

Introduction
Small RNAs are <200 nucleotide in length and usually do not translate into a protein. A major function of small RNAs is gene silencing. There are three families of small RNAs that regulate gene expression in animals; micro RNAs (miRNAs), small interfering RNAs (siRNAs), and piwi-interacting RNAs (piRNAs). miRNAs are single-stranded RNAs, ~20–22 nucleotides in length, and play a pivotal role in the regulation of protein-coding genes. siRNAs are ~21 nucleotides long and mediate post-transcriptional suppression of transcripts and transposons and contribute to antiviral defense. piRNAs are 24–30 nucleotides long and their main function is to silence transposable elements in germline cells1. The current miRBase sequence database (released March 2018; http://www.mirbase.org/), a searchable online repository of published miRNA sequences and associated annotations maintained by researchers at the University of Manchester, reported 2,654 mature miRNAs in the human genome. miRNAs influence the development and function of immune cells, modulate innate and adaptive immune responses, and are emerging as robust biomarkers in transplantation as well as targets for therapeutic intervention. Herein, we briefly review the biogenesis and function of the miRNAs and provide an overview of the tools to quantify miRNAs in tissues and body fluids. We then summarize data regarding miRNA expression patterns in kidney transplant recipients.
Nomenclature
Human miRNAs are designated by the suffix ‘hsa’. Mature miRNA is designated as ‘miR’, while the gene is designated as ‘mir’2. The numbering of genes is sequential (e.g., mir-22 was the next miRNA published after mir-21). Mature sequences that are identical but originate from distinct precursor sequences and genomic loci are designated with numbered suffixes (e.g., hsa-miR-121–1 and hsa-miR-121–2). Closely related mature sequences that are near-identical except for one or two nucleotides are designated by letter suffixes (e.g., has-miR-121a and has-miR-121b). When cloning studies identify two miRNAs which originate from the same predicted precursor, the mature sequence that is less abundant is designated with a star symbol (e.g., hsa-miR-21*). When there is insufficient data to determine which mature sequence is the predominant one, the miRNAs are designated based on their 3’ arm origin (e.g., hsa-miR-142–3p) or 5’ arm origin (hsa-miR-142–5p)2,3. The first miRNA discovered in humans, let-7, does not follow the naming convention for historical reasons2.
Biogenesis
Based on their genomic location, miRNA genes can be intergenic or intronic. Intergenic miRNAs are found in genomic regions that are distinct from known transcription units. These can be monocistronic with their own promoters or polycistronic, where more than one miRNA is transcribed as a cluster with a shared promoter. Intronic miRNAs are found in the introns of annotated genes, both protein-coding and noncoding. They use the same promoter as their host genes. Rarely, genes encoding miRNAs are located in the exon. miRNAs are transcribed in the nucleus by RNA polymerase II. Initially, the transcription results in a ~80 nucleotide long nascent transcript called pri-miR, which is 5’ capped and 3’ polyadenylated4. pri-miR is processed in the nucleus by an RNAse III-like enzyme, Drosha, in concert with DGCR8 (DiGeorge Syndrome Critical Region 8), to a ~65 nucleotide long nascent transcript termed pre-miR. Exportin 5, a nuclear export factor, exports the pre-miR into the cytoplasm5, where it is cleaved by another RNAse III-like enzyme, Dicer, to form short (~22 nt) ds-RNA duplexes. One of the strands of the duplex is incorporated into the RNA-induced silencing complex (RISC) and guided to the target mRNAs. The other strand of the duplex is degraded. Pairing between the mature single stranded miRNA and its target mRNA takes place in association with the Argonaute family of proteins in the RISC6.
In addition to the canonical biogenesis described above, there are several non-canonical pathways of miRNA biogenesis: (i) a 7-methylguanosine(m7G)-capped pre-miR can be generated directly through transcription, bypassing Drosha processing, and exported to cytoplasm; (ii) a small RNA precursor (mitron) can be generated through mRNA splicing and debranching, also bypassing the Drosha-mediated processing step, folded into pre-miR and exported to the cytoplasm; (iii) some small nucleolar RNAs (snoRNAs) can be cleaved to produce pre-miR; (iv) terminal uridylyl transferase (TUTase)-dependent group II pri-miR produces pre-miR with a shorter 3′ overhang that undergoes monouridylation in the cytoplasm for Dicer processing; and (v) a Dicer-independent pathway in which a short pre-miR is produced by Drosha and is exported directly and loaded to Argonaute protein without Dicer processing1.
Function
The core of the RISC is comprised of an Argonaute protein (Ago4 in mammals) and GW182 protein (TNRC6A-C in mammals). The seed sequence of a miRNA, a stretch of 6 nucleotides spanning nucleotide 2–7, determines the mRNA target recognition. Base-pairing between the miRNA seed sequence and complementary sequence in the 3‵ UTR or coding region of the target mRNA is responsible for mRNA degradation or inhibition of translation7. miRNA bound to target mRNA directs cleavage at the site resulting in mRNA degradation. miRNA bound to target mRNA at multiple sites causes translational repression8. A single miRNA can target multiple protein-coding mRNAs and many miRNAs can work synergistically or competitively to target a single mRNA. Thus, miRNAs regulate diverse physiological processes such as cell development, differentiation, proliferation, and apoptosis, as well as pathological processes such as oncogenesis and immune rejection9.
In addition to the intracellular regulatory function, the presence of nuclease-resistant extracellular miRNAs has been identified in all biological fluids. While spiked-in synthetic miRNAs are susceptible to quick degradation in the body fluids, extracellular miRNAs resist such degradation. The extracellular miRNAs are protected from RNase degradation by packaging them into membrane vesicles including apoptotic bodies, microvesicles, and exosomes, or by complexing with argonaute protein-positive ribonucleoprotein particles or, rarely, with high-density lipoproteins. It is not clear whether these secreted miRNAs simply represent cellular byproducts or participate in cell-to-cell communication10.
Quantification
Several inherent characteristics of mature miRNAs makes their detection with high sensitivity and specificity technically demanding11,12. For example, mature miRNAs lack 5’ cap and 3’ poly A tail, features that would facilitate their selective purification. The small size of mature miRNAs makes conventional polymerase chain reaction (PCR) assays difficult because of the inability of primers to bind to such small templates. Moreover, miRNAs are heterogeneous in their GC content, which results in a relatively large interval of melting temperatures of nucleic acid duplexes for the population of miRNAs. The target sequence in the mature miRNA is also present in the pri-miR and pre-miR. Finally, miRNAs within the same family may differ by a single nucleotide. There are three common methods for the detection and quantification of specific individual miRNAs in tissues, cells, or fluids including circulating body fluids: (i) Real time quantitative PCR, (ii) Northern blot, and (iii) In situ hybridization. There are four common methods for global profiling of miRNAs: (i) Microarrays, (ii) TaqMan™ Low Density Arrays, (iii) nCounter® miRNA expression assay, and (iv) RNA sequencing.
Real time quantitative PCR:
The stem-loop primer design is the main ingredient that overcomes the challenge of amplifying ~22 nucleotide miRNAs by PCR (e.g., Megaplex™ miRNA assay, ThermoFisher Scientific). The stem-loop primer consists of a constant portion that forms a stem-loop and extends the ~22 nucleotide miRNA to more than ~60 nucleotides to allow for subsequent traditional PCR assay13. The stem-loop primer also consists of a variable 6-nucleotide extension that is the reverse complement of the last 6 nucleotides on the 3’ end of miRNA of interest. The stacked bases in the stem-loop design, compared to a linear design, provide thermal stability and spatial constraint, thereby minimizing errors in primer binding. During amplification, the miRNA is reverse transcribed using the stem-loop primer. Next, a miRNA specific forward primer, a universal reverse primer that is specific for the stem-loop portion, and a labelled probe is used to initiate a typical real-time PCR assay. In addition to the stem-loop primer design a poly(T) adaptor PCR can also be used for quantifying miRNAs. In this method, total RNAs, including miRNAs, are extended by a poly(A) tailing reaction using poly(A) polymerase14. The miRNA with a poly(A) tail is converted into cDNA and then PCR-amplified using a miRNA-specific forward primer and a universal poly(T) adaptor reverse primer (e.g., miScript II™, Qiagen).
Detection of the PCR product is achieved by fluorescent probes-based TaqMan™ method or DNA binding cyanine dye-based SYBR Green method. In the TaqMan™ method, an oligonucleotide probe is constructed containing a reporter fluorescent dye on the 5′end and a quencher dye on the 3′end. In an intact probe the proximity of the quencher dye greatly reduces the fluorescence emitted by the reporter dye by fluorescence resonance energy transfer through space. In the presence of target sequence, the probe anneals downstream from one of the primer sites and is cleaved by the 5′nuclease activity of Taq DNA polymerase as this primer is extended. The cleavage of the probe separates the reporter dye from the quencher dye, increasing the reporter dye signal, and removes the probe from the target strand, allowing primer extension to continue to the end of the template strand. In the SYBR Green method, the dye immediately binds to all double-stranded DNA present in the sample and to each new copy of double-stranded DNA during PCR amplification. The increase in fluorescence intensity is proportionate to the amount of PCR product produced. TaqMan™ probes have higher sensitivity and specificity for the target miRNA but must be designed for each miRNA of interest.
Quantification of the amplified PCR product is achieved by relative quantification (comparative CT method) or by absolute quantification (standard curve method)15. The quantitative endpoint for real-time PCR is the threshold cycle (CT), defined as the PCR cycle at which the fluorescent signal of the reporter dye crosses an arbitrarily placed threshold. The numerical value of the CT is inversely related to the amount of amplicon in the reaction. In the comparative CT method, the data is presented as fold change (FC) in expression, where FC =2−ΔΔ CT. To compare a gene of interest in a given sample, FC = 2−ΔΔ CT = [(CT gene of interest - CT internal control)sample A – (CT gene of interest - CT internal control)sample B)]. The comparative CT method makes several assumptions, including that the efficiency of the PCR is close to 1 and the PCR efficiency of the target gene is similar to the internal control gene15. When performing statistical analysis, statistical tests should not be run on the raw CT data and standard deviation should always be calculated after 2−ΔΔ CT, 2−Δ CT, or 2−CT.
In the standard curve method, the absolute quantity is determined using an external calibration curve and the data is presented relatively compared to a defined unit of interest (e.g., copies per μg of total RNA). We have developed a standard curve method, using a PCR generated 73-bp mouse Bak amplicon as the standard, for the absolute quantification of mRNAs and miRNAs (Figure 1)16–18. To use a single Bak standard curve to quantify all genes of interest, we ensure that the amplification efficiency of target genes is >90% so that there is minimal bias due to different amplification efficiencies of target genes and the Bak amplicon.
Figure 1. Development of a Bak amplicon based standard curve for absolute quantification of mRNAs and miRNAs by real time quantitative polymerase chain reaction assay.
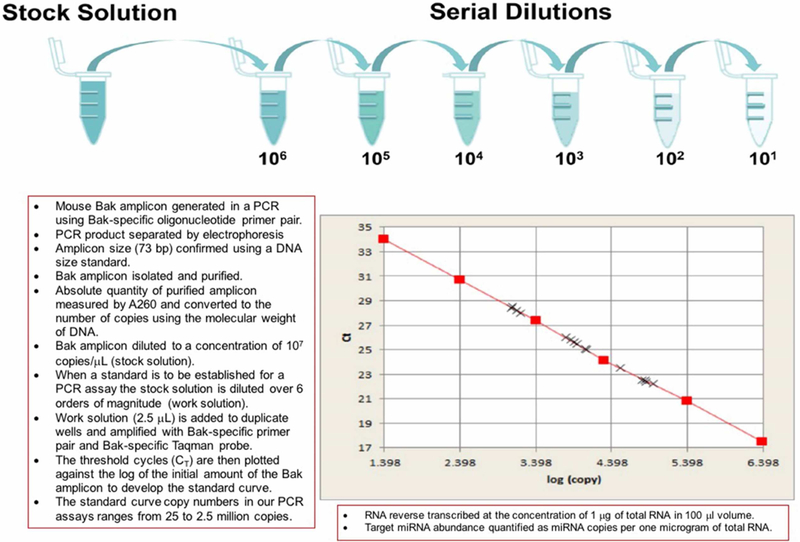
A universal standard curve for absolute quantification of mRNAs and miRNAs by quantitative polymerase chain reaction assay was developed in our laboratory by synthesizing and quantifying a customized Bak amplicon template.
In droplet digital PCR™ method (Bio-Rad Laboratories), the sample for PCR is partitioned into thousands of nanoliter-size samples encapsulated into oil droplets prior to amplification19. At the end of the PCR reaction, PCR-positive and PCR-negative droplets are counted. Each droplet either contains or does not contain the nucleic acid of interest. Poisson distribution is assumed to estimate the number of molecules in the reaction. Droplet digital PCR provides absolute quantification based on the principles of sample partitioning and Poisson statistics, thus overcoming the normalization and calibrator issues. It is relatively insensitive to potential PCR inhibitors and directly provides the copies of target per microliter of reaction. Droplet digital PCR is an end-point analysis, and the absolute quantification relies on the presence or absence of fluorescence in each droplet, rather than the fluorescence levels during the reaction. These factors offer the advantage of direct and independent quantification of DNA without standard curves19. The current droplet digital PCR™ system (QX100™ and QX200™) is compatible with TaqMan™ probes but not with SYBR Green dye.
Northern blot:
RNA is denatured and separated by urea polyacrylamide gel electrophoresis, transferred to nylon membrane, fixed, and hybridized with labelled DNA or RNA probes. Target miRNAs with sequences complementary to the probe are detected. Locked-nucleic acid probes are widely used as these are stable, specific and have no radioactive contamination (e.g., miRCURY LNA™ miRNA detection probes, Qiagen). These are nucleotides in which the furanose ring of the ribose sugar is chemically locked in a 3′-endo (North) conformation, with an extra methylene bridge connecting the 2′ oxygen and 4′ carbon, thus increasing its hybridization properties20.
In situ hybridization:
This technology allows for detection of specific nucleic acid sequences in tissue samples at the cellular level. Because of their increased thermal stability, locked-nucleic acids are used to generate short nucleotide probes with high melting temperature required for in situ hybridization. miRNA in situ hybridization is technically challenging and various modifications for fixation and permeabilization, hybridization, washing, and sequence amplification and detection are available21.
Microarrays:
Modified oligonucleotide probes antisense to miRNAs are anchored on microscopic glass slides. miRNAs in a given sample are biotinylated and are captured on the microarray by the oligonucleotide probes in hybridization. Streptavidin-Phycoerythrin is then used for detecting the biotinylated miRNA (e.g., GeneChip™ miRNA array, ThermoFisher Scientific). The hybridized microarray slide is scanned to measure the relative gene expression of each miRNA captured on the slide. Several technical variants of miRNA arrays have been independently developed. These variants differ in oligonucleotide probe design, probe immobilization chemistry, sample labeling and microarray chip signal-detection methods22.
TaqMan™ Low Density Arrays:
TaqMan™ Low Density Arrays (TLDA, ThermoFisher Scientific) which are based on TaqMan™ stem-loop PCR, are available in three forms: 96-well pates, 384-well microfluidic cards, and OpenArray® plates. The wells in all three formats are preloaded with miRNA primers and TaqMan™ fluorogenic probes. After the cDNA sample and TaqMan™ Master Mix are loaded on to the wells, the plate/card is placed in a thermal cycler for PCR. miRNA expression level is reported using the comparative CT (ΔΔCT) method. TaqMan™ OpenArray® contains 754 human miRNA sequences and enables generation of miRNA profile of up to 48 samples in a single working day.
nCounter® miRNA expression assay:
The NanoString technology is based on direct molecular barcoding and digital detection of target molecules using a pair of color-coded probes and requires minimal sample intervention. The ‘direct’ refers to the process of counting individual tagged nucleic acids without any need for amplification and the ‘digital’ refers to the capacity to use absolute and specific quantification, independent of relative measures like intensity or amplification cycles. The miRNAs are ligated with unique oligonucleotide tags. The probe pair consists of a Reporter Probe, which carries a signal on its 5’ end, and a Capture Probe, which carries biotin on its 3’ end. During hybridization, probe pairs are used in large excess, such that each miRNA with oligonucleotide tag finds a probe pair. The hybridization mixture containing miRNA/probe complexes is allowed to bind to magnetic beads complementary to sequences on the Capture Probe. After washing, the Capture Probes and miRNA/probe complexes are eluted off the beads and are hybridized to magnetic beads complementary to sequences on the Reporter Probe. The purified target/probe complexes are eluted off the beads and immobilized on the cartridge for data collection. The digital images are processed and the barcode counts are tabulated.
RNA sequencing:
Next-generation sequencing methods offer several advantages over microarray or PCR assays, including discovery of novel small RNAs, detection of single nucleotide polymorphisms, distinguishing of different isoforms, a better signal to noise ratio than microarrays, and a higher throughput than PCR or Northern blotting. Sequencing also allows comprehensive quantification of gene expression over a dynamic range (no upper or lower limit) compared to other techniques23. The steps involved in sequencing include small RNA isolation, cDNA library preparation, and sequencing.
In a typical library preparation protocol, adapters are ligated to the RNAs followed by reverse transcription and PCR amplification24. Serious biases may be introduced during the adapter ligation steps due to RNA sequence/structure effects resulting in the preferential ligation of certain small RNAs with a given adapter sequence25. An approach to neutralize this effect and improve the fidelity of sequencing results is randomization of adapter sequences close to the ligation junction. Instead of modifying the adapters, bias suppression has been attempted also through optimizing reaction conditions. In one such approach, polyethylene glycol, a macromolecular crowding agent known to increase ligation efficiency and reduce bias is used. Another concern is the formation of adapter dimers. Currently available library preparation kits use either strategies to eliminate excess 3′ adapter before 5′ adapter ligation, including purification steps, or complementary oligonucleotides that inactivate the 3′ adapter. Some protocols use polyadenylation instead of ligation for 3′ adapter addition. Multiple A residues are added to the 3′ end using poly(A) polymerase. Then, a 5′ adapter is ligated either directly to the RNA or to the nascent cDNA after reverse transcription. Certain newer protocols eliminate ligation bias by avoiding adapter ligation altogether through adding 3′ adapter by polyadenylation and 5′ adapter by reverse transcriptase template-switching.
A robust protocol for small RNA sequencing using barcoded 3’ adapters (multiple unique sequences at the 5’ end of the 3’ adapter oligonucleotides), that allows for pooling of multiple RNA samples, was initially reported a decade ago24,26,27. TruSeq (Illumina) small RNA library preparation, which allows for pooling of multiple RNA samples, is a commonly used protocol used for small RNA sequencing. After isolation of total RNA from the sample, synthetic oligonucleotide adapters of known sequence are ligated to the 3′ and 5′ ends of the small RNA pool using T4 RNA ligases. The adapters introduce primer-binding sites for reverse transcription and PCR-amplification. The amplification is performed with two primers that anneal to the adapter ends. This step selectively enriches RNA fragments with adapter molecules on both ends. Subsequently, the cDNA construct is purified, and the library is normalized prior to sequencing. Using unique index adapter sequences, samples can be multiplexed, with up to 48 libraries combined in a single lane. The index is added at the amplification step following reverse transcription. Libraries are pooled immediately before gel purification or after gel purification. Other small RNA library preparation protocols that are compatible with Illumina sequencing platform include BIOO Scientific NEXTflex® small RNA sequencing kit (PerkinElmer), NEBNext® small RNA library prep set (New England BioLabs), SMARTer® smRNA-seq kit (TaKaRa), and CATS small RNA seq kit (Diagenode)25. In the Illumina sequencing platform, the library is loaded into a flow cell with a lawn of surface-bound oligonucleotides complementary to the adapters. The 3’ and 5’ adapters of each cDNA library fragment bind to complementary oligonucleotides forming a ‘bridge’ that allows priming and PCR amplification of the cDNA fragment sandwiched between the two adapters28. Millions of copies of unique sequence clusters are produced which serve as single stranded templates for sequencing. Based on the sequence of the template, fluorescently labeled dNTPs are added to the growing chain. The addition of a single nucleotide leads to emission of a characteristic fluorescent signal. The wavelength and intensity of emission identifies the base. The length of the read depends on the number of sequencing cycles. The reads are sorted based on the index sequence and aligned to a reference transcript to produce a transcription map with the level of expression of each gene.
miRNA Expression Patterns in Kidney Transplant Recipients
Kidney transplantation has evolved from a risky experimental procedure to a treatment of choice for patients with end-stage kidney disease. However, allograft destructive immune response may occur at any time during the life span of the kidney transplant despite the use of potent immunosuppressive drugs. The mechanistic pathways of rejection, both acute and chronic, are being resolved. The Banff classification of allograft pathology captures well the multiple pathways of immune rejection in the kidney allograft29.
We have proposed a time-line model to illustrate immune rejection of kidney allograft as a continuum, with initial events identified by molecular perturbations prior to the late histological changes30. In this conceptualization, molecular biomarkers serve not only a diagnostic role but also an anticipatory, treatment response-predictive, and prognostic role. The hypothesis that early intervention is efficacious is an important rationale for the development of molecular monitoring strategies. miRNAs play a pivotal role in the development, maturation, and function of the cells of the innate and adaptive immune system. As miRNAs are stable and present in high abundance they have emerged as attractive biomarkers to assess kidney allograft status.
Table 1 is a summary of published studies on miRNA expression patterns observed in human kidney transplant recipients. Recent reviews have addressed the role of miRNAs in other solid organ transplants and in hematopoietic cell transplantation31–33. Our first study of global profiling of mature human miRNAs in kidney allograft recipients with or without acute rejection (AR) in the allografts used TLDA. Among the 365 mature human miRNAs analyzed in the training set (4 normal biopsies and 3 AR biopsies), 174±7 miRNAs (48%) were expressed in the biopsy samples17. Unsupervised hierarchical clustering of miRNA expression patterns correctly classified the normal allograft biopsies and the AR biopsies (Figure 2). In a validation set of 26 kidney allograft biopsies (9 AR and 17 normal allograft biopsies), we validated a subset of differentially expressed miRNAs using targeted PCR assays. Our analysis involving receiver operating characteristic curve showed that AR can be predicted accurately using intragraft levels of miR-142–5p or miR-155 (area under the curve 0.99 and 0.98, respectively, P<0.0001). We found a positive association between intragraft levels of CD3 (T-cell co-receptor) mRNA or CD20 (B cell molecule) mRNA and intragraft levels of miR-142–5p, miR-155, or miR-223, miRNAs that were overexpressed in AR. We also found a positive association between intragraft levels of NKCC2 (tubular co-transporter) mRNA and intragraft levels of miR-10b, miR-30a-3p, or let-7c, miRNAs that were underexpressed in AR (Figure 3). To address whether the altered expression of miRNAs in AR biopsies is due to relative proportions of immune cells and kidney parenchymal cells or due to altered regulation of miRNAs within the cells themselves, we quantified the abundance of differentially expressed miRNAs in normal human peripheral blood mononuclear cells (PBMC) and in normal human renal tubular epithelial cells (HREC). We also investigated whether stimulation of PBMCs or HRECs altered the level of expression of miRNAs. We found that the miRNAs overexpressed in AR biopsies (miR-142–5p, miR-155, and miR-223) were all expressed at a higher level in normal human PBMCs compared to miRNAs underexpressed in AR biopsies (miR-30a-3p, miR-10b, or let-7c). Stimulation of PBMCs with the mitogen phytohemagglutinin (PHA) resulted in an increase in the abundance of miR-155 and a decrease in miR-223, let-7c, or miR-142–5p. Quantification of miRNAs in primary cultures of HRECs showed that miR-30a-3p, miR-10b, or let-7c are expressed at a higher level in HRECs compared to PBMCs, and that stimulation of HRECs with cell-free supernatants of PHA-activated PBMCs results in a decrease in the abundance of miR-30a-3p. Our in vitro studies suggest the possibility that the altered expression of miRNAs in AR biopsies may be due to the altered regulation of miRNAs within the infiltrating immune cells and the resident kidney parenchymal cells themselves17.
Table 1.
Published studies on miRNA expression in human kidney transplants
| PMID (Journal) | First Author (Year) | Specimen | Profiling technique | Targeted or global profiling | Platform | Company | Patients/Diagnosis | Major Findings |
|---|---|---|---|---|---|---|---|---|
| 18346642 (Transpl Immunol) | Sui (2008) | Biopsy tissue | Microarray PCR |
Global Targeted |
LNA-based miChip - |
University of Heidelberg, Germany - |
3 AR & 3 Native kidney control (RNA pooled in each group) | AR: 8 miRNAs Up & 12 miRNAs Down PCR for 2 miRNAs AR: Up-miR-320 Down-miR-324–3p |
| 19289845 (Proc Natl Acad Sci USA) | Anglicheau (2009) | Biopsy tissue (RNAlater) | PCR (TLDA) PCR |
Global Targeted |
Taqman miRNA Taqman miRNA |
Applied Biosystems/ ThermoFisher | 3 AR & 4 Normal 9 AR & 17 Normal |
ACR: 10 miRNAs Up & 43 miRNAs Down PCR done for 6 miRNAs AR: Up-miR-142–5p, miR-155 & miR-223 Down-miR-10b, miR-30a-3p & let-7c |
| 21794090 (Am J Transplant) | Scian (2011) | Biopsy tissue (RNAlater) Urine |
Microarray PCR PCR |
Global Targeted Targeted |
Sentrix Universal BeadChip array Taqman miRNA Taqman miRNA |
Illumina Applied Biosystems/ ThermoFisher Applied Biosystems/ThermoFisher |
13 IFTA & 5 Normal 32 IFTA & 13 Normal 7 IFTA & 7 Normal |
Differential expression: 56 miRNAs IFTA: Up-miR-32 & miR-142–3p. Down-miR-107, miR-204 & miR-211 IFTA: Up-miR-142–3p. Down-miR-204 & miR-211 |
| 21812927 (Am J Transplant) | Lorenzen (2011) | Urine | PCR | Targeted | Taqman miRNA | Applied Biosystems/ThermoFisher | 68 ACR & 20 Stable | ACR: Up-miR-10a Down-miR-10b & miR-210 |
| 23131772 (Transplantation) | Ben-Dov (2012) | Biopsy tissue (RNAlater) | RNA Seq | Global | Sequencing by synthesis Taqman miRNA | Illumina Applied Biosystems/ThermoFisher | 3 IFTA & 4 Normal 10 IFTA & 8 Normal | IFTA: 9 miRNAs Up & 0 miRNAs Down IFTA Up: miR-21, miR-21* & miR-142–3p |
| 23469132 (PLoS One) | Glowacki (2013) | Serum | PCR | Targeted | Taqman miRNA | Applied Biosystems/ThermoFisher | 42 IFTA | Severe IFTA: Up-miR-21 |
| 23511211 (Transplantation) | Wilflingseder (2013) | Biopsy tissue (FFPE) | Microarray | Global | GeneChip | Affymetrix/ThermoFisher | 10 Stable, 14 DGF, 30 ACR, 11 ABMR |
DGF vs Stable: 7 miRNAs Up & 0 Down ACR vs Stable: 4 miRNAs Up & 18 Down ABMR vs Stable: 6 miRNAs Up & 0 Down |
| 24025639 (Kidney Int) | Maluf (2014) | Urine cells | Microarray PCR PCR |
Global Targeted Targeted |
GeneChip Taqman miRNA Taqman miRNA |
Affymetrix/ThermoFisher Applied Biosystems/ThermoFisher Applied Biosystems/ThermoFisher | IFTA 10 & 12 Normal IFTA 7 & Normal 10 Longitudinal: 41 good graft function & 25 poor graft function |
Differential expression: 22 miRNAs IFTA: Up-miR-125b, Down-miR-125b, miR-203, miR-204 & miR-211 Differential expression: miR-99a, miR-140–3p, miR-142–3p, miR-200* & miR-200b |
| 24731148 (Am J Transplant) | Li 2014) | Plasma | PCR | Targeted | Taqman miRNA | Applied Biosystems/ThermoFisher | 31 BK viremia & 15 Normal | BK viremia: Up-bkv-miR-B1–5p & bkv- miR-B1–3p |
| 25659925 (Exp Cell Res) | Liu (2015) | Biopsy tissue | RNA-Seq | Global Targeted |
Pyrosequencing Taqman miRNA |
454 Life Sciences/Roche Applied Biosystems/ThermoFisher | 15 AR & 15 Normal | Differential expression: 75 miRNAs PCR done for miR-10b. AR: Down-miR-10b |
| 26002284 (Transp Immunol) | Soltaninejad (2015) | Biopsy tissue (RNAlater) PBMC |
PCR PCR |
Targeted Targeted |
Taqman miRNA Taqman miRNA |
Applied Biosystems/ThermoFisher |
17 TCMR & 18 No rejection |
TCMR: Up- miR-142–5p, miR-142–3p, miR-155 & miR-223 TCMR: Up-miR-142–3p & miR-223 |
| 26154388 (Transplantation) | Vitalone (2015) | Biopsy tissue | PCR | Targeted | Taqman miRNA | ThermoFisher | 29 AR & 68 No AR | AR: Up-miR-25, miR-142–3p & miR-342- 3p. Down-miR-10b, miR-181a, miR-192, miR-204, miR-215 & miR-615–3p |
| 26734715 (PLoS One) | MgGuinness (2016) | Biopsy tissue (pre-transplant, RNAlater) | PCR | Targeted | Taqman miRNA | Applied Biosystems/ThermoFisher | 27 DGF 67 No DGF | DGF: Down-miR-125b & miR-217 |
| 26444957 (Transplantation) | Matz (2016) | Whole blood (PAXgene tube) | Microarray PCR | Global Targeted | miRXplore microarray Taqman miRNA |
Miltenyi Biotec Life Technologies/ThermoFisher |
4 TMVR & 4 Stable PCR: 24 TMVR & 137 all other diagnosis |
Differential expression: 23 miRNAs TMVR: Down-miR-15a, miR-15b, miR-16, miR-103a, miR-106a & miR-107 |
| 27663089 (Transpl Immunol) | Matz (2016) | Plasma | PCR | Targeted | Taqman miRNA | Life Technologies/ThermoFisher | 39 TCMR & 40 Stable | TCMR: Down-miR-15b, miR-103a & miR-106a |
| 27521993 (Clin Biochem) | Vahed (2017) | Urine cells | PCR | Targeted | Stem loop primers / iCycler iQ system | Bio-Rad | 23 IFTA, 24 Stable & 15 non-Tx healthy | IFTA (vs. non-Tx healthy): Up-miR-21 & miR-142–3p Down-miR-200b |
| 27323802 (Biomarkers) | Iwasaki (2017) | PBMC | Microarray | Global Targeted Targeted |
miRCURY LNA microarray Taqman miRNA Taqman miRNA |
Exiqon/Qiagen Applied Biosystems/ThermoFisher Applied Biosystems/ThermoFisher |
11 Normal, 11 DSA+normal, 6 DSA+w/graft dysfunction (RNA pooled in each group) 22 Normal, 10 subclinical cABMR, 13 DSA+ w/no graft dysfunction w/other histology, 9 clinical cABMR | Differential expression: 9 miRNAs Normal: Up-miR-142–5p Clinical cABMR: Up-miR-486–5p |
| 28380212 (Braz J Med Biol Res) | Domenico (2017) | PBMC Urine cells | PCR | Targeted Targeted | Taqman miRNA Taqman miRNA | Applied Biosystems/ThermoFisher | 23 AR, 18 ATN, 8 Stable | ATN: Up-miR-142–3p ATN: Up-miR-142–3p |
| 28455659 (Int Urol Nephrol) | Vahed (2017) | Plasma | PCR | Targeted | miRCURY LNA | Exiqon/Qiagen | 26 IFTA & 27 Stable | IFTA: Up-miR-21, miR-142–3p & miR-155 |
| 28880456 (Br J Clin Pharmacol) | Milan (2017) | Urine cells | PCR | Targeted | miRCURY LNA Roche LightCycler 480 | Exiqon/Qiagen Roche | 8 AR & 72 No AR | AR: Up-miR-142–3p & miR-155–5p. Down-miR-210–3p |
| 29267352 (PLoS One) | Kim (2017) | Urine exosomes | PCR | Targeted | Taqman miRNA | Applied Biosystems/ThermoFisher | 13 BK virus nephropathy & 67 Other histology | BK virus nephropathy: Up-bkv-miR-B1–5p |
| 29518695 (J Clin Virol) | Virtanen (2018) | Plasma | PCR | Targeted | Taqman miRNA | Applied Biosystems/ThermoFisher | 9 BK viremia (serial samples) & 2 Normal | BK viremia: Up-bkv-miR-B1–5p & bkv-miR-B1–3p |
AR: Acute rejection
ABMR: Antibody mediated rejection
ACR: Acute cellular rejection
cABMR: Chronic antibody mediated rejection
DGF: Delayed graft function
DSA: Donor specific antibodies
FFPE: Formalin fixed paraffin embedded
IFTA: Interstitial fibrosis and tubular atrophy
PMID: PubMed identifier
TCMR: T-cell mediated rejection
TMVR: T-cell mediated vascular rejection
Tx: Transplant
Figure 2: Clustering analysis of miRNA expression differentiate acute rejection biopsies from normal allograft biopsies of human kidney allografts.
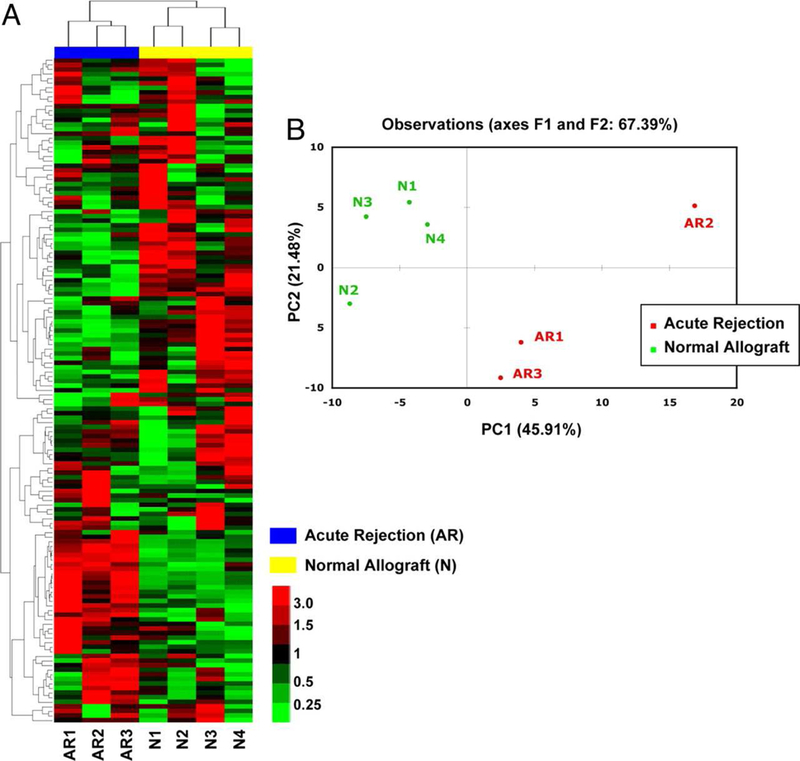
Unsupervised hierarchical clustering (A) and principal component analysis (B) of miRNA expression in 7 human kidney allograft biopsies (3 AR and 4 normal) examined using TLDA. Two major clusters accurately divided the AR biopsies from normal allograft biopsies. Principal component analysis confirmed the separation of AR samples from normal allograft biopsies (From Anglicheau D, Sharma VK, Ding R, et al. MicroRNA expression profiles predictive of human renal allograft status. Proc Natl Acad Sci U S A. 2009;106(13):5331; with permission).
Figure 3. Positive association between miRNAs and mRNAs in human kidney allograft biopsies.
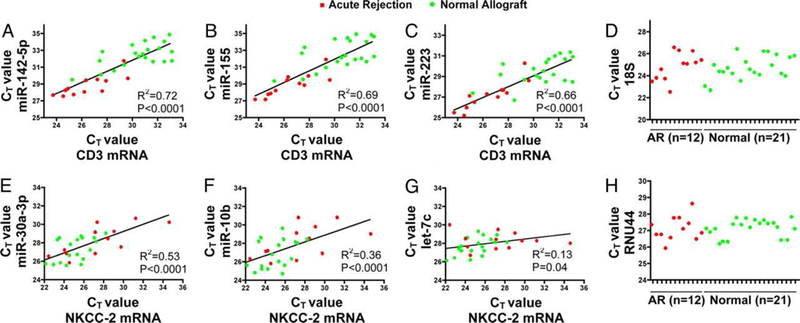
The relationship between the intragraft levels of miRNA and mRNA is shown. A strong positive association between the levels of CD3 mRNA and the levels of miRNAs overexpressed in AR biopsies was found: (A) miR-142–5p; (B) miR-155; or (C) miR-223. A positive association between kidney tubule specific NKCC-2 mRNA and miRNAs underexpressed in AR biopsies was also observed: (E) miR-30a-3p; (F) miR-10b; or (G) let-7c. The mean (±SD) CT values of the endogenous control for mRNAs (18S rRNA) (D) and for miRNAs (RNU44 small nucleolar RNA) (H) were similar between the groups (From Anglicheau D, Sharma VK, Ding R, et al. MicroRNA expression profiles predictive of human renal allograft status. Proc Natl Acad Sci U S A. 2009;106(13):5332; with permission).
In the first study of human kidney allograft biopsy miRNA sequencing18, we characterized the miRNA expression profiles of 3 biopsies with fibrosis and 4 normal allograft biopsies using barcoded deep-sequencing of a cDNA library prepared from multiplexed RNA by the method developed by Hafner et al. 26,27. Among the differentially expressed miRNAs, two sequence families (miR-21 and miR-142–3p) showed expression exceeding 0.1% of total miRNA, suggesting that they may be causally involved in the fibrotic process18. Inspection of kidney miRNA profiles in relation to profiles obtained from kidney cell lines, PBMCs, and cultured fibroblasts suggested that occurrence of miR-142–3p in fibrosis samples is due to leukocyte infiltration, whereas miR-21 is likely upregulated in kidney parenchymal cells (Figure 4). We used an independent cohort of 18 kidney-transplant recipients (10 fibrosis and 8 normal) to validate, by targeted PCR assay, a subset of differentially expressed miRNAs identified by global profiling.
Figure 4. MicroRNA profiles generated by small RNA sequencing distinguish human kidney allograft biopsies with fibrosis from normal allograft biopsies.
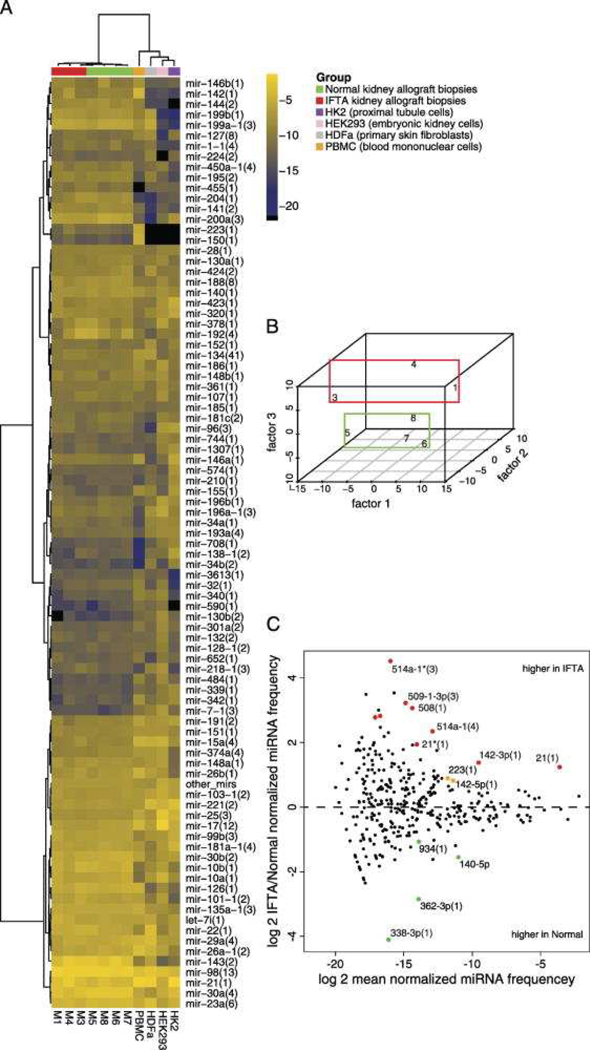
This Figure depicts the miRNA profiles generated by small RNA sequencing. A, Hierarchical clustering and heatmap representation of kidney allograft biopsies with fibrosis (IFTA, Interstitial fibrosis and tubular atrophy) and normal allograft biopsies, human peripheral blood mononuclear cells (PBMC), HDFa (primary skin fibroblast), HEK293 cells (human embryonic kidney cells), and HK2 cells (kidney proximal tubule cells) according to merged miRNA profiles. B, Multidimensional scaling showing separation of IFTA biopsies from Normal biopsies. C, MA plot showing the differentially expressed miRNA sequence families. Colored data points represent P<0.05 (red points signify in addition FDR<0.1) (From Ben-Dov IZ, Muthukumar T, Morozov P, et al. MicroRNA sequence profiles of human kidney allografts with or without tubulointerstitial fibrosis. Transplantation. 2012;94(11):1089; with permission).
Identifying miRNA Targets
There are two ways in which targets of miRNAs are identified; computational methods and experimental methods. Computational methods rely on the identification of phylogenetically conserved matches to the miRNA seed sequence34. Different miRNA-target prediction algorithms predict targets with different techniques and criteria such as base pairing, target accessibility, and evolutionary conservation of target site35. Once targets are predicted the next step is to infer the miRNA functions. Computational methods to reveal functions of miRNAs involve annotating the functions of miRNA through functional enrichment analysis using their target mRNA; identifying the co-expressed miRNA/mRNA groups, either at the sequence level or by integrating sequence and expression profiles of miRNAs and mRNAs; and inferring functional miRNA-mRNA regulatory modules, which are regulatory networks of miRNAs and their target mRNAs in specific biological processes35. Experimental methods are based on immunoprecipitation of the miRNA effector complex, followed by sequencing-based identification of its interacting RNAs. Crosslinking immunoprecipitation (CLIP) methods involve ultraviolet irradiation of tissues, organisms, or cells, for covalently crosslinking miRNA targets to the Argonaute proteins. The crosslinked RNAs are reduced in size, amplified by PCR and then sequenced for the identification of Argonaute tags that contain miRNA binding sites on target mRNAs. One such method for human cell lines, photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP), is based on the incorporation of photoreactive ribonucleoside analogs into nascent RNA transcripts by living cells. Irradiation of the cells induces crosslinking of photoreactive nucleoside-labeled cellular RNAs to interacting RNA-binding proteins. Immunoprecipitation of RNA-binding proteins of interest is followed by isolation of the crosslinked and coimmunoprecipitated RNA, which is sequenced 36.
In our miRNA sequencing of kidney allograft biopsies with fibrosis, we focused on miR-21 and searched for possible targets among transcripts that were coimmunoprecipitated with Argonaute 1–4 (EIF2C1–4) proteins. We queried a published dataset of mRNA clusters from pooled PAR-CLIP experiments on FLAG/HA-tagged Argonaute proteins in human embryonic kidney HEK293 cells for miR-21 binding sites. We found enrichment for 7-mer (ATAAGCT) and 8-mer (GATAAGCT) seed-complementary sequences of miR-21, and identified SMAD7, as a miR-21 target18 (Figure 5). SMAD7 has been shown to inhibit the fibrotic effect of TGFβ, a potent fibrogenic cytokine involved in repair following tissue injury, by blocking SMAD2 activation. This finding provides further credence to a TGFβ-centric hypothesis in the pathogenesis of chronic nephropathy/rejection (manifested histologically as interstitial fibrosis and tubular atrophy) of the kidney allograft 37,38. In this hypothesis, which integrates immune and non-immune events in the pathogenesis of allograft fibrosis, TGFβ represents a critical and self-perpetuating event for the progressive damage to the kidney allograft and attendant decline in graft function37,38.
Figure 5. miRNA target identification.
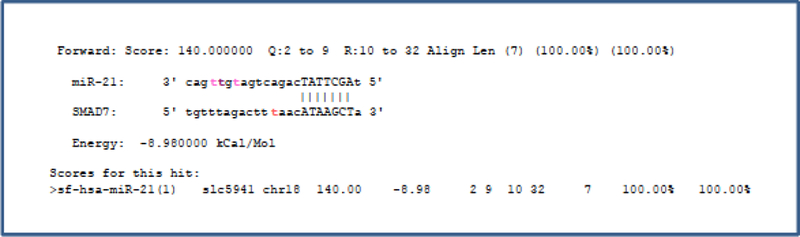
SMAD7 was identified as an miR-21 target by querying a published Argonaute PARCLIP Dataset; aligned and scored by the miRanda algorithm (microrna.org). Colored “t” represents a position found to be crosslinked according to PAR-CLIP T to C transition signature (From Ben-Dov IZ, Muthukumar T, Morozov P, et al. MicroRNA sequence profiles of human kidney allografts with or without tubulointerstitial fibrosis. Transplantation. 2012;94(11):1086–94; with permission).
Conclusion
miRNAs have emerged as robust molecular markers for assessing human kidney allograft status. In addition, miRNA expression patterns have provided mechanistic insights for kidney allograft dysfunction and are attractive targets for intervention. However, several challenges remain before they can be applied for managing kidney transplant recipients. Similar to the multicenter Clinical Trials in Organ Transplantation 04 that validated urinary cell mRNA profiles for diagnosing acute rejection of kidney allograft39, large, well-designed, multicenter trials are needed to assess the usefulness of miRNAs in clinical organ transplantation. We are optimistic of the progress thus far and anticipate clinical trials for testing miRNA-based management strategies in kidney transplant recipients.
KEY POINTS.
MicroRNAs (miRNAs) are small, non-coding, single stranded RNAs that regulate protein coding genes.
Common methods for detecting and quantifying specific individual miRNAs include: Real time quantitative polymerase chain reaction, Northern blot, and In Situ hybridization. Common methods for global profiling of miRNAs include: Microarrays, TaqMan™ Low Density Arrays, nCounter® miRNA expression assay, and RNA sequencing.
Cellular and extracellular miRNAs are emerging as robust biomarkers of allograft status.
SYNOPSIS.
miRNAs, ~20–22 nucleotide single stranded RNA species that play a pivotal role in the regulation of protein-coding genes, are emerging as robust biomarkers for assessing allograft status. Herein, we briefly review the biogenesis and function of the miRNAs, and provide an overview of the tools to quantify miRNAs in tissues and body fluids. We then review our studies of discovery and validation of alterations in miRNA expression within kidney allografts with or without acute rejection, as well as with or without fibrosis, and summarize published data on miRNA expression patterns in kidney transplant recipients.
Footnotes
DISCLOSURE STATEMENT
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014;15(8):509–524. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNA annotation. RNA 2003;9(3):277–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desvignes T, Batzel P, Berezikov E, et al. miRNA Nomenclature: A View Incorporating Genetic Origins, Biosynthetic Pathways, and Sequence Variants. Trends Genet 2015;31(11):613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004;10(12):1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 2003;17(24):3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta 2010;1803(11):1231–1243. [DOI] [PubMed] [Google Scholar]
- 7.Vidigal JA, Ventura A. The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol 2015;25(3):137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004;5(7):522–531. [DOI] [PubMed] [Google Scholar]
- 9.Anglicheau D, Muthukumar T, Suthanthiran M. MicroRNAs: small RNAs with big effects. Transplantation 2010;90(2):105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turchinovich A, Tonevitsky AG, Burwinkel B. Extracellular miRNA: A Collision of Two Paradigms. Trends Biochem Sci 2016;41(10):883–892. [DOI] [PubMed] [Google Scholar]
- 11.Benes V, Castoldi M. Expression profiling of microRNA using real-time quantitative PCR, how to use it and what is available. Methods 2010;50(4):244–249. [DOI] [PubMed] [Google Scholar]
- 12.de Planell-Saguer M, Rodicio MC. Analytical aspects of microRNA in diagnostics: a review. Anal Chim Acta 2011;699(2):134–152. [DOI] [PubMed] [Google Scholar]
- 13.Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 2005;33(20):e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi R, Chiang VL. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 2005;39(4):519–525. [DOI] [PubMed] [Google Scholar]
- 15.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols 2008;3(6):1101–1108. [DOI] [PubMed] [Google Scholar]
- 16.Muthukumar T, Dadhania D, Ding R, et al. Messenger RNA for FOXP3 in the urine of renal-allograft recipients. N Engl J Med 2005;353(22):2342–2351. [DOI] [PubMed] [Google Scholar]
- 17.Anglicheau D, Sharma VK, Ding R, et al. MicroRNA expression profiles predictive of human renal allograft status. Proc Natl Acad Sci U S A 2009;106(13):5330–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Dov IZ, Muthukumar T, Morozov P, Mueller FB, Tuschl T, Suthanthiran M. MicroRNA sequence profiles of human kidney allografts with or without tubulointerstitial fibrosis. Transplantation 2012;94(11):1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hindson CM, Chevillet JR, Briggs HA, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods 2013;10(10):1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat Protoc 2007;2(6):1508–1514. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen BS. MicroRNA in situ hybridization. Methods Mol Biol 2012;822:67–84. [DOI] [PubMed] [Google Scholar]
- 22.Liu CG, Calin GA, Volinia S, Croce CM. MicroRNA expression profiling using microarrays. Nature protocols 2008;3(4):563–578. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hafner M, Landgraf P, Ludwig J, et al. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008;44(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dard-Dascot C, Naquin D, d’Aubenton-Carafa Y, Alix K, Thermes C, van Dijk E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genomics 2018;19(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hafner M, Renwick N, Brown M, et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 2011;17(9):1697–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hafner M, Renwick N, Farazi TA, Mihailovic A, Pena JT, Tuschl T. Barcoded cDNA library preparation for small RNA profiling by next-generation sequencing. Methods 2012;58(2):164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Illumina. In-depth NGS Introduction https://www.illumina.com/science/technology/next-generation-sequencing.html.
- 29.Haas M, Loupy A, Lefaucheur C, et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant 2018;18(2):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anglicheau D, Suthanthiran M. Noninvasive prediction of organ graft rejection and outcome using gene expression patterns. Transplantation 2008;86(2):192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamdorf M, Kawakita S, Everly M. The Potential of MicroRNAs as Novel Biomarkers for Transplant Rejection. J Immunol Res 2017;2017:4072364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomuleasa C, Fuji S, Cucuianu A, et al. MicroRNAs as biomarkers for graft-versus-host disease following allogeneic stem cell transplantation. Ann Hematol 2015;94(7):1081–1092. [DOI] [PubMed] [Google Scholar]
- 33.Mas VR, Dumur CI, Scian MJ, Gehrau RC, Maluf DG. MicroRNAs as biomarkers in solid organ transplantation. Am J Transplant 2013;13(1):11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seitz H Issues in current microRNA target identification methods. RNA Biol 2017;14(7):831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu B, Li J, Cairns MJ. Identifying miRNAs, targets and functions. Brief Bioinform 2014;15(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hafner M, Landthaler M, Burger L, et al. PAR-CliP--a method to identify transcriptome-wide the binding sites of RNA binding proteins. J Vis Exp 2010(41). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma VK, Bologa RM, Xu GP, et al. Intragraft TGF-beta 1 mRNA: a correlate of interstitial fibrosis and chronic allograft nephropathy. Kidney Int 1996;49(5):1297–1303. [DOI] [PubMed] [Google Scholar]
- 38.Muthukumar T, Lee JR, Dadhania DM, et al. Allograft rejection and tubulointerstitial fibrosis in human kidney allografts: interrogation by urinary cell mRNA profiling. Transplant Rev (Orlando) 2014;28(3):145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suthanthiran M, Schwartz JE, Ding R, et al. Urinary-cell mRNA profile and acute cellular rejection in kidney allografts. N Engl J Med 2013;369(1):20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]