

Abstract
Exendin-4 is a glucagon-like peptide-1 (GLP-1) receptor agonist and potent insulinotropic agent for type 2 diabetes patients; however, its therapeutic utility is limited due to the frequent injections required. Long-acting agonists reduce the number of injections, but they can compromise potency. In this study, chondroitin sulfate-g-glycocholic acid-coated and Exendin-4 (Ex-4)-loaded liposomes (EL-CSG) were prepared for oral administration of Ex-4. The Ex-4 loading efficiency was 77% and the loading content in the nanoparticles was 1 wt-%. In rat models, a single oral dose (200 μg/kg) of EL-CSG showed a relative oral bioavailability of 19.5%, compared with subcutaneous administration (20 μg/kg), and sustained pharmacokinetics for up to 72 h. The overall long-term pharmacodynamic effects, assessed by hemoglobin A1c (HbA1c), body weight, and blood lipid concentrations, of daily oral EL-CSG (300 μg/kg) for four weeks were equivalent to or better than daily subcutaneous injections of free Ex-4 solution (20 μg/kg).
Keywords: Exendin-4, oral drug delivery, bile acid, liposome, liposome intestinal absorption, Type 2 diabetes
1. Introduction
Type 2 diabetes mellitus (T2DM) with impaired insulin secretion is in part caused by decreased secretion of glucagon-like peptide-1 (GLP-1) [1]. Exendin-4 (Ex-4) is a GLP-1 receptor agonist comprised of 39 amino acids that has been used in the treatment of T2DM. Ex-4 has a significantly longer half-life in vivo compared to that of endogenous GLP-1 [2,3], while possessing GLP-1-like glucoregulatory activities, such as glucose-dependent enhanced insulin secretion, suppressed glucagon secretion, reduced gastric mobility, and decreased food intake [4,5]. Exenatide, a synthetic version of Ex-4, has been approved as an adjunctive therapy for patients with T2DM who fail to achieve glycemic control with oral antidiabetic agents [4,6]. Ex-4 has been widely used for T2DM and has great potential as a future therapeutic for neurodegenerative diseases, such as Alzheimer’s and Parkinson’s disease, which are linked to insulin resistance in the brain [7,8]. However, patients are administered frequent subcutaneous (SC) injections, resulting in high costs and poor patient compliance in addition to side effects, such as injection site infection [6,9]. Despite injectable long-acting GLP-1 receptor agonists, peroral administration of such active pharmaceutical ingredients may represent a more convenient and comfortable alternative for diabetic patients [10].
Various attempts have been made to develop oral delivery systems for Ex-4 [11–13]. However, the oral bioavailability, particularly in humans, of protein/peptide drugs such as Ex-4 is limited due to enzymatic inactivation and absorption barriers present in the gastrointestinal tract (GIT) [10]. These typically include physical barriers of viscous mucous layers and tight junctions of aligned enterocytes in the GIT, chemical barriers of low stomach pH, and biological barriers such as enzymatic degradation. Overcoming these problems is essential for enhancing absorption of orally administered peptide-based drugs [10,14]. Consequently, the development of a delivery vehicle for oral administration of protein/peptide drugs presents an interesting challenge. Recent reports have indicated promising oral absorption results of Fc-modified poly(lactic-co-glycolic acid) nanoparticles loaded with Ex-4 for targeting Fc receptors [15]. However, the binding capacity in adults of Fc receptors in the GIT is low and differences in the intestinal environment between rodents and humans are likely to confound extrapolation of animal model data to the clinic. Since it seems that problems still remain in clinical applications, we focused on the enterohepatic circulation of bile acid and started developing an oral drug delivery carrier modified with bile acid.
Enterohepatic circulation of bile acids is composed of two major processes: secretion from the liver and absorption in the intestines. Bile acids are secreted into the duodenum, where they emulsify water-insoluble nutrients to facilitate absorption [16]. In the distal small intestine, bile acids are absorbed by both passive and active mechanisms. Passive absorption occurs in the proximal regions of the small intestine and colon, while active absorption is restricted to the ileum [17]. In humans and other vertebrates examined to date, the ileal epithelium has developed efficient transport mechanisms for the active reclamation of bile acids [18]. This enterohepatic circulation is an efficient process; only 5–10% of the intestinal bile acids are eliminated in the feces [19] and as much as 30 g/day and 20 mg/day of intestinal bile acids are recycled in humans [20] and mice [21], respectively. This capacity is several orders greater in magnitude than the reabsorption capacity of gastric leptin (its secreted amount is unknown, but very limited [22]) or vitamin B12 (cyanocobalamin, 2–4 μg/day in humans [23]) mediated by oral absorption. A recent study revealed that low-molecular-weight heparin modified with deoxycholic acid enters distal ileocytes using the apical sodium-dependent bile acid transporter (ASBT) [24]. The observed entry mechanism of this modified heparin was clearly distinguished from the pumping mechanism for small-molecule bile acids, of which details were recently elucidated. While the specific entry and transport mechanisms are largely unknown and widely open for investigation, it was reported that nanoparticles, self-assembled from heparin-taurocholic acid-docetaxel conjugates, were orally absorbed to elicit an antitumor effect [25]. It was also shown that intact nanoparticles were orally absorbed, using anionic probe polystyrene nanoparticles with glycocholic acid (GCA)-decorated surfaces [26].
In addition, micro- to nano-sized drug delivery carriers are available, each having distinct pharmaceutical characteristics. For micro-sized formulations, high encapsulation rates are expected due to their large physical dimensions, and their release behavior can be characterized as slow kinetics [27]. For nano-sized formulations, the encapsulation rate is generally low, and their release behavior is comparatively fast. Various nano-sized pharmaceutical formulations have been developed, and preparations to improve absorbability have been studied extensively [28]. When considering GIT transit time in humans, especially through the small intestine, a sustained release of ~24 h deemed sufficient for a formulation to act as a drug carrier. One absorption mechanism is the transcellular pathway that involves either passive transport or endocytosis of nanoparticles through epithelial cells. The cut-off size for nanoparticles to be taken up by epithelial cells has been reported as 300 nm [29]; maintaining nanoparticles below this cut-off size is expected to improve intestinal absorption.
In the present study, we provide new evidence for enhanced oral delivery of Ex-4 using liposome-based nanocarriers and surface GCA. GCA was selected because it is the richest component in human bile salts [30] with a relatively low logP value [31], which allows more exposure to the water phase, and requires a relatively straightforward conjugation chemistry that simplifies analysis. Ex-4-loaded cationic liposomes (EL) comprised of 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were fabricated by conventional methods. The resulting liposomes were further coated with chondroitin sulfate to yield EL-CS or GCA-conjugated chondroitin sulfate to yield EL-CSG. Using a T2DM rat model, the biological efficacy of orally administered EL-CSG was compared with that of EL-CS, diabetes and non-diabetes controls, and SC-injected free solution by using oral glucose tolerance tests (OGTTs) and analyzing the pharmacodynamic effects after 4 weeks of treatment.
2. Materials and Methods
2.1. Materials
GCA, dimethylformamide, ethylenediamine (EDA), dicyclohexylcarbodiimide, N-hydroxysuccinimide, ethyl acetate, acetonitrile, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride, diethyl ether, streptozocin, and 2-ethanesulfonic acid (MES) buffer were purchased from Sigma-Aldrich (St. Louis, MO, USA). Potassium sodium-encapsulated Preyssler-type phosphotungstate was purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Exenatide (BYETTA®) was purchased from AstraZeneca (Cambridge, United Kingdom). DOPC was purchased from Echelon Bioscience (Salt Lake City, UT, USA) and DOTAP was purchased from CordenPharma (Boulder, CO, USA). CS (molecular weight approximately 25 kDa, injectable grade) was obtained from Yantai Dongcheng Biochemicals Co., Ltd. (Shandong, China). Fasted State Simulated Gastric Fluid (FaSSGF, FFF01) and Fasted State Simulated Intestinal Fluid (FaSSIF-V2, V2FAS01) were purchased from Biorelevant.com Ltd. (Surrey, United Kingdom). Dialysis membrane (Biotech Cellulose Ester Membrane, molecular weight cut-off 50 kDa) was purchased from Repligen (Waltham, MA, USA). High fat diet (TD 96132) was purchased from Envigo (Cambridgeshire, United Kingdom). The Diazyme Direct HbA1c Assay kit (DZ168A-K) was purchased from Diazyme Laboratories (San Diego, CA, USA). The Cholesterol Assay Kit (ab65390) was purchased from Abcam (Cambridge, MA, USA).
2.2. Synthesis of glycocholic acid -conjugated chondroitin sulfate (CSG)
The procedure for the synthesis of CSG is presented in Fig. S1. To synthesize GCA-EDA, GCA (644 µmol), dicyclohexylcarbodiimide (838 µmol), N-hydroxysuccinimide (838 µmol), and EDA (32.2 mmol) were dissolved in 3 mL of dimethylformamide. The reaction mixture was stirred for 24 h at room temperature. The reacted solution was filtered and precipitated in ethyl acetate. The collected pellet was dried in vacuo for 24 h. The dried powder was dissolved in 10 mL of distilled water, filtered, and lyophilized [32]. To synthesize CSG, the GCA-EDA (425 µmol), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (550 µmol), N-hydroxysuccinimide (550 µmol), and CS (8 µmol) were dissolved in 3 mL of 0.1 M MES buffer, pH 6.0. The reaction mixture was stirred for 24 h at room temperature. The reacted solution was filtered and precipitated three times in 60% cold ethanol. The collected pellet was dried in vacuo for 24 h. The synthesized CSG was characterized using a Mercury 400 1H NMR spectrophotometer (Fig. S1).
2.3. Preparation of EL
EL were prepared as outlined in a previous report [33]. In brief, DOPC (38 mg) and DOTAP (39 mg) were dissolved in 15 mL of diethyl ether and 6 mL Ex-4 solution (0.5 mg/mL) was added. To produce an emulsified mixture, the solution was placed in a VWR Scientific Aquasonic 75T Ultrasonic cleaner for 5 min. After sonicating the flask, the solvent was removed by rotary evaporation at 60 °C. The obtained EL was then purified via dialysis (Spectra/Por® molecular weight cut-off 50 kDa membrane) against distilled water for 24 h to remove unloaded Ex-4. The purified liposome solution was passed through a 200-nm filter (Whatman Nuclepore, Maidstone, UK) at 60 °C using a membrane extruder LiposomeFast® LF-50 (Avestin, Inc., ON, Canada).
The Ex-4 loading efficiency was estimated using reversed-phase high-performance liquid chromatography (HPLC) [13,33]. Specifically, the DIONEX UltiMate 3000 serial HPLC system consisted of a quaternary pump, degasser, autosampler, column oven, UV detector, and HP ChemStation Software (all from Thermo Scientific, USA). The chromatographic conditions were optimized with a Kromasil® C4 column (4.6 mm × 250 mm, i.d., 5 μm particles, AkzoNobel, USA). The column temperature was maintained at 40 °C. Mobile phase A was potassium dihydrogen phosphate (KH2PO4, 20 mmol/L) adjusted to pH 2.5 with phosphoric acid, and mobile phase B was acetonitrile. The mobile phase was filtered through a 0.45-μm membrane filter and degassed with an ultrasonic water bath prior to use. The following gradient conditions were used for Ex-4 determination: 30–45% mobile phase A over 30 min and then returned to 30% mobile phase A over 10 min. The flow rate was 1.0 mL/min and the injection volume was 50 μL. The UV signal was detected at 214 nm because the absorption of the amide group was more sensitive than that of Phe, Tyr, and Trp at 280 nm.
The Ex-4 loading efficiency and loading capacity in the nanoparticles were calculated using the equations (1) and (2) listed below [34,35].
| (1) |
| (2) |
The EL-CSG and EL-CS were prepared by electrostatic interaction by mixing cationic liposomes with anionic CS or CSG complexes. In brief, 1 mL of EL was mixed with 1.0 mL of CS or CSG (10 mg/mL) and then stirred for 2 min.
2.4. Size and zeta-potential measurements
The size and zeta-potential of each liposomal formulation were measured using dynamic light scattering (Zetasizer Nano ZS, Malvern Instruments, USA). Prior to measurement, the liposome solution was stabilized at room temperature for 2 h. The average of three measurements was used.
2.5. In vitro Ex-4 release study
In vitro Ex-4 release was measured by a dialysis method [36]. In brief, the EL, EL-CS, and EL-CSG were dispersed in distilled water. The suspension was placed into a dialysis membrane bag (Spectra/Por® molecular weight cut-off 50 kDa). The bag was sealed and subsequently immersed in a vial containing fresh release medium, such as Simulated Intestinal Fluid, pH 6.81; USP 26 [37]. The release of Ex-4 from the liposomes was performed with mechanical mixing (100 revolutions/min) at 37 °C. Two hundred microliters of the outer solution was sampled and replaced with fresh buffer at predetermined time intervals (0–48 h). The Ex-4 concentration in each sample (n=3) was measured by HPLC, as indicated in section 2.3.
2.6. Particle degradation in solutions, including simulated GIT fluids
To prepare freeze-dried EL-CSG (from 1 mL), 0.8 mL trehalose solution (50 mg/mL) was used as a cryoprotectant during the freeze-drying process [38,39]. To evaluate the particle stability of EL-CSG, 1.0 mL of test media (distilled water, phosphate-buffered saline (PBS, pH 7.4), FaSSGF, or FaSSIF-V2) was mixed with the freeze-dried powder. After reconstitution, the nanoparticles were characterized using the Zetasizer Nano ZS.
2.7. Ex vivo nanoparticle transport efficacy in the small intestine
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Utah. To perform the everted gut sac (EGS) assay, the small intestines were isolated from 200–225 g male Sprague-Dawley rats (Charles River Laboratories International, Inc.). The distal ileum was isolated at a length of 5 cm and each segment was inverted lumen-side out. After one end of the segment was tied, the inner space was filled with oxygenated culture medium (800 µL) and the other end was then tied to make a 4-cm-long EGS. The EGS was placed in Krebs-Ringer solution (J67591, Alfa Aesar, USA) containing 0.05 mg/mL free Ex-4 (BYETTA® appropriately diluted with PBS), EL, EL-CS, or EL-CSG at 37 °C; the amount of Ex-4 transported into each sac was measured after 90 min [40]. The amount of Ex-4 transported per unit tissue area (calculated by tissue dimension and not surface area expanded by villi and microvilli) was analyzed by an Ex-4 ELISA kit (EK-070–94, Phoenix Pharmaceuticals Inc., Mountain View, CA, USA). Cellular integrity and tissue viability over 90 min were confirmed by monitoring lactate dehydrogenase (LDH) concentration per unit area of tissue [41].
2.8. In vivo blood Ex-4 level and bioavailability
To evaluate the blood insulin level and bioavailability of EL-CS and EL-CSG, 200–225 g male Sprague-Dawley rats (Charles River Laboratories International, Inc.) were randomly divided into three groups (n=6 per group). The rats were water fasted overnight prior to nanoparticle administration and the fasting continued for 12 h thereafter. The following formulations were administered to rats individually: oral EL-CS (200 μg/kg), oral EL-CSG (200 μg/kg), and SC injection of free Ex-4 solution (20 μg/kg). Blood samples (200 µL) were collected from the jugular vein, centrifuged (1,800 ×g, 4 °C, 10 min), and subsequently quantified for plasma Ex-4 concentration using the Ex-4 ELISA kit. Before in vivo experiments, we examined whether the applied ELISA kit shows selectivity for exendin-4 and confirmed that there is no cross-reactivity between Ex-4 and GLP-1. The relative bioavailability (BAR) was calculated using equation (3) [42], where AUC is the area under the plasma concentration vs. time curve.
| (3) |
2.9. In vivo blood glucose level
To establish the T2DM model, 150–170 g male Sprague-Dawley rats were divided into five groups (n=6 per group). The rats were fed a high fat diet consisting of 18 %kcal protein, 40 %kcal carbohydrate, and 40 %kcal fat (TD 96132, Envigo) for 2 weeks. After 2 weeks, the rats were subjected to an overnight fast and injected SC with streptozocin (35 mg/kg body weight in 0.1 M citrate buffer, pH 4.5). The rats had free access to food and water after the streptozocin injection. After a 12-h fast, the rats with fasting glucose levels >200 mg/dL were considered diabetic and were used for further study [43]. The biological efficacy of orally administered EL-CSG was compared with that of EL-CS, and SC-injected free Ex-4 solution, and with diabetes control (DC) and non-diabetic control (NC, normal control) rats, using an OGTT as previously reported [44,45]. Specifically, the rats were fasted overnight and then administered enteric-coated capsules containing EL-CS (300 μg/kg) or EL-CSG (300 μg/kg), or they were injected SC with free Ex-4 solution (20 μg/kg). To initiate the OGTT, an oral dose of glucose (1 g/kg) was administered to the animals 2 h after receiving the Ex-4 treatment in order to imitate the postprandial state, which is characterized by a rapid and significant increase in blood glucose after a meal [46]. Blood samples were collected from the tail veins of the test rats, and their blood glucose levels were evaluated using a glucose meter (Contour Next EZ, Bayer).
2.10. In vivo pharmacodynamic study
To evaluate therapeutic effects, the T2DM rats were randomly divided into four groups (n=6 per group). Over a period of 4 weeks, free Ex-4 solution (20 µg/kg, daily) was administered by SC injection, while EL-CS (300 µg/kg, daily) and EL-CSG (300 µg/kg, daily) were administered by gavage at 6 PM, one hour prior to the first meal of the day. After 4 weeks administration, blood samples (200 µL) were collected from the jugular vein, centrifuged (1,800 ×g, 4 °C, 10 min), and the content of Ex-4 (EK-070–94, Phoenix Pharmaceuticals Inc.), hemoglobin A1c (HbA1c, Diazyme Lab., San Diego, CA, USA), total cholesterol (T-Chol, ab65390, Abcam), high-density lipoprotein (HDL, ab65390, Abcam), and low-density lipoprotein (LDL, ab65390, Abcam) in the plasma was quantified using each assay kit.
2.11. Statistical analysis
Each result from the in vitro studies is expressed as the mean ± standard deviation (SD). Each result from the ex vivo and in vivo studies is expressed as the mean ± standard error of the mean (SEM). Differences between the values were assessed using Student’s t-tests.
3. Results and Discussion
3.1. Synthesis and characterization of liposome coating materials
In order to stabilize cationic liposomes in the GIT environment by anionic polymer coating and provide a surface to enhance intestinal absorption, we synthesized CSG. The procedure for the synthesis of CSG is presented in Fig. S1. The carboxyl group of GCA was modified to yield a primary amine by reaction with EDA. The modified GCA was coupled to the carboxylic groups of CS through a conventional carbodiimide coupling reaction. Successful synthesis of CSG was confirmed by the appearance of the −CH3 peaks of GCA at 0.5–1 ppm using 1H-NMR. The degree of GCA conjugation on CS was estimated by comparing the intensity of the −CH3 peak of CS at 1.8 ppm to that of GCA at 0.5–1 ppm. The degree of GCA conjugation was determined to be ~18 units of GCA per 100 units of CS. Varying amount of CSG was also added to Caco-2 cell culture medium to optimize its concentration; in this cell culture model, the non-cytotoxic concentration was estimated as 1 mg/mL (Fig. S2).
3.2. Preparation and characterization of EL-CSG
Three liposomal formulations, shown in Table 1, were prepared according to the method outlined in a previous report [47]. The conventional liposomes were prepared with a Reverse-Phase Evaporation technique, using DOPC and DOTAP. This method has been reported to be capable of encapsulating relatively water soluble drugs in the inner aqueous phase of liposomes at high concentrations [48]. The size, size distribution, and zeta potential of the Ex-4 loaded liposomes are summarized in Table 1. The average size of the EL-CS (205 nm) and EL-CSG (230 nm) was larger than that of the corresponding liposomes without CS or CSG coating (154 nm). The size changes were more than anticipated, which is probably due to alterations in electrostatic repulsion forces and surface properties by GCA modification. The zeta potential of the EL-CS and EL-CSG changed from cationic (+57 mV) to anionic as the Ex-4-loaded cationic liposomes were coated with the anionic CS or CSG (−33 mV and −31 mV, respectively). The Ex-4 loading efficiencies of EL, EL-CS, and EL-CSG were 77.2, 72.8, and 74.2%, respectively, and the content of EL-CSG in the nanoparticles was 1.2 wt-%, which is high enough to meet the therapeutic window of extremely potent Ex-4 (minimum therapeutic concentration (MTC) of Ex-4 is 50–350 pg/mL in humans [49], 5–10 μg/SC injection) after oral administration. Due to a large water phase volume used in this experiment and electrostatic interactions between cationic lipid and anionic Ex-4, it is assumed that Ex-4 is located on the inner and outer surfaces of the resulting liposomes. The low loading content of Ex-4 in the liposomes still resulted in a strong cationic surface for polymer coating. The formulations were immediately used for oral uptake studies or freeze-dried in the presence of trehalose for storage. When the lyophilized powder was reconstituted, the particle properties were equivalent to those of the fresh formulations.
Table 1.
Characteristics of CS and CSG-coated Ex-4-loaded liposomes
| Formulation | Ex-4 (mg) | Liposome composition | Coating materials | Ex-4 loading efficiency (%)a | Ex-4 content (%)a | Size (nm)a | Zeta potential (mV)a | ||
|---|---|---|---|---|---|---|---|---|---|
| DOPC (mg) | DOTAP (mg) | CS (mg) | CSG (mg) | ||||||
| Blank liposome | - | 38 | 39 | - | - | - | - | 136 ± 1.8 | +61 ± 0.4 |
| EL | 3 | 38 | 39 | - | - | 77.2 ± 3.4 | 2.9 ± 0.1 | 154 ± 0.2 | +57 ± 0.6 |
| EL-CS | 3 | 38 | 39 | 100 | - | 72.8 ± 2.2 | 1.2 ± 0.1 | 205 ± 2.6 | − 33 ± 0.2 |
| EL-CSG | 3 | 38 | 39 | - | 100 | 74.2 ± 2.5 | 1.2 ± 0.1 | 229 ± 4.0 | − 31 ± 0.2 |
Each value represents the mean ± SD, n=3
The size and shape of blank liposomes, EL, EL-CS, and EL-CSG in distilled water was measured by transmission electron microscopy after negative staining, such as potassium sodium-encapsulated Preyssler-type phosphotungstate (Fig. 1c–f). Negative staining usually retains the concavo-convex structure of the surface, which often collapses during drying, and allows observation of the liposome multilayer structure [50]. Compared with blank liposomes, these nanostructures were also observed in a previous report [51] where cationic liposomes were prepared with a single-stranded oligodeoxynucleotide. The addition of oligodeoxynucleotide to the cationic liposomes induced both liposome aggregation and the formation of a novel condensed lamellar phase. This phase was proposed to be stabilized by anionic single-stranded oligodeoxynucleotide molecules intercalated between cationic bilayers. The layered structure observed in EL suggests that the interaction between the cationic surface of the liposomes and Ex-4 (anionic single-chain peptide) in the multilayered structure allows high encapsulation efficiency of Ex-4 into the liposomes.
Fig. 1.
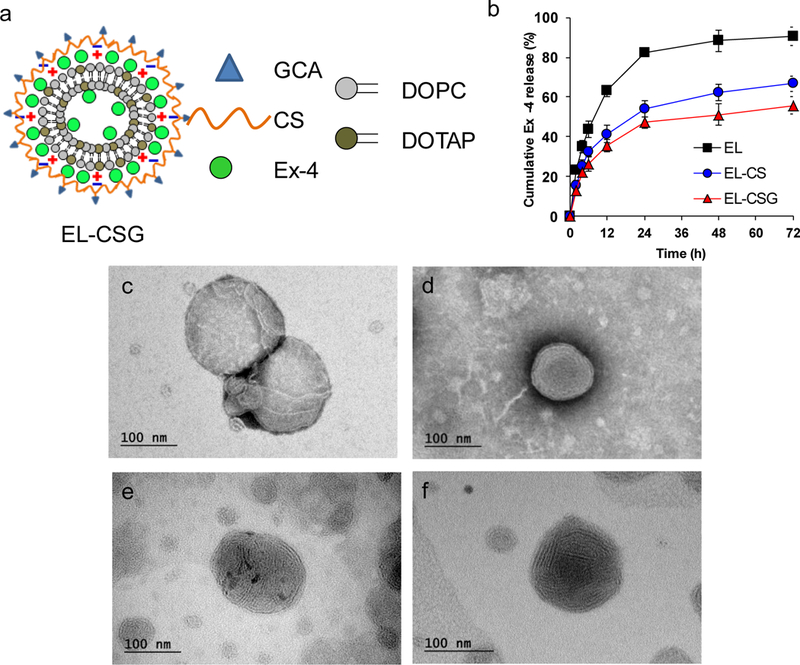
a) Schematic representation of EL-CSG. b) Drug release profile of EL, EL-CS, and EL-CSG in Simulated Intestinal Fluid (pH 6.8) (mean ± SD, n=3). Transmission electron micrographs of the freeze-dried nanoparticles c) Blank liposome, d) EL, e) EL-CS, and f) EL-CSG in the presence of trehalose after being resuspended in distilled water.
3.3. Ex-4 release profile and protection from intestine-simulated fluid
Ex-4 release profiles under GIT-simulated pH conditions were measured. EL, EL-CS, and EL-CSG showed 88, 62, and 51% cumulative release over 24 h, respectively (Fig. 1b), indicating that the coating layers delayed the release rates over 72 h. At the end point (~72 h), the release efficiencies of EL, EL-CS, and EL-CSG were 90.5, 66.7, and 55.7 %, respectively. These results suggest that these liposomes are generally stable and will lose a limited amount of Ex-4 during transit time in the GIT. To demonstrate further the particle stability under GIT conditions, the particle properties were evaluated in simulated fluid (Table 2). PBS was used as a negative control. EL-CSG was rapidly reconstituted within a few seconds under the four media conditions. In addition, the initial particle properties after reconstitution with all media were maintained for at least 1 day at 25 °C (data not shown). However, liposome aggregation in the simulated gastric fluid under fasting conditions (i.e., FaSSGF) was slightly higher than that in simulated intestinal fluid under fasting conditions (i.e., FaSSIF-V2). Ex-4 is comparatively stable within a pH range of 1.2–8 [52] and CSG-coated liposomes are stable in the GIT after oral administration; these features would play an important role in increased Ex-4 absorption because they increase the opportunity for the Ex-4-loaded nanoparticles to reach the distal small intestine.
Table 2.
Stability of EL-CSG in simulated gastrointestinal fluids
| Formulation | Reconstitution media | Size (nm) | PDIa | |
|---|---|---|---|---|
| Initial | - | 181± 1.4 | 0.12 ± 0.04 | |
| EL-CSG | Freeze drying | Distilled water | 175 ± 2.0 | 0.13 ± 0.03 |
| PBS | 195 ± 1.3 | 0.19 ± 0.03 | ||
| FaSSGF | 201 ± 3.9 | 0.23 ± 0.05 | ||
| FaSSIF-V2 | 171 ± 1.4 | 0.13 ± 0.03 | ||
PDI: polydispersity index
Each value represents the mean ± SD, n=3
3.4. Ex vivo nanoparticle transport efficacy using the EGS model
We used the EGS model due to the unavailability of an acceptable monolayer cell culture model that mimics ileocyte absorption at a tissue level. The Ex-4 transport of EL, EL-CS, and EL-CSG was compared with that of free solution (Fig. 2a). The control cationic EL was 2.1 times less permeable than was anionic EL-CSG at the same time point, which was considered non-specific transport. All results support the supposition that the transport is rather specific at the distal ileum, despite a certain degree of non-specific transport that is observed in the EGS model. The cellular integrity and viability of the tissue over 90 min was confirmed by monitoring LDH concentration normalized per unit area of the tissue (Fig. 2b). All concentrations of LDH released were below 40 U/L/cm2, which indicates that the tissue remained viable during the test period [41].
Fig. 2.
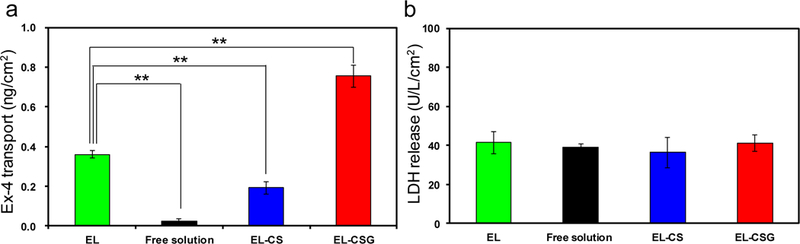
a) Nanoparticle transport into EGS tissue in Krebs-Ringer solution containing EL, EL-CS, EL-CSG, or free Ex-4 solution (0.05 mg/mL, 37 °C, 90 min incubation). b) LDH release from 50 µg/mL EL-CS-, EL-CSG-, or free solution-treated EGS tissue of the distal ileum after a 90-min incubation (mean ± SD, n=4). **p < 0.01.
3.5. In vivo pharmacokinetic study and absorption pathway
To demonstrate the Ex-4 absorption efficacy of EL-CSG in the GIT, we measured time-dependent plasma Ex-4 levels and their related pharmacokinetic parameters (Table 3). The plasma Ex-4 levels of the SC-injected free Ex-4 solution increased and decreased rapidly within 12 h post-administration, while those of EL-CSG gradually increased for 12 h, with an early peak at 3 h, and then slowly decreased over the next 60 h (Fig. 3). We hypothesized that EL-CSG is absorbed into the intestine with bile acids via ASBT, and then transferred to the bloodstream via the enterohepatic circulation, similar to the mechanism recognized with other drugs, such as meloxicam [53]. The temporary increase in plasma concentration at 3 h seems triggered by reabsorption or recycling event. It is difficult to reach a solid conclusion regarding the contribution of enterohepatic circulation because these data were derived from only a peripheral pharmacokinetic study.
Table 3.
Pharmacokinetic analysis of free Ex-4 solution (SC administration), EL-CS, and EL-CSG (oral administration) in normal rats
| Formulation | Admin route | Dose volume (μL) | Dose (μg/kg) | Tmax(h)a | Cmax (ng/mL) | AUC(0–72h) (ng·h/mL) | BAR (%) |
|---|---|---|---|---|---|---|---|
| Free Ex-4 solution | SC | 20 | 20 | - | - | 111 ± 5 | 100 |
| EL-CS | Oral | 500 | 200 | 12 | 1.3 ± 0.2 | 46 ± 8 | 4.1 ± 0.7 |
| EL-CSG | Oral | 500 | 200 | 3 | 7.8 ± 1.1 | 216 ± 23 | 19.5 ± 2.0 |
Cmax: maximum plasma concentration; Tmax: time at which Cmax occurs; T1/2: terminal half-life; AUC(0–72h): area under the plasma concentration vs. time curve from 0 to 72 h; BAR : relative bioavailability.
Where indicated, each value represents the mean ± SEM, n=6
Fig. 3.
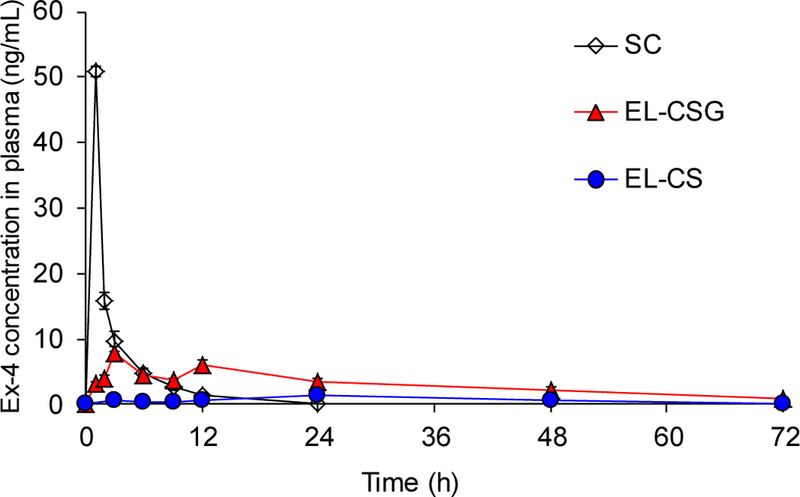
Plasma Ex-4 level versus time profiles of Sprague-Dawley rats. Free Ex-4 solution (20 µg/kg) was administered by SC injection. EL-CS and EL-CSG (200 µg/kg) were administered by oral gavage. (mean ± SEM, n=6)
In order to understand these contributions, we will need to cannulate the portal vein and compare the pharmacokinetic profiles between the portal and peripheral circulation after feeding with EL-CSG; this will be the focus of future studies. However, considering the current data, these observations are probably related to the transit time profiles of individual nanoparticles in the GIT. A certain fraction of the nanoparticles may migrate along the GIT with water, while the remainder may interact with the mucus layer, slowing its movement. The sustained pharmacokinetic profiles produce beneficial effects, mimicking a long-acting drug, which could be more important than oral bioavailability. For instance, to meet the minimum therapeutic concentration of Ex-4, it is injected twice a day in clinical settings due to rapid wash-out 12 h after SC injection (Fig. 3). Although a 10 times higher oral dose was used in this study (20 μg/kg SC vs. 200 μg/kg Oral), the blood concentration of the drug was maintained for 3 days after this single oral dose. The Ex-4 therapeutic range, expressed as the MTC, is well-known to be 50–350 pg/mL in human clinical use. These results strongly suggest that this liposome has sufficient potency to be used as an oral antidiabetic therapy.
For comparing oral bioavailability, EL-CS was used as a negative control group. The rats orally administered EL-CSG exhibited a 4.7-fold higher AUC than the group orally administered EL-CS. EL-CSG exhibited an enhanced oral bioavailability (oBA; 19.5%) despite a series of barriers encountered with the intestinal absorption of Ex-4. These results demonstrate that the EL-CSG formulation supports ASBT-mediated, sustained absorption and release of a significant fraction of Ex-4 in the plasma. The Ex-4 formulation is likely influenced also by the stomach conditions. The oBA can be affected by various parameters, such as physiological factors (GIT movement and pH), drug characteristics (vulnerability to pH and digestive enzymes, and stability during formulation), and formulation characteristics (release rate and protection capacity)[54,55]. The oBA was remarkably high, considering that the formulation is exposed first to gastric conditions without enteric protection and Ex-4 is then lost by release until it reaches the absorption site.
3.6. In vivo pharmacodynamics with T2DM rats
To ascertain the biological efficacy of EL-CSG after oral administration, OGTT was conducted on diabetic rats treated with EL-CSG, free Ex-4 solution, EL-CS, or PBS (DC), as well as on normal rats (NC). When T2DM rats treated with PBS, EL-CS, or free Ex-4 solution were given an oral solution of glucose (1 g/kg body weight), their blood glucose levels increased immediately, reaching a maximum of 238, 213, and 194%, respectively, after 30 min. Moreover, the blood glucose levels of EL-CSG-treated diabetic rats increased less (maximum 171%, P <0.01) than the other groups, indicating that oral EL-CSG improved their glycemic response (Fig. 4a and 4b). These results show that the experimental T2DM rats exhibited a hyperglycemic state in response to oral glucose and that these rats were valid as a pathological model for pharmacodynamic studies of T2DM. EL-CSG also can be used as an appropriate oral T2DM treatment. Extrapolation of pharmacological effects in rat models to those in human patients is still debatable. Regardless, the pharmacological effect of EL-CSG was positive in the rat diabetes model of high fat diet combined with streptozocin [56], which is an accepted model of T2DM in humans.
Fig. 4.
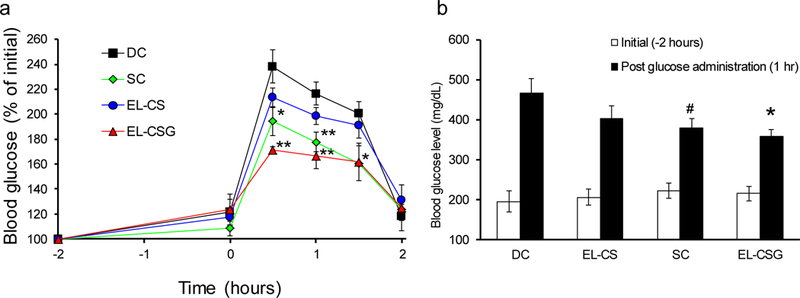
a) Blood glucose level versus time profiles of T2DM rats. Glucose (1 g/kg) was administered orally at 0 h. Two hours prior to glucose administration, 20 µg/kg of free Ex-4 solution was injected SC, while 300 µg/kg of EL-CS or EL-CSG was administered orally. b) Comparison of blood glucose concentrations 1 h post-glucose administration. (mean ± SEM, n=6) #p < 0.1, *p < 0.05, **p < 0.01 compared to DC group.
To evaluate therapeutic effects and safety, high doses of free Ex-4 solution, EL-CS, and EL-GCS were administered daily to T2DM rats for 4 weeks. After the 4-week administration, body weights, as well as HbA1c, T-Chol, LDL cholesterol, and HDL cholesterol levels in blood plasma were measured as T2DM-treatment indicators. There are many metabolic disorders in T2DM that are based on decreased insulin action, such as chronic hyperglycemia, abnormal serum lipid metabolism, and hypertension. As a diabetic treatment, there are three well-known pharmacological effects of Ex-4, namely (1) promoting insulin secretion from pancreatic β cells depending on blood glucose levels, (2) delaying gastric emptying and suppressing appetite and food intake, and (3) protecting the function of β cells. These are important parameters for controlling the diabetic condition. With respect to blood glucose management, it is important to reduce HbA1c levels while minimizing blood glucose fluctuations without causing hypoglycemia, in order to suppress vascular complications.
The change in body weight from 0 to 4 weeks, as well as the HbA1c, T-Chol, and LDL cholesterol levels significantly decreased (Fig. 5a–d), while the HDL cholesterol levels increased (Fig. 5e) in the EL-CSG-treated T2DM rats compared to the corresponding values in the DC and NC rats. Ileum tissue morphology after 4 weeks of EL-CS and EL-CSG oral administration was also assessed by hematoxylin and eosin staining and no morphological abnormalities were observed (Fig. S2). The side effects of Ex-4 in human clinical use are well-known to be milder than those of insulin, the latter including severe hypoglycemia. For example, the main side effects of Ex-4 are gastrointestinal in nature, including acidic or sour stomach, belching, diarrhea, heartburn, indigestion, nausea, and vomiting. During the 4-week administration period, the rats appeared healthy with respect to general appearance, behavior, and excretion, thus no side effects were noted in this study.
Fig. 5.
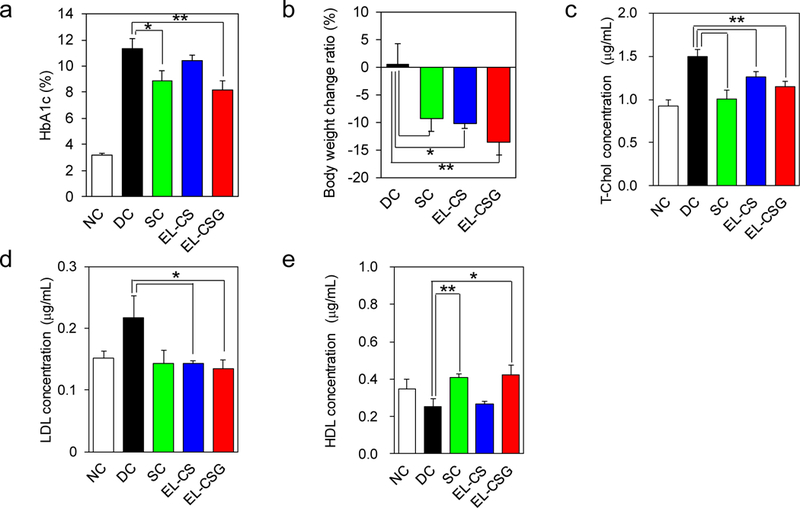
Comparison of a) HbA1c, b) body weight change, c) T-Chol, d) LDL, and e) HDL levels after 4 weeks of treatment with Free Ex-4 solution (20 µg/kg, daily SC injection), EL-CS, and EL-CSG (300 µg/kg, daily, oral) into T2DM rats (mean ± SEM, n=6). DC is the group of PBS-treated diabetic rats. NC is the group of PBS-treated normal rats. **p < 0.01, *p < 0.05.
In addition to its action on pancreatic β cells to promote insulin secretion, Ex-4 has a variety of other effects, such as suppressing glucagon secretion from pancreatic α cells, suppressing appetite through the central nervous system, and delaying gastric emptying. Intestinal absorption of Ex-4 after oral administration of EL-CSG is believed to reduce HbA1c (−3.1 %) and lead to weight loss (−13.6 %) via these mechanisms. With regard to cholesterol levels, T-Chol decreased by 22.8%, HDL increased by 66.3%, and LDL decreased by 38.1% in the EL-CSG administration group, showing the same trends as in the group administered Ex-4 free solution by SC injection. It is known that GLP-1 decreases chylomicron absorption from the small intestine, inhibits fat synthesis through activation of 5’ adenosine monophosphate-activated protein kinase in the liver, accumulates fat in the liver, and decreases triglycerides in the blood [57]. It has also been reported to suppress cholesterol absorption in the small intestine and enhance expression of LDL receptors in the liver [58].
These results suggest that intestinal absorption of Ex-4 via ASBT is unlikely to significantly affect cholesterol homeostasis. Regarding diabetes treatment, it is important to comprehensively manage blood sugar, blood lipids, and blood pressure. The Ex-4 formulation has a multifaceted action in addition to its hypoglycemic effect, and is expected to inhibit diabetic complications. Therefore, this Ex-4 formulation has potential as a future antidiabetic drug. In addition, the present pharmaceutical technology, which can extend drug absorption after oral administration, is a promising preparation for the oral delivery of many drugs, including those targeting diabetes.
4. Conclusions
To evaluate the translational potential of oral nanoparticle absorption utilizing bile acid transporters, a new cationic liposomal preparation loaded with Exendin-4 and coated with anionic chondroitin sulfate-g-glycocholic acid was developed. The observations of oral bioavailability, sustained pharmacokinetics, good glucose response rates, and long-term pharmacodynamic effects provide support that the formulation and delivery route are safe and effective for treating type 2 diabetes in rats.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was partially supported by the National Institutes of Health (NIH DK114015-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no competing financial interest.
Additional information
Supplementary information is available in the online version of the paper.
Correspondence and requests for materials should be addressed to Y.H.B (you.bae@utah.edu).
References
- [1].Todd JF, Bloom SR, Incretins and other peptides in the treatment of diabetes, Diabet. Med 24 (2007) 223–232. [DOI] [PubMed] [Google Scholar]
- [2].Drucker DJ, Biologic actions and therapeutic potential of the proglucagon-derived peptides., Nat. Clin. Pract. Endocrinol. Metab 1 (2005) 22–31. [DOI] [PubMed] [Google Scholar]
- [3].Holst JJ, Gromada J, Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans., Am. J. Physiol. Endocrinol. Metab 287 (2004) E199–206. [DOI] [PubMed] [Google Scholar]
- [4].Davidson MB, Bate G, Kirkpatrick P, Exenatide, Nat. Rev. Drug Discov 4 (2005) 713–714. [DOI] [PubMed] [Google Scholar]
- [5].Nielsen LL, Young AA, Parkes DG, Pharmacology of exenatide (synthetic exendin-4): A potential therapeutic for improved glycemic control of type 2 diabetes, Regul. Pept 117 (2004) 77–88. [DOI] [PubMed] [Google Scholar]
- [6].Gedulin BR, Smith PA, Jodka CM, Chen K, Bhavsar S, Nielsen LL, Parkes DG, Young AA, Pharmacokinetics and pharmacodynamics of exenatide following alternate routes of administration, Int. J. Pharm 356 (2008) 231–238. [DOI] [PubMed] [Google Scholar]
- [7].Talbot K, Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs, Neurodegener. Dis. Manag 4 (2014) 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund P, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T, Exenatide and the treatment of patients with Parkinson’s disease, J. Clin. Invest 123 (2013) 2370–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Simonsen L, Holst JJ, Deacon CF, Exendin-4, but not glucagon-like peptide-1, is cleared exclusively by glomerular filtration in anaesthetised pigs, Diabetologia 49 (2006) 706–712. [DOI] [PubMed] [Google Scholar]
- [10].Hamman JH, Enslin GM, Kotzé AF, Oral delivery of peptide drugs: Barriers and developments, BioDrugs 19 (2005) 165–177. [DOI] [PubMed] [Google Scholar]
- [11].Su YC, Jin CH, Han JS, Yu SY, Lee S, Kang CL, Preparation, characterization, and application of biotinylated and biotin-PEGylated glucagon-like peptide-1 analogues for enhanced oral delivery, Bioconjug. Chem 19 (2008) 334–341. [DOI] [PubMed] [Google Scholar]
- [12].Jin CH, Chae SY, Son S, Kim TH, Um KA, Youn YS, Lee S, Lee KC, A new orally available glucagon-like peptide-1 receptor agonist, biotinylated exendin-4, displays improved hypoglycemic effects in db/db mice, J. Control. Release 133 (2009) 172–177. [DOI] [PubMed] [Google Scholar]
- [13].Nguyen HN, Wey SP, Juang JH, Sonaje K, Ho YC, Chuang EY, Hsu CW, Yen TC, Lin KJ, Sung HW, The glucose-lowering potential of exendin-4 orally delivered via a pH-sensitive nanoparticle vehicle and effects on subsequent insulin secretion in vivo, Biomaterials 32 (2011) 2673–2682. [DOI] [PubMed] [Google Scholar]
- [14].Goldberg M, Gomez-Orellana I, Challenges for the oral delivery of macromolecules, Nat. Rev. Drug Discov 2 (2003) 289–295. [DOI] [PubMed] [Google Scholar]
- [15].Shi Y, Sun X, Zhang L, Sun K, Li K, Li Y, Zhang Q, Fc-modified exenatide-loaded nanoparticles for oral delivery to improve hypoglycemic effects in mice, Sci. Rep 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Malagelada JR, DiMagno EP, Summerskill WHJ, Go VLW, Regulation of pancreatic and gallbladder functions by intraluminal fatty acids and bile acids in man, J. Clin. Invest 58 (1976) 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schiff ER, Small NC, Dietschy JM, Characterization of the kinetics of the passive and active transport mechanisms for bile acid absorption in the small intestine and colon of the rat., J. Clin. Invest 51 (1972) 1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Inagaki T, Moschetta A, Lee Y-K, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA, Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor, Proc. Natl. Acad. Sci 103 (2006) 3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Alrefai WA, Gill RK, Bile acid transporters: Structure, function, regulation and pathophysiological implications, Pharm. Res 24 (2007) 1803–1823. [DOI] [PubMed] [Google Scholar]
- [20].Hofmann AF, The enterohepatic circulation of bile acids in man, Clin Gastroenterol 6 (1977) 3–24. [PubMed] [Google Scholar]
- [21].Hofmann AF, Hofmann F, Hofmann Alan, A.F., The enterohepatic circulation of bile acids in mammals: form and functions, Front. Biosci Volume (2009) 2584. [DOI] [PubMed]
- [22].Cammisotto PG, Renaud C, Gingras D, Delvin E, Levy E, Bendayan M, Endocrine and exocrine secretion of leptin by the gastric mucosa, J. Histochem. Cytochem 53 (2005) 851–860. [DOI] [PubMed] [Google Scholar]
- [23].Carmel R, How I treat cobalamin (vitamin B12) deficiency, Blood 112 (2008) 2214–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Al-Hilal TA, Chung SW, Alam F, Park J, Lee KE, Jeon H, Kim K, Kwon IC, Kim IS, Kim SY, Byun Y, Functional transformations of bile acid transporters induced by high-affinity macromolecules, Sci. Rep 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Khatun Z, Nurunnabi M, Reeck GR, Cho KJ, Lee YK, Oral delivery of taurocholic acid linked heparin-docetaxel conjugates for cancer therapy, J. Control. Release 170 (2013) 74–82. [DOI] [PubMed] [Google Scholar]
- [26].Kim KS, Suzuki K, Cho H, Youn YS, Bae YH, Oral Nanoparticles Exhibit Specific High-Efficiency Intestinal Uptake and Lymphatic Transport, ACS Nano 12 (2018) 8893–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhu C, Huang Y, Zhang X, Mei L, Pan X, Li G, Wu C, Comparative studies on exenatide-loaded poly (D,L-lactic-co-glycolic acid) microparticles prepared by a novel ultra-fine particle processing system and spray drying, Colloids Surfaces B Biointerfaces 132 (2015) 103–110. [DOI] [PubMed] [Google Scholar]
- [28].Griffin BT, Guo J, Presas E, Donovan MD, Alonso MJ, O’Driscoll CM, Pharmacokinetic, pharmacodynamic and biodistribution following oral administration of nanocarriers containing peptide and protein drugs, Adv. Drug Deliv. Rev 106 (2016) 367–380. [DOI] [PubMed] [Google Scholar]
- [29].He C, Yin L, Tang C, Yin C, Size-dependent absorption mechanism of polymeric nanoparticles for oral delivery of protein drugs, Biomaterials 33 (2012) 8569–8578. [DOI] [PubMed] [Google Scholar]
- [30].Rees DO, Crick PJ, Jenkins GJ, Wang Y, Griffiths WJ, Brown TH, Al-Sarireh B, Comparison of the composition of bile acids in bile of patients with adenocarcinoma of the pancreas and benign disease, J. Steroid Biochem. Mol. Biol 174 (2017) 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Roda a, Minutello a, Angellotti M. a, Fini a, Bile acid structure-activity relationship: evaluation of bile acid lipophilicity using 1-octanol/water partition coefficient and reverse phase HPLC., J. Lipid Res 31 (1990) 1433–1443. [PubMed] [Google Scholar]
- [32].Suzuki AZ, Watanabe T, Kawamoto M, Nishiyama K, Yamashita H, Ishii M, Iwamura M, Furuta T, Coumarin-4-ylmethoxycarbonyls as Phototriggers for Alcohols and Phenols, Org. Lett 5 (2003) 4867–4870. [DOI] [PubMed] [Google Scholar]
- [33].Sarmento B, Ribeiro A, Veiga F, Ferreira D, Development and validation of a rapid reversed-phase HPLC method for the determination of insulin from nanoparticulate systems, Biomed. Chromatogr 20 (2006) 898–903. [DOI] [PubMed] [Google Scholar]
- [34].Lin YH, Chen CTCH, Liang HF, Kulkarni AR, Lee PW, Chen CTCH, Sung HW, Novel nanoparticles for oral insulin delivery via the paracellular pathway, Nanotechnology 18 (2007). [Google Scholar]
- [35].Chen MC, Wong HS, Lin KJ, Chen HL, Wey SP, Sonaje K, Lin YH, Chu CY, Sung HW, The characteristics, biodistribution and bioavailability of a chitosan-based nanoparticulate system for the oral delivery of heparin, Biomaterials 30 (2009) 6629–6637. [DOI] [PubMed] [Google Scholar]
- [36].Pandya NT, Jani P, Vanza J, Tandel H, Solid lipid nanoparticles as an efficient drug delivery system of olmesartan medoxomil for the treatment of hypertension, Colloids Surfaces B Biointerfaces 165 (2018) 37–44. [DOI] [PubMed] [Google Scholar]
- [37].Plasencia AMA, Mowry J, Smith J, Quigley K, In vitro release of fentanyl from transdermal patches in gastric and intestinal fluid, Clin. Toxicol 52 (2014) 945–947. [DOI] [PubMed] [Google Scholar]
- [38].Abdelwahed W, Degobert G, Stainmesse S, Fessi H, Freeze-drying of nanoparticles: Formulation, process and storage considerations, Adv. Drug Deliv. Rev 58 (2006) 1688–1713. [DOI] [PubMed] [Google Scholar]
- [39].Chang L, Shepherd D, Sun J, Ouellette D, Grant KL, Tang X, Pikal MJ, Mechanism of protein stabilization by sugars during freeze-drying and storage: Native structure preservation, specific interaction, and/or immobilization in a glassy matrix?, J. Pharm. Sci 94 (2005) 1427–1444. [DOI] [PubMed] [Google Scholar]
- [40].Li M, Si L, Pan H, Rabba AK, Yan F, Qiu J, Li G, Excipients enhance intestinal absorption of ganciclovir by P-gp inhibition: Assessed in vitro by everted gut sac and in situ by improved intestinal perfusion, Int. J. Pharm 403 (2011) 37–45. [DOI] [PubMed] [Google Scholar]
- [41].Surampalli G, Nanjwade BK, Patil PA, Safety evaluation of naringenin upon experimental exposure on rat gastrointestinal epithelium for novel optimal drug delivery, Drug Deliv 23 (2016) 512–524. [DOI] [PubMed] [Google Scholar]
- [42].Teply BA, Tong R, Jeong SY, Luther G, Sherifi I, Yim CH, Khademhosseini A, Farokhzad OC, Langer RS, Cheng J, The use of charge-coupled polymeric microparticles and micromagnets for modulating the bioavailability of orally delivered macromolecules, Biomaterials 29 (2008) 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Khan HBH, Vinayagam KS, Moorthy BT, Palanivelu S, Panchanatham S, Anti-inflammatory and anti-hyperlipidemic effect of semecarpus anacardium in a high fat diet: STZ-induced type 2 diabetic rat model, Inflammopharmacology 21 (2013) 37–46. [DOI] [PubMed] [Google Scholar]
- [44].Stern MP, Williams K, Haffner SM, Identification of persons at high risk for type 2 diabetes mellitus: Do we need the oral glucose tolerance test?, Ann. Intern. Med 136 (2002) 575–581. [DOI] [PubMed] [Google Scholar]
- [45].Chuang EY, Nguyen GTH, Su FY, Lin KJ, Chen CT, Mi FL, Yen TC, Juang JH, Sung HW, Combination therapy via oral co-administration of insulin- and exendin-4-loaded nanoparticles to treat type 2 diabetic rats undergoing OGTT, Biomaterials 34 (2013) 7994–8001. [DOI] [PubMed] [Google Scholar]
- [46].Avignon A, Radauceanu A, Monnier L, Nonfasting plasma glucose is a better marker of diabetic control than fasting plasma glucose in type 2 diabetes, Diabetes Care 20 (1997) 1822–1826. [DOI] [PubMed] [Google Scholar]
- [47].Cortesi R, Esposito E, Gambarin S, Telloli P, Menegatti E, Nastruzzi C, Preparation of liposomes by reverse-phase evaporation using alternative organic solvents, J. Microencapsul 16 (1999) 251–256. [DOI] [PubMed] [Google Scholar]
- [48].Szoka F, Papahadjopoulos D, Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation., Proc. Natl. Acad. Sci 75 (1978) 4194–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Taylor K, Kim D, Nielsen LL, Aisporna M, Baron AD, Fineman MS, Day-long subcutaneous infusion of exenatide lowers glycemia in patients with type 2 diabetes, Horm. Metab. Res 37 (2005) 627–632. [DOI] [PubMed] [Google Scholar]
- [50].Almeida JD, Edwards DC, Brand CM, Heath TD, Formation of virosomes from influenza subunits and liposomes, Lancet 306 (1975) 899–901. [DOI] [PubMed] [Google Scholar]
- [51].Weisman S, Hirsch-Lerner D, Barenholz Y, Talmon Y, Nanostructure of cationic lipidoligonucleotide complexes, Biophys. J 87 (2004) 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sun Y, Wang M, Sun B, Li F, Liu S, Zhang Y, Zhou Y, Chen Y, Kong W, An investigation into the gastrointestinal stability of exenatide in the presence of pure enzymes, everted intestinal rings and intestinal homogenates, Biol. Pharm. Bull 39 (2016) 42–48. [DOI] [PubMed] [Google Scholar]
- [53].Busch U, Schmid J, Heinzel G, Schmaus H, Baierl J, Huber C, Roth W, Pharmacokinetics of meloxicam in animals and the relevance to humans, Drug Metab. Dispos 26 (1998) 576–584. [PubMed] [Google Scholar]
- [54].Martinez MN, Amidon GL, A mechanistic approach to understanding the factors affecting drug absorption: A review of fundamentals, J. Clin. Pharmacol 42 (2002) 620–643. [DOI] [PubMed] [Google Scholar]
- [55].Wilkinson GR, The effects of diet, aging and disease-states on presystemic elimination and oral drug bioavailability in humans, Adv. Drug Deliv. Rev 27 (1997) 129–159. [DOI] [PubMed] [Google Scholar]
- [56].Skovsø S, Modeling type 2 diabetes in rats using high fat diet and streptozotocin, J. Diabetes Investig 5 (2014) 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ben-Shlomo S, Zvibel I, Shnell M, Shlomai A, Chepurko E, Halpern Z, Barzilai N, Oren R, Fishman S, Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase, J. Hepatol 54 (2011) 1214–1223. [DOI] [PubMed] [Google Scholar]
- [58].Li L, Miao Z, Liu R, Yang M, Liu H, Yang G, Liraglutide prevents hypoadiponectinemia-induced insulin resistance and alterations of gene expression involved in glucose and lipid metabolism., Mol. Med 17 (2011) 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.