbons from Pyrolysis Oil. Angew. Chem., Int. Ed. 2017, 56, 2334–2339. 10.1002/anie.201610405. [DOI] [PubMed] [Google Scholar]
- Mao J.; Zhou J.; Xia Z.; Wang Z.; Xu Z.; Xu W.; Yan P.; Liu K.; Guo X.; Zhang Z. C. Anatase TiO2 Activated by Gold Nanoparticles for Selective Hydrodeoxygenation of Guaiacol to Phenolics. ACS Catal. 2017, 7, 695–705. 10.1021/acscatal.6b02368. [DOI] [Google Scholar]
- Cai Z.; Wang F.; Zhang X.; Ahishakiye R.; Xie Y.; Shen Y. Selective Hydrodeoxygenation of Guaiacol to Phenolics over Activated Carbon Supported Molybdenum Catalysts. Molecular Catalysis 2017, 441, 28–34. 10.1016/j.mcat.2017.07.024. [DOI] [Google Scholar]
- Verboekend D.; Liao Y.; Schutyser W.; Sels B. F. Alkylphenols to Phenol and Olefins by Zeolite Catalysis: a Pathway to Valorize Raw and Fossilized Lignocellulose. Green Chem. 2016, 18, 297–306. 10.1039/C5GC01868D. [DOI] [Google Scholar]
- Liao Y.; d’Halluin M.; Makshina E.; Verboekend D.; Sels B. F. Shape Selectivity Vapor-phase Conversion of Lignin-derived 4-ethylphenol to Phenol and Ethylene over Acidic Aluminosilicates: Impact of Acid Properties and Pore Constraint. Appl. Catal., B 2018, 234, 117–129. 10.1016/j.apcatb.2018.04.001. [DOI] [Google Scholar]
- Zhang J.; Lombardo L.; Gözaydın G.; Dyson P.; Yan N. Single-step Conversion of Lignin Monomers to Phenol: Bridging the Gap between Lignin and High-value Chemicals. Chin. J. Catal. 2018, 39, 1445–1452. 10.1016/S1872-2067(18)63132-8. [DOI] [Google Scholar]
- Arias K. S.; Climent M. J.; Corma A.; Iborra S. Synthesis of High Quality Alkyl Naphthenic Kerosene by Reacting an Oil Refinery with a Biomass Refinery Stream. Energy Environ. Sci. 2015, 8, 317–331. 10.1039/C4EE03194F. [DOI] [Google Scholar]
- Huber G. W.; Corma A. Synergies between Bio- and Oil Refineries for the Production of Fuels from Biomass. Angew. Chem., Int. Ed. 2007, 46, 7184–7201. 10.1002/anie.200604504. [DOI] [PubMed] [Google Scholar]
- Huber G. W.; O’Connor P.; Corma A. Processing Biomass in Conventional Oil Refineries: Production of High Quality Diesel by Hydrotreating Vegetable Oils in Heavy Vacuum Oil Mixtures. Appl. Catal., A 2007, 329, 120–129. 10.1016/j.apcata.2007.07.002. [DOI] [Google Scholar]
- Asmadi M.; Kawamoto H.; Saka S. Thermal Reactions of Guaiacol and Syringol as Lignin Model Aromatic Nuclei. J. Anal. Appl. Pyrolysis 2011, 92, 88–98. 10.1016/j.jaap.2011.04.011. [DOI] [Google Scholar]
- Lan X.; Hensen E. J. M.; Weber T. Hydrodeoxygenation of Guaiacol over Ni2P/SiO2–reaction Mechanism and Catalyst Deactivation. Appl. Catal., A 2018, 550, 57–66. 10.1016/j.apcata.2017.10.018. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.