

Abstract
The proteasome is an ATP-dependent, 2.5-megadalton machine responsible for selective protein degradation in eukaryotic cells. Here we present cryo-EM structures of the substrate-engaged human proteasome in seven conformational states at 2.8-3.6 Å resolution, captured during breakdown of a polyubiquitylated protein. These structures visualize a continuum of dynamic substrate-proteasome interactions from ubiquitin recognition to substrate translocation, during which ATP hydrolysis sequentially navigates through all six ATPases. Three principal modes of coordinated hydrolysis are observed, featuring hydrolytic events in two oppositely positioned ATPases, in two adjacent ATPases, and in one ATPase at a time. These hydrolytic modes regulate deubiquitylation, translocation initiation and processive unfolding of substrates, respectively. ATP hydrolysis powers a hinge-like motion in each ATPase that regulates its substrate interaction. Synchronization of ATP binding, ADP release and ATP hydrolysis in three adjacent ATPases drives rigid-body rotations of substrate-bound ATPases that are propagated unidirectionally in the ATPase ring and unfold the substrate.
The ubiquitin-proteasome pathway (UPP) plays a central role in selective protein degradation in eukaryotic cells. It regulates myriad cellular processes, such as cell cycle, apoptosis, immune response, inflammation, and the response to proteotoxic stress1–3. Ubiquitylated substrates are recognized and degraded by the 2.5-megadalton 26S proteasome holoenzyme3. The holoenzyme is assembled from a barrel-shaped, proteolytically active core particle (CP) and two 19S regulatory particles (RP) capping both ends of the CP cylinder. The RP controls substrate access into the CP and is formed from the lid and base subcomplexes. Recognition of ubiquitylated substrates is mediated by ubiquitin receptors Rpn1, Rpn10 and Rpn132–4. Once substrate is captured by the RP, its globular domains are mechanically unfolded by a ring-like heterohexameric adenosine triphosphatase (ATPase) motor in the base. The motor module consists of six distinct subunits, Rpt1−6, from the ATPases-associated-with-diverse-cellular-activities (AAA) family1–3, and regulates the engagement, Rpn11-catalyzed deubiquitylation5 and degradation of substrates in an ATP-dependent manner6 through hitherto unknown mechanisms.
Previous cryo-electron microscopy (cryo-EM) analyses have revealed the architecture of the substrate-free holoenzyme in six distinct states3,7–10. However, it remains unclear how the conformations of the substrate-free holoenzyme are related to their functional states in the presence of substrate. In this work, we have determined atomic structures of the substrate-engaged human proteasome in seven conformational states. Our analysis reveals mechanisms by which the substrate is engaged, deubiquitylated, unfolded and translocated by the human proteasome.
Overview of seven conformational states
To capture the human proteasome in the action of substrate processing, we used the model substrate Sic1PY and a novel nucleotide-substitution strategy11. In brief, the purified holoenzyme was first primed with polyubiquitylated Sic1PY in stoichiometric excess in the presence of 1 mM ATP for a short interval of time, then supplied with 1 mM ATPγS before being vitrified into cryo-EM samples (see Methods). The slowly hydrolyzed ATPγS is expected to compete with ATP in occupying nucleotide-binding pockets in the AAA-ATPases, thus potentially pausing the substrate-engaged proteasome in any possible intermediate states before the completion of substrate degradation. Through this approach we determined cryo-EM structures of the substrate-engaged proteasome in seven distinct conformational states, designated EA1, EA2, EB, EC1, EC2, ED1 and ED2, to nominal resolutions of 2.8-3.6 Å (Fig. 1, Extended Data Figs. 1–3, Extended Data Table 1).
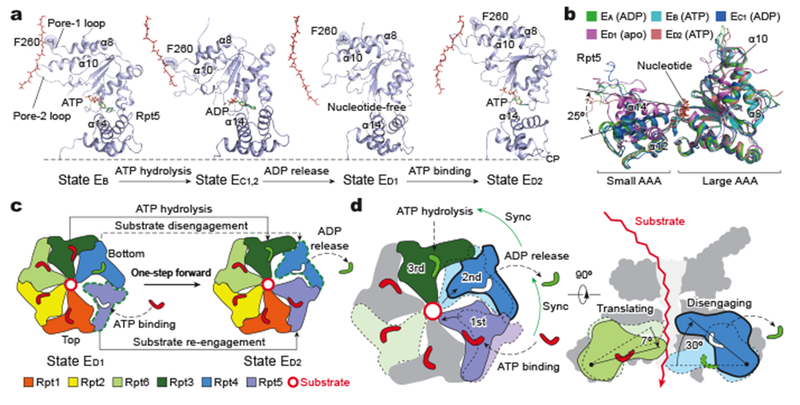
Figure 1. Cryo-EM structures of the substrate-bound human proteasome in distinct states.
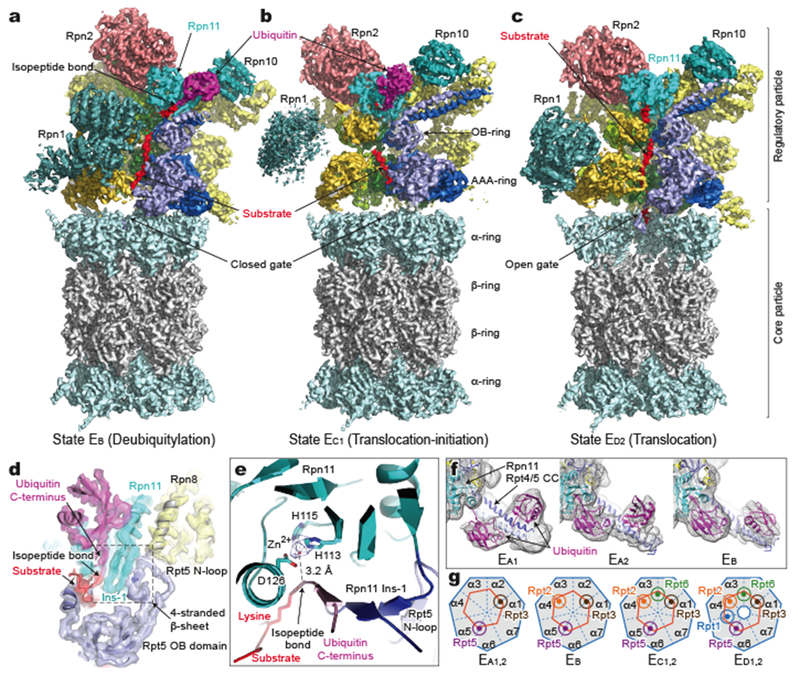
a-c, Cryo-EM density maps of substrate-bound human proteasome in state EB at 3.3 Å (a), in state EC1 at 3.5 Å (b), and in state ED2 at 3.2 Å (c). The Rpt1 density is omitted in a-c to show the substrate density inside the ATPase ring. Two α-subunits are omitted to show substrate density inside the CP gate in c. d, A close-up view of the quaternary interface around the scissile isopeptide bond between ubiquitin and the substrate lysine in EB. The cryo-EM density is rendered as a transparent surface, superimposed with the cartoon representation of the atomic model. e, A close-up view of the zinc ion (hotpink sphere) closely approached by the isopeptide bond. The zinc ion density is shown as a blue mesh at 10σ level. Side chains of Rpn11 coordinating with the zinc ion are labeled. f, Comparison of two ubiquitin moieties between Rpn11 and Rpt4/5 CC among EA1, EA2 and EB. The cryo-EM densities rendered as grey mesh representations are low-pass filtered to 8 Å. The atomic model of ubiquitin is shown as a magenta cartoon representation. g, A schematic diagram summarizes the Rpt C-terminal insertion to the α-pockets of the CP and the state of CP gate in all states (Extended Data Fig. 5). The CP is represented as a heptagon, the ATPase ring as a hexagon, and the Rpt C-tail insertion as a colored sphere in a circle.
States EA1 and EA2 presumably represent conformations of initial ubiquitin recognition, and capture snapshots before and after one ubiquitin near Rpn10 is bound to Rpn11, respectively12 (Fig. 1f). State EB reveals a novel deubiquitylation-compatible complex, in which the isopeptide bond between substrate lysine and ubiquitin is visible in the vicinity of the zinc-binding site of Rpn11 (Fig. 1a, d)8. States EC1 and EC2 present two successive conformations that are compatible with the initiation of substrate translocation, whereas states ED1 and ED2 capture two consecutive conformations of processive substrate translocation (Fig. 1b, c).
Key structural features of the seven states demonstrate remarkable spatiotemporal continuity (Fig. 2, Extended Data Figs. 4–8, Extended Data Table 2, Supplementary Video 1). Foremost, in both states EA1 and EA2, a ubiquitin density is observed on the T1 site of Rpn1, and two ubiquitin densities, presumably linked via Lys63, abut the Rpt4-Rpt5 N-terminal coiled coil (CC) domain next to Rpn10 (Fig. 1f, Extended Data Fig. 4a)4. One ubiquitin is tightly bound to Rpn11 throughout states EA2, EB and EC1, but interestingly it is released in EC2, ED1 and ED2. Notably, two, three, four and five C-terminal tails of the ATPases are inserted in the inter-subunit surface pockets on the α-ring of the CP in EA, EB, EC and ED, respectively7,8 (Fig. 1g, Extended Data Fig. 5). The CP gate is thus closed in EA1,2, EB and EC1,2, but open in ED1,2 (Extended Data Fig. 5a)7,8. States EA1,2 also present 13 potential substrate densities in the chamber of the closed CP, including one in contact with the proteolytic site Thr1 of the β2 subunit, which is found in all states (Extended Data Fig. 6a, Extended Data Table 2). More details regarding the spatiotemporal continuity in substrate binding and nucleotide states are described in the following sections. Taken together, these observations indicate that the seven states are on the pathway of substrate processing by the holoenzyme.
Figure 2. Dynamic substrate-proteasome interactions.
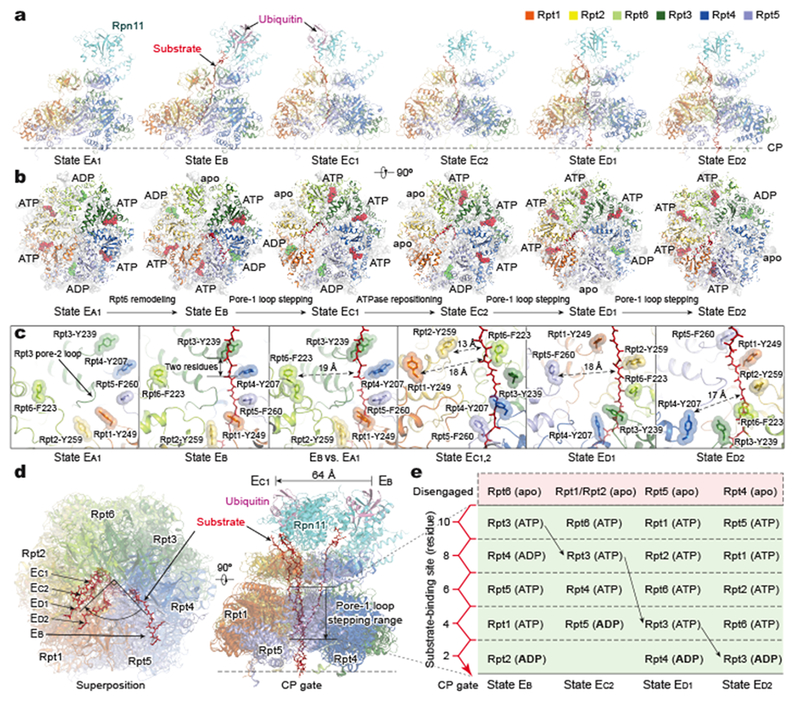
a, Side views of the ATPase-Rpn11 subcomplex interacting with substrate in five states (EB, EC1,2 and ED1,2) in comparison with state EA1. The substrate is modelled as a polypeptide backbone structure and is represented as red sticks. Ubiquitin, Rpn11 and ATPases are rendered as transparent cartoons to show the substrate translocating inside the axial channel. The relative location of the CP is marked by the horizontal dashed line. b, Top views of the ATPase motors of distinct states. Nucleotides are shown in stick representation. The sphere representation of ADP is in green. The CP is rendered as a grey surface representation. The structures are aligned altogether using their CP components. c, Varying architecture of pore-1 loop staircase interacting with substrate in five states (EB, EC1,2 and ED1,2) as compared to that in state EA1. Aromatic residues in the pore-1 loops are labeled and shown in stick representation superimposed with transparent sphere representation for highlighting. The distances from disengaged pore-1 loops to the substrate are marked. The third panel shows the superposition of state EB with state EA. d, Top view (left) and side view (right) of all substrate-bound ATPase-Rpn11 structures superimposed together based on structural alignment against the CP. e, A diagram summarizing the axial stepping of the substrate-contacting pore-1 loops and their correlations with nucleotide states. The vertical axis shows the relative location of pore-1 loops interacting the substrate, with the CP positioned at the bottom. Numbers label the relative distance from the lowest substrate-pore loop contact, using the number of residues as a metric. State EC1 is omitted here as its AAA-ATPase structure is identical to that of EC2. The color code of subunits used in all panels is shown in the upper right inset.
Quaternary rearrangement for substrate deubiquitylation
A prominent feature in state EB is the formation of a quaternary subcomplex involving substrate-ubiquitin-bound Rpn11, Rpn8 and the N-loop (residue 99-119) of Rpt5, which emanates from the top of its oligonucleotide-/oligosaccharide-binding (OB) domain13 (Fig. 1d-f). To facilitate such a quaternary rearrangement, the lid is rotated outward away from the axis of the OB ring relative to state EA, resulting in a broader entrance to the ATPase axial channel (Fig. 2a, Extended Data Fig. 4f, g).
The ubiquitin-bound Rpn11-Rpn8-Rpt5 subcomplex starts to form in state EA2, although the ATPase ring is not yet engaged with substrate (Extended Data Fig. 4a, b). Rpn11-bound ubiquitin also contacts the Rpt5 CC domain (at residue 61-64) in EA2 and resides midway between its positions in states EA1 and EB (Fig. 1f). In state EA1, this ubiquitin abuts the Rpt4-Rpt5 CC domain in the vicinity of Rpn11, but does not contact Rpn11 (Fig. 1f). It is presumably covalently conjugated to substrate and linked to Rpn10-bound ubiquitin moieties via Lys63. The Rpt4-Rpt5 CC domain is shifted up during the EA1-to-EA2 transition, shrinking the gap between Rpn11 and Rpt4-Rpt5 CC. These observations suggest that EA2 captures an intermediate state where a ubiquitin is being transferred from Rpt4-Rpt5 CC to Rpn11.
The Rpn11-ubiquitin interface is centered on a hydrophobic pocket around Trp111 and Phe133 of Rpn11, with the positioning of ubiquitin being comparable to that in the crystal structure of the ubiquitin-bound yeast Rpn11-Rpn8 complex14. The Insert-1 (Ins1) loop of Rpn11 assumes a β-hairpin conformation, and pairs on one side with the C-terminal strand of ubiquitin and on the other side with a segment of the Rpt5 N-loop, forming a four-stranded β-sheet (Fig. 1d). This β-sheet directs the ubiquitin C-terminus toward the catalytic zinc-binding site in Rpn11 and places the isopeptide bond within 3.2 Å of the zinc ion, which has a strong density in our cryo-EM maps (Fig. 1e). Compared to the crystal structure of ubiquitin-bound Rpn11 and the Rpn11 structure in state EC1, the Ins1 β-hairpin is rotated outward by 5 Å in state EB, allowing proper coordination of the isopeptide bond with the zinc ion14 (Extended Data Fig. 4d, e). This finding suggests that the conserved Rpt5 N-loop stabilizes the Rpn11-ubiquitin contact and optimizes the orientation of the scissile isopeptide bond for efficient deubiquitylation. By contrast, the Rpt5 N-loop is disordered in most other states (EA1, EC1,2 and ED1,2) and the isopeptide bond is not observed between the Rpn11-bound ubiquitin and substrate in EC1 (Fig. 1b).
The Ins1 region of Rpn11 alternates among three distinct configurations (Extended Data Fig. 4c). It is a large open loop in state EA1, folds into a β-hairpin throughout states EA2, EB and EC1 whenever ubiquitin is bound, and is converted into a smaller, tighter loop in states EC2 and ED1,2. Taken together, the quaternary organization surrounding the zinc-binding site appears to explain why Rpn11 exhibits much higher deubiquitylation activity in the context of the proteasome than in its non-proteasomal forms15–18.
Substrate interactions with the holoenzyme
In the progression from states EB to ED1,2, substrate contact with Rpn11 is consistently centered around a hydrophobic groove at Phe118 and Trp121 of Rpn11. In state EB, this binding site faces the Rpt3-Rpt4 OB interface and the substrate extends straight from this site to Val125 of Rpn11, beneath which the isopeptide bond linking the ubiquitin to the substrate is held. In states EC1,2 and ED1,2, the substrate closely approaches Phe118 of Rpt1 in the interior of the OB ring.
Within the axial channel of the ATPase ring, the substrate is threaded into a right-handed spiral staircase architecture in contact with the aromatic residues of pore-1 loops (Fig. 2c, Extended Data Fig. 6b-d). The aromatic side-chains intercalate with the zigzagging main chain of substrate through hydrophobic interactions. In addition, the main chains of the pore-1 loops potentially form hydrogen bonds with the main chain of substrate. Substrate-contacting pore-1 loops are evenly distributed along the substrate, with two adjacent pore-1 loops spanning two amino acid residues in the substrate. The topology of this substrate-pore loop staircase architecture is nearly invariant from states EB to ED2. In contrast, the overall path of the translocating substrate is significantly changed from EB to EC/D (Fig. 2d).
The pore-1 loops of Rpt3, Rpt6, Rpt1 and Rpt5 reside at the highest position in contact with the substrate in EB, EC, ED1 and ED2, respectively. Notably, the pore-1 loop of Rpt3 moves from the highest position to the lowest position in the substrate-pore loop staircase from EB to ED2 (Fig. 2e). Meanwhile, the pore-2 loops support the opposite side of the substrate through their charged acidic residues, forming another, somewhat shorter staircase beneath that of the pore-1 loop. In states EB, EC, ED1 and ED2, the pore loops from Rpt6, the Rpt1-Rpt2 dimer, Rpt5 and Rpt4 are disengaged from the substrate, respectively (Fig. 2c).
A continuum of nucleotide states
The current resolution allows us to confidently distinguish ADP from ATP in the nucleotide-binding pockets of the ATPases (Fig. 2b, Extended Data Figs. 3c, 7). However, it is not possible to differentiate between ATP and ATPγS, as a mixture of both ATP and ATPγS was present in our buffer (see Methods). Thus, it remains possible that the nucleotide-binding sites competent for ATP hydrolysis are occupied by ATPγS in our structures. Previous studies have shown that ATPγS binding largely emulates ATP binding and may not necessarily change ATPase conformations8. For simplicity, we will refer to nonhydrolyzed nucleotides as ATP hereafter.
Notably, the ADP-bound states navigate counterclockwise sequentially from Rpt6 to Rpt3 throughout all six ATPase subunits, indicating a full cycle of coordinated ATP hydrolysis throughout the AAA-ATPase ring from state EA to ED2 (Fig. 2b). The nucleotide states of the ATPases are strongly correlated with their substrate-pore loop interactions (Fig. 2e). Foremost, ATP is invariably found to bind the substrate-engaged ATPases at the middle or top positions in the pore-loop staircase, except in the early states EA and EB. Except for EA, at least one ATPase in each conformational state exhibits a very weak or partial density in its nucleotide-binding pocket. We refer to the nucleotide state of these ATPases as an apo-like state. The apo-like state is always observed in the ATPases whose pore loops are disengaged from substrate. Thus, all apo-like subunits form prominent gaps at the interfaces with their nearest neighbors on both sides, resembling in this respect Rpt6 in state EB (Fig. 3a).
Figure 3. Structural basis for nucleotide-driven substrate engagement in the AAA-ATPase channel.
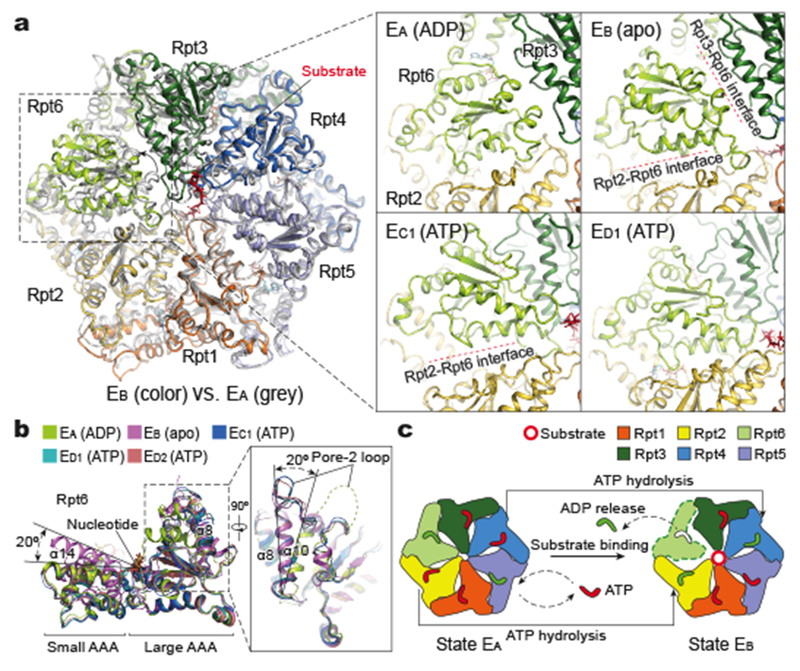
a, Superposition of the AAA-ring structures of states EA (grey) and EB (color). The insets show side-by-side comparison of Rpt6 conformations in the four most distant states. Interfacial gaps are marked by red dashed lines. b, Superposition of the Rpt6 AAA domain structures from five distinct states aligned against the large AAA subdomain shows that Rpt6 assumes three major conformations. Transition from EA to EB involves both refolding of the pore-2 loop, shown in the right insert, and a 20° rigid-body rotation between the large and small AAA subdomains. c, Schematic representation of ATP hydrolysis and nucleotide exchange required to implement the transition from EA to EB.
Adjacent to the apo-like subunit, the ATPase whose pore-1 loop resides at the lowest position in contact with substrate is always bound to ADP (Fig. 2b, e). The apo-like subunits and their counterclockwise adjacent ADP-bound subunits consistently exhibit a relatively wide nucleotide-binding pocket, with the arginine fingers from the clockwise adjacent subunit displaced more than 10 Å away from the Walker A motif (Extended Data Fig. 8a). In contrast, whenever the ATPases engaged with substrate have their pore-1 loops located in the middle or top register in the substrate-pore loop staircase, the pocket is always tightly packed, with the arginine fingers residing within 3-4 Å away from either γ-phosphate or β-phosphate, indicating potential competence for ATP hydrolysis in these sites.
ATP-dependent substrate engagement
The organization of the substrate-engaged pore-1 loop staircase in EB is highly similar to that of the substrate-free pore-1 loop staircase in EA, suggesting that state EB reflects substrate engagement prior to translocation (Fig. 2c). Structural comparison of states EA and EB clarifies how ATP hydrolysis regulates substrate engagement for deubiquitylation (Fig. 3). In state EA, the AAA channel is too narrow to engage substrate, suggesting that it is in a closed state. To open the channel for the substrate engagement, the AAA-ATPase ring must rearrange its quaternary organization.
Indeed, the AAA domain of Rpt6 undergoes dramatic structural changes from EA to EB. A ~40° rotation out of the plane of the ATPase ring is observed in the large AAA subdomain of Rpt6, creating prominent gaps at the Rpt2-Rpt6 and Rpt3-Rpt6 interfaces (Fig. 3a). By contrast, the AAA domains of other ATPases mostly move as a single rigid body with subtle changes restricted to the pore loops. In state EA, the pore-2 loop of Rpt6 is largely disordered. Markedly, this loop refolds into an ordered structure in EB, spanning residues 251-266 (Fig. 3b). Consistent with this observation, the ADP bound to Rpt6 in state EA is released in EB. Thus, ATP hydrolysis and ADP release in Rpt6 are programmed to trigger an iris-like movement in the ATPase ring that opens the axial channel (Fig. 2a, d). The coordinated ATP hydrolysis in Rpt5 directly opposite from Rpt6 in EA, and in Rpt4 opposite from Rpt2 in EB, is expected to increase the conformational flexibility of the ATPase ring required to open the AAA channel, thus allowing substrate engagement (Fig. 3d). Indeed, a greater degree of pore-1 loop movement is observed in Rpt3, Rpt4 and Rpt5 than in Rpt1 and Rpt2 during the EA-to-EB transition (Fig. 3a).
Initiation of substrate translocation
From state EA to ED1, the Rpt1-Rpt2 dimer undergoes a complete cycle of ATP hydrolysis and nucleotide exchange that initiates substrate translocation (Fig. 4). During the EB-to-EC1 transition, a ~40° vertical rotation of the Rpt1-Rpt2 dimer moves the corresponding pore loops to almost the highest altitude but 13-18 Å away from the substrate (Figs. 2c, 4a, Extended Data Fig. 4i). Concomitantly, ATP binding to Rpt6 promotes engagement of the Rpt6 pore-1 loop with substrate. Together, these conformational changes result in one-step forward translocation of the substrate by a distance of two residues (Figs. 2e, 4b). ADP release in Rpt1 during the EC1-to-EC2 transition does not trigger obvious conformational changes (Fig. 4a). However, during the EC2-to-ED1 transition, ATP binding to both Rpt1 and Rpt2 drives their substrate re-engagement at the top of the substrate-pore loop staircase and, together with ADP release from Rpt5, results in two-step forward translocation of the substrate (Fig. 4b). The total distance of substrate translation over the first three steps is comparable to that from the lowest substrate-bound pore loop to the CP gate.
Figure 4. ATP hydrolysis drives initial three steps of substrate translocation through the AAA-ATPase channel.
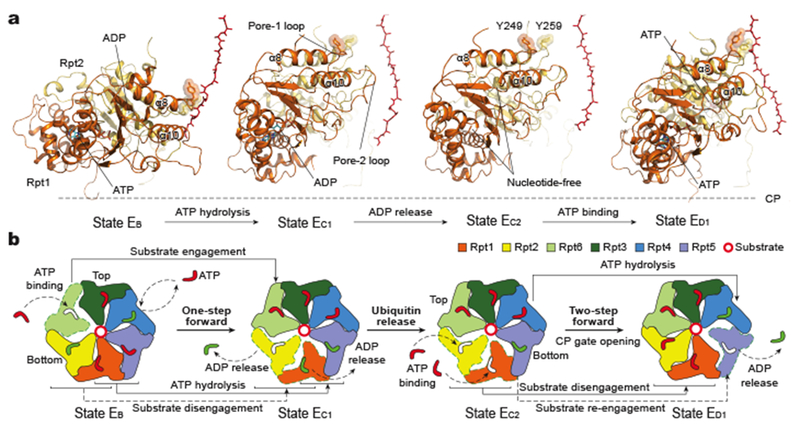
a, Side-by-side comparison of the Rpt1-Rpt2 dimer conformations in four sequential states that undergo a complete cycle of ATP hydrolysis and exchange in the Rpt1-Rpt2 dimer. Structures are aligned against the CP to show their conformational changes relative to the CP gate. Tyrosine residues in the pore-1 loops are shown in stick representation and highlighted with transparent sphere representation. Substrate is shown in red in stick representation. b, Schematic representation of the cascade of ATP hydrolysis and nucleotide exchange required to implement the transition from EB to ED1.
Interestingly, the ATPase ring conformation is nearly identical between states EC1 and EC2, although multiple events are associated with the EC1-to-EC2 transition, including ubiquitin dissociation from Rpn11, ADP release from Rpt1, conformational changes in the lid, and repositioning of the ATPase ring above the CP (Fig. 2a, b, Extended Data Fig. 4g, h). Thus, the initiation of substrate translocation is extensively coordinated with other regulatory events preparing the proteasome for processive substrate degradation (Fig. 1g, Extended Data Table 2).
Substrate unfolding and translocation
Systematic structural alignment defines a generic hinge-like rotation of 20-25° in each ATPase between its small and large AAA subdomains upon nucleotide binding or release (Figs. 3b, 5b, Extended Data Fig. 8b-e). By contrast, the dihedral angle between the small and large AAA subdomains remains nearly invariant between the ADP-bound and ATP-bound states during substrate translocation, suggesting that nucleotide binding locks the AAA domain into a single rigid body. Hence, the release of γ-phosphate after ATP hydrolysis appears insufficient to immediately trigger intrinsic motion in the AAA domain; instead, it potentiates such conformational changes, which can be triggered later by either nucleotide exchange or changes in inter-ATPase interactions (Fig. 5a-c).
Figure 5. Mechanism for processive substrate translocation driven by a complete cycle of ATP hydrolysis.
a, Side-by-side comparison of Rpt5 conformations in four sequential states that cover a complete cycle of ATP hydrolysis and exchange in Rpt5. The structures are aligned against the CP to show their conformational changes relative to the CP gate. Phenylalanine-260 of the pore-1 loop is shown in stick representation and highlighted with transparent sphere representation. Substrate is shown in red in stick representation. b, Superposition of Rpt5 structures from five distinct states aligned against the large AAA subdomain shows that Rpt5 assumes two major conformations between apo-like and nucleotide-bound states. c, Schematic representation of ATP hydrolysis and nucleotide exchange required to implement the transition from ED1 to ED2. d, Schematic illustrates mechanism for processive substrate translocation. Synchronization of nucleotide processing in three adjacent ATPases, i.e., ATP binding, ADP release and ATP hydrolysis (left), creates differential vertical rotations in each ATPase that cooperatively translate the substrate (right).
Structural comparison of ED1 and ED2 provides insight into the mechanism of substrate unfolding and translocation (Fig. 5d). To establish processivity in substrate translocation, at least three adjacent ATPases must synchronize their nucleotide processing: the first binding an ATP, the second releasing an ADP, and the third hydrolyzing an ATP (Fig. 5d, left). In the second ATPase, ADP release allows conversion of potential energy harvested from ATP hydrolysis into kinetic energy, which powers the most prominent vertical rotation (30-40°) in the ATPase ring and is transferred to adjacent ATPases from both sides along the ATPase ring (Fig. 5d, right; Extended Data Fig. 4i-l). On the clockwise side, the vertical rotation of the second ATPase is synchronized with conformational changes in the first ATPase during its binding of ATP. On the counterclockwise side, the rotation facilitates ATP hydrolysis in the third ATPase, likely through repositioning of the arginine fingers coordinating the neighboring ATP. Following ATP hydrolysis, γ-phosphate release from the third ATPase abolishes the interaction between γ-phosphate and the trans-acting arginine fingers from the second ATPase, thus destabilizing their inter-subunit association and promoting substrate disengagement from the second ATPase. Such coordinated hydrolysis is essential for unidirectionally propagating conformational changes in the ATPase ring (Fig. 5c).
The large vertical rotation of the second ATPase away from the lowest register of the substrate-pore loop staircase makes room at the bottom, and via inter-subunit interactions, drives each of the four substrate-bound ATPases to rotate downward as a single rigid body with a small differential angle (5-10°) (Fig. 5d, right). Together, these substrate-bound ATPases collectively translate the substrate toward the CP via the pore-loop staircase. Through such a highly concerted process, the chemical energy of ATP hydrolysis is converted into the mechanical work of substrate unfolding (Supplementary Video 2).
Long-range quaternary allosteric regulation
In both states ED1 and ED2, the Rpn1 toroidal domain forms a surface cavity with the Rpt1-Rpt2 CC domain, into which a short helix of Rpn2 is inserted (Extended Data Fig. 9a-c). This helix resides in the middle of a long loop (residue 820-871) emanating from the Rpn2 toroidal domain. This long-range association of Rpn1-Rpn2, which is not observed in other states (EA-C), seems to stabilize a larger interface formed between Rpn1-Rpn2 and Rpt1-Rpt2, and may thus regulate substrate translocation allosterically. Furthermore, the Rpt5 CC domain switches its interaction between Rpn9 and Rpn10 in states ED1 and ED2, which also seems to regulate substrate processing in a long-range fashion, consistent with a recent biochemical study19 (Extended Data Fig. 9d, e).
Insights into the complete cycle of substrate processing
Our data suggest that ATP hydrolysis in the proteasome holoenzyme follows distinct modes at different stages of substrate processing (Fig. 6, Extended Data Fig. 10). The first mode features coordinated ATP hydrolysis in a pair of oppositely positioned ATPases. We speculate that this is orchestrated to promote initial substrate recognition and deubiquitylation. This mode is reminiscent of the nucleotide-binding pattern observed in state SD2 of the substrate-free human proteasome8 and the hexameric ClpX protease of E. coli20. The second mode features coordinated ATP hydrolysis in two adjacent ATPases and is used to initiate substrate translocation and to coordinate CP gating. This mode seems compatible with previous studies suggesting that binding of four nucleotides promotes substrate engagement6,21. Substrate processing culminates in a third hydrolytic mode, which is by comparison greatly simplified, as it features sequential hydrolysis of one nucleotide at a time. The third mode is likely the most efficient for maintaining processivity in substrate translocation. In both the second and third modes, synchronized ATP binding and ADP release in at least two adjacent ATPases convert the chemical energy of ATP hydrolysis into differential rigid-body rotations in the ATPase ring that mechanically unfold the substrate and are propagated unidirectionally through coordinated ATP hydrolysis in the counterclockwise adjacent ATPase. The third mode is consistent with the proposed ATP hydrolysis mechanism of several other hexameric ATPase motors22–25.
Figure 6. Model of the complete cycle of substrate processing by the human 26S proteasome.
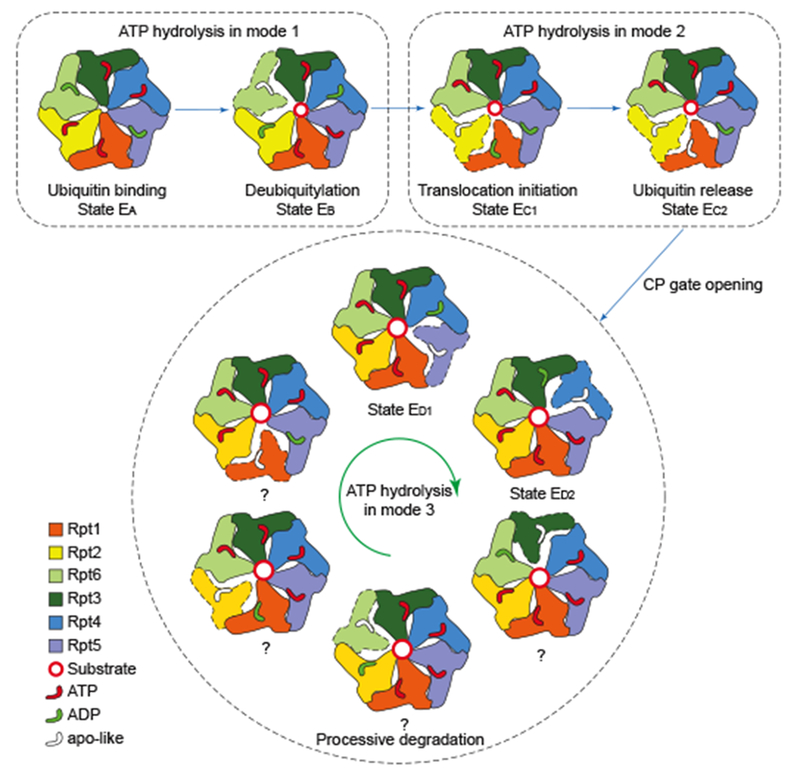
The cartoon summarizes the concept of three principal modes of coordinated ATP hydrolysis observed in the seven states and our proposal of how they regulate the complete cycle of substrate processing by the proteasome holoenzyme.
During transitions between consecutive states of the proteasome, the multiplicity of nucleotide processing events in distinct ATPases implies that fast steps and sparsely populated intermediate states might have been missed in our cryo-EM reconstructions. Future studies identifying these missing intermediates will be required to clarify how ATP hydrolytic events and nucleotide exchange are coordinated with each other and allosterically linked to substrate translocation.
In summary, we have determined the atomic structures of substrate-engaged human proteasome in seven native states during degradation of a polyubiquitylated substrate. These structures establish a foundation for understanding dynamic substrate-proteasome interactions during the complete cycle of substrate processing, and provide a wealth of information accounting for several decades of biochemical studies of proteasome function1–3. A plethora of potential substrate-binding sites revealed in this study may facilitate future development of drugs that modulate proteasome functions implicated in various diseases, such as multiple myeloma and neurodegenerative diseases.
Methods
Preparation of polyubiquitylated Sic1PY.
PY motif-inserted Sic1 (Sic1PY) and WWHECT were chosen as the model substrate and the E3 ubiquitin ligase, respectively11. The PY motif is recognized by WW domains the Rsp5 family of ligases. In the Sic1PY construct used in this study, a PY motif of Pro-Pro-Pro-Ser was inserted to the N-terminus (MTPSTPPSRGTRYLA) of the Cdk inhibitor Sic126,27, resulting in a modified N-terminus of MTPSTPPPPPSSRGTRYLA, where the first residue to be ubiquitylated is likely Lys18 of Sic1PY. The WWHECT was derived from wildtype Rsp5 by deletion of its N-terminal 220 amino acids, and conjugates ubiquitin chains via Lys63 linkage11. Both proteins were expressed in Escherichia coli and purified as previously reported11. Plasmids expressing Sic1PY and WWHECT were gifts from Dr. Y. Saeki (Tokyo Metropolitan Institute of Medical Science). After plasmid transformation into BL21 (DE3) cells, cultures were grown to OD600 of 0.7 in LB medium with 50 μg/ml ampicillin. Cultures were cooled to 30 °C, 0.5 mM IPTG was added to 0.5 mM, and incubation proceeded for 3 h. After being harvested (3000 × g, 10 min), cells were suspended and lysed by sonication in 50 mM PBS (pH 7.0) containing 300 mM NaCl, 10% glycerol, 1 mM DTT, 0.2% Triton X-100 and 1×protease inhibitor cocktail. The supernatant was recovered after centrifugation (15,000 × g, 30 min), then incubated with pre-equilibrated TALON resin for 2 h at 4°C. After this binding step, the resin was washed with 20 column volumes of 50 mM Tris-HCl (pH 7.5) containing 100 mM NaCl, 10% glycerol, and 1 mM DTT. Sic1PY was then eluted with the same buffer containing 150 mM imidazole. The eluted sample was further purified by FPLC (Superdex 75; 0.25 ml/min), using a buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 10% glycerol.
To purify WWHECT, plasmid-transformed BL21 (DE3) cells were grown to OD600 of 0.5 in LB medium with 50 μg/ml ampicillin. The culture was then cooled to 20 °C and WWHECT synthesis induced by the addition of IPTG to 0.2 mM. Cells were harvested 15 h after induction and lysed through the same procedure as used for the Sic1PY. A 15,000 × g supernatant was incubated with pre-equilibrated glutathione sepharose resin for 2 h at 4°C. The resin was washed by 20 column volumes of washing buffer (50 mM Tris-HCl [pH 7.5] containing 100 mM NaCl, 10% glycerol and 1 mM DTT), then incubated with the same buffer containing PreScission protease for 12 h at 4°C. The resin was removed by centrifugation and the supernatant was then applied to FPLC (Superdex 75) as described above.
To ubiquitinate the Sic1PY, 40 μg/ml Sic1PY, 500 nM Ube1 (Boston Biochem), 2 μM UbcH5a (Boston Biochem), 100 μg/ml WWHECT and 1 mg/ml ubiquitin (Boston Biochem) were incubated in reaction buffer (50 mM Tris-HCl [pH 7.5],100 mM NaCl, and 10% glycerol, 2 mM ATP, 10 mM MgCl2, and 1 mM DTT) for 3 h at room temperature. Pre-equilibrated TALON resin was then incubated with the this sample for 1 h at 4°C. After the resin was washed with 20 column volumes of the wash buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 10% glycerol), the polyubiquitinated Sic1PY (PUb-Sic1PY) was eluted with the same buffer containing 150 mM imidazole. The elution was applied to an Amicon ultrafiltration device with 30K molecular cut-off for removal of imidazole. The ubiquitination reaction was examined by Western blotting with anti-T7 antibody (Extended Data Fig. 1f).
Purification of the human 26S proteasome.
Human proteasomes were affinity-purified on a large scale from a stable HEK293 cell line harboring HTBH (hexahistidine, TEV cleavage site, biotin, and hexahistidine) tagged hRPN11 (a gift from L. Huang, University of California, Irvine)28. The cells were Dounce-homogenized in a lysis buffer (50 mM PBS [pH 7.5], 10% glycerol, 5 mM MgCl2, 0.5% NP-40, 5 mM ATP and 1 mM DTT) containing protease inhibitors. Lysates were cleared by centrifugation (20,000 × g, 30 min), then incubated with NeutrAvidin agarose resin (Thermo Scientific) for 3 h at 4 °C. The beads were washed with excess lysis buffer followed by wash buffer (50 mM Tris-HCl [pH 7.5], 1 mM MgCl2 and 1 mM ATP). 26S proteasomes were cleaved from the beads by TEV protease (Invitrogen). The resin was removed by centrifugation and the supernatant was then further purified by gel filtration on a Superose 6 10/300 GL column at a flow rate of 0.15 ml/minute in buffer (30 mM Hepes [pH 7.5], 60 mM NaCl, 1 mM MgCl2, 10% glycerol, 0.5 mM DTT, 0.6 mM ATP). Gel-filtration fractions were concentrated to about 2 mg/ml and the buffer was exchanged to 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM ATP and 10% glycerol (Extended Data Fig. 1a-c).
Biochemical verification of the substrate-bound human proteasome.
To verify the preparation of the PUb-Sic1PY, we performed degradation assays on the PUb-Sic1PY using our purified human 26S proteasome. 100 nM proteasome was incubated with 20 μg/ml PUb-Sic1PY for 10 min at 37°C in a buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10% glycerol, 5 mM ATP. The reaction was stopped by adding SDS loading buffer and 100 mM DTT. Samples were collected at the time points of 2, 5, and 10 min. Sic1PY degradation reaction was followed by Western blotting with the anti-T7 antibody (Extended Data Fig. 1h).
To verify the formation of proteasome-substrate complex in our cryo-EM imaging experiments, we crosslinked the proteasome-substrate complexes and examined them by native gel electrophoresis. However, crosslinking was not used for the sample preparation for cryo-EM data collection in order to preserve the native states of substrate interactions with the proteasome. Before crosslinking, proteasome and PUb-Sic1PY samples were first exchanged to a buffer containing 50 mM PBS (pH 7.5), 100 mM NaCl, 10% glycerol, 1 mM ATP using Zeba™ Micro Spin Desalting Columns (7K, Thermo Fisher). Then 1 μl of 2 mg/ml PUb-Sic1 and 1 μl of 1 mg/ml proteasome were mixed with 17 μl of the same buffer for 30s. After 1mM ATPγS was added, 1 µl of 2.3% freshly prepared solution of glutaraldehyde was added and incubated for 15 minutes at 37°C. The cross-linked complex was then examined by the native gel electrophoresis (Extended Data Fig. 1g).
Cryo-EM imaging and data collection.
10 μl of 2 mg/ml proteasome was incubated with 9 μl of 2 mg/ml PUb-Sic1PY (molar ratio ~3:1 for substrate vs proteasome) for 30 s (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 10% Glycerol and 1 mM ATP) at room temperature and 1 μl 20 mM ATPγS was then immediately added into the solution. To remove the glycerol, the complex system was applied to Zeba™ Micro Spin Desalting Columns (7K, Thermo Fisher), exchanging the buffer to 50 mM Tris-HCl (pH 7.5) containing 100 mM NaCl, 1mM ATP and 1 mM ATPγS. The glycerol removal process usually took about 10 min before cryo-plunging. 0.005% NP-40 was added to the proteasome solution immediately before cryo-plunging. Cryo-EM grids were prepared with FEI Vitrobot Mark IV. C-flat grids (R1/1 and R1.2/1.3, 400 Mesh, Protochips, CA, USA) were glow-discharged before a 2.5-μl drop of 1.5 mg/ml substrate-engaged proteasome solution was applied to the grids in an environmentally-controlled chamber with 100% humidity and temperature fixed at 4 °C. After 2 sec of blotting, the grid was plunged into liquid ethane and then transferred to liquid nitrogen. The cryo-grids were initially screened at a nominal magnification of 235,000 times in an FEI Tecnai Arctica microscope, equipped with an Autoloader and an acceleration voltage of 200 kV. Good quality grids were transferred to an FEI Titan Krios G2 microscope equipped with the post-column Gatan BioQuantum energy filter connected to Gatan K2 Summit direct electron detector. Coma-free alignment was manually optimized and parallel illumination was verified prior to data collection. Cryo-EM data were collected semi-automatically by Leginon29 version 3.1 and SerialEM30 with the Gatan K2 Summit operating (Gatan Inc., CA, USA) in a super-resolution counting mode and with the Gatan BioQuantum operating in the zero-loss imaging mode (10 μm energy slit). A total exposure time of 10 second with 250 ms per frame resulted in a 40-frame movie per exposure with an accumulated dose of 44 electrons/Å2. The calibrated physical pixel size and the super-resolution pixel size are 1.37 Å and 0.685 Å, respectively. The raw data were saved at the pixel size of 0.685 Å. The defocus in data collection was set in the range of −0.7 to −3.0 μm. A total of 44,664 movies were collected throughout eight sessions of data collection.
Cryo-EM data processing and reconstruction.
The micrograph frames of 44,664 raw movies were aligned and averaged with MotionCor2 program31 at the super-resolution pixel size 0.685 Å. Each drift-corrected micrograph was used for the determination of the micrograph CTF parameters with program Gctf32. 2,669,687 particles of 26S proteasome were picked using program deepEM33. Reference-free 2D classification and 3D classification were carried out with two-fold binned data with the pixel size of 1.37 Å in both RELION 2.134 and ROME, which combined maximum-likelihood based image alignment and statistical machine-learning based classification35. Focused 3D classification, which we used in the later stage of data processing, and high-resolution refinement, were mainly done with RELION 2.1. Map reconstruction and local resolution calculation were finished with programs in both RELION 2.1 and ROME. A significant part of data processing, mostly 2D/3D classification, was performed with a 1024-core CPU cluster equipped with 64 Intel Xeon Gold 6142 (2.6 GHz 16-core) CPUs, a NVIDIA DGX-1 supercomputing system equipped with 8 Tesla V100 GPUs or a 10-node GPU cluster equipped with 40 Tesla V100 GPUs.
We applied a hierarchical 3D classification strategy to analyze the very large dataset (Extended Data Fig. 2). The entire data-processing procedure consisted of four steps. In the first step, we separated doubly capped proteasome particles from singly-capped ones through several rounds of 2D and 3D classification. This resulted in 1,552,828 doubly-capped particles and 478,919 singly-capped ones. These particles were aligned to the consensus models of doubly- and singly-capped proteasomes to get their approximate shift and angular parameters. With these parameters, each complete doubly-capped particle was split into two pseudo-single-cap particles by re-centering the box onto the RP-CP subcomplex. Then the box size of pseudo-singly-capped particles and true singly-capped particles was shrunk to 600×600. This is an effective way to reduce irrelevant heterogeneity due to conformational variations, and improve map resolution8,36. There were 3,584,040 particles in the dataset chosen for the following steps of analysis. In the second step, we focused on the gate of the CP. Several rounds of 2D and 3D classification were done to distinguish the states of the CP gate, that is, separating the SA-like closed-gate states from those open-gate or namely SD-like states. It was obvious that the RP subcomplex of the SD-like states rotates by a large angle compared to the SA-like states7,8. The RP-CP subcomplex was masked during the 3D classification. There were 732,666 particles in the SA-like states and 2,521,686 particles in the SD-like states left after this step7,8. In the third step, we used focused 3D classification34 to further classify within these two different states. Since the CP is structurally stable, we did refinement with the CP masked, so that we can determine the x-y shift and angular parameters of all particles when they were aligned against the reference of the CP. Using these parameters, we continued 3D classification with the RP masked and with alignment skipped. This means we only classified images based on structural changes in the RP relative to the CP. After the classification, we clearly saw that the RP dramatically swings and rotates against the CP. By focusing on the variation of substrate interactions with the AAA-ATPase and Rpn10/11, we classified these particles into 5 major states, designated EA, EB, EC, ED1, ED2, respectively, accounting for 7.8%, 14.8%, 9.9%, 27.2%, and 40.1% of the particles. In the final step, we used focused classification to further detect significant structural changes within each of these five states. After auto-refinement with the RP masked, we continued skip-alignment classification with the lid, the AAA-ATPase, or certain combination of RP subunits masked. Application of differential masks depended on specific structural characteristics of different states. For example, Rpn1 in state EB state was partially blurred without further 3D classification. We therefore masked Rpn1 together with ATPase in 3D classification, which resulted in improvement of its density quality in certain 3D classes. The whole RP complex is highly dynamic in state EC. Thus, we performed further classification with the whole RP masked, resulting in two distinct states, named EC1 and EC2. EC1 showed a clear ubiquitin density, which is absent in EC2. Similarly, we also obtained an intermediate state from initial EB dataset, named EA2, which showed a ubiquitin-binding mode different from that in EA.
The final refinement of each state was done using data with a pixel size of 1.37 Å that were binned by two folds from the raw data in the super-counting mode. Based on the in-plane shift and Euler angle of each particle from the last iteration of refinement, we reconstructed the two half-maps of each state using raw single-particle images at the super-counting mode with a pixel size of 0.685 Å. To enhance the local density quality for each state, we applied two types of local mask in the last several iterations of refinement, one focusing on the complete RP and the other focusing on the CP and ATPase components, which yielded two maps for each state that showed improved local resolution in the lid and CP, respectively. For each state, the maps refined by differential masking were merged in Fourier space into a single map. This procedure was also applied for the half maps prior to FSC calculation. Because states EA1 and EA2 exhibit identical structures in their CP and AAA-ATPase components, we combined them together and refined the combined dataset by applying the CP/ATPase mask. The final reconstructions of the combined EA, EA1, EA2, EB, EC1, EC2, ED1 and ED2 datasets gave overall resolutions of 2.8 Å, 3.0 Å, 3.2 Å, 3.3 Å, 3.5 Å, 3.6 Å, 3.3 Å and 3.2 Å, respectively, measured by the gold-standard FSC at 0.143-cutoff on two separately refined and merged half maps. Prior to visualization, all density maps were sharpened by applying a negative B-factor. Local resolution variations were further estimated using ResMap on the two half maps refined independently37.
Atomic model building and refinement.
The higher-resolution cryo-EM maps allowed us to refine atomic models with improved quality and to extend sequence register beyond the published structures of the substrate-free proteasomes through de novo modeling (Extended Data Table 1). Given that we did not stall the substrates in a homogeneous location during their degradation, and also that substrate translocation through the proteasome is not sequence specific, the substrate densities were modelled using polypeptide chains without assignment of amino acid sequence (except for the lysine residue forming a visible isopeptide bond with ubiquitin in state EB).
To build the initial atomic model of the substrate-bound 26S proteasome complex, we used previously published human proteasome structures8 as starting models and rebuilt each atomic model in Coot38 for each of the seven conformational states. In states ED1,2, many residues at the N-terminus of Rpt1 and Rpt2 coiled coil domain and the C-terminal toroidal domain of Rpn2 that were missing in other states and in the previously published substrate-free structures7–10,36,39–45 were shown as reliable densities with flanking of large side chains. These high-resolution features allowed us to conduct de novo tracing of these previously missing elements, including a newly identified helix of Rpn2 residing in a long loop (residue 820-871) emanating from the Rpn2 toroidal domain (Extended Data Fig. 9). In all previously published cryo-EM structures of human 26S proteasomes, the local resolution of the lid subcomplex was generally worse than 4.9 Å and was insufficient to ensure the correct register of the side chains. Our density maps of all states, particularly EB and ED1,2, exhibit significantly improved local resolution in the lid subcomplex (Extended Data Table 1, Extended Data Fig. 3), allowing us to rebuild the majority of the lid subcomplex. The local resolution of Rpn1 in states EB and ED1,2 also reached 4-5 Å, allowing us to improve the backbone model and make partial side-chain register. Rpn1 has very poor local resolution in states EC1,2 precluding de novo atomic modelling. Thus, we used the improved atomic model of Rpn1 from states EB and ED1,2 to fit the poor Rpn1 densities of EC1,2 as a rigid body.
The nucleotide densities are of sufficient quality for differentiating ADP from ATP, which allowed us to build the atomic models of ADP and ATP into their densities. A resolution of no worse than 3.6 Å may be required to distinguish ADP from ATP, because ATP adds an extra size of 2.46 Å with its γ-phosphate and three additional oxygens atoms relative to ADP. The magnesium ion bound to ATP was well resolved in state EA (Extended Data Fig. 3c). By contrast, no magnesium ion density was observed around the ADP-assigned nucleotide density. Except state EA1,2, at least one of the ATPases in each state has a very poor nucleotide density quality in its nucleotide-binding site. Although there are visible extra densities in the nucleotide-binding site at a low contour level in most of the apo-like ATPase subunits after the protein structures are in place, these weak extra densities are insufficient for even fitting a complete ADP with good confidence. For instance, some of them may allow fitting of ribose and/or α-phosphate but then β-phosphate is totally out of density. To avoid over-interpretation and to practice prudence in high-quality atomic modeling, we avoided building atomic models of nucleotides into these poor densities at all and referred to the corresponding ATPases as the “apo-like state” throughout this study. The poor extra densities in the nucleotide-binding sites of these apo-like ATPases most likely reflect partial or low occupancy or unstable binding of nucleotide, which is expected when the nucleotide-binding site undergoes nucleotide exchange.
Atomic model refinement was conducted in Phenix46 with its real-space refinement program. We used both simulated annealing and global minimization with NCS, rotamer and Ramachandran constraints. Partial rebuilding, model correction and density-fitting improvement in Coot38 were iterated after each round of atomic model refinement in Phenix46. The improved atomic models were then refined again in Phenix, followed by rebuilding in Coot38. The refinement and rebuilding cycle was repeated until the model quality reached expectation (Extended Data Table 1).
Structural analysis and visualization.
All figures of structures were plotted in Chimera47, PyMOL48, or Coot38. Structural alignment and comparison were performed in both PyMOL and Chimera. Interaction analysis between adjacent subunits was performed using PISA49.
Data availability.
Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-9215 (the combined EA refined with CP-ATPase mask), EMD-9216 (whole EA1), EMD-9217 (whole EA2), EMD-9218 (whole EB), EMD-9219 (whole EC1), EMD-9220 (whole EC2), EMD-9221 (whole ED1), EMD-9222 (whole ED2), EMD-9223 (RP of EA1), EMD-9224 (RP of EA2), EMD-9225 (RP of EB), EMD-9226 (RP of EC1), EMD-9227 (RP of EC2), EMD-9228 (RP of ED1) and EMD-9229 (RP of ED2). Coordinates are available from the RCSB Protein Data Bank under accession codes 6MSB (whole EA1), 6MSD (whole EA2), 6MSE (whole EB), 6MSG (whole EC1), 6MSH (whole EC2), 6MSJ (whole ED1), 6MSK (whole ED2). Raw data are available from Y.M.
Extended Data
Extended Data Figure 1. Characterization and structure determination of the substrate-engaged human proteasome.
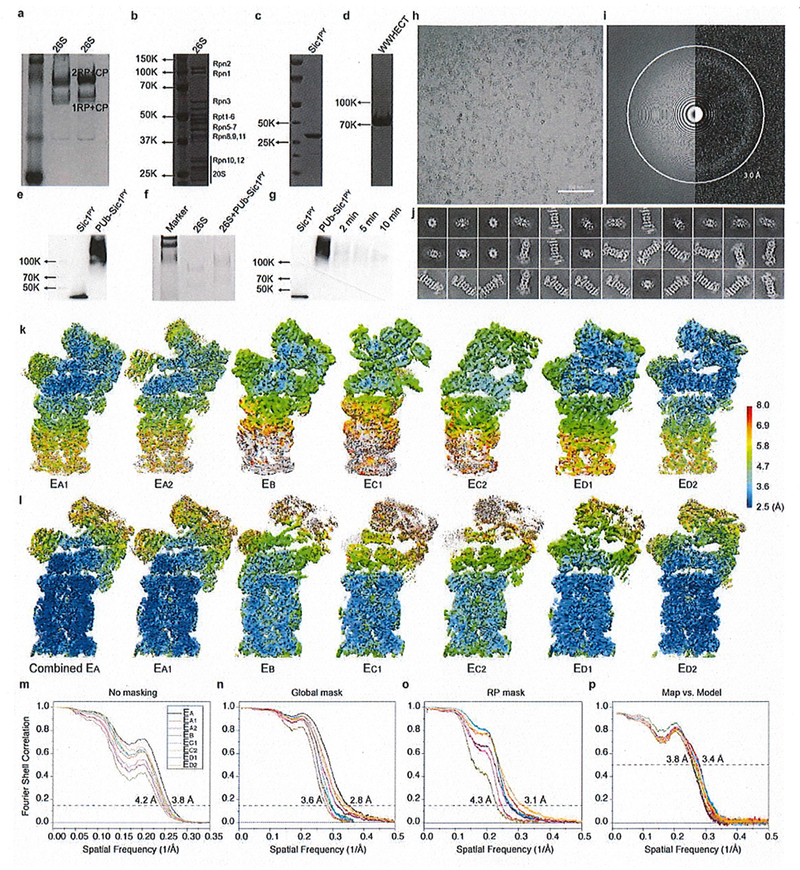
(a) Native PAGE analysis of proteasome purified through Superose 6 10/300 GL column. (b) SDS-PAGE results of the 26S proteasome after the FPLC. (c) SDS-PAGE analysis of the Sic1PY purified through Superdex 75 column. (d) SDS-PAGE analysis of the WWHECT purified through Superdex 75 column. All PAGE from a to d were stained by coomassie blue. (e) Result of polyubiquitinated Sic1PY. After the ubiquitination, the samples were applied to SDS gel. Then the gel was examined by western-blot with anti-T7 antibody. The result suggests that almost all Sic1PY has been ubiquitinated. (f) Native PAGE analysis of the crosslink between the proteasome and the substrate. As a control, 26S without substrate was also applied to the same crosslinking assay. With the addition of the PUb-Sic1PY, the proteasome was observed to run slower than those without PUb-Sic1PY. This result verifies that the substrate has been captured by the proteasome but not totally degraded yet at the time point when they were prepared for cryo-EM experiments. However, the crosslinking assay, designed only to verify the substrate-bound state of the proteasome, was not used in our cryo-EM sample preparation for data collection (see Methods). (g) SDS-PAGE analysis of the degradation of PUb-Sic1PY. The gel was examined by western-blot with anti-T7 antibody. Obvious degradation of the PUb-Sic1PY has been observed, which suggests that both the proteasome and substrate were suitable for the degradation study. (h) A typical ctyo-EM micrograph of the substrate-engaged human proteasome after motion correction. (i) The power spectmm evaluation of the micrograph shown in h. (j) A gallery of unsupervised class averages calculated by ROME55 using machine-learning-based clustering. (k) The local resolution estimation calculated by ResMap56 on seven maps refined by focusing the mask on the RP component. (l) The local resolution estimation on seven maps refined by focus the mask on the CP and ATPase components. (m) The gold-standard FSC plots of eight maps calculated without masking the raw half maps. (n) The gold-standard FSC plots of the maps refined with focusing on the CP and ATPase components, calculated with masking the raw half maps. (o) The gold-standard FSC plots of the maps refined with focusing on the RP subcomplex, calculated with masking the raw half maps. (p) The cross-map FSC plots calculated by Phenix between the each refined map and its corresponding atomic model. For each state, the maps refined by differential masking were merged in Fourier space into a single map, which was used for the cross-map FSC calculation. The same color code is used in panels m-p.
Extended Data Figure 2. Focused classification to sort out the seven conformational states.
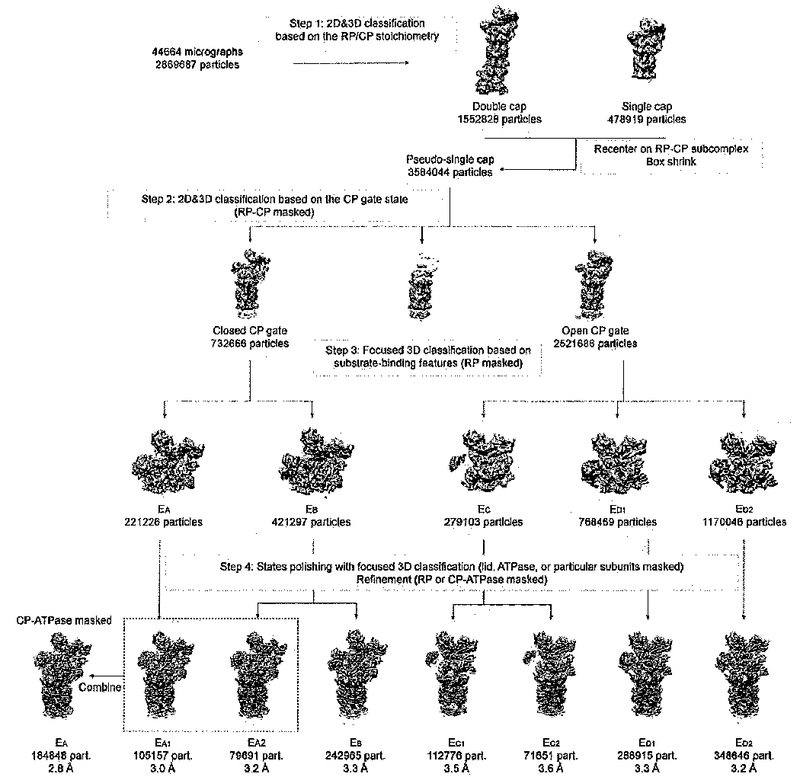
The diagram illustrates the major four steps of our hierarchical focused classification strategy. Further detailed iterations of classification in each step are omitted for clarify.
Extended Data Figure 3. The cryo-EM maps and quality assessment.
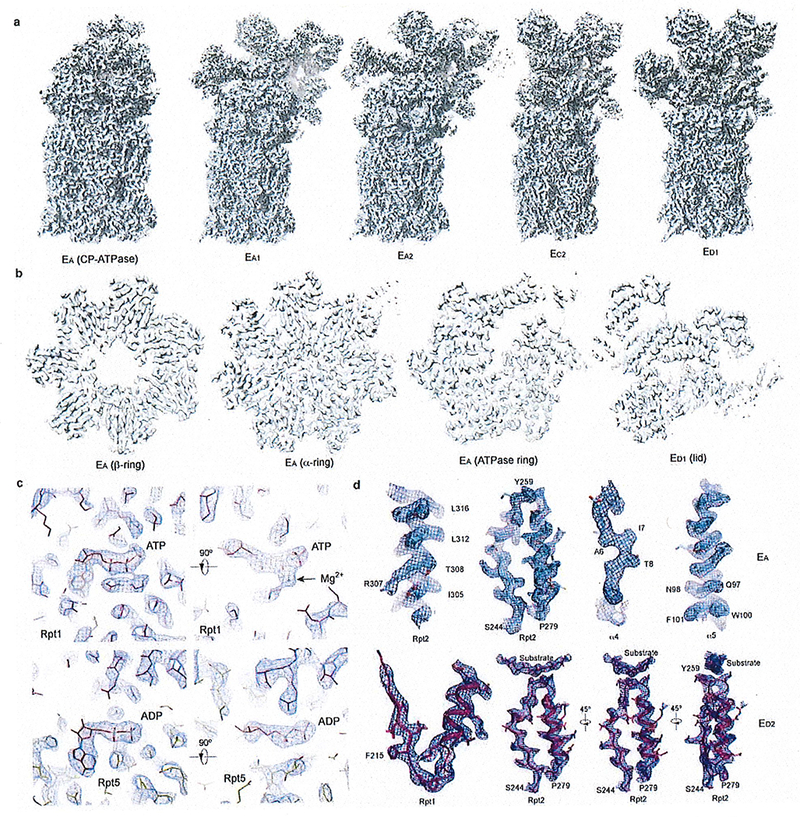
(a) The other five refined cryo-EM maps that are not shown in the main figures. (b) Typical central cross-sections in the density maps for each of the four subcomplexes, the lid in state ED1, the ATPase ring, the α ring and the β ring in state EA. (c) Typical nucleotide-binding pocket densities from the EA map. The ATP density is compared with the ADP density in two orthogonal perspectives. The magnesium density next to the ATP is labeled, which is absent in the ADP-bound pocket. (d) Typical densities of second my structures and substrate in the proteasome superimposed with their atomic models.
Extended Data Figure 4. Key structural features and comparisons that help sort out the sequence of the seven conformational states.
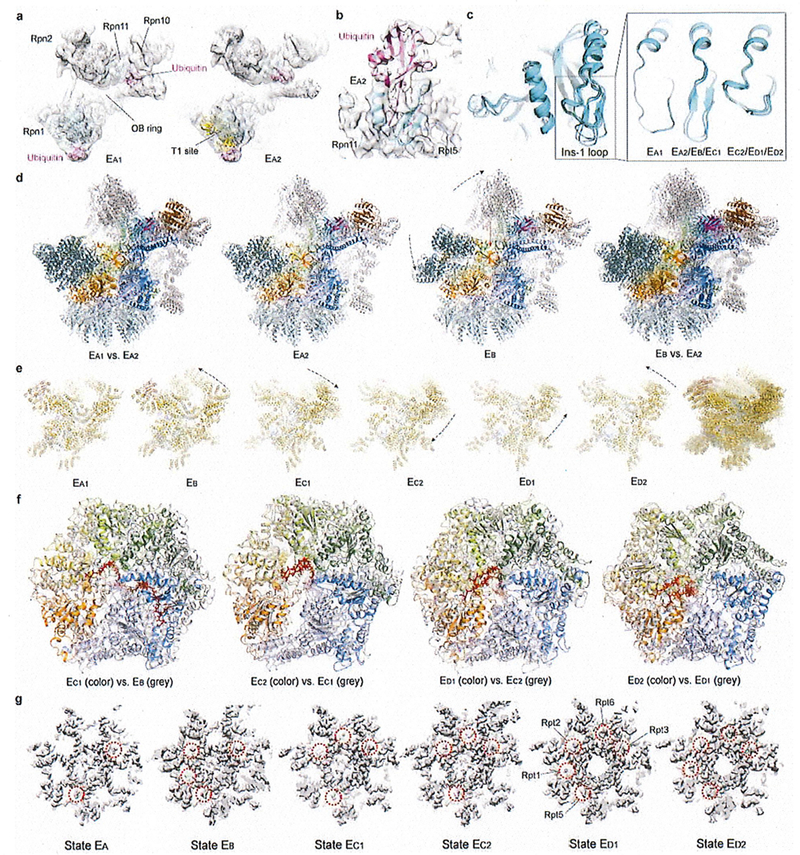
(a) The ubiquitin densities in state EA1 (left) and EA2 (right). The T1 site is labelled by fitting the yellow cartoon representation of the NMR structure (PDB ID 2n3u) of Rpn1 T1 element in complex with two ubiquitins into our density, showing the ubiquitin on Rpn1 is bound to a site very close to the T1 site17. The density maps are low-pass filtered to 8 Å to show the ubiquitin features clearly, due to the lower local resolution of the ubiquitin density in these maps. (b) The ubiquitin-Rpn11-Rpt5 interface observed at high resolution in state EB is also observed in state EA2, albeit at somewhat lower local resolution. The EA2 density is shown as a transparent surface. (c) Comparison of the Insertion-1 loop of Rpn11 in different states. (d) Comparison of the RP structures between EA and EB. (e) Comparison of the lid subcomplex conformations among all states. (f) Comparison of ATPase ring structures between two successive states. The structures are aligned together against their CP in d-f. (g) Comparison of the RP-CP interface and the Rpt C-tail insertions into the CP surface pockets in different states. The ctyo-EM densities of the RP-CP interfaces are shown as a grey surface representation.
Extended Data Figure 5. Substrate densities in different states.
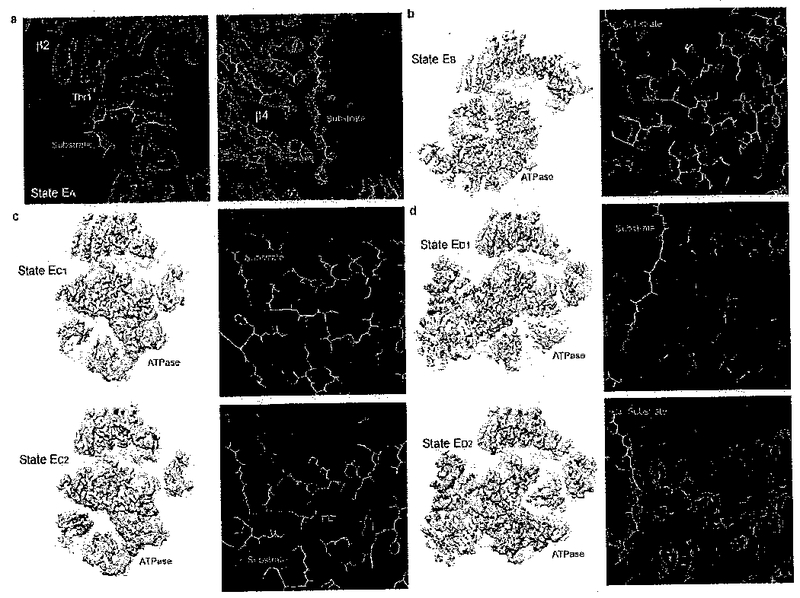
(a) Close-up views of two typical substrate densities observed in the CP chamber in state EA. The left panel shows the substrate density directly contacting the proteolytically active Thr1 in the β2 subunit. The right panel shows a long substrate density at the seam between two β4 subunits inside the CP. (b) The overall ATPase ring density of state EB (left) and a close-up view of the substrate density (right). (c) The overall ATPase ring density of state EC1,2 (left) and a close-up view of the substrate density (right). (d) The overall ATPase ring density of state ED1,2 (left) and a close-up view of the substrate density (right). All close-up views were directly screen-copied from Coot58 after atomic modelling into the density maps without furher modification or beautification.
Extended Data Figure 6. The nucleotide densities in all states.
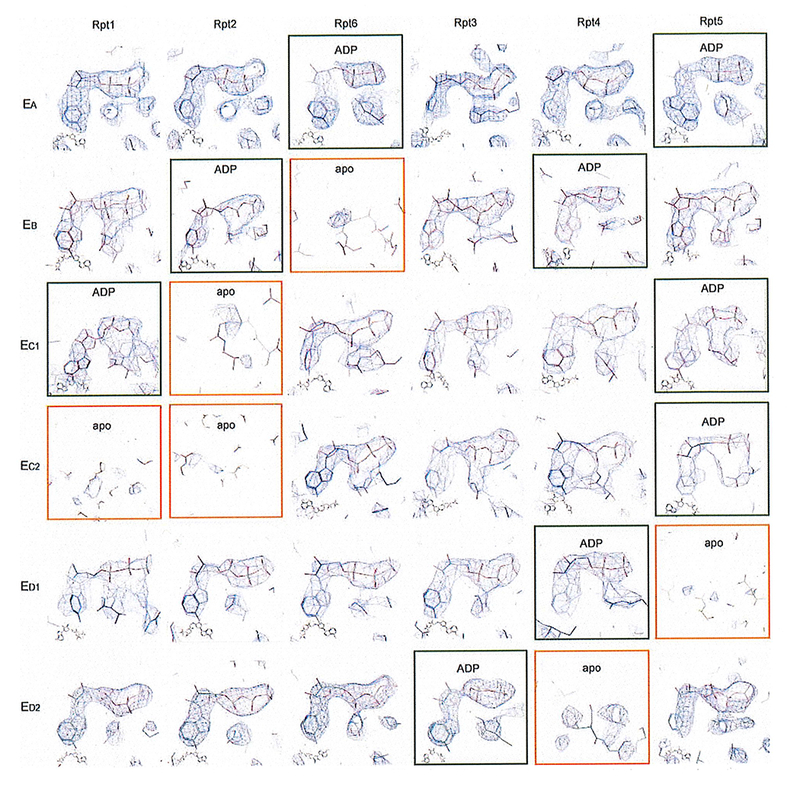
The nucleotide densities fitting with atomic models are shown with blue meshes. All close-up views were directly screen-copied from Coot58 after atomic modelling into the density maps without further modification or beautification. At the common contour level used for atomic modelling, the potential nucleotide densities in the apo-like subunits are mostly out, albeit they can show up partial nucleotide shapes at much lower contour level.
Extended Data Figure 7. Long-range regulation of the AAA-ATPase by the lid-base interactions.
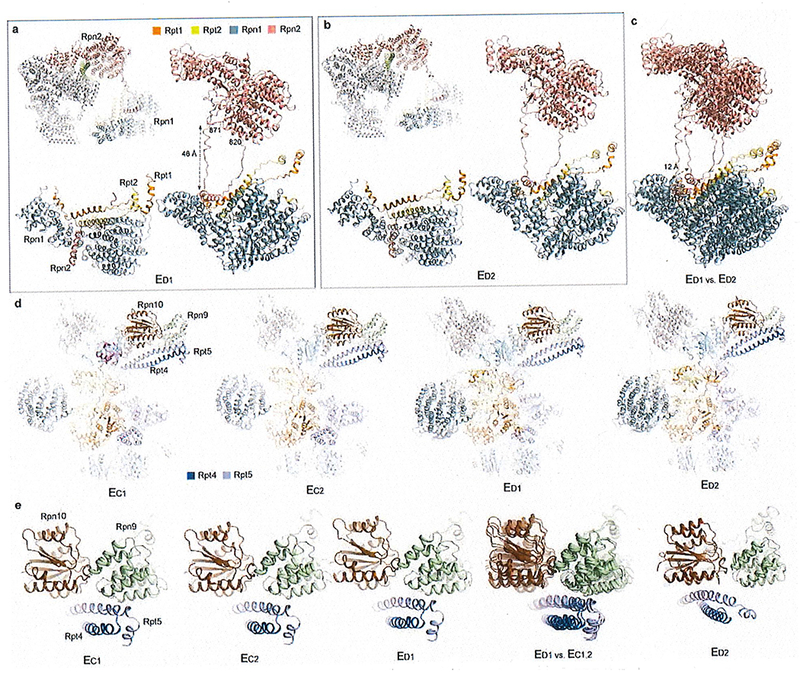
(a) and (b) The long-range association of the Rpn1-Rpn2 through a looping structure from Rpn2 (residue 820-871) observed in state ED1 (a) and ED2 (b). (c) Comparison of the Rpn1-Rpn2 long-range association between the two states shows a marked movement of 12 Å. (d) The comparison of the Rpt4-Rpt5 CC interactions with Rpn9 and Rpn10 in states EC1,2 and ED1,3. (e) Closeup views of the Rpt4-Rpt5 CC contacts with Rpn9 in states EC1,2 and ED1, and its contact switching to Rpn10 in state ED2.
Extended Data Figure 8. Geometries of nucleotide-binding pockets and nucleotide-driven intrasubunit conformational changes of AAA domains.

a, Comparison of the nucleotide-binding pockets of six ATPases in all states illustrates a common pattern in the geometry of the nucleotide-binding sites. Each row shows the geometry of the nucleotide-binding pocket of one ATPase in all six states. In each panel showing an ATP or ADP-bound state, one red dashed line marks the distance from β/γ-phosphate of nucleotide to the arginine finger of the adjacent ATPase, while the other line marks the distance from the same phosphate to the Walker B motif. In the case of apo-like states, the red lines extend to the proline of the Walker A motif rather than to the phosphate groups. These geometries indicate the potential reactivity of these sites49. When the ATPase is positioned in the middle of pore-loop staircase, but not at the lowest position, the nucleotide-binding pockets are tightly packed whether ATP or ADP is bound. By contrast, when the ATPase is either in the lowest position of the substrate-pore loop staircase or disengaged from the substrate, the nucleotide-binding pocket is open regardless of whether it is ADP-bound or free of nucleotide. b-e, Superposition of the AAA domain structure of Rpt1 (b), Rpt2 (c), Rpt3 (d), or Rpt4 (e) structures from six distinct states aligned against their large AAA subdomains. Rpt1 assumes two major conformations and Rpt2 assumes three.
Extended Data Figure 9. Changes in lid-base interactions are associated with ATP hydrolysis events through long-range allosteric regulation.
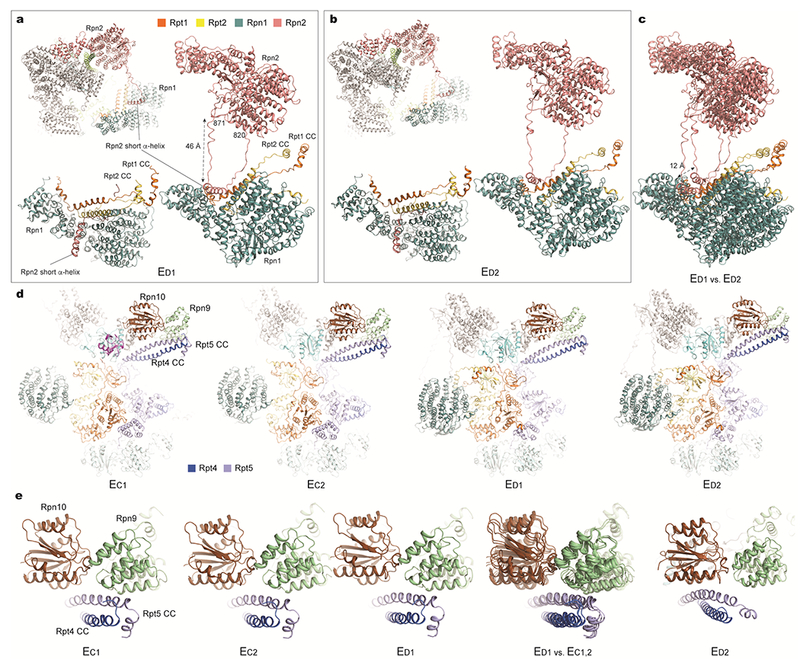
a and b, Long-range association between Rpn1 and Rpn2 through a looping structure from Rpn2 (residue 820-871) observed in state ED1 (a) and ED2 (b). c, Comparison of the Rpn1-Rpn2 long-range association between these two states shows a marked, 12 Å movement of Rpn1 relative to the CP. In both states ED1 and ED2, the Rpn1 toroidal domain and the CC domain of the Rpt1-Rpt2 dimer together form a surface cavity in which a short helix from Rpn2 is inserted50. This helix resides in the middle of a long loop (residue 820-871) emanating from the Rpn2 toroidal domain. The long-range association of Rpn1-Rpn2 seems to stabilize a larger interface formed between Rpn1-Rpn2 and Rpt1-Rpt2. However, such a quaternary architecture is not observed in other states (EA-C). In states EC1,2, the Rpn1 density is considerably blurred, reflecting strong motions potentially breaking the long-range Rpn1-Rpn2 association (Fig. 1b, Extended Data Fig. 3a). Thus, the specific Rpn1 conformation in each state appears to be highly coordinated with the hydrolytic cycle of the ATPase ring, and is controlled by Rpn1’s interactions with Rpn2 in a long-range fashion. d, Comparison of the interactions of the Rpt4-Rpt5 CC with Rpn9 and Rpn10 in states EC1,2 and ED1,3. e, Closeup views of the Rpt4-Rpt5 CC in contact with Rpn9 in states EC1,2 and ED1, and of this CC’s contact switching to Rpn10 in state ED2. These observations are consistent with a recent study51.
Extended Data Figure 10. Expanded model of the complete cycle of substrate processing by the human 26S proteasome.
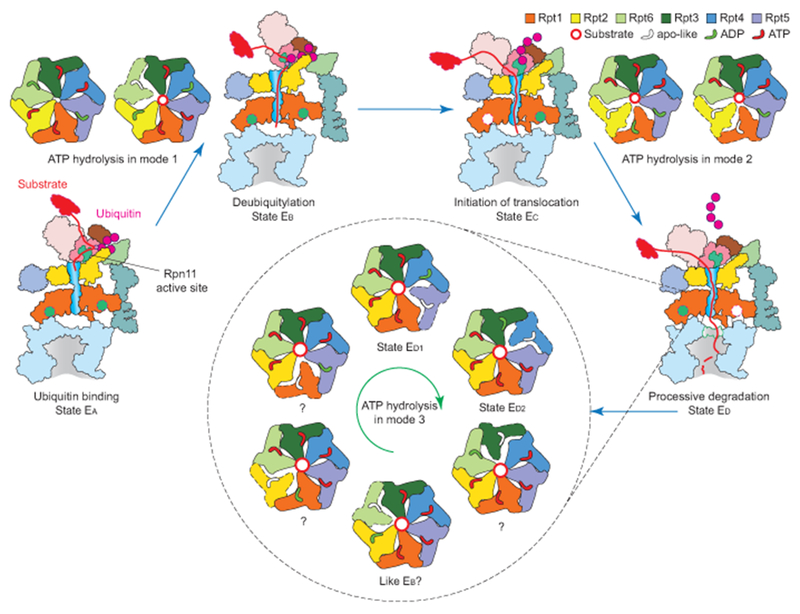
The cartoon summarizes the concept of three principle modes of coordinated ATP hydrolysis observed in the seven states and our proposal of how they regulate the complete cycle of substrate processing by the proteasome holoenzyme. Coordinated ATP hydrolysis in modes 1, 2 and 3 features hydrolytic events in two oppositely positioned ATPases8,18, in two consecutive ATPases6,19,52, and in only one ATPase at a time20–23,53–56, respectively. Substrate processing undergoes three major steps before CP gate opening for processive translocation: (1) ubiquitin recognition; (2) simultaneous deubiquitylation and substrate engagement with the AAA-ATPase ring; and (3) translocation initiation, which involves multiple simultaneous events, including ubiquitin release, ATPase repositioning and switching of Rpt C-tail insertion pattern. In some cases, the initiation of translocation may precede deubiquitylation. In steps 1 and 2, the ATPases follow the mode-1 ATP hydrolysis. In step 3, they follow the mode-2 ATP hydrolysis. After the gate is open, the AAA-ATPases hydrolyze ATP in mode 3, in which only one nucleotide is hydrolyzed at a time.
Extended Data Table 1. Cryo-EM data collection, refinement and validation statistics.
Note: Atomic model refinement with state EA map was done only for the CP and ATPase using part of the full atomic model from state EA1. The final refined atomic model, 6MSB, includes the model components of CP and ATPase refined from the combined EA map and the rest of holoenzyme from the EA1 map. 5VFO was only used for the CP component for the initial atomic model of ED2.
| EA (EMDB-9215) (PDB 6MSB) | EA1 (EMDB-9216,9223) (PDB 6MSB) | EA2 (EMDB-9217,9224) (PDB 6MSD) | EB (EMDB-9218,9225) (PDB 6MSE) | EC1 (EMDB-9219,9226) (PDB 6MSG) | EC2 (EMDB-9220,9227) (PDB 6MSH) | ED1 (EMDB-9221,9228) (PDB 6MSJ) | ED2 (EMDB-9222,9229) (PDB 6MSK) | |
|---|---|---|---|---|---|---|---|---|
| Data collection and processing | ||||||||
| Magnification | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e–/Å2) | 44 | 44 | 44 | 44 | 44 | 44 | 44 | 44 |
| Defocus range (μm) | −0.6 to −3.5 | −0.6 to −3.5 | −0.6 to −3.5 | −0.6 to −3.5 | −0.6 to −3.5 | −0.6 to −3.5 | −0.6 to −3.5 | −0.6 to −3.5 |
| Pixel size (Å) | 0.685 | 0.685 | 0.685 | 0.685 | 0.685 | 0.685 | 0.685 | 0.685 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 3,584,040 | 3,584,040 | 3,584,040 | 3,584,040 | 3,584,040 | 3,584,040 | 3,584,040 | 3,584,040 |
| Final particle images (no.) | 184,848 | 105,157 | 79,691 | 242,965 | 112,776 | 71,651 | 288,915 | 348,646 |
| Map resolution (Å) | 2.8 | 3.0 | 3.2 | 3.3 | 3.5 | 3.6 | 3.3 | 3.2 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range(Å) | 2.5-5.8 | 2.5-5.8 | 2.5-6.9 | 2.5-4.7 | 2.8-5.8 | 3.0-6.9 | 2.5-5.8 | 2.5-4.7 |
| Refinement | ||||||||
| Initial model used | 5VFS | 5VFO(partly) | ||||||
| Model resolution (Å) | 3.2 | 3.4 | 3.8 | 3.5 | 3.7 | 3.8 | 3.4 | 3.4 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Model resolution range (Å) | 2.5-5.8 | 2.5-5.8 | 2.5-6.9 | 2.5-4.7 | 2.8-5.8 | 3.0-6.9 | 2.5-5.8 | 2.5-4.7 |
| Map sharpening B factor (Å2) | −50 | −50 | −46 | −35 | −30 | −50 | −35 | −30 |
| Model composition | ||||||||
| Non-hydrogen atoms | 104400 | 104400 | 104400 | 105147 | 104415 | 103777 | 105336 | 105244 |
| Protein residues | 13180 | 13180 | 13180 | 13400 | 13305 | 13328 | 13403 | 13395 |
| Ligands | 11 | 11 | 11 | 10 | 6 | 5 | 10 | 10 |
| B factors (Å2) | ||||||||
| Protein | 98.31 | 120.31 | 195.53 | 117.64 | 220.54 | 280.72 | 187.13 | 197.24 |
| Ligand | 89.88 | 112.21 | 191.81 | 118.98 | 217.88 | 261.40 | 181.50 | 192.26 |
| R.m.s. deviations | ||||||||
| Bond lengths (Å) | 0.007 | 0.01 | 0.012 | 0.006 | 0.005 | 0.004 | 0.007 | 0.007 |
| Bond angles (°) | 0.925 | 1.075 | 1.013 | 1.025 | 0.871 | 0.901 | 1.266 | 1.187 |
| Validation | ||||||||
| MolProbity score | 1.5 | 1.69 | 1.77 | 1.83 | 1.65 | 1.71 | 1.86 | 1.88 |
| Clashscore | 2.69 | 2.91 | 5.02 | 5.63 | 4.00 | 4.2 | 5.87 | 5.83 |
| Poor rotamers (%) | 0.34 | 0.42 | 0.42 | 0.66 | 0.28 | 0.19 | 0.45 | 0.5 |
| Ramachandran plot | ||||||||
| Favored (%) | 93.07 | 92.45 | 91.59 | 90.83 | 92.56 | 91.46 | 90.13 | 89.61 |
| Allowed (%) | 6.76 | 7.33 | 8.15 | 8.83 | 7.21 | 8.31 | 9.58 | 10.09 |
| Disallowed (%) | 0.17 | 0.22 | 0.26 | 0.34 | 0.23 | 0.23 | 0.29 | 0.3 |
Extended Data Table 2.
Summary of key structural features.
| Key structural features connecting the seven states into a continuum of dynamic structural changes | |||||||
| States | EA1 | EA2 | EB | EC1 | EC2 | ED1 | ED2 |
| Locations of ubiquitin densities | Rpn1, Rpn10, Rpt4/5 CC | Rpn11, Rpn10, Rpn1, Rpt4/5 CC | Rpn11, Rpn10, Rpt4/5 CC | Rpn11 | None observed | None observed | None observed |
| Rpn11 Ins-1 conformation | Large loop | β-hairpin | β-hairpin | β-hairpin | Retracted small loop | Retracted small loop | Retracted small loop |
| Pore-loop staircase contacting substrate | None | None | Rpt3 at the top, followed by Rpt4/5/1/2 | Rpt6 at the top, followed by Rpt3/5/1 | Rpt6 at the top, followed by Rpt3/5/1 | Rpt1 at the top, followed by Rpt2/6/3/4 | Rpt5 at the top, followed by Rpt1/2/6/3 |
| Rpt subunits disengaged from pore loop staircase | Rpt6 | Rpt6 | Rpt6 | Rpt1 and Rpt2 | Rpt1 and Rpt2 | Rpt5 | Rpt4 |
| Rpt subunits with apo-like state | None | None | Rpt6 | Rpt2 | Rpt1 and Rpt2 | Rpt5 | Rpt4 |
| Rpt subunits with ADP bound | Rpt6 and Rpt5 | Rpt6 and Rpt5 | Rpt2 and Rpt4 | Rpt1 and Rpt5 | Rpt5 | Rpt4 | Rpt3 |
| Rpt C-tails insertion into α-pockets | Rpt3 and Rpt5 | Rpt3 and Rpt5 | Rpt2/3/5 | Rpt2/3/5/6 | Rpt2/3/5/6 | Rpt1/2/3/5/6 | Rpt1/2/3/5/6 |
| CP gate state | Closed | Closed | Closed | Closed | Closed | Open | Open |
| Resemblance to the substrate-free contormations 7-10, 36, 39-45 | SA | SA (in ATPase and CP) | None | SB (in overall lid-base relationship) | SC (in overall lid-base relationship) | SD (in overall lid-base relationship, RP-CP interface and CP gate) | SD2 (in pore-loop staircase, overall lid-base relationship, RP-CP interface and CP gate) |
| Functional step | Ubiquitin recognition | Ubiquitin transfer | Deubiquitylati on | Initiation of translocation | Priming for CP gate opening | Processive translocation | Processive translocation |
| Potential substrate-binding sites observed in the CP chamber | |||||||
| State | Key contacts at the binding site | Features | Number of substrate residues | ||||
| EA, EB, EC1,2, ED1,2 | Thr1, Cys31 of β2, Cys129 of β3 | Symmetric | 6 | ||||
| EA | Asn24, Tyr134, Phe137 of β4 | At the seam between two β4 subunits | 10 | ||||
| EA | Tyr103 of α1, Tyr61, Tyr90 of β1, and Phe88 of β2 | Symmetric, at the inter-subunit interface | 5 | ||||
| EA | Asn90 of α5, Tyr90 of β5, Phe101 of β6 | Symmetric, at the inter-subunit interface | 3 | ||||
| EA, EB, EC1,2, ED1,2 | Tyr105, Arg117 of α1, His88 of α2 | Symmetric | 3 | ||||
| EA, EB, EC1,2, ED1,2 | Tyr59, Cys91, Tyr98 of β4 | Symmetric | 3 | ||||
| EA | Phe69, Cys91 and Tyr98 of β3 | Asymmetric, only present in the chamber with the CP gate open | 3 | ||||
| EA | Ile3, Tyr6, Tyr104 of β3, Tyr120 of β4 | Asymmetric, only present in the chamber with the CP gate open | 3 | ||||
| EB, EC1,2, ED1,2 | Tyr30 of β7 | Symmetric | 4 | ||||
Supplementary Material
Acknowledgments
We apologize for not citing many relevant original research papers due to space limit of the print version of this manuscript. References are significantly expanded on the online-only contents. We thank Y. Saeki for gifting the plasmids expressing Sic1PY and WWHECT, H. Huang for gifting proteasome-expressing cell lines, D. Yu, J. Xu, Y. Ma, C. Fan, J. Jackson for technical support, and S. Elsasser for critical reading of the manuscript. This work was funded in part by an Intel Corporation academic grant, the Thousand Talents Plan of China, National Natural Science Foundation of China grant no. 11774012 and 91530321, Peking-Tsinghua Center for Life Sciences (Y.M.), the NIH grant GM43601 (D.F.) and an Edward Mallinckrodt, Jr. Foundation award (Y.L.). The cryo-EM data were collected from the Cryo-EM Platform at Peking University. Initial cryo-EM screening was performed in part at the Center for Nanoscale Systems at Harvard University supported by the National Science Foundation under NSF award no. 1541959 and the NIH grant AI100645. The data processing was performed in part in the Sullivan cluster, which is funded in part by a gift from Mr. and Mrs. Daniel J. Sullivan, Jr., and in the Weiming No.1 and Life Science No. 1 High-Performance Computing Platform at Peking University.
Footnotes
Competing interests The authors declare no competing financial interests.
Supplementary Information is available in the online version of the paper.
References
- 1.Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N & Ciechanover A The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 26, 869–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finley D, Chen X & Walters KJ Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci 41, 77–93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bard JAM et al. Structure and Function of the 26S Proteasome. Ann. Rev. Biochem 87, 697–724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi Y et al. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351, aad9421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verma R et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Smith DM, Fraga H, Reis C, Kafri G & Goldberg AL ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144, 526–538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S et al. Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. USA 113, 12991–12996 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Y et al. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat. Commun 9, 1360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisele MR et al. Expanded Coverage of the 26S Proteasome Conformational Landscape Reveals Mechanisms of Peptidase Gating. Cell Rep. 24, 1301–1315 e1305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wehmer M et al. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. USA 114, 1305–1310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saeki Y, Isono E & Toh EA Preparation of ubiquitinated substrates by the PY motif-insertion method for monitoring 26S proteasome activity. Methods Enzymol. 399, 215–227 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Lee BH, King RW, Finley D & Kirschner MW Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science 348, 1250834 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang F et al. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol. Cell 34, 473–484 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worden EJ, Dong KC & Martin A An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome. Mol. Cell 67, 799–811 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Pathare GR et al. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc. Natl. Acad. Sci. USA 111, 2984–2989 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Worden EJ, Padovani C & Martin A Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat. Struct. Mol. Biol 21, 220–227 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Dambacher CM, Worden EJ, Herzik MA, Martin A & Lander GC Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. Elife 5, e13027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Y et al. Conformational Landscape of the p28-Bound Human Proteasome Regulatory Particle. Mol. Cell 67, 322–333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snoberger A, Brettrager EJ & Smith DM Conformational switching in the coiled-coil domains of a proteasomal ATPase regulates substrate processing. Nat. Commun 9, 2374 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glynn SE, Martin A, Nager AR, Baker TA & Sauer RT Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139, 744–756 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YC, Snoberger A, Schupp J & Smith DM ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat. Commun 6, 8520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puchades C et al. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358, aao0464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ripstein ZA, Huang R, Augustyniak R, Kay LE & Rubinstein JL Structure of a AAA+ unfoldase in the process of unfolding substrate. Elife 6, e25754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gates SN et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han H, Monroe N, Sundquist WI, Shen PS & Hill CP The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. Elife 6, e31324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma R, McDonald H, Yates JR 3rd & Deshaies RJ Selective degradation of ubiquitinated Sic1 by purified 26S proteasome yields active S phase cyclin-Cdk. Mol. Cell 8, 439–448 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Verma R, Oania R, Graumann J & Deshaies RJ Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell 118, 99–110 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Wang X et al. Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Suloway C et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang K Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y, Ouyang Q & Mao Y A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy. BMC Bioinformatics 18, 348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimanius D, Forsberg BO, Scheres SH & Lindahl E Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5, e18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J et al. Massively parallel unsupervised single-particle cryo-EM data clustering via statistical manifold learning. PLoS One 12, e0182130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schweitzer A et al. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc. Natl. Acad. Sci. USA 113, 7816–7821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kucukelbir A, Sigworth FJ & Tagare HD Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Lasker K et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. USA 109, 1380–1387 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lander GC et al. Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.da Fonseca PC, He J & Morris EP Molecular model of the human 26S proteasome. Mol. Cell 46, 54–66 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Huang X, Luan B, Wu J & Shi Y An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol 23, 778–785 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Luan B et al. Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proc. Natl. Acad. Sci. USA 113, 2642–2647 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beck F et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proc. Natl. Acad. Sci. USA 109, 14870–14875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sledz P et al. Structure of the 26S proteasome with ATP-gammaS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc. Natl. Acad. Sci. USA 110, 7264–7269 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 48.The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC. [Google Scholar]
- 49.Krissinel E & Henrick K Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Zhang X & Wigley DB The ‘glutamate switch’ provides a link between ATPase activity and ligand binding in AAA+ proteins. Nat. Struct. Mol. Biol 15, 1223–1227 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He J et al. The structure of the 26S proteasome subunit Rpn2 reveals its PC repeat domain as a closed toroid of two concentric alpha-helical rings. Structure 20, 513–521 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Horwitz AA et al. ATP-induced structural transitions in PAN, the proteasome-regulatory ATPase complex in Archaea. J. Biol. Chem 282, 22921–22929 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Deville C et al. Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci. Adv 3, e1701726 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monroe N, Han H, Shen PS, Sundquist WI & Hill CP Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. Elife 6, e24487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Enemark EJ & Joshua-Tor L Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Thomsen ND & Berger JM Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-9215 (the combined EA refined with CP-ATPase mask), EMD-9216 (whole EA1), EMD-9217 (whole EA2), EMD-9218 (whole EB), EMD-9219 (whole EC1), EMD-9220 (whole EC2), EMD-9221 (whole ED1), EMD-9222 (whole ED2), EMD-9223 (RP of EA1), EMD-9224 (RP of EA2), EMD-9225 (RP of EB), EMD-9226 (RP of EC1), EMD-9227 (RP of EC2), EMD-9228 (RP of ED1) and EMD-9229 (RP of ED2). Coordinates are available from the RCSB Protein Data Bank under accession codes 6MSB (whole EA1), 6MSD (whole EA2), 6MSE (whole EB), 6MSG (whole EC1), 6MSH (whole EC2), 6MSJ (whole ED1), 6MSK (whole ED2). Raw data are available from Y.M.