

Abstract
Protein disulfide isomerases (PDIs) catalyze disulfide bond formation between protein cysteine residues during protein folding in the endoplasmic reticulum (ER) lumen and are essential for maintaining ER homoeostasis. The life cycle of the hepatitis C virus (HCV) is closely associated with the ER. Synthesis and maturation of HCV proteins occur in the ER membrane and are mediated by multiple host cell factors that include also PDI. Here, we present a study investigating the effect of PDI inhibition on Huh7 human hepatoma cells harboring an HCV subgenomic replicon using the abscisic acid-derived PDI inhibitor origamicin. Transcriptional profiling shows that origamicin changed the expression levels of genes involved in the oxidative and ER stress responses and the unfolded protein response, as indicated by the upregulation of antioxidant enzymes and chaperone proteins, the downregulation of cell-cycle proteins, and induction of apoptosis-associated genes. Our data suggest that origamicin negatively impacts HCV replication by causing an imbalance in cellular homoeostasis and induction of stress responses. These insights suggest that inhibition of PDIs by low-molecular-weight inhibitors could be a promising approach to the discovery of novel antiviral compounds.
Introduction
A cell is a spatially limited environment of tightly regulated biological processes that ensure cell viability under varying external conditions. Cells respond to stress conditions by activating various counter measures, a prominent example being the endoplasmic reticulum (ER) stress response.1,2 The ER plays a key role in the proteosynthesis and folding of secretory and transmembrane proteins.3 Perturbation of ER homeostasis impairs these processes and leads to accumulation of misfolded and unfolded proteins in the ER lumen,1,2 triggering the unfolded protein response (UPR), which includes the upregulation of molecular chaperones and the protein degradation machinery.4 Another well-known example of a cellular stress condition is oxidative stress, which usually results from the increased concentration of reactive oxygen species (ROS).5,6 Examples of ROS are hydrogen peroxide (H2O2), hypochlorous acid (HOCl), or superoxide anion (O2–). ROS cause oxidative stress by oxidatively modifying proteins, impair their function, and induce protein unfolding and aggregation.5,7,8 ROS also play an important role in several physiological processes provided their location and activity are within a narrow range and the overall cellular redox homeostasis is not disrupted.9 For instance, hydrogen peroxide can act as a regulator of cell proliferation, signal transduction, apoptosis, and is involved in the formation of disulfide bonds in proteins.6,9−11
The protein disulfide bond formation in the ER is mediated by sequential action of oxidoreduction 1 (Ero1α and Ero1β) and protein disulfide isomerase (PDI).11−13 The cascade is initiated by catalytic transfer of electrons from molecular oxygen to two cysteines in Ero1 that form a disulfide bond.14,15 The oxidized Ero1 interacts with PDI and catalyzes formation of a disulfide bond in PDI while being reduced. Finally, oxidized PDI is primed to catalyze the oxidation of a cysteine pair in a client protein, creating either a de novo disulfide bond or rearranging a pre-existing one.16
Although neither the X-ray nor the nuclear magnetic resonance structure of full-length human PDI has been determined, structural information derived from crystal structures of PDI fragments and PDI functional homologs has been obtained and suggests that PDI has a horseshoe shape consisting of four thioredoxin domains and a linker region termed x.17,18 These structural domains are arranged in the order a, b, b′, x, and a′ thereby creating the overall architecture of PDI. Both the a and a′ thioredoxin domains have an active site that include a pair of cysteines within a −CXXC– motif. The b and b′ domains, however, lack catalytic activity. The linker connecting the domains b′ and a′ provides structural flexibility and modulates access to substrate proteins.19,20 The catalytically active cysteine pairs present in the a and a′ domains represent the molecular basis of PDI’s redox activity.21 Notably, PDI also possesses an additional chaperone activity.22 Apparently, this chaperone activity functions independently of the redox activity;23 however, recent experiments cast doubt on this assumption.24 The chaperone activity of PDI plays a role in protein folding and acts to avoid protein misfolding and protein aggregation.25 The PDI chaperone function is important for cell survival under conditions of elevated temperature or oxidative stress. In summary, PDI plays a crucial role in oxidative protein folding and ER homeostasis because of its dual role as an oxidoreductase and molecular chaperone.
In this study, we investigated how PDI inhibition affects replication of the hepatitis C virus (HCV), a member of the hepacivirus genus.26 The positive-sense single-stranded RNA genome of HCV encodes three virion structural proteins E1, E2, and core protein, as well as the nonstructural proteins p7, NS2, NS3, NS4A, NS4B, and NS5A.27 The virus predominantly infects hepatocytes where it causes substantial rearrangement of the ER membrane architecture and induces formation of the “membranous web”.28 The altered ER membrane serves as the locations of viral replication and virion assembly.29 HCV is translated on the ER into a polyprotein that undergoes processing by host and viral proteins including proteases, kinases, and PDIs.30,31 These host–virus interactions represent potential targets for antiviral drug discovery.
Previously, we demonstrated that origamicin, an abscisic acid (ABA)-derived inhibitor binding to human PDIs, has an inhibitory effect on HCV replication, but the underlying mechanism of the inhibition is not well understood.32 Here, we investigated the effects of origamicin on cellular metabolism by conducting Affymetrix microarray mRNA profiling, and found that origamicin induces expression of proteins linked to ER and oxidative stress response. These findings suggest that PDI inhibition leads to an imbalance in cellular homeostasis and to induction of stress responses that negatively affect HCV replication.
Results
Study Design
The PDI inhibitor origamicin is derived from the plant hormone ABA (Figure 1A, top),32 but its effect on the physiology of human hepatocytes is largely unknown. The chemical modifications of ABA that create origamicin comprise a methyl esterification of the acid, a chlorination of the methyl group on the third position of the cyclohexenone ring, and an R-alkyne handle on the fifth position of the cyclohexenone ring (Figure 1A, bottom), allowing bio-orthogonal labeling.33 We investigated the effect of origamicin treatment on global mRNA levels by using a hepatocyte cell line (Huh7) harboring the HCV subgenomic replicon pFK-I389neo/luc/NS3-3′/5.1 (Figure 1B). This replicon system, termed E9 cells, is widely used to study the replication of HCV in a cell culture model.34 We treated the replicon cells with two concentrations of origamicin (i.e., mock, 25 and 50 μM) for 24 h. To confirm that PDI inhibition impairs HCV replication in our model system, we also tested the effect of two well-described PDI inhibitors, 16F16 and securinine35 (Supporting Information, Figure S1), which showed similar antiviral activity, further supporting a role for PDI in the viral life cycle. After the treatment, we extracted the total RNA and performed an Affymetrix microarray RNA quantification to identify genes with changed mRNA expression level. The in silico analysis of the expression profile also included pathway and gene ontology (GO) analyses, and was subsequently validated by quantitative real-time polymerase chain reaction (qRT-PCR) on selected differentially expressed mRNAs. An overview of the experimental workflow is depicted in Figure 1C.
Figure 1.
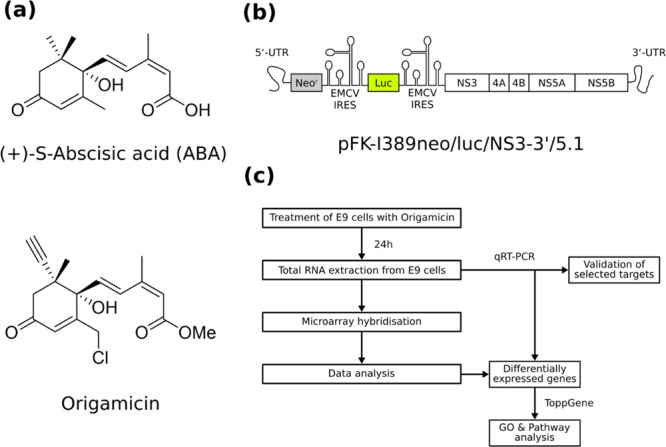
Study design. Chemical structures of (a) (+)-S-ABA (top) and origamicin (bottom). (b) Schematic representation of the subgenomic HCV replicon (pFK-I389neo/luc/NS3-3′/5.1) used in this study. (c) Overview of the experimental workflow.
Origamicin Induces Changes in the mRNA Profile in HCV Replicon Cells
We performed an Affymetrix microarray hybridization experiment to identify the gene expression profile of origamicin-treated Huh7 cells harboring the HCV subgenomic replicon. After the data preprocessing, we performed a hierarchical cluster analysis as well as a principal component analysis (PCA) to determine the overall similarity among mRNA profiles under the tested conditions (Supporting Information, Figure S2). We found that the mRNA profile of cells treated with 25 μM origamicin, which corresponds to the origamicin HCV replicon IC50Replicon value,32 was similar to that of the mock-treated cell. However, when Huh7 cells were treated with 50 μM origamicin concentration, which was reported to have a strong inhibitory effect on the HCV replicons,32 we observed pronounced changes in the mRNA profile. Volcano plots, which relate the fold change of gene expression to the p-value of the corresponding change, nicely illustrate this observation (Supporting Information, Figure S3). The volcano plots indicate that only a few genes showed statistically significant expression changes at the lower origamicin concentration, whereas the higher concentration caused expression changes in a substantially larger number of genes.
In order to further study the origamicin-induced changes in mRNA levels, we removed low-intensity mRNA signals from the data set and also excluded mRNAs showing an expression change of less than two, and finally performed correction for the false discovery rate (FDR) using the Benjamin–Hochberg procedure.36 We found that 25 μM origamicin induced the differential expression of 165 genes of which 88 were upregulated, whereas the treatment with 50 μM origamicin increased the number of differentially expressed genes (DEGs) more than 13-fold to 2196 with a ratio of upregulated versus downregulated of nearly 2:1 (Figure 2A). Interestingly, almost all DEGs at the lower concentration of 25 μM origamicin are part of the DEGs at 50 μM origamicin (Figure 2B). A heat map of all the 157 DEGs shared between both treatment conditions revealed similarities in the gene expression pattern. In addition, the expression changes of these selected DEGs showed the same pattern as the hierarchical cluster analysis and the PCA of the global gene expression.
Figure 2.

DEGs induced by origamicin. (a) Numbers of DEGs [log2 FC > |1|, FDR (Benjamini–Hochberg) >0.05] upon treatment of E9 cells with 25 or 50 μM origamicin. (b) Venn diagram of DEGs from both treatments. (c) Heatmap and hierarchical clustering of DEGs found in both treatment conditions.
Origamicin Affects Cellular Redox Homeostasis and Stress Responses
We focused on the upregulated genes in the subsequent analysis. The lists of the top 25 upregulated genes in the 25 μM origamicin treatment (Table 1) and the ones in the 50 μM origamicin treatment (Table 2) show a similar pattern of upregulated genes.
Table 1. Top 25 Genes Upregulated Upon Treatment with 25 μM Origamicin.
| HGNC gene title | gene symbol | log2 FC | adj. p val |
|---|---|---|---|
| lactate dehydrogenase B | LDHB | 4.1614 | 0.0007 |
| 2.9311 | 0.0026 | ||
| prostaglandin reductase 1 | PTGR1 | 3.8695 | 0.0008 |
| 2.7341 | 0.0002 | ||
| 2.4668 | 0.0008 | ||
| 2.4607 | 0.0040 | ||
| TIMP metallopeptidase inhibitor 4 | TIMP4 | 2.6268 | 0.0020 |
| heme oxygenase 1 | HMOX1 | 2.4266 | 0.0014 |
| solute carrier family 22 member 18 | SLC22A18 | 2.2615 | 0.0028 |
| secreted phosphoprotein 1 | SPP1 | 2.1407 | 0.0020 |
| aldo-keto reductase family 1 member C2 | AKR1C2 | 2.0914 | 0.0009 |
| 1.9536 | 0.0014 | ||
| 1.9984 | 0.0071 | ||
| Kruppel-like factor 4 | KLF4 | 2.0655 | 0.0368 |
| SEL1L family member 3 | SEL1L3 | 1.9494 | 0.0024 |
| 1.5055 | 0.0020 | ||
| S100 calcium binding protein P | S100P | 1.9409 | 0.0005 |
| aldo-keto reductase family 1 member C1 | AKR1C1 | 1.8081 | 0.0020 |
| 1.7625 | 0.0016 | ||
| biliverdin reductase B | BLVRB | 1.7900 | 0.0008 |
| microtubule-associated protein 1A | MAP1A | 1.6942 | 0.0115 |
| tumor necrosis factor superfamily member 9 | TNFSF9 | 1.6447 | 0.0092 |
| coagulation factor II thrombin receptor like 2 | F2RL2 | 1.6105 | 0.0133 |
| complement C1s | C1S | 1.6080 | 0.0080 |
| 1.4128 | 0.0076 | ||
| cell cycle progression 1 | CCPG1 | 1.5958 | 0.0165 |
| 1.4043 | 0.0086 | ||
| 1.1542 | 0.0119 | ||
| 1.0644 | 0.0206 | ||
| small integral membrane protein 14 | SMIM14 | 1.5908 | 0.0016 |
| 1.3124 | 0.0117 | ||
| 1.1323 | 0.0042 | ||
| neuronal cell adhesion molecule | NRCAM | 1.5658 | 0.0096 |
| asparagine synthetase (glutamine-hydrolyzing) | ASNS | 1.5463 | 0.0007 |
| G protein subunit α L | GNAL | 1.5241 | 0.0225 |
| 1.2939 | 0.0133 | ||
| 1.0122 | 0.0206 | ||
| kynureninase | KYNU | 1.4894 | 0.0122 |
| uncharacterized LOC105377348 | LOC105377348 | 1.4676 | 0.0118 |
| cystatin A | CSTA | 1.4530 | 0.0042 |
| nuclear protein 1, transcriptional regulator | NUPR1 | 1.4346 | 0.0196 |
Table 2. Top 25 Genes Upregulated upon Treatment with 50 μM Origamicin.
| HGNC gene title | gene symbol | log2 FC | adj. p val |
|---|---|---|---|
| prostaglandin reductase 1 | PTGR1 | 5.9905 | 0.0000 |
| 4.3559 | 0.0000 | ||
| 3.9627 | 0.0000 | ||
| 3.7137 | 0.0000 | ||
| family with sequence similarity 129 member A | FAM129A | 5.0422 | 0.0002 |
| TIMP metallopeptidase inhibitor 4 | TIMP4 | 5.0177 | 0.0000 |
| heme oxygenase 1 | HMOX1 | 4.7228 | 0.0000 |
| nuclear protein 1, transcriptional regulator | NUPR1 | 4.5687 | 0.0000 |
| lactate dehydrogenase B | LDHB | 4.5585 | 0.0000 |
| 3.2221 | 0.0001 | ||
| coagulation factor II thrombin receptor like 2 | F2RL2 | 4.4213 | 0.0000 |
| Kruppel-like factor 4 | KLF4 | 3.9982 | 0.0003 |
| microtubule-associated protein 1A | MAP1A | 3.9962 | 0.0001 |
| hydroxysteroid 17-β dehydrogenase 14 | HSD17B14 | 3.8868 | 0.0000 |
| 3.8402 | 0.0000 | ||
| 3.7008 | 0.0000 | ||
| 3.5317 | 0.0002 | ||
| S100 calcium-binding protein P | S100P | 3.8383 | 0.0000 |
| prion protein | PRNP | 3.6150 | 0.0001 |
| oxidative stress-induced growth inhibitor 1 | OSGIN1 | 3.5927 | 0.0001 |
| solute carrier family 22 member 18 | SLC22A18 | 3.5476 | 0.0000 |
| poly(A)-binding protein cytoplasmic 1 like | PABPC1L | 3.5040 | 0.0001 |
| SEL1L family member 3 | SEL1L3 | 3.4650 | 0.0000 |
| 3.1717 | 0.0000 | ||
| tumor necrosis factor superfamily member 9 | TNFSF9 | 3.3751 | 0.0001 |
| solute carrier family 16 member 14 | SLC16A14 | 3.3624 | 0.0001 |
| biliverdin reductase B | BLVRB | 3.2877 | 0.0000 |
| growth differentiation factor 15 | GDF15 | 3.2408 | 0.0000 |
| cell cycle progression 1 | CCPG1 | 3.1793 | 0.0002 |
| 3.1367 | 0.0001 | ||
| 2.8356 | 0.0001 | ||
| 2.7128 | 0.0003 | ||
| 2.5277 | 0.0001 | ||
| cystathionine γ-lyase | CTH | 3.1504 | 0.0000 |
| 2.8430 | 0.0000 | ||
| aldo-keto reductase family 1 member C2 | AKR1C2 | 3.1462 | 0.0001 |
| 2.6607 | 0.0000 | ||
| 2.4592 | 0.0000 | ||
| spexin hormone | SPX | 3.0124 | 0.0000 |
| Metastasis-associated lung adenocarcinoma transcript 1 (nonprotein coding) | MALAT1 | 3.0044 | 0.0004 |
| 2.8543 | 0.0002 | ||
| 2.5933 | 0.0001 | ||
| 2.5909 | 0.0009 | ||
| 2.5584 | 0.0008 | ||
| 2.4961 | 0.0008 | ||
| 2.0539 | 0.0001 |
Interestingly, we noticed the enrichment of several genes that are involved in cellular stress responses, for example, PTGR1, HMOX1, and S100P. PTGR1 acts as an antioxidant enzyme37 and HMOX1 is a well-established component of the oxidative stress response.38 S100P was described as upregulated during the ER stress response and as being involved in apoptosis.39 To verify our initial conclusions, we performed pathway analysis and GO classification analysis on the identified DEGs. However, we were not able to find a clear pattern among the upregulated genes for the 25 μM origamicin treatment. As described above, the general mild effect of this treatment condition is likely to have only a small effect on the gene expression profile. The absence of an obvious pattern among the downregulated genes at this treatment condition supports this assumption (Supporting Information, Table S1). By contrast, pathway analysis of the identified upregulated genes in the 50 μM origamicin treatment regimen indicated that these genes are all involved in the UPR and in ER-related pathways (Figure 3A). Furthermore, GO classification analysis on the high concentration treatment regime shows an accumulation of genes involved in the ER and oxidative stress response, the unfolding protein response, and apoptosis-related processes (Figure 3B). Interestingly, a majority of all genes downregulated by 50 μM origamicin (Supporting Information, Table S2) are part of the cell cycle and cell division pathways (Supporting Information, Figure S4). This observation is in agreement with previous reports of initiation of cell cycle arrest in the G1 phase mediated by the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) receptor.40 The cell cycle arrest is caused by the phosphorylation of eIF2, which also causes translational attenuation to prevent the ER from further protein loading,41 consistent with the changes in mRNA levels that we observe related to cell cycle arrest.
Figure 3.

GO and pathway analyses. (a) Pathway enrichment analysis and (b) GO analysis classifying genes by the biological process (showing only the top 20 hits) were performed using ToppGene and selecting genes upregulated by >1.0 log2 fold in origamicin-treated E9 cells. The bars represent the ratio of identified genes versus all annotated genes in the corresponding pathway or biological classification, respectively. The red curve represents the adjusted p-value corrected for FDR using the procedure by Bonferroni.
Experimental Validation of the in Silico Analysis by qRT-PCR
We validated our computational analysis on selected candidate genes that were previously identified as upregulated in both treatment conditions and showed a broad range of expression changes (Supporting Information, Table S3). Therefore, we repeated the treatment of E9 cells with 25 μM origamicin, extracted the RNA, and performed qRT-PCR quantification of mRNAs for selected candidate genes: PTGR1,37 AKR1C1,42 HMOX1,38 BLVRB,43 S100P,39 CSTA,44 G6PD,45 CBR1,46 and NQO1.47 All of these genes showed significant upregulation as expected (Figure 4A). Moreover, the magnitude of the expression change was in agreement with our microarray data indicated by a Pearson’s product-moment correlation coefficient of r = 0.81 (Figure 4B).
Figure 4.
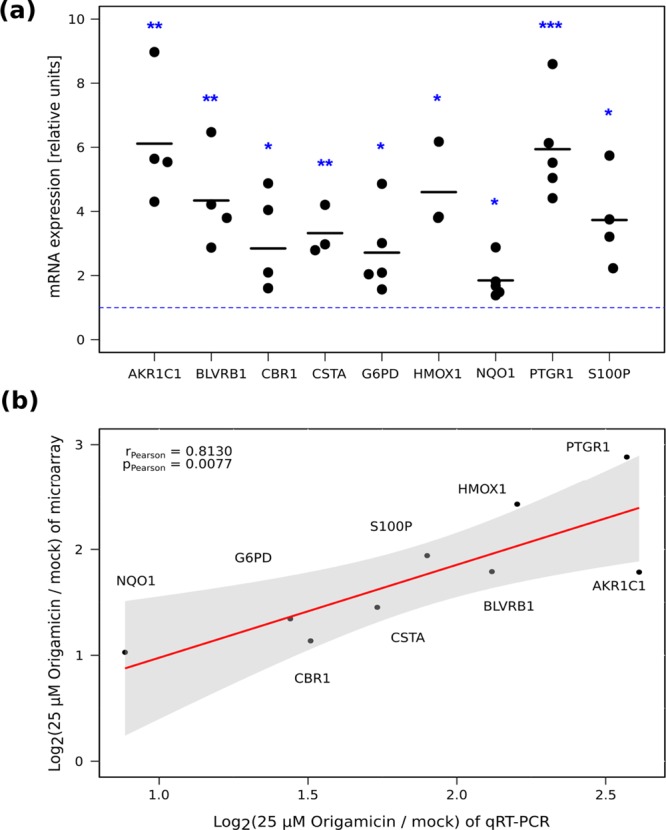
Quantification and validation of gene expression. (a) qRT-PCR validation of selected upregulated genes. The blue dotted line indicates the threshold for expression change. At least three independent experiments per gene were performed and all data are normalized to 18S. Statistical significance was evaluated by using an unpaired two-sample Student’s t-test (* for p-value < 0.05, ** for p-value < 0.01, and *** for p-value < 0.001). (b) Correlation analysis between genes that were identified via microarray and qRT-PCR validation using Pearson’s product-moment correlation coefficient. A linear regression curve is shown in red; the 95% confidence interval is depicted as a gray area.
It should be noted that several of these selected genes (e.g. HMOX1, S100P, CSTA) are known for their inhibitory role in HCV replication.48−50
Discussion
Here, we present new evidence for PDI inhibition by origamicin, a relatively uncharacterized inhibitor of PDIs, originally derived from the plant hormone ABA.32 Unlike ABA, this synthetic small molecule inhibits the activity of PDI.32 This indicates that the introduced chemical modifications converted ABA into a new type of PDI inhibitor.
To our knowledge, this is the first study focusing on the effect of PDI inhibition on cellular metabolism using comparative gene expression analysis. We identified a large number of DEGs upon origamicin treatment. Remarkably, all upregulated genes among these DEGs are linked to protein unfolding response, redox homeostasis, and ER stress. As PDI is directly involved in oxidative protein folding in the ER, we conclude that exposure of cells to this PDI inhibitor impairs protein folding in the ER lumen and induces ER stress (Figure 5). The cellular counter measure for ER stress is the UPR, which can induce apoptosis as a long-term response.51 The immediate response, however, is the PERK-mediated translation attenuation and cell cycle arrest as well as upregulation of proteins involved in assisting protein folding, that is, chaperones.52 A common example of such a chaperone in the context of ER stress is HSPA5 (also referred to as BiP or GRP78).53,54 We were not able to detect upregulation of HSPA5 in our study; however, it has been shown that hepatocytes Hsp40 and Hsp70 are potent modulators of the UPR.55 This is in agreement with our finding that several HSP40 or HSP70 chaperones are significantly upregulated (e.g., Hsp40B4, Hsp40B9, Hsp70A1, Hsp70B1).
Figure 5.

Suggested mechanism of inhibition of HCV.
HCV replication critically depends on optimal metabolic conditions that include an efficiently working protein biosynthesis machinery.56 In addition, HCV is heavily dependent on a proper ER environment because of the location of HCV’s viral factories in the ER and the dependency on the ER for viral egress.57,58 Hence, it is expected that perturbation of the ER homeostasis through PDI inhibition causes disruption of the HCV life cycle but the exact mechanism has to be elucidated in further studies.
Moreover, the formation of protein disulfide bonds in the glycoproteins E1 and E2 is a prerequisite for the assembly of infectious viral particles.31,59 Consequently, the failure of the disulfide bond formation system, mainly constituted by PDIs, might substantially affect the production of infectious viral particles. We are aware of the limitations of a subgenomic replicon system lacking these envelope proteins and, hence, HCV replicon models are not a suitable choice to study viral assembly and egress. Further research using infectious virus is needed to determine the antiviral effect of PDI inhibition on HCV’s life cycle. There are other (flavi)viruses—such as the dengue virus or Zika virus—which similar to HCV are also strongly dependent on ER homeostasis and PDI activity.60 Thus, they are interesting targets for further investigating the impact of PDI inhibition on viral life cycles.
Conclusions
Our findings have implications for the role of PDI in the life cycle of HCV and potentially for other members of the flaviviridae family. We conclude that chemical modification of naturally occurring molecules is a promising approach to provide new antivirals and novel drugs for other diseases, which are desperately needed.
Materials and Methods
Tissue Culture
Huh-7 human hepatoma cells, a kind gift from Dr. Lubica Supekova (The Scripps Research Institute, La Jolla, CA), harboring the HCV subgenomic replicon plasmid pFK-I389neo/luc/NS3-3′/5.1, obtained as previously described,33 were grown in Dulbecco’s modified Eagle medium (Gibco-Invitrogen, Burlington, ON, Canada) supplemented with 100 nmol nonessential amino acids/L (Gibco, Burlington, ON, Canada), 50 U penicillin/mL, 50 μg/mL streptomycin, 10% fetal bovine serum (Cansera International, Rexdale, ON; PAA Laboratories, Etobicoke, ON, Canada), and 250 μg/mL G418 Geneticin (Gibco, Burlington, ON, Canada). The pFK-I389neo/luc/NS3-3′/5.1 plasmid that contains HCV subgenomic replicons and expresses HCV nonstructural proteins (NS3 to NS5B) from the encephalomyocarditis virus internal ribosome entry site was utilized as previously described.32,60,61 Cells were either mock-treated with methanol or treated with origamicin (provided by Suzanne Abrams) with a final concentration of 25 or 50 μM, respectively, for 24 h and then harvested.
Total RNA Preparation for Microarray Experiments
Total RNA was prepared using the RNeasy Mini kit (Qiagen, Mississauga, Ontario) as per the manufacturer’s protocols. The RNA was quantified using the NanoDrop ND-1000 spectrophotometer, and the quality of the RNA was verified using the RNA 6000 Pico LabChip kit (Agilent Technologies, Mississauga, Ontario).
Gene Expression Analysis
Cellular RNA samples were hybridized to high-density oligonucleotide arrays (Affymetrix GeneChip HG-U133 Plus2). Primary image analysis was performed by using GeneChip version 3.1 (Affymetrix, Santa Clara, CA). The data sets were processed as previously described62 and subsequently converted to log2 transformed data. A first analysis for DEGs (log2 fold change cut-off > 2) was performed using a two-sided t-test for unpaired data with equal variances (p-value cut-off < 0.05). The R packages “genefilter” and “limma” were used to further analyze and filter the data set.63,64 Genes with expression values lower than the mean in at least three microarray samples were deemed “absent” and excluded from the subsequent analysis. DEGs were determined using the empirical Bayes approach in combination with multiple hypothesis testing based on the methods of Benjamini and Hochberg.36 An adjusted FDR p-value of <0.05 was set as the threshold for significance. Genes were annotated using the Affymetrix annotation files (HG-U133_Plus_2; release 36) in conjunction with GeneAnnot 2.2.65 Pathway and GO analyses were performed using the ToppGene Suite.66 Affymetrix raw data are provided in the Supporting Information.
Luciferase Reporter Assay and Total Protein Quantification
Huh-7 cells stably expressing HCV subgenomic replicons were treated with PDI inhibitors (i.e., 16F16 and securinine, Sigma-Aldrich) for 24 h as described previously.32 Briefly, cells were washed twice in PBS and lysed with cell culture lysis buffer (Promega, Madison, WI). Luciferase assay substrate was added, and expression of luciferase was determined by bioluminescence measurement on an Lmax luminometer (Molecular Devices Corporation, Sunnyvale, CA). Total protein concentration was determined by Bio-Rad DC protein assay (Bio-Rad, Mississauga, Canada) according to the manufacturer’s protocol. EC50 values were determined by plotting luminescence activity against the concentration values and fit using a four-parameter equation.
Quantitative Real-Time Polymerase Chain Reaction
RNA isolation and quantification was performed as described above. The RNA integrity was confirmed by electrophoresis on 1.0% agarose gel in 1 × TBE (Ambion). Reverse transcription of 500 ng of total RNA using the Superscript II RT kit (Invitrogen) according to the manufacturer’s protocols was performed on a T100 thermal cycler (Bio-Rad). Quantitative PCR was subsequently performed on a CFX thermal cycler (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad), as per the manufacturer’s protocol. Primer sequences are listed in the Supporting Information, Table S4. Fold changes in expression relative to mock-treated samples were calculated with the 2–ΔΔCq method using 18S rRNA levels for normalization.67 Data are presented as the mean of replicates. Statistical significance was evaluated using an unpaired two-tailed Student’s t-test. Correlation analysis was performed using Pearson’s product-moment correlation coefficient.
Statistical Analysis
We used the auxiliary R packages “gplots”,68 “plotrix”,69 “ggplot2”,70 and “Venn diagram”,71 and functions such as cor.test() for data analysis and drawing figures.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) and Natural Sciences and Engineering Research Council (NSERC) of Canada. D.Ö. was supported by a postdoctoral fellowship from the CIHR. The authors would like to thank Prof. Dr. Suzanne Abrams for providing origamicin and Sylvie Bélanger for technical assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02676.
HCV replication inhibition by PDI inhibitors, clustering of microarray data, volcano plots of gene expression, GO and pathway analyses, top 25 genes downregulated genes, selected genes, oligonucleotides used in this study (PDF)
Author Present Address
∥ Department of Pathology & Laboratory Medicine, Faculty of Medicine, BC Children’s Hospital, 4480 Oak Street, Vancouver, BC, Canada, V6H 3V4.
Author Contributions
D.Ö. contributed by conception and design, collection and assembly of data, data analysis and interpretation, and article writing. J.P.P. contributed by conception and design, and article writing. F.S. contributed by collection and assembly of data, and article writing. A.S., B.R., and A.F. contributed by collection and assembly of the data.
The authors declare no competing financial interest.
Supplementary Material
References
- Ron D.; Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Kaufman R. J. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 2006, 66, S102–S109. 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- Hebert D. N.; Molinari M. In and Out of the ER: Protein Folding, Quality Control, Degradation, and Related Human Diseases. Physiol. Rev. 2007, 87, 1377–1408. 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- Gardner B. M.; Pincus D.; Gotthardt K.; Gallagher C. M.; Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harbor Perspect. Biol. 2013, 5, a013169. 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay J. A. Pathways of oxidative damage. Annu. Rev. Microbiol. 2003, 57, 395–418. 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Suzuki Y. J.; Forman H. J.; Sevanian A. Oxidants as stimulators of signal transduction. Free Radical Biol. Med. 1997, 22, 269–285. 10.1016/S0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- Winter J.; Ilbert M.; Graf P. C. F.; Özcelik D.; Jakob U. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell 2008, 135, 691–701. 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J. D.; Kaufman R. J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword?. Antioxid. Redox Signaling 2007, 9, 2277–2294. 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Nakamura H.; Nakamura K.; Yodoi J. Redox regulation of cellular activation. Annu. Rev. Immunol. 1997, 15, 351–369. 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- Pierce G. B.; Parchment R. E.; Lewellyn A. L. Hydrogen peroxide as a mediator of programmed cell death in the blastocyst. Differentiation 1991, 46, 181–186. 10.1111/j.1432-0436.1991.tb00880.x. [DOI] [PubMed] [Google Scholar]
- Ali Khan H.; Mutus B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2, 70. 10.3389/fchem.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier C. S.; Kaiser C. A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 549–556. 10.1016/j.bbamcr.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Benham A. M.; van Lith M.; Sitia R.; Braakman I. Ero1-PDI interactions, the response to redox flux and the implications for disulfide bond formation in the mammalian endoplasmic reticulum. Philos. Trans. R. Soc., B 2013, 368, 20110403. 10.1098/rstb.2011.0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B. P.; Weissman J. S. The FAD- and O2-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 2002, 10, 983–994. 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- Gross E.; et al. Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 299–304. 10.1073/pnas.0506448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatahet F.; Ruddock L. W. Protein Disulfide Isomerase: A Critical Evaluation of Its Function in Disulfide Bond Formation. Antioxid. Redox Signaling 2009, 11, 2807–2850. 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- Tian G.; Xiang S.; Noiva R.; Lennarz W. J.; Schindelin H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 2006, 124, 61–73. 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- Tian G.; et al. The catalytic activity of protein-disulfide isomerase requires a conformationally flexible molecule. J. Biol. Chem. 2008, 283, 33630–33640. 10.1074/jbc.m806026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V. D.; et al. Alternative Conformations of the x Region of Human Protein Disulphide-Isomerase Modulate Exposure of the Substrate Binding b’ Domain. J. Mol. Biol. 2008, 383, 1144–1155. 10.1016/j.jmb.2008.08.085. [DOI] [PubMed] [Google Scholar]
- Wang C.; et al. Plasticity of Human Protein Disulfide Isomerase. J. Biol. Chem. 2010, 285, 26788–26797. 10.1074/jbc.m110.107839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby N. J.; Creighton T. E. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry 1995, 34, 11725–11735. 10.1021/bi00037a009. [DOI] [PubMed] [Google Scholar]
- Wilson R.; Lees J. F.; Bulleid N. J. Protein disulfide isomerase acts as a molecular chaperone during the assembly of procollagen. J. Biol. Chem. 1998, 273, 9637–9643. 10.1074/jbc.273.16.9637. [DOI] [PubMed] [Google Scholar]
- Quan H.; Fan G.; Wang C.-c. Independence of the Chaperone Activity of Protein Disulfide Isomerase from Its Thioredoxin-like Active Site. J. Biol. Chem. 1995, 270, 17078–17080. 10.1074/jbc.270.29.17078. [DOI] [PubMed] [Google Scholar]
- Wang C.; et al. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signaling 2013, 19, 36–45. 10.1089/ars.2012.4630. [DOI] [PubMed] [Google Scholar]
- Ngiam C.; Jeenes D. J.; Punt P. J.; Van Den Hondel C. A. M. J. J.; Archer D. B. Characterization of a Foldase, Protein Disulfide Isomerase A, in the Protein Secretory Pathway of Aspergillus niger. Appl. Environ. Microbiol. 2000, 66, 775–782. 10.1128/aem.66.2.775-782.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton J. T.; Foung S.; Muerhoff A. S.; Bukh J.; Simmonds P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011, 92, 233–246. 10.1099/vir.0.027490-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed K. E.; Rice C. M. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 2000, 242, 55–84. 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- Hsu N.-Y.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass V.; Gosert R.; Moradpour D. Investigation of the hepatitis C virus replication complex. Methods Mol. Biol. 2009, 510, 195–209. 10.1007/978-1-59745-394-3_15. [DOI] [PubMed] [Google Scholar]
- Dubuisson J. Hepatitis C virus proteins. World J. Gastroenterol. 2007, 13, 2406. 10.3748/wjg.v13.i17.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson J.; Rice C. M. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol. 1996, 70, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]; https://jvi.asm.org/content/70/2/778.long.
- Rakic B.; et al. A Small-Molecule Probe for Hepatitis C Virus Replication that Blocks Protein Folding. Chem. Biol. 2006, 13, 1051–1060. 10.1016/j.chembiol.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann V.; et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- Hoffstrom B. G.; Kaplan A.; Letso R.; Schmid R. S.; Turmel G. J.; Lo D. C.; Stockwell B. R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Am. Stat. Assoc. 1995, 57, 289–300. [Google Scholar]; http://www.jstor.org/stable/2346101
- Na H.-K.; et al. Tobacco smoking-response genes in blood and buccal cells. Toxicol. Lett. 2015, 232, 429–437. 10.1016/j.toxlet.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Poss K. D.; Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 10919–10924. 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba T.; et al. Up-regulation of S100P expression by non-steroidal anti-inflammatory drugs and its role in anti-tumorigenic effects. J. Biol. Chem. 2009, 284, 4158–4167. 10.1074/jbc.m806051200. [DOI] [PubMed] [Google Scholar]
- Ozcan U.; et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Bernales S.; Papa F. R.; Walter P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- Burczynski M. E.; Sridhar G. R.; Palackal N. T.; Penning T. M. The Reactive Oxygen Species- and Michael Acceptor-inducible Human Aldo-Keto Reductase AKR1C1 Reduces the α,β-Unsaturated Aldehyde 4-Hydroxy-2-nonenal to 1,4-Dihydroxy-2-nonene. J. Biol. Chem. 2001, 276, 2890–2897. 10.1074/jbc.m006655200. [DOI] [PubMed] [Google Scholar]
- Wu S.; et al. BLVRB redox mutation defines heme degradation in a metabolic pathway of enhanced thrombopoiesis in humans. Blood 2016, 128, 699–709. 10.1182/blood-2016-02-696997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerink W. M. A.; Stevenson J. C. R.; Horbach G. J.; Schoonen W. G. E. J. The development of RAD51C, Cystatin A, p53 and Nrf2 luciferase-reporter assays in metabolically competent HepG2 cells for the assessment of mechanism-based genotoxicity and of oxidative stress in the early research phase of drug development. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2010, 696, 21–40. 10.1016/j.mrgentox.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Ho H.-y.; Cheng M.-l.; Chiu D. T.-y. Glucose-6-phosphate dehydrogenase—from oxidative stress to cellular functions and degenerative diseases. Redox Rep. 2007, 12, 109–118. 10.1179/135100007x200209. [DOI] [PubMed] [Google Scholar]
- Kim Y. N.; et al. Neuroprotective effects of PEP-1-carbonyl reductase 1 against oxidative-stress-induced ischemic neuronal cell damage. Free Radical Biol. Med. 2014, 69, 181–196. 10.1016/j.freeradbiomed.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova A. T.; Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y.; Zheng J.; Lambrecht R. W.; Bonkovsky H. L. Reciprocal effects of micro-RNA-122 on expression of heme oxygenase-1 and hepatitis C virus genes in human hepatocytes. Gastroenterology 2007, 133, 1166–1174. 10.1053/j.gastro.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani J.; et al. Ca2+/S100 proteins regulate HCV virus NS5A-FKBP8/FKBP38 interaction and HCV virus RNA replication. Liver Int. 2013, 33, 1008–1018. 10.1111/liv.12151. [DOI] [PubMed] [Google Scholar]
- Gerold G.; et al. Quantitative Proteomics Identifies Serum Response Factor Binding Protein 1 as a Host Factor for Hepatitis C Virus Entry. Cell Rep. 2015, 12, 864–878. 10.1016/j.celrep.2015.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R.; Reed J. C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman R. J. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Hendershot L. M.; Valentine V. A.; Lee A. S.; Morris S. W.; Shapiro D. N. Localization of the gene encoding human BiP/GRP78, the endoplasmic reticulum cognate of the HSP70 family, to chromosome 9q34. Genomics 1994, 20, 281–284. 10.1006/geno.1994.1166. [DOI] [PubMed] [Google Scholar]
- Mayer M. P.; Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker S. A.; Robinson P.; Abel A.; Tweardy D. J. Modulation of the unfolded protein response during hepatocyte and cardiomyocyte apoptosis in trauma/hemorrhagic shock. Sci. Rep. 2013, 3, 1187. 10.1038/srep01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393–403. 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- Inoue T.; Tsai B. How Viruses Use the Endoplasmic Reticulum. Cold Spring Harbor Perspect. Biol. 2013, 5, a013250. 10.1101/cshperspect.a013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahid A.; et al. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J. Virol. 2013, 87, 1605–1617. 10.1128/jvi.02659-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez A. B.; Escribano-Romero E.; Merino-Ramos T.; Saiz J. C.; Martín-Acebes M. A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. 10.3389/fmicb.2014.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagan S. M.; et al. The influence of cholesterol and lipid metabolism on host cell structure and hepatitis C virus replication. Biochem. Cell Biol. 2006, 84, 67–79. 10.1139/o05-149. [DOI] [PubMed] [Google Scholar]
- Rakic B.; et al. Peroxisome Proliferator-Activated Receptor α Antagonism Inhibits Hepatitis C Virus Replication. Chem. Biol. 2006, 13, 23–30. 10.1016/j.chembiol.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Su A. I.; et al. Nonlinear partial differential equations and applications: Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 15669–15674. 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E.; et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.; Carey V.; Huber W.; Hahne F.. Genefilter Methods for Filtering Genes From High-throughput Experiments. R Package, version 1.56.0, 2016, 10.18129/B9.bioc.genefilter. [DOI]
- Ferrari F.; et al. Novel definition files for human GeneChips based on GeneAnnot. BMC Bioinf. 2007, 8, 446. 10.1186/1471-2105-8-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Bardes E. E.; Aronow B. J.; Jegga A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J.; Schmittgen T. D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Warnes G. R.; et al. gplots: Various R programming tools for plotting data. R Package, version 2.11.3, 2016.
- Lemon J. Plotrix: a package in the red light district of R. R. News 2006, 6, 8–12. [Google Scholar]
- Wickham H.ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, 2009. [Google Scholar]
- Chen H.; Boutros P. C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinf. 2011, 12, 35. 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.