

Abstract
Selective detection and staining of toxic amyloid plaques, a potential biomarker present in the Alzheimer’s disease (AD) brain is crucial for both clinical diagnosis and monitoring AD disease progression. Herein, we report a coumarin-quinoline (CQ) conjugate-based turn-on near-infrared (NIR) fluorescence probe for specific detection of β-amyloid (Aβ) aggregates. CQ probe is highly sensitive and exhibits ~100-fold fluorescence enhancement in vitro upon binding Aβ aggregates with enhanced quantum yield. Furthermore, the probe has ~10-fold higher binding affinity towards Aβ aggregates (86 nM) compared to commonly used Thioflavin T. Most importantly, CQ probe displays unambiguous selectivity towards Aβ aggregates compared to other toxic protein aggregates such as tau, α-synuclein (α-Syn) and islet amyloid polypeptide (IAPP). In addition, CQ is nontoxic to neuronal cells and shows significant blood brain barrier permeability. Remarkably, CQ stains Aβ plaques in human brain tissue over co-existing tau aggregates and neurofibrillary tangles (NFTs), which are associated in AD and tauopathies. This is a highly desirable attribute to distinguish AD from tau pathology and mixed dementia.
Keywords: NIR fluorescence probes, Selective detection of Aβ42 plaques, Alzheimer’s disease, Neurofibrillary tangles (NFTs), Tauopathies, Mixed dementia
1. Introduction
Protein misfolding and aggregation initiate several neurodegenerative diseases. For instance, misfolding and accretion of Aβ, α-Syn, tau, IAPP, polyglutamine and superoxide dismutase are considered as causative factors for Alzheimer’s, tauopathies, diabetes Huntington’s, Parkinson’s and amyotrophic lateral sclerosis diseases, respectively (Knowles et al., 2014; Soto et al., 2003; Rajasekhar et al., 2015). AD is the most common and dominant form of dementia, which affects millions of people worldwide (Prince et al., 2013). Misfolded Aβ peptide (predominantly Aβ42) and hyperphosphorylated tau protein undergo aggregation to form insoluble Aβ plaques and NFTs, respectively in the brain, which are the hallmarks of AD (Hamley et al., 2012; Nisbet et al., 2015; Iqbal et al., 2014). However, tau and NFTs are independently considered as causative factors in many other neurological disorders and tauopathies that include progressive supranuclear palsy, and frontotemporal dementia among others (Wang and Mandelkow, 2016). Therefore, selective detection of Aβ plaques over NFTs and other similar protein aggregates is critical for detecting, monitoring and distinguishing AD from tau-related neurological disorders and mixed dementia (Spires-Jones et al., 2014; DeToma et al., 2012). Currently, AD diagnosis is mostly based on the assessment of cognitive state of the patients, which means the disease is already in an advanced stage, and the brain is irreversibly degenerated (McKhann et al., 1984; Yang et al., 2013). The protein aggregates implicated in various diseases mostly share a common β-sheet structure, which makes it difficult to develop selective probes for specific bio-structures (Ross et al., 2005; Hermona et al., 2008). Many of the reported probes bind to different protein aggregates without specificity, and are not suitable for selectively detecting and diagnosing an individual disease. There is an immense need to develop toxic aggregate-specific imaging agents or tracers, which offers realistic avenues for developing diagnostics to understand the disease progression and study the effect of therapeutic agents on a specific disease condition. In recent times, PET, SPECT and MRI-based techniques have been developed for detecting and imaging Aβ plaques in the brain (Adlard et al., 2014). While PET imaging is an important tool to image protein aggregates, it is expensive and hazardous to human health due to the use of radio-labeled nuclei (Zhu et al., 2014). Consequently, much attention has been focused on the development of NIR fluorescence probes owing to their easy synthesis, non-invasive nature, low cost, long shelf-life, minimal interference from auto-fluorescence and greater penetration depth to extract information from deep inside the specimen; these attributes make them ideal candidates as diagnostic and imaging agents for toxic Aβ aggregates (Staderini et al., 2015; Narayanaswamy et al., 2016, 2015; Rajasekhar et al., 2016a, 2016b; Li et al., 2016; Ren et al., 2016; Kim et al., 2015; Cui et al., 2014; Hintersteiner et al., 2005; Cao et al., 2012; Lv et al., 2016; Hatai et al., 2017; Gao et al., 2017; Yin et al., 2015; Han et al., 2016; Yu et al., 2015; Xie et al., 2015). Herein, we report a low molecular weight ( > 600 Da) coumarin-quinoline (CQ) conjugate-based NIR fluorescence probe (Zhu et al., 2013) for selective detection of Aβ fibrillar aggregates, and its preferential staining of Aβ plaques in the human brain tissue over NFTs (Fig. 1).
Fig. 1.
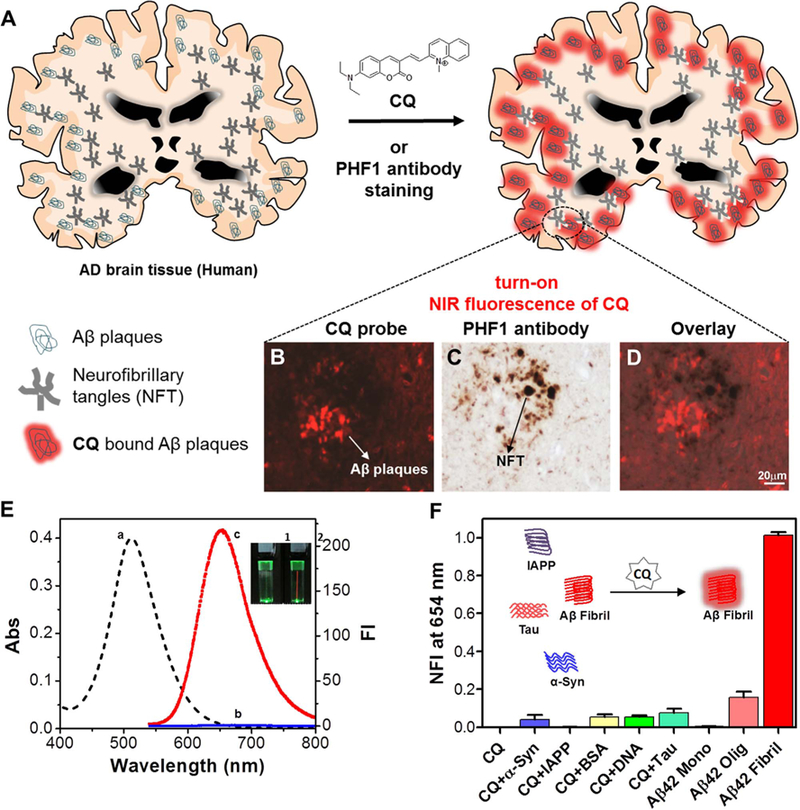
A) Molecular structure of CQ and selective staining of Aβ plaques in human brain tissue. CQ stains Aβ plaques in the AD brain tissue while NFTs of tau corresponding to tau pathology were not detected (B), the neuritic component in the same plaque is only stained with PHF1 antibody (C). The merged image shows there is no colocalization of PHF1 and CQ compound (D). E) Absorption (a) and emission (λex = 521 nm) spectra of CQ (2 µM) in the absence (b) and presence (c) of Aβ42 fibrillar aggregates (10 µM) (λem= 654 nm). Inset: Photographs of cuvettes containing solutions of CQ (2 µM) (1) and CQ (2 µM) + Aβ42 fibrillar aggregates (20 µM) (2), respectively upon illuminating with laser pointer emitting green light, which showed a red beam in cuvette containing CQ + Aβ42 aggregates. F) Normalized fluorescence intensity (NFI) of CQ upon interaction with α-Syn aggregates (50 µM), IAPP aggregates (50 µM), HSA (50 µM), calf thymus DNA (50 µM), tau aggregates (50 µM), Aβ42 monomer (50 µM), Aβ42 oligomers (50 µM) and Aβ42 aggregates (10 µM). Inset: Selective turn-on fluorescence of CQ upon binding to Aβ42 fibrillar aggregates. Abs = absorbance, NFI = Normalized fluorescence intensity.
2. Experimental details
2.1. Materials and methods
All reagents or solvents were obtained from sigma Aldrich and used without further purification. All air and moisture sensitive reactions were carried out under an argon atmosphere. Absorption spectra were recorded with Perkin Elmer Model Lambda 900 spectrophotometer. Fluorescence spectral measurements were carried out by using Perkin Elmer Model LS 55 fluorescence spectrophotometer.
2.2. Preparation of Aβ42 oligomers and fibrillar aggregates
Aβ42 peptide (0.25 mg) (Merck, calbiochem) was dissolved in hexafluoro-2-propanol (HFIP, 0.2 mL) and incubated at room temperature for 1 h. HFIP was then removed by a flow of nitrogen and further dried under vacuum. HFIP-treated Aβ42 was then dissolved in DMSO to a final concentration of 1 mM and diluted to 200 μM with 10 mM PBS (pH 7.4). The solution was incubated at 37°C for 48 h with gentle and constant shaking. The formation of Aβ42 fibrils was confirmed by Thioflavin T (ThT) assay. For the oligomer preparation, Aβ42 was dissolved in DMSO to a final concentration of 1 mM and diluted to 100 μM in PBS buffer (10 mM, pH 7.4). The solution was incubated at 37 °C for 1 h, after which the sample was incubated for 24 h at 4°C. The obtained sample was centrifuged, and the supernatant with oligomers was used for further experiments (Walsh et al., 1997; Stine et al., 2011; Rajasekhar et al., 2016a, 2016b).
2.3. Measuring binding constant of CQ bound to Aβ Aggregates in vitro
A mixture containing Aβ42 fibrillar aggregates (2 μM) was titrated with increasing concentration of probe CQ (0 −1.15 μM) and fluorescence intensity at 654 nm was recorded (λex = 521 nm). The Kd binding curve was generated by GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA) by using the following equation
Where, X is concentration of probe CQ
Y is change in fluorescence intensity
Bmax is the maximum specific binding has the same units as Y.
Kd is the equilibrium binding constant.
2.4. Molecular docking
The geometries of ligands namely, ThT and CQ were optimized using Gaussian09 software by employing density functional theory at the level of B3LYP/6–311++G (d,p). We used dodecamer of amyloid fibril as the target structure which was based on the reported structure for the pentamer of amyloid peptide as in protein data base (PDB code is 2BEG). Since the population of dodecamer has been directly correlated to the dementia, we have chosen it as the target biostructure. The molecular docking studies for two ligands with dodecamer target structure was carried out using AutoDock 4.2. The grid box dimension has been chosen as 150 × 100 × 190 Å using a 0.375 grid step such that it covers the entire fibril and incorporates surface sites as well. The Lamarckian Genetic Algorithm was used for ligand conformation search and was run 500 times, which would generate 500 possible protein-ligand complexes. All other parameters were left to have default values. The resulting ligand conformers were clustered by using root mean square deviation values. The lowest energy amyloid fibril-ligand structure in each binding site was used as the starting configuration for subsequent MD simulations. The binding free energy corresponding to most stable fibril-ligand structure was used to calculate the binding constant and has been compared to experimental data.
2.5. Brain tissue staining
For staining human AD samples a 1 mM CQ stock and a 0.5% (g/mL)/1.57 mM ThT solution were prepared in water. The CQ stain working concentration was 100 nM in PBS (calcium free) whereas; ThT was filtered and used without any further dilution. Slides were deparaffinized and rehydrated by soaking as follows: 10 min xylene I, 10 min xylene II, 10 min 100% ethanol, 5 min 100% ethanol, 5 min 95% ethanol, 3 min 75% ethanol, 3 min 50% ethanol, 3 min 30% ethanol, 3 min 0.85% NaCl, 3 min PBS. Either 100 nM CQ stain or 0.5% (g/mL) ThT were added dropwise and incubated for 2–3 min at room temperature. Slides were then flushed with PBS and immediately submerged in PBS for 10 min. The PBS solution was changed every 2 min. The slides were then washed sequentially in 30% ethanol (1–2 min), 50% ethanol (1–2 min), 30% ethanol (1–2 min) and submerged in water for 10 min. For the final wash, slides were washed in 0.5 × PBS for 20 min with stirring. Tissues were visualized using a Nikon TE2000 inverted confocal microscope (Nikon, Tokyo, Japan) with a Radiance 2100MP Rainbow Laser (Bio-Rad Laboratories).
3. Results and discussion
3.1. Photophysical and molecular docking studies of CQ with Aβ fibrillar aggregates
The photophysical properties of CQ (2 µM) were investigated in PBS buffer (10 mM, pH = 7.4) (Fig. 1E). The absorption spectrum showed a broad absorption band with absorption maxima (λex) at 516 nm. Upon excitation at 516 nm, CQ exhibited a very weak fluorescence band with emission maxima (λem) at 664 nm. In the presence of Aβ fibrillar aggregates (10 µM), CQ exhibited a red shift of 5 nm in the absorption maxima (521 nm) and a blue shift of 10 nm in the emission maxima (654 nm). Remarkably, CQ (2 µM) exhibited 100-fold fluorescence enhancement in the presence of Aβ fibrillar aggregates (10 µM). The quantum yield of CQ in PBS buffer (10 mM, pH = 7.4) was 0.08, which increased to 0.36 upon binding to Aβ42 fibrillar aggregates. The observed fluorescence enhancement of CQ can be attributed to the restricted torsional motion of the probe upon binding to Aβ fibrillar aggregates that restricts molecular relaxation, enhances molecular planarity and decreases non-radiative decay rate, resulting in CQ fluorescence enhancement (Yu et al., 2016). To validate the role of restricted molecular motion on the fluorescence enhancement of probe CQ, we recorded the fluorescence spectra of CQ (5 µM, 10 mM PBS) as a function of increased glycerol percentage in the sample solution (Fig. S1). A linear increment in the fluorescence of CQ was observed with increase in glycerol percentage, which suggests that increase in viscosity of the solution has restricted molecular motion of CQ. Thus, we postulate that restricted molecular motion and attaining molecular planarity together account for the observed enhanced fluorescence of CQ in the presence of Aβ fibrillar aggregates.
A significant red shift in absorption spectra has been reported in the case of ThT and Congo red, upon binding to protein aggregates, due to fibril-induced molecular planarization of the probe and subsequent increase in hyperconjugation (Murugan et al., 2013). However, CQ only exhibited a small red shift (5 nm), which suggests that Aβ42 fibrillar environment does not affect its ground state geometry significantly. The time-dependent density functional/molecular mechanics (TD-DFT/MM) study incorporating polarizable electrostatic embedding scheme reproduce the observed red shift for CQ bound to Aβ42 fibril (Supplementary material, Section 1.10).
3.2. CQ selectivity towards Aβ fibrillar aggregates
Next, we studied the emission behaviour of CQ with polymorphic forms of Aβ peptide (monomer, oligomers and fibrillar aggregates) (Fig. 1F). Maximum fluorescence enhancement was observed with Aβ fibrillar aggregates, followed by oligomers and monomeric forms. Further, we investigated the selective fluorescence enhancement of CQ with macrobiomolecules and protein aggregates implicated in other neurodegenerative diseases (Fig. 1F). In the presence of human serum albumin (HSA, 50 µM), CQ showed a red shift of ~34 nm in emission (λex = 521 nm); however, fluorescence enhancement was not observed, as seen in the case of CQ with Aβ fibrillar aggregates. Similarly, CQ showed minimal fluorescence enhancement in the presence of calf thymus DNA (50 µM) (Fig. 1F and Fig. S3). α-Syn and IAPP are responsible for Parkinson’s disease and type-2 diabetes, respectively, and form fibrillar aggregates similar to Aβ (Maries et al., 2013; Jaikaran et al., 2001). The preformed fibrillar aggregates of α-Syn (50 µM) and IAPP (50 µM) did not enhance the fluorescence of CQ (2 µM), which confirms the selectivity of the probe towards Aβ fibrillar aggregates. Molecular dynamics and molecular docking studies, followed by free energy calculations using Molecular Mechanics/Generalized Born Surface Area (MM/GBSA), were performed on CQ at various binding sites of Aβ, IAPP and α-Syn protofibrils (Fig. S4). The binding affinity exhibited by CQ at various binding sites of Aβ fibrillar aggregates was several orders higher compared to that of IAPP and α-Syn fibrillar aggregates (Table S1–S3), which explained the observed selective binding of CQ to Aβ fibrillar aggregates. Neurofibrillary tangles (NFTs) comprised mainly of tau protein coexist with Aβ plaques in the brain and play a crucial role in the toxicity observed in AD; however, tau aggregates are independently involved in several other neurodegenerative disorders (Wang and Mandelkow, 2016). Therefore, selective detection of Aβ plaques over NFTs in AD is critical for diagnosis of AD in the case of mixed dementia. Remarkably, CQ (2 µM) treated with Tau fibrillar aggregates (50 µM) showed minimal fluorescence enhancement, which suggests that the probe selectively detects and differentiates fibrillar aggregates of Aβ from tau. Subsequently, we performed competition binding affinity experiments where fluorescence of CQ (2 µM) was recorded in the presence of Aβ42 (10 µM) aggregates containing α-Syn fibrillar aggregates (20 µM), IAPP fibrillar aggregates (20 µM), DNA (20 µM) or HSA (20 µM) (Fig. S3). In all the cases, the observed fluorescence enhancement of CQ was comparable to that of the probe treated with Aβ fibrillar aggregates alone. This reveals that probe CQ selectively binds and detects Aβ fibrillar aggregates in the presence of other protein aggregates or macrobiomolecules.
3.3. Calculation of binding parameters, binding site determination and FRET studies of CQ bound to Aβ fibrillar aggregates
Binding saturation assay was performed to extract the binding affinity of CQ towards Aβ fibrillar aggregates. In this assay, emission spectra were recorded upon addition of increasing concentration of CQ (0.052, 0.078, 0.102, 0.210, 0.5, 1 and 5 µM) to a fixed concentration of Aβ fibrillar aggregates (10 µM). The fluorescence intensity at 654 nm was plotted as a function of CQ concentration, and the data was fitted using single binding site equation to obtain the dissociation constant (Kd). The Kd value determined thus was found to be 86 ± 6.3 nM (binding constant Ka, 12.14 ± 0.43 µM), which is much lower than the Kd of ThT (~0.8 µM) and Congo red (~1.1 µM) for Aβ aggregates. Molecular docking study revealed the free energy of binding for CQ-Aβ42 fibril complex as −12.2 kcal/mol, which translates into nanomolar binding affinity (1.2 nM) of the probe; this is in agreement with the experimentally extracted value of Kd. To gain further insights into the binding sites and affinity of the probe, displacement assay was carried out by titrating CQ against ThT (10 µM) bound Aβ42 fibrillar aggregates (20 µM). Remarkably, gradual addition of CQ (0.02–20 µM) to the ThT/Aβ42 fibrillar aggregate complex resulted in steady decay in fluorescence of ThT at 483 nm (λex = 450 nm) and enhancement in the emission intensity of CQ at 654 nm (λex = 521 nm). The decrease in emission at 483 nm and corresponding increase in emission at 654 nm suggest effective displacement of ThT by CQ, driven by the formation of high affinity CQ/Aβ42 aggregate complex (Fig. 2B and S5). An interesting observation was made during this titration study, where the emission at 654 nm corresponding to CQ was observed when the sample (CQ/ThT/Aβ fibrillar aggregates) was excited at 450 nm (ThT excitation maxima). These changes in fluorescence intensity of CQ upon ThT excitation can be attributed to FRET between the Aβ42 fibrillar aggregates bound to ThT and CQ (Jares-Erijman et al., 2003). Initially during titration, CQ bound to the ThT/Aβ42 fibrillar complex by forming a FRET pair with ThT bound to Aβ42 fibrils (Figs. S6 and S7).
Fig. 2.
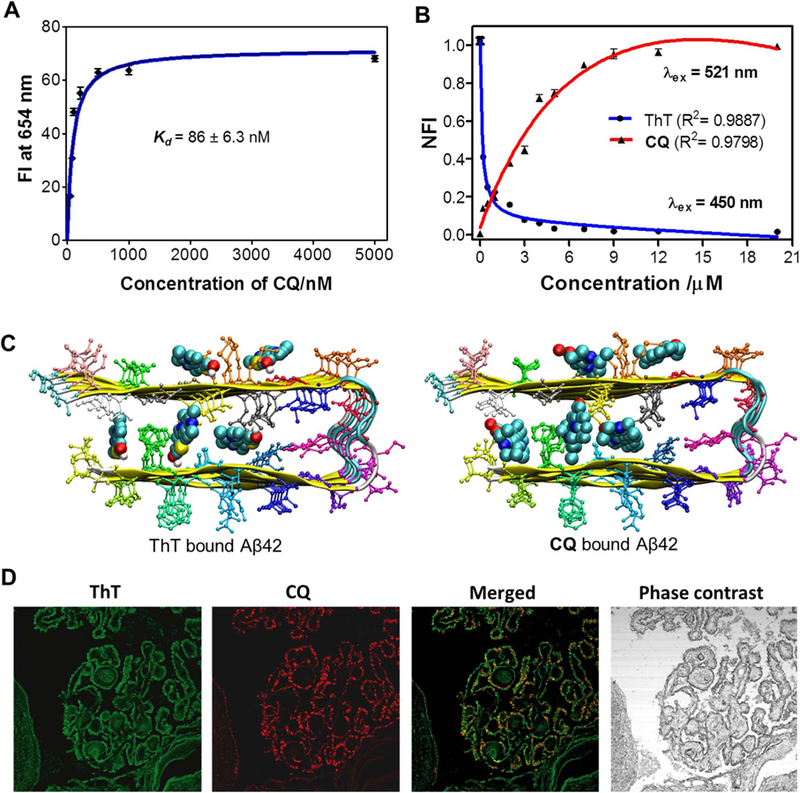
A) Plot of the fluorescence intensity (FI) as a function of the concentration of CQ(0.052, 0.078, 0.102, 0.210, 0.5, 1 and 5 mM) in the presence of Aβ42fibrillar aggregate (10 μM) in PBS (10 mM, pH = 7.4). B) Change in fluorescence of ThT (λex at 450 nm; fluorescence measured at 483 nm) and CQ (λex at 521 nm; fluorescence measured at ~654 nm) upon titrating CQ to ThT/Aβ42fibrillar aggregate complex (ThT, 10 µM/Aβ42fibrils, 20 µM). C) Binding sites available for ThT and CQ within Aβ42 fibril (12 mer assembly). D) CQ stains multiple amyloid plaque structures with specificity. Dual staining of human brain cross sections using CQ and ThT (100 nM CQ: 1.57 mM ThT). While the ThT appears to stain many different amyloid aggregates, the CQ produces lower background fluorescence and shows extreme selectivity for amyloid angiopathy. This is evident by the ring shaped structures localized at the outer wall of the tissue. Each experiment was repeated three times (n = 3) and error bars represent the standard deviation (SD) of the fluorescence measurement. NFI = Normalized fluorescence intensity.
Further increase in concentrations of CQ ( > 0.2 µM) displaced the ThT bound to Aβ42 fibrils, which resulted in decreased FRET-fluorescence of CQ (Fig. S6). However, FRET-based fluorescence at 654 nm was totally quenched at > 15 µM concentration owing to complete displacement of ThT bound to Aβ42 fibrillar aggregates. The complete quenching of fluorescence intensity of ThT (at 483 nm) clearly indicated that CQ had similar binding pockets as ThT on Aβ42 fibrillar aggregates, which was further confirmed by molecular docking studies (Fig. 2C). On the other hand, excitation of CQ (521 nm) showed a gradual increase in fluorescence intensity even after complete displacement of ThT, which suggests the presence of multiple binding sites for CQ on Aβ42 fibrillar aggregates (Fig. 2C).
The molecular docking studies carried out for CQ and ThT on Aβ42 fibrils showed that these probes have 4 different binding sites within the fibril, namely (i) entry cleft site (ii) core-1 site (iii) core-2 site (iv) surface site (Fig. 2C). The data on ThT probe were in agreement with earlier reports, which have also suggested multiple binding sites (Kuang et al., 2015). The entry cleft is the site that is associated with the most stable Aβ42 fibril:CQ complex formation. The calculated/determined binding affinity value for ThT was found to be much lower compared to that of CQ for Aβ42 fibril. The empirical docking free energy score included van der Waals, electrostatic, hydrogen bonding and solvation contributions to binding free energy. The electrostatic interaction between the Aβ42 fibril:CQ complex was observed to be negligible, suggesting that the van der Waals interactions are the driving force for complex formation. However, molecular docking assumed a rigid target structure and the final free energy was obtained for a single configuration of Aβ42 fibril:CQ complexes. In order to gain detailed insights, we analyzed the binding free energy results of Aβ42 fibril:CQ complexes obtained from MM/GBSA approaches; this also accounted for sampling effect. In particular, the free energy from this approach included van der Waals, electrostatic, polar, and non-polar solvation contributions along with entropic contributions obtained using normal mode analysis. Although the electrostatic contributions are supposed to be dominant compared to van der Waals, they are nullified by the polar solvation free energy. Therefore, van der Waals contribution became the driving force for the formation of stable Aβ42 fibril:CQ complexes. Interestingly, depending on the spatial location of the probe on Aβ42 fibril, the van der Waals contribution varied and this, in turn, affected the overall binding free energy of CQ. The binding free energy for CQ at all four sites of Aβ42 fibril was relatively higher compared to the reported values of ThT, and this clearly established the fact that CQ efficiently displaced bound ThT from Aβ42 fibril; this observation was in agreement with experimental FRET data.
3.4. Determination of stability and BBB permeability of CQ
Next, we evaluated log P values for probe CQ using the flask shake method and found this to be 2.4, which suggests that CQ possesses the desirable lipophilicity to cross the blood brain barrier (BBB). In vitro stability studies indicated that CQ is stable in human blood serum (HBS), as more than 97% of the probe was found intact after 60 min incubation in HBS at 37 °C (Fig. S8). Cytotoxicity of probe CQ was studied in PC12 cells at varying concentrations. Probe CQ was nontoxic until 10 μM, which is much higher than the working concentrations (2 μM) used in our experiments (Fig. S9). Subsequently, we studied the BBB permeability of CQ in mice. The probe CQ was injected intraperitoneally, and the mice were sacrificed after 1 h. The brain was homogenized and analyzed through absorption and mass spectrometry (Supplementary Material, Section 1.10). The brain lysate showed presence of CQ probe, which underscored the BBB crossing ability of CQ (Dickstein et al., 2006).
3.5. Selective detection of Aβ plaques in human brain tissue
The specificity and sensitivity of CQ towards Aβ plaques were evaluated using thioflavin and immunohistochemistry in AD patient brain tissues. ThT was used as positive control for the detection of amyloid plaques. The concentration of CQ required for visualization of Aβ plaques was 15,700-fold less than that of ThT [CQ (100 nM): ThT (1.57 mM)]. Notably, the CQ stain responded to destaining better than ThT, which resulted in a significant decrease in background fluorescence (Fig. 2D). Furthermore, the CQ stain was found to be more selective for Aβ plaques while ThT resulted in increased background staining. This superior fluorescence staining by CQ with high signal-to-noise ratio is likely due to its high selectivity and sensitivity towards Aβ plaques. We further analyzed the specificity of CQ using age-matched controls, where the ThT analog thioflavin S (ThS) was used as control to stain Aβ plaques [Fig. 3A (ThS, 1.57 mM) & 3B (CQ, 100 nM)]. In the AD brain tissues, although ThS stained Aβ plaques, it also co-stained many other regions [Fig. 3C (ThS, 1.57 mM) & 3D (CQ, 100 nM)]. In contrast, CQ stained specifically the Aβ plaques with high signal-to-noise ratio. Furthermore, CQ stained the same Aβ plaques and congophilic angiopathy as Congo Red [Fig. 3E (Congo Red) & 3F (CQ, 100 nM)]. Several forms of Aβ plaques were observed, including amyloid angiopathy (Fig. S11A and S11B) and core/diffused plaques (Fig. S12A-C). The CQ stain appeared to be selective for amyloid angiogenesis, which is evident by the green background fluorescence in the dual staining of tissue sections and independent staining of identical cross-sections (Fig. S11). Notably, ThS labeled neurofibrillary tangles (NFT) significantly more than the CQ stain (Fig. S12A and S12C). To further investigate the selectivity of CQ, we co-stained AD brain specimens with PHF1 antibody, which labels NFTs. As shown in Fig. 1B, CQ labeled Aβ plaque, while PHF1 antibody stained NFTs (Fig. 1C). The merged image confirmed the specificity of CQ, as no overlapping staining was observed (Fig. 1D). We also analyzed another tissue section that was rich in NFTs, as visualized with PHF1 antibody (Fig. 4B); however, the serial adjacent section when stained with CQ (100 nM) revealed no such aggregates (Fig. 4A). Finally, we investigated CQ specificity in supranuclear palsy (PSP), which is a known tauopathy. Similar to the results obtained with AD tissue specimens, CQ revealed no staining, whereas PHF1 antibody readily detected the tau-positive PSP pathology in the brainstem (Fig. 4C and D). Together, these results confirmed that CQ is highly specific for Aβ plaques, unlike ThT or ThS, which stain both Aβ plaques and NFTs.
Fig. 3.
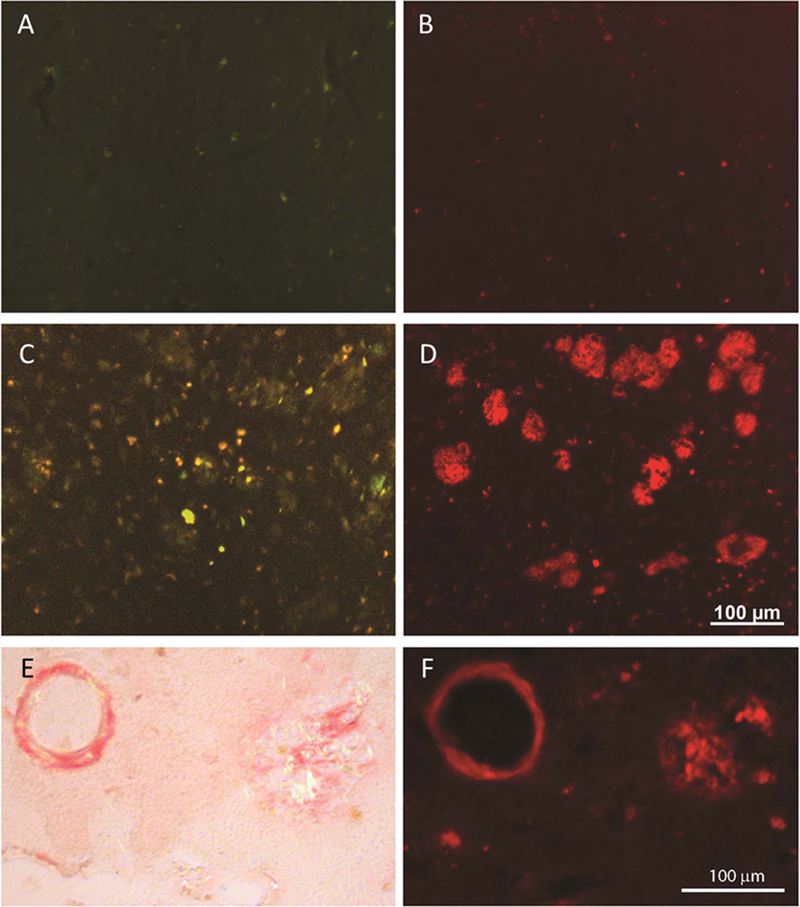
In an aged control individual, the staining with ThS (1.57 mM) (A) and CQ (100 nM) (B) shows very little staining (fluorescence) signal. In AD brain tissue, ThS stains Aβ plaques yet many other fluorescent structures are apparent (C), however, CQ labels the Aβ plaques with greater specificity in the adjacent section from the same case (D). CQ (F) labels the same Aβ plaques and congophilic angiopathy as Congo Red (E).
Fig. 4.
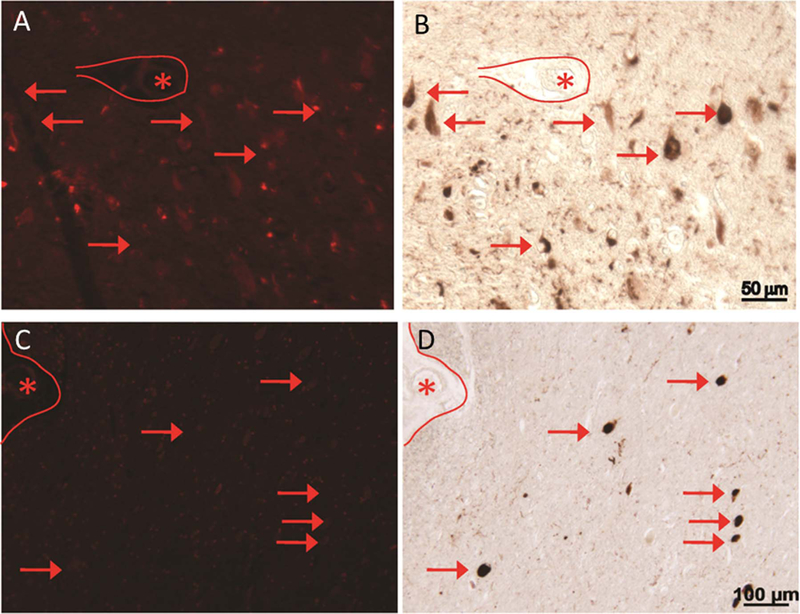
CQ (100 nM) clearly stains Aβ plaques in the AD brain tissue while NFTs of tau corresponding to Tau pathology were not detected. In a field with many neurofibrillary tangles stained by PHF1, the serial section stained with CQ compound does not reveal the NFTs (A and C). Some NFTs may be seen at near background level, but do not fluoresce at nearly the same intensity as Aβ plaques. Further, in specific taupathy, progressive supranuclear palsy (PSP) samples, the tau-positive PSP tangles in the brainstem are not detected by CQ (A-D). *marking landmark vessels.
4. Conclusion
We have developed a coumarin-quinoline (CQ) conjugate-based turn-on NIR fluorescence probe for selective detection of Aβ42 aggregates involved in AD pathology. Probe CQ was found to exhibit high serum stability and binding affinity (86 nM), and fluorescence enhancement (100-fold) when bound to Aβ42 fibrillar aggregates with enhanced quantum yield (0.36). CQ specifically bound to Aβ42 aggregates and exhibited fluorescence selectively over other biomacromolecules (HSA and DNA) and toxic protein aggregates of tau, α-Syn and IAPP, all of which possesses β-sheet structure similar to Aβ42 aggregates. The differential fluorescence staining ability of CQ, i.e., distinguishing Aβ42 aggregates from tau aggregates is crucial for clinical diagnosis of AD from tauopathies. Molecular dynamics and free energy calculations of CQ with various protein aggregates showed high binding affinity of CQ towards Aβ42 aggregates, which is key to its selective nature. Probe CQ bound to Aβ42 aggregates through multiple binding sites, which was determined by molecular docking and FRET studies. We further demonstrated the high specificity of CQ for Aβ plaques in the AD brain tissue, unlike commonly used ThT or ThS, which stain both Aβ plaques and NFTs. All these remarkable characteristics suggest that CQ probe is a promising candidate for developing specific in vitro and in vivo methods for clinical diagnosis. Currently, work is in progress in our laboratory to develop CQ-based fluorescence and PET methods for possible early diagnosis of AD.
Supplementary Material
Acknowledgements
We thank Prof. C. N. R. Rao FRS for constant support and encouragement, JNCASR, TRC TRC (TRC-JNC/TG/4426 (TRC‐JNC/TG/4426)), Science and Engineering Research Board (SERB), the Department of Science and Technology (DST) [Research Grant: SB/S1/OC-47/2103SB/S1/OC‐47/2103], National], National Institute on Aging, National Institutes of Health Institute (R21-AG 47447 to KS) for financial support. Dr. Robert Tycko, NIH for providing the model structure for IAPP fibril which has been used in the investigation of interaction of CQ probe with IAPP fibril.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.bios.2017.06.030.
References
- Adlard PA, Tran BA, Finkelstein DI, Desmond PM, Johnston LA, Bush AI, Egan GF, 2014. Front. Neurosci 8, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Farahi M, Dakanali M, Chang WM, Sigurdson CJ, Theodorakis EA, Yang J, 2012. J. Am. Chem. Soc 134, 17338–17341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Ono M, Watanabe H, Kimura H, Liu B, Saji H, 2014. J. Am. Chem. Soc 136, 3388–3394. [DOI] [PubMed] [Google Scholar]
- DeToma AS, Salamekh S, Ramamoorthy A, Lim MH, 2012. Chem. Soc. Rev 41, 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA, 2006. FASEB J 20, 426–433. [DOI] [PubMed] [Google Scholar]
- Gao M, Yu F, Lv C, Choo J, Chen L, 2017. Chem. Soc. Rev 46, 2237–2271. [DOI] [PubMed] [Google Scholar]
- Hamley IW, 2012. Chem. Rev 112, 5147–5192. [DOI] [PubMed] [Google Scholar]
- Han X, Yu F, Song X, Chen L, 2016. Chem. Sci 7, 5098–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatai J, Motiei L, Margulies D, 2017. J. Am. Chem. Soc 139, 2136–2139. [DOI] [PubMed] [Google Scholar]
- Hermona S, Ehud G, 2008. Curr. Alzheimer Res 5, 232.18537539 [Google Scholar]
- Hintersteiner M, Enz A, Frey P, Jaton A-L, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold K-H, Gremlich H-U, 2005. Nat. Biotechnol 23, 577–583. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong C-X, 2014. Biochem. Pharmacol 88, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaikaran ETAS, Clark A, 2001. Biochim. Biophys. Acta 1537, 179–203. [DOI] [PubMed] [Google Scholar]
- Jares-Erijman EA, Jovin TM, 2003. Nat. Biotechnol 21, 1387–1395. [DOI] [PubMed] [Google Scholar]
- Kim D, Moon H, Baik SH, Singha S, Jun YW, Wang T, Kim KH, Park BS, Jung J, Mook-Jung I, Ahn KH, 2015. J. Am. Chem. Soc 137, 6781–6789. [DOI] [PubMed] [Google Scholar]
- Knowles TPJ, Vendruscolo M, Dobson CM, 2014. Nat. Rev. Mol. Cell Biol 15, 384–396. [DOI] [PubMed] [Google Scholar]
- Kuang G, Murugan NA, Tu Y, Nordberg A, Ãgren H, 2015. J. Phys. Chem. B 119, 11560–11567. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu D, Ho S-L, Li H-W, Yang R, Wong MS, 2016. Biomaterials 94, 84–92. [DOI] [PubMed] [Google Scholar]
- Lv G, Sun A, Wei P, Zhang N, Lan H, Yi T, 2016. Chem. Commun 52, 8865–8868. [DOI] [PubMed] [Google Scholar]
- Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K, 2003. Nat. Rev. Neurosci 4, 727–738. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM, 1984. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- Murugan NA, Olsen JGMH, Kongsted J, Rinkevicius Z, Aidas K, Ãgren H, 2013. J. Phys. Chem. Lett 4, 70–77. [DOI] [PubMed] [Google Scholar]
- Narayanaswamy N, Das S, Samanta PK, Banu K, Sharma GP, Mondal N, Dhar SK, Pati SK, Govindaraju T, 2015. Nucleic Acids Res 43, 8651–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanaswamy N, Narra S, Nair RR, Saini DK, Kondaiah P, Govindaraju T, 2016. Chem. Sci 7, 2832–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisbet RM, Polanco JC, Ittner LM, Gotz J, 2015. Acta Neuropathol 129, 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri C,P, 2013. Alzheimer’s Dement 9, 63–75. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K, Chakrabarti M, Govindaraju T, 2015. Chem. Commun 51, 13434–13450. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K, Madhu C, Govindaraju T, 2016a. ACS Chem. Neurosci 7, 1300–1310. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K, Narayanaswamy N, Murugan NA, Kuang G, Ãgren H, Govindaraju T, 2016b. Sci. Rep 6, 23668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Xu M, Liang SH, Xiang H, Tang L, Zhang M, Ding D, Li X, Zhang H, Hu Y, 2016. Biosens. Bioelectron 75, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA, 2005. Nat. Rev. Mol. Cell Biol 6, 891–898. [DOI] [PubMed] [Google Scholar]
- Soto C, 2003. Nat. Rev. Neurosci 4, 49–60. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, Hyman BT, 2014. Neuron 82, 756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staderini M, Martin MA, Bolognesi ML, Menendez JC, 2015. Chem. Soc. Rev 44, 1807–1819. [DOI] [PubMed] [Google Scholar]
- Stine WB, Jungbauer L, Yu C, LaDu M, 2011. Preparing synthetic Aβ in different aggregation states. In: Roberson ED (Ed.), Alzheimer’s Disease and Frontotemporal Dementia 670 Humana Press, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB, 1997. J. Biol. Chem 272, 22364–22372. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E, 2016. Nat. Rev. Neurosci 17, 5–21. [DOI] [PubMed] [Google Scholar]
- Xie JY, Li CY, Li YF, Fei J, Xu F, Ou-Yang J, Liu J, 2015. Anal. Chem 88, 9746–9752. [DOI] [PubMed] [Google Scholar]
- Yang Z, Slavin MJ, Sachdev PS, 2013. Nat. Rev. Neurol 9, 382–393. [DOI] [PubMed] [Google Scholar]
- Yin K, Yu F, Zhang W, Chen L, 2015. Biosens. Bioelectron 74, 156–164. [DOI] [PubMed] [Google Scholar]
- Yu W-T, Wu T-W, Huang C-L, Chen IC, Tan K-T, 2016. Chem. Sci 7, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Gao M, Li M, Chen L, 2015. Biomaterials 63, 93–101. [DOI] [PubMed] [Google Scholar]
- Zhu L, Ploessl K, Kung HF, 2014. Chem. Soc. Rev 43, 6683–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Lin W, Yuan L, 2013. Dyes Pigment 99, 465–471. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.