

Abstract
Preeclampsia (PE) is a disorder specific to pregnancy characterized by new-onset hypertension and proteinuria after 20 weeks of gestation. There is no definite treatment for PE except delivery of the placenta. The purpose of this study was to elucidate the biological pathways involved in the development of PE and to discover a novel biomarker for PE by performing global gene expression analysis of amniotic fluid cell-free RNA.
The participants were recruited from the Department of Obstetrics and Gynecology of CHA Gangnam Medical Center (Seoul, Korea) between March 2014 and February 2015. Eight samples were collected from 8 subjects at second trimester who were later diagnosed with PE. From the amniotic fluid samples, cell-free RNA extraction was performed and gene expression was analyzed using the GeneChip PrimeView Array. Transcriptome data previously analyzed by our group from 9 euploid mid-trimester amniotic fluid samples were used as the control for comparative analysis. Functional analysis of the probe sets was performed using the online Database for Annotation, Visualization, and Integrated Discovery (DAVID) toolkit 6.7.
We identified 1841 differentially expressed genes (DEGs) between the PE group and the control. Of these, 1557 genes were upregulated in the PE group, while 284 genes were upregulated in the control. The functional annotation of DEGs identified specific enriched functions such as “transport,” “signal transduction,” and “stress response.” Functional annotation clustering with enriched genes in the PE group revealed that translation-related genes, cell–cell adhesion genes, and immune-related genes were enriched. KEGG pathway analysis showed that several biological pathways, including the ribosome pathway and various immune pathways, were dysregulated. Several genes, including RPS29, IGF-2, and UBC, were significantly upregulated in PE, up to tenfold.
This study provides the first genome-wide expression analysis of amniotic fluid cell-free RNA in PE. The results showed that gene expression involving the ribosome pathway and immunologic pathways are dysregulated in PE. Our results will aid in understanding the underlying pathogenesis of PE.
Keywords: amniotic fluid, fetal development, microarray analysis, preeclampsia, transcriptome
1. Introduction
Preeclampsia (PE) is a disorder specific to pregnancy characterized by new-onset hypertension and proteinuria after 20 weeks of gestation. PE adversely affects multiple organs, including the liver, kidney, lung, and brain, and can lead to serious complications if left untreated. This systematic disorder affects 3% to 5% of all pregnancies and remains one of the main causes of maternal and perinatal morbidity and mortality.[1]
The treatment of PE is symptomatic, and the purpose of treatment is to maintain the pregnancy until fetal lung maturation completes.[2] The ultimate treatment for PE is preterm delivery of the placenta. Therefore, the placenta is crucial for the development of PE. Multiple pathogenic mechanisms have been implicated in PE. However, further understanding of the pathogenesis of PE is required to prevent and predict the development of PE.
Amniotic fluid cell-free RNA (AF-cf-RNA) has been reported to reflect fetal organ development and amniotic fluid (AF) composition.[3] Several studies have been performed to elucidate novel pathogenesis for fetal aneuploidy by using AF-cf-RNA.[4–6] Slonim et al[6] compared the differences in gene expression of AF supernatant samples and suggested the importance of oxidative stress and ion transport in the trisomy 21 fetuses. In trisomy 18, AF cell-free transcriptome (AF-cf-transcriptome) analysis revealed that genes involved in adrenal development are significantly downregulated, which may explain the pre- and postnatal growth restriction of trisomy 18.[4] In addition, gene expression involving neurological and cardiovascular pathways in AFs are significantly different between twin–twin transfusion syndrome (TTTS) recipients and singleton controls.[7] This result suggests that AF-cf-transcriptome reflects the long-term complications of TTTS survivors.
However, there has been no report analyzing the AF transcriptome in PE. Therefore, this study was performed to characterize the AF-cf-transcriptome during second trimester pregnancy destined to develop PE and to provide novel biological pathways involved in the pathogenesis of PE by performing global gene expression analysis using AF-cf-RNA.
2. Materials and methods
AF samples were obtained from 82 pregnant women who underwent amniocentesis for fetal chromosomal analysis after finding a soft marker for Down syndrome through ultrasonography screening in the Department of Obstetrics and Gynecology, CHA Gangnam Medical Center (Seoul, Korea) between March 2014 and May 2015. This study was reviewed and approved by the institutional review board of CHA Gangnam Medical Center in 2014 (GCI-14–11). All participants provided written informed consent to participate in the study before AF was obtained. Initially, 82 participants were enrolled and 68 subjects receiving early second trimester amniocentesis before 22 gestational weeks were included. Among these, 16 participants were diagnosed with PE later in the pregnancy. Diagnosis of PE was based on the general diagnostic criteria of PE, such as new-onset hypertension with proteinuria after 20 gestational weeks (systolic blood pressure of 140 mm Hg or higher, or diastolic blood pressure of 90 mm Hg or higher on 2 occasions at least 4 hours apart and urine dipstick test of 1+ or higher). We excluded 4 participants with twin pregnancy, 2 with gestational diabetes in later pregnancy, 1 subject who was diagnosed with Turner syndrome, and 1 who failed to follow-up. Karyotyping confirmed that all 8 fetuses were euploid. For comparative analysis, we used gene expression datasets from mid-trimester euploid AF samples that were produced in our previous studies.[8] We calculated appropriate sample size with the model that is provided by the department of bioinformatics and computational biology of MD Anderson Cancer Center https://bioinformatics.mdanderson.org/main/SampleSizes:Overview#Sample_Sizes). Using this model, we set the number of differentially expressed genes (DEGs): 2000; acceptable number of false positives:1; desired fold differences: 3; desired power 0.8; standard deviation 0.7. With per-gene value of alpha is 0.0050, the result of the computation was 8 in each group.
2.1. Cell-free RNA extraction
AF was centrifuged at 350 × g for 10 minutes at room temperature to remove amniocytes. The supernatants were stored at −80°C. The cell-free RNA (cfRNA) was extracted from 5 to 10 mL of the AF supernatant. To extract cfRNA, the QIAamp Circulating Nucleic Acid (Qiagen, Germany) kit with an on-column DNase digestion step to remove genomic DNA was used according to the manufacturer's instructions. The RNA was purified and concentrated with the RNeasy MinElute Cleanup kit (Qiagen, Germany) and eluted with RNase-free water. The UV absorbance at 260 nm (A260 value) was measured, and samples with A260:A280 ratios greater than 1.8 were stored at −80°C for further analyses.
2.2. Microarray
Gene expression was analyzed with the GeneChip PrimeView array (Affymeterix, Santa Clara, CA). Biotinylated aRNA was prepared according to the standard Affymetrix protocol (Expression Analysis Technical Manual, 2001; Affymetrix) from 100 ng total RNA using the GeneChip 3’IVT Express Kit (Affymetrix). Following fragmentation, 12 μg of aRNA was hybridized for 16 hours at 45°C on a GeneChip Human array. GeneChips were washed and stained in the Affymetrix Fluidics Station 450. GeneChips were scanned using the Affymetrix GeneChip Scanner 3000 7G. The data were analyzed with Robust Multi-array Analysis (RMA) using the Affymetrix default analysis settings and global scaling as a normalization method. The trimmed mean target intensity of each array was arbitrarily set to 100. The normalized and log-transformed intensity values were then analyzed using GeneSpring 12.5 (Agilent Technologies, CA).
2.3. Data analysis
The data were imported into the GeneSpring GX 7.3 software (Agilent Technologies) for filtering and basic statistical analysis. Signals were considered “detected” when their value was larger than the median value of the control probe signal. We identified DEGs in PE, control, and both samples if they had a Benjamini–Hochberg adjusted P value < .05. Genes were considered significantly differentially expressed in both PE and control if they had a fold-change of at least 1.5 according to Welch t test. Unsupervised hierarchical clustering analyses and principal component analysis were performed to visualize the overall expression characteristics of all samples used in the study. Biological function and pathway analyses were performed using the online Database for Annotation, Visualization and Integrated Discovery (DAVID) toolkit 6.8, an ontology-based webtool (http://david.abcc.ncifcrf.gov/). DAVID provides typical batch annotation and gene ontology (GO) term enrichment was ascertained by computing the EASE score (modified Fisher exact test) for each GO classification. We uploaded the gene lists defined as DEG for each group using the official gene symbols to identify enriched gene ontologies for gene expression, specific tissue expression, and functional pathway analysis. The biological functions of selected genes were analyzed using information from the GO database,[9] the Kyoto Encyclopedia of Genes and Genomes (KEGG),[10] and the Online Mendelian Inheritance in Man (OMIM) database.[11]
3. Results
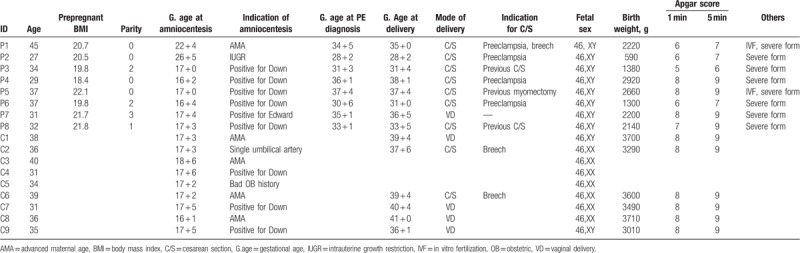
In the study, we evaluated DEGs in the AF-cf-transcriptome between samples from PE and control. Table 1 presents the characteristics of the participants. The median age, median gestational ages of delivery, and median birth weight of babies in the PE group were 33 years (27–45 years), 34 + 3 weeks (28 + 2–38 + 1 weeks), and 2170 g (590–2920 g), respectively. On the contrary, the median age, median gestational ages, and median birth weight of babies in the control group were 36 years (31–40 years), 39 + 4 weeks (36 + 1–41 + 0 weeks), and 3545 g (3010–3710 g), respectively.
Table 1.
Subject characteristics.
There were 1841 genes that were significantly differentially regulated in PE compared with the control; 1557 genes were upregulated in PE and 284 were downregulated. Hierarchical clustering analyses were performed to visualize the overall expression characteristics of all samples used in the study (Fig. 1A). The changes between PE and control were distinct at a global level (Fig. 1B). The analyses revealed that the overall patterns of gene expression in PE were distinctly different from those in the control.
Figure 1.
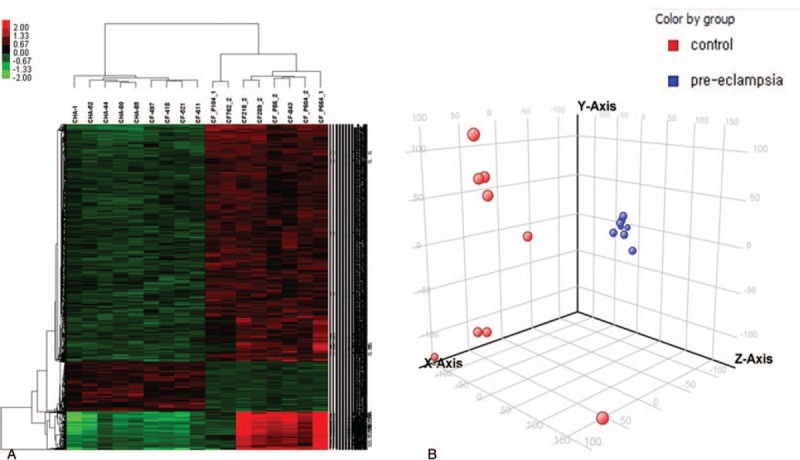
(A) Heatmap produced by hierarchical clustering of genes and samples, presenting differentially expressed genes in preeclampsia versus control. (B) Principal component analysis (PCA) of the top 3 identified components of transcriptome presenting the separation by disease. Control (red) and preeclampsia (blue).
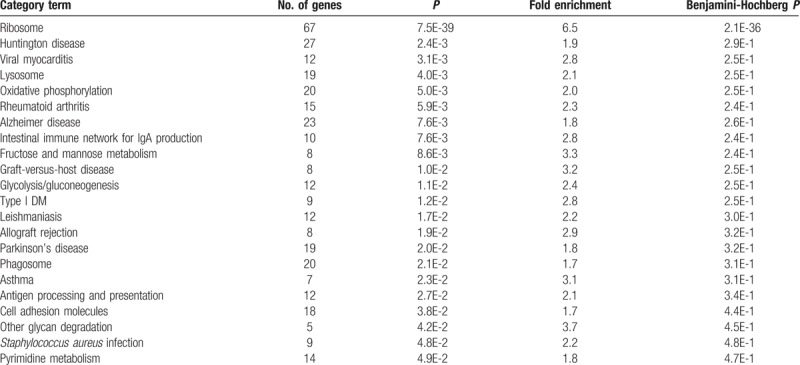
Functional classification of DEGs using DAVID showed that genes associated with stress response, signal transduction, and cellular transport are differentially expressed in PE (Table 2). It is important to identify dysregulated biological process to suggest potential underlying mechanisms of PE. KEGG pathway analysis with 1557 upregulated genes in PE provided insight into the aberrantly regulated signaling pathways, including the ribosomal pathway, viral myocarditis, rheumatoid arthritis, intestinal immune network for IgA production, graft-versus-host disease, Leishmaniasis, allograft rejection, asthma, and antigen processing and presentation (Table 3). Functional annotation clustering analysis with upregulated genes in PE using DAVID showed that several functional annotation groups, such as ribosome and protein translation, cell–cell adhesion, and immune response, were significantly enriched.
Table 2.
Functional annotation of differentially regulated genes in preeclampsia.

Table 3.
KEGG pathway analysis with enriched genes in preeclampsia.
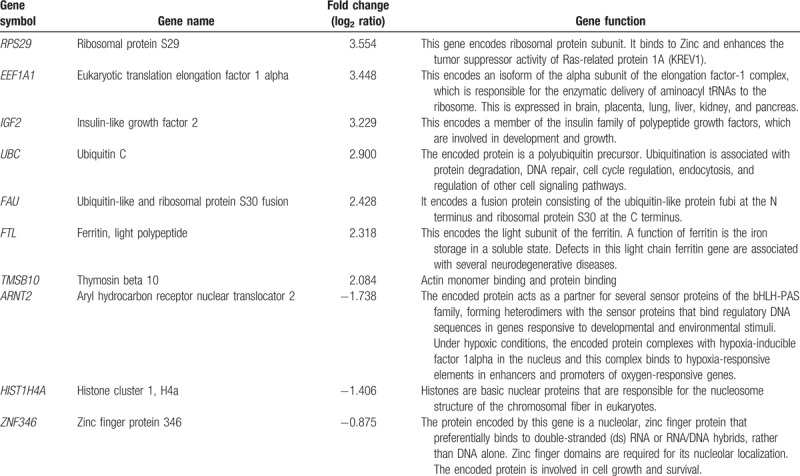
We analyzed general annotations of upregulated genes in PE and identified several chromosomes, including chromosomes 1, 12, 16, 17,19, 20, and 22. To provide potential candidates for predictive biomarkers of PE, we selected 7 upregulated genes and 3 downregulated genes based on fold-change value in PE (Table 4). Most of the upregulated genes were associated with the ribosome pathway.
Table 4.
Selected differentially regulated genes of interest as potential biomarkers of preeclampsia.
4. Discussion
AF is a complex and dynamic solution that provides valuable information on fetal health status, such as fetal karyotyping, fetal lung maturity testing, and diagnosis of intra-amniotic infection. The presence of cell-free NAs (cf-NAs) in AF has been reported.[12] Although several studies have examined how the fetal status affects the expression of cf-NAs in the AF, as well as used AF-cf-NAs for discovering novel biomarkers for complications during pregnancy, including fetal aneuploidy, TTTS, and maternal obesity,[4–6,13,14] there has been no study on the pathogenesis of PE using AF-cf-NAs. In the present study, we analyzed the differences in global gene expression in the AF-cf-RNAs between PE and normal pregnancy to provide valuable information for the pathogenesis of PE. Our results showed that immune-related pathways and the ribosome pathway were enriched in the early second trimester AF-cf-transcriptome of PE.
The current proposed model for the pathophysiology of PE is a placental dysfunction, which results from defective deep placentation.[1] Several causative factors of defective placentation have been suggested, including maternal-fetal genotype incompatibility, preconception exposure to a paternal antigen that disrupts pregnancy-induced immunomodulation, and abnormal trophoblast decidual interaction. For successful pregnancy, embryo implantation and placental development require immune escape mechanisms from maternal immune attack against semi-allogenic fetal tissue.[15] Enrichment in the several autoimmune-associated pathways, such as the graft-versus-host disease, rheumatoid arthritis, and allograft rejection pathways in the PE group support the importance of maternal-fetal immune recognition.
The seasonal variation of PE incidence has been observed. Magnus and Eskild [16]conducted a cross-sectional population-based study with 1,869,388 deliveries in Norway between 1967 and 1998. The authors reported mothers of children born in August had the lowest risk for PE, while risk was highest in the winter months. Phillips et al[17] also identified the seasonal variation in the incidence of PE, which was more strongly associated with the timing of conception than the timing of delivery. The highest incidence of PE was associated with conception during the summer. However, there was no known exact mechanism of pathogenesis to explain the seasonal variation of PE development. One of the potential causes of seasonality is the seasonal variation in infectious diseases.[18] Interestingly, KEGG pathway analysis revealed that functional pathways associated with the immune response to infections, including viral myocarditis, Leishmaniasis, Staphylococcus aureus infection, and intestinal immune network for IgA production pathways, were enriched in PE in addition to the autoimmune-related pathway. A significant enrichment in pathways involved in various infections may support the role of infection in the development of PE. This result may provide clues for the role of infection in the development of PE.
Our study revealed that the ribosome pathway was enriched in the AF cell-free transcriptome of PE. Ribosomes are complex molecular machineries that consist of ribosomal RNAs (rRNAs) and proteins and they are responsible for the synthesis of protein during mRNA translation in the cell cytoplasm. In addition, ribosomal proteins (RPs) have several extraribosomal functions in addition to constituting the ribosome complex. Extraribosomal functions of RPs include regulating cell cycle and apoptosis, defending against viral infection, and repairing DNA damage.[19] We propose several hypotheses as to why the ribosome pathways were enriched in the PE group. The release of NAs into the extracellular fluid is thought to be related to cellular apoptosis, necrosis, and active secretion of cells.[20] rRNAs, which are involved in the ribosome pathway, comprise 80% of total intracellular RNAs.[21] Therefore, increased placental apoptosis in PE increase the abundance of rRNA in cf-NA.[22] Another hypothesis is regarding its role in stress response. Various cellular stresses, such as oxidative stress, inflammatory cytokines, and autoimmunity, have been reported in PE. To adapt to those stresses, cells synthesize proteins, thus activating ribosome pathways in PE.
There are few studies elucidated the pathophysiology of PE by analyzing proteome and transcriptome changes in the placenta.[23,24] Our research group conducted genome-wide expression profiling for 10 samples of placenta from pregnant women with PE and found a common factor in the dysregulation of ribosome, proteasome, and oxidative phosphorylation pathways.[24] van Uitert et al[23] performed meta-analysis with the placenta transcriptome data of 11 studies representing 116 PE pregnancies and 139 normotensive controls. The authors showed the involvement of the hypoxia/HIF1A pathway in gene expression profiling of PE and presented CREBBP/EP300, which was involved in the pathological processes in PE, as a novel regulator. However, those studies analyzed transcriptome expression in placenta obtained after delivery, and therefore, the results did not reflect the defect in early placental development. We collected AF during early second trimester before PE developed.
Our results demonstrated that RNA associated with the ribosome pathway, insulin-like growth factor B, and ubiquitin C were upregulated more than 10-fold in PE compared with the control. These genes may be predictive biomarkers for PE during early pregnancy. We isolated cf-RNA from AF obtained through amniocentesis, which is an invasive procedure. For clinical application, further validation studies using maternal blood should be performed.
There are several limitations in this study. First, we performed this study with transcriptome data obtained from 8 PE samples and 9 controls. This sample size is relatively small to draw comprehensive conclusions. Second, we did not conduct a functional study to support our hypothesis based on bioinformatic analysis. Third, there may have been annotation biases in our bioinformatic analysis, especially as inequality across gene annotation resources has been reported.[25] The researchers formed hypotheses based on the genes involved in the enriched processes and selected the genes that had previous experimental evidence, leading to annotation bias. Lastly, we did not use controls for subject characteristics in this study, which may have affected the gene expression patterns we were able to observe.
Despite these limitations, there are several strengths to this study. This study is the first to examine AF-cf-transcriptomes in PE. In addition, AF samples were obtained during early second trimester before the subjects developed PE. It is difficult to obtain fetal developmental information during early pregnancy due to the onset of hypertension and significant proteinuria after 20 weeks of pregnancy.
Our study demonstrated the comparable differences in early second trimester AF-cf-transcriptomes between the PE and control groups. In PE, the ribosome pathway and immune-associated pathways were enriched, which may reflect the importance of stress response and infection and immune-tolerance mechanisms in the pathogenesis of PE, respectively. Further studies are required to clarify and validate our results.
Author contributions
Formal analysis: Yong Wook Jung, Jung In Shim, Sung Han Shim.
Funding acquisition: Dong Hyun Cha.
Investigation: So Hyun Shim, Yun-jeong Shin, Sung Han Shim, Dong Hyun Cha.
Methodology: Yong Wook Jung, Jung In Shim, So Hyun Shim, Yun-jeong Shin, Sung Han Shim, Dong Hyun Cha.
Project administration: Dong Hyun Cha.
Resources: Sung Woon Chang, Dong Hyun Cha.
Software: Sung Han Shim.
Supervision: Sung Woon Chang.
Validation: Sung Han Shim.
Writing – original draft: Yong Wook Jung.
Writing – review & editing: Sung Woon Chang, Dong Hyun Cha.
Yong Wook Jung orcid: 0000-0003-2098-8143.
Footnotes
Abbreviations: AF-cf-RNA = amniotic fluid cell-free RNA, AF-cf-transcriptome = amniotic fluid cell-free transcriptome, DAVID = The online Database for Annotation, Visualization, and Integrated Discovery, DEGs = differentially expressed genes, GO = Gene ontology, KEGG = The Kyoto Encyclopedia of Genes and Genomes, OMIM = The Online Mendelian Inheritance in Man, PE = preeclampsia, RPs = ribosomal proteins, rRNAs = ribosomal RNAs, TTTS = twin--twin transfusion syndrome.
SWC and DHC equally contributed to this work as the co-corresponding authors.
Funding/support: This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI18C1839).
The authors declared that there are no conflicts of interest regarding the publication of the paper.
References
- [1].Chaiworapongsa T, Chaemsaithong P, Yeo L, et al. Pre-eclampsia part 1: current understanding of its pathophysiology. Nat Rev Nephrol 2014;10:466–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chaiworapongsa T, Chaemsaithong P, Korzeniewski SJ, et al. Pre-eclampsia part 2: prediction, prevention and management. Nat Rev Nephrol 2014;10:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jung YW, Shim SS, Park JE, et al. Analysis of the cell-free amniotic fluid transcriptome expressed during the euploid mid-trimester of pregnancy. Eur J Obstet Gynecol Reprod Biol 2016;203:94–8. [DOI] [PubMed] [Google Scholar]
- [4].Koide K, Slonim DK, Johnson KL, et al. Transcriptomic analysis of cell-free fetal RNA suggests a specific molecular phenotype in trisomy 18. Hum Genet 2011;129:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Massingham LJ, Johnson KL, Scholl TM, et al. Amniotic fluid RNA gene expression profiling provides insights into the phenotype of Turner syndrome. Hum Genet 2014;133:1075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Slonim DK, Koide K, Johnson KL, et al. Functional genomic analysis of amniotic fluid cell-free mRNA suggests that oxidative stress is significant in Down syndrome fetuses. Proc Natl Acad Sci U S A 2009;106:9425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hui L, Wick HC, Moise KJ, Jr, et al. Global gene expression analysis of amniotic fluid cell-free RNA from recipient twins with twin-twin transfusion syndrome. Prenat Diagn 2013;33:873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jang JH, Jung YW, Shim SH, et al. Global gene expression changes of amniotic fluid cell free RNA according to fetal development. Eur J Obstet Gynecol Reprod Biol 2017;216:104–10. [DOI] [PubMed] [Google Scholar]
- [9].Harris MA, Clark J, Ireland A, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 2004;32(Database issue):D258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 2006;34(Database issue):D354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hamosh A, Scott AF, Amberger J, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2002;30:52–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bianchi DW, LeShane ES, Cowan JM. Large amounts of cell-free fetal DNA are present in amniotic fluid. Clin Chem 2001;47:1867–9. [PubMed] [Google Scholar]
- [13].Larrabee PB, Johnson KL, Lai C, et al. Global gene expression analysis of the living human fetus using cell-free messenger RNA in amniotic fluid. JAMA 2005;293:836–42. [DOI] [PubMed] [Google Scholar]
- [14].Edlow AG, Vora NL, Hui L, et al. Maternal obesity affects fetal neurodevelopmental and metabolic gene expression: a pilot study. PLoS One 2014;9:e88661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Triggianese P, Perricone C, Chimenti MS, et al. Innate immune system at the maternal-fetal interface: mechanisms of disease and targets of therapy in pregnancy syndromes. Am J Reprod Immunol 2016;76:245–57. [DOI] [PubMed] [Google Scholar]
- [16].Magnus P, Eskild A. Seasonal variation in the occurrence of pre-eclampsia. BJOG 2001;108:1116–9. [DOI] [PubMed] [Google Scholar]
- [17].Phillips JK, Bernstein IM, Mongeon JA, et al. Seasonal variation in preeclampsia based on timing of conception. Obstet Gynecol 2004;104:1015–20. [DOI] [PubMed] [Google Scholar]
- [18].TePoel MR, Saftlas AF, Wallis AB. Association of seasonality with hypertension in pregnancy: a systematic review. J Reprod Immunol 2011;89:140–52. [DOI] [PubMed] [Google Scholar]
- [19].Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell 2009;34:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pos O, Biro O, Szemes T, et al. Circulating cell-free nucleic acids: characteristics and applications. Eur J Hum Genet 2018;26:937–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Eun H-M. Enzymology Primer for Recombinant DNA Technology. San Diego, CA: Academic Press; 1996. [Google Scholar]
- [22].Hahn S, Rusterholz C, Hosli I, et al. Cell-free nucleic acids as potential markers for preeclampsia. Placenta 2011;32Suppl:S17–20. [DOI] [PubMed] [Google Scholar]
- [23].van Uitert M, Moerland PD, Enquobahrie DA, et al. Meta-analysis of placental transcriptome data identifies a novel molecular pathway related to preeclampsia. PLoS One 2015;10:e0132468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kang JH, Song H, Yoon JA, et al. Preeclampsia leads to dysregulation of various signaling pathways in placenta. J Hypertens 2011;29:928–36. [DOI] [PubMed] [Google Scholar]
- [25].Haynes WA, Tomczak A, Khatri P. Gene annotation bias impedes biomedical research. Sci Rep 2018;8:1362. [DOI] [PMC free article] [PubMed] [Google Scholar]