

Abstract

The first example of near-room-temperature α-arylation of benzo[b]thiophenes is reported. The discovery rests on the observation of a switch in α-/β-regioselectivity at different loadings of Pd2(dba)3·CHCl3 in the coupling between benzo[b]thiophene and 4-iodotoluene. We show that this unprecedented regioselectivity switch is driven by a Ag(I)-mediated C–H activation at the α-C–H position, which becomes the dominant mode of reactivity at low concentrations of Pd. Competition experiments, kinetic studies, KIE, and D/H scrambling experiments have been carried out supporting this mechanism.
1. Introduction
The widespread presence of arylated benzo[b]thiophenes and thiophenes in biological compounds, pharmaceuticals, and material sciences makes these scaffolds attractive targets for synthetic methodologies (Scheme 1).1 While conventional cross-couplings are still widely used,2 direct C–H arylation has emerged over the last two decades as a powerful approach that eliminates the need for prefunctionalization, thus leading to shorter synthetic routes. The direct C2-arylation of benzo[b]thiophene was first demonstrated by Ohta in 1990,3 and over the past decade several further examples have been reported using Pd,4 Cu,5 or Ir6 as catalysts.7 However, all the methodologies reported require the use of elevated temperatures (100–150 °C), which limits their functional group compatibility. Furthermore, high catalyst loadings are typically required with a few exceptions.4g,4i,4j,4m The development of mild conditions for the C2-arylation of benzo[b]thiophenes is therefore a highly desirable synthetic target, which would allow a significant expansion of the functional group tolerance and applicability of the methodology. Herein we report studies leading to the discovery of a novel Ag(I)–C–H activation-based methodology that overcomes the aforementioned limitations and affords the first near-room-temperature C2-arylation of benzo[b]thiophenes.8 This new methodology offers wide functional group tolerance, operates at near room temperature, and requires only 0.4 mol % Pd-catalyst loading.
Scheme 1. Examples of 2-Arylbenzo[b]thiophenes.

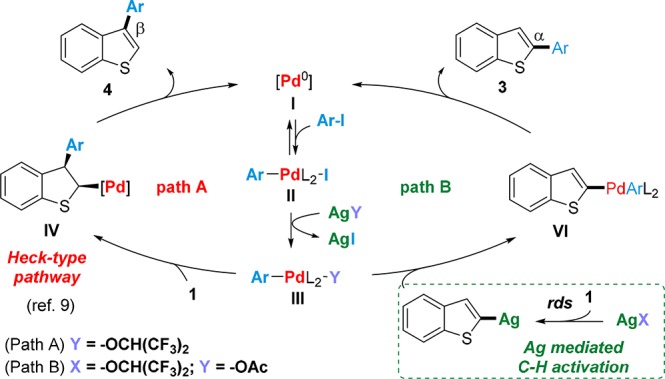
Our investigation began with an unexpected observation during the development of our recently reported C3-selective C–H arylation of benzo[b]thiophenes (1a) with aryl iodides (2a, Scheme 2).9 During optimization of the Pd-catalyst loading of this process we observed that the ratio C2/C3 showed a marked dependence on the concentration of Pd catalyst used in the experiment (Scheme 2). Indeed, while 1:>99 selectivity was obtained when using 2.5 mol % Pd2dba3·CHCl3 this ratio was eroded when decreasing the catalyst loading and eventually reversed at loadings as low as 0.05 mol %. To the best of our knowledge this is the first time a change in the regioselectivity of a C–H functionalization process has been shown to originate in a change in catalyst concentration. Driven by the potential mechanistic implications and the possibility to develop unprecedentedly mild conditions for C2-arylation, we proceeded to investigate the origin of this regioselectivity switch. Given that changes in catalyst concentration should not affect the ratios of the different in-cycle catalytic species in a typical catalytic cycle (Scheme 3, Path A),10 we hypothesized that this switch could originate in the existence of an alternative pathway involving a cocatalyzed process.
Scheme 2. Dependence on the Catalyst Loadings of the Regioselectivity of Coupling between 1a and 2a,

Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard.
1a (0.5 mmol), 2a (0.75 mmol), Ag2CO3 (0.375 mmol), and HFIP (0.5 mL).
Scheme 3. Proposed Mechanism for Formation of 4 (Path A) and 3 (Path B).
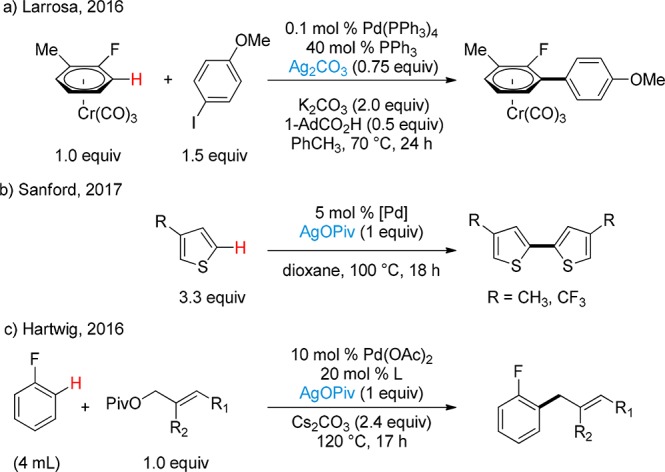
Through a combination of stoichiometric and kinetic studies, we have recently demonstrated that Ag(I) salts are able to carry out C–H activation of electron-deficient arenes.11 This allows for a C–H arylation process that occurs with very low catalyst loadings of Pd (Scheme 4a). Concurrently, Sanford and co-workers established the same prominent role of Ag(I) salts on the C–H activation of both polyfluoroarenes and thiophenes at 100 °C (Scheme 4b).12 Additionally, the investigation of a selective allylation of aryl C–H bonds catalyzed by Pd and mediated by AgOPiv led Hartwig and co-workers to the isolation of the first phosphine-ligated arylsilver(I) complex, which was shown to react with an allyl–Pd complex (Scheme 4c).13
Scheme 4. Previous Methodologies Reporting Experimental Evidence of Ag(I)-Mediated C–H Activation.
On the basis of these precedents, we proposed that a Ag(I)-mediated C2-selective C–H activation process could be responsible for the observed regioselectivity switch on the arylation of benzo[b]thiophene, via competitive Path B (Scheme 3). If the Ag(I)-mediated C–H activation is the rate-determining step of the process leading to C2-arylation, then a lowering of Pd catalyst loading could lead to the observed switch by disproportionately slowing down the C3-arylation process.
2. Results and Discussion
2.1. Reaction Optimization and Scope
Inspired by this mechanistic hypothesis, we decided to probe whether a low-temperature Ag(I)-mediated C2-selective C–H activation would be feasible. We studied the D/H scrambling of 2-d-benzo[b]thiophene (d1-1a) in the presence of different silver additives in hexafluoro-2-propanol (HFIP) (Table 1). Gratifyingly, 10% D/H scrambling was observed in the presence of Ag2O (Table 1, entry 3), increasing to 34% when NaOAc was added as an additive.14
Table 1. D/H Scrambling Studies to Test for Ag(I)–C–H Activationa.

| silver additive | ratio d1-1a:1a |
|---|---|
| Ag2CO3 | >99:1 |
| AgOAc | >99:1 |
| Ag2O | 99:10 |
| Ag2O + NaOAcb | 66:34 |
Ratio determined by quantitative 1H NMR.
NaOAc (0.5 equiv).
Encouraged by these results we then explored the development of a low-temperature C2-arylation protocol based on the observed Ag(I)–C–H activation. Remarkably, reaction of 1a with 2a proceeded at near room temperature in 45% yield when carried out in the presence of 0.75 equiv of Ag2O and 0.5 equiv of NaOAc and only 0.2 mol % Pd2dba3·CHCl3 (Table 2, entry 2). A switch of catalyst to 0.4 mol % Pd(OAc)2 further increased the yield of product 3aa to 54% (Table 2, entry 3). Inverting the stoichiometry of the two coupling partners increased the yield to 73% (Table 2, entry 4), consistently with the proposed rate-limiting C–H activation. Finally, increasing the amount of Ag2O to 1 equiv afforded 3aa in 83% yield (Table 2, entry 5).15,16
Table 2. Optimization of Reaction Conditionsa,b.

| entry | [Pd] | additive | 3aa (%) | 4aa (%) |
|---|---|---|---|---|
| 1 | Pd2(dba)3·CHCl3 | - | 26 | 10 |
| 2 | Pd2(dba)3·CHCl3 | NaOAc | 45 | 1 |
| 3 | Pd(OAc)2 | NaOAc | 54 | 6 |
| 4c | Pd(OAc)2 | NaOAc | 73 | 5 |
| 5c,d | Pd(OAc)2 | NaOAc | 83 | 3 |
Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard.
1a (0.25 mmol).
1a (2 equiv), 2a (1 equiv, 0.25 mmol).
Ag2O (1 equiv).
With the optimized conditions in hand, we proceeded to investigate the scope of the reaction (Table 3). Iodoarenes bearing either electron-donating (2a–2d) or electron-withdrawing groups (2e–2o) in para-position reacted in good to excellent yields. Remarkably, the mild reaction conditions enabled compatibility with alcohol (3ad), aldehyde (3ae), and ketone (3af) substituents, which often suffer from issues of chemoselectivity when harsher conditions are employed. Halogen substituents were also tolerated, giving the possibility to further functionalize the products through traditional cross-coupling (3ah–3aj).2 Highly electron-poor iodoarenes showed modest reactivity, although higher yields can be obtained by increasing the temperature to 50 °C (3ak–3al). While 4-iodoaniline was incompatible with the system, probably due to inhibiting coordination of the lone pair of the nitrogen (2m) to the catalyst,17 amide- and cyano-substituted iodoarenes reacted to generate the α-arylated products in yields of 38% (3an) and 63%, respectively (3ao). The methodology also exhibited compatibility with meta (3ap–3aq) and ortho-substituted iodoarenes (3ar–3ax), albeit with lower α:β regioselectivity in the latter case.18 The reactivity of heterocyclic iodoarenes was also investigated: in particular, 2,6-substituted pyridine 2y was found to react to a small extent (3ay), while N-tosyl-5-iodoindole generated the desired α-functionalized product in excellent regioselectivity and 67% yield (3az). To further highlight the mild conditions afforded by this protocol, we tested the coupling between 1a and (S)-N-boc-4-iodo-phenylalanine 2aa′ obtaining the desired product 3aa′ in 83% yield without observing racemization at the chiral center.19 Finally, the reaction is amenable to scaling up: the arylation of 1a with 2a was performed on a 20 mmol scale, obtaining the desired arylated product in 71% yield and 97:3 (C2/C3) regioselectivity.
Table 3. Direct C–H Arylation of Unsubstituted Benzo[b]thiophene 1a with Iodoarenes 2a−2aa′a.
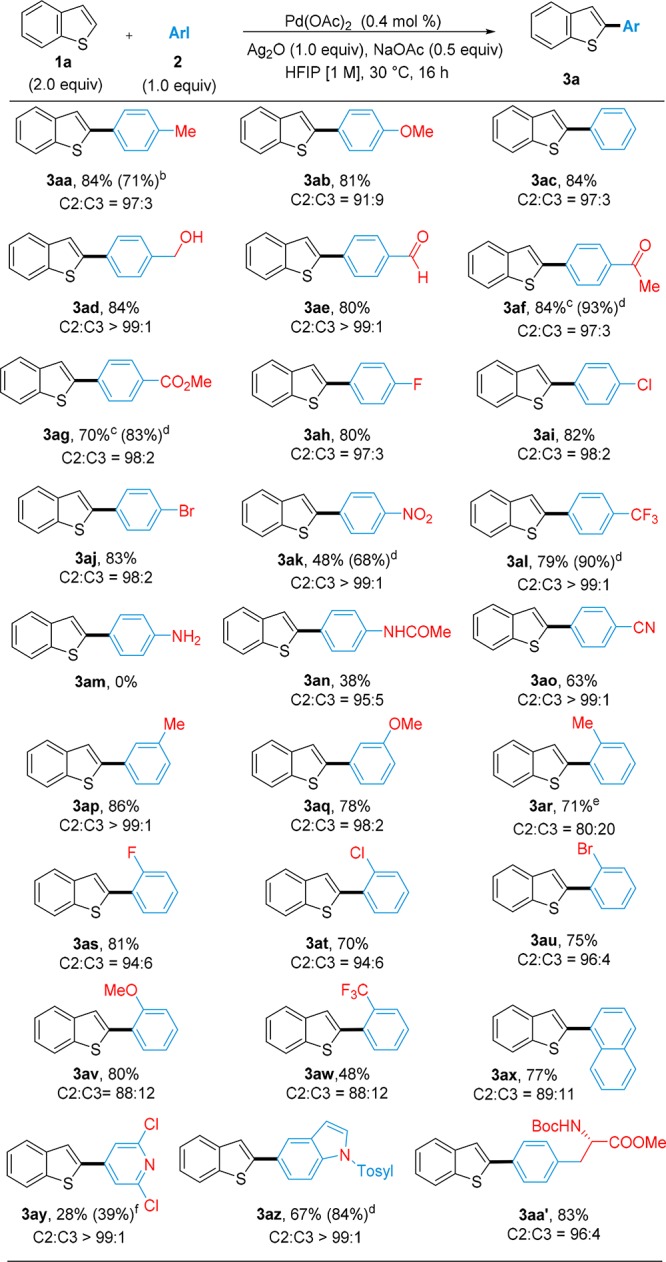
Reaction carried out on a scale of 0.75 mmol of 2. Isolated yields of C2 products are given. C2/C3 ratios were determined by GC-MS analysis of the crude reaction mixtures. Yields in brackets were determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard.
Performed on a 20 mmol scale of 2a.
Performed on a 0.25 mmol scale of 2a.
Performed at 50 °C.
Isolated as an inseparable mixture 4:1 of C2/C3 arylated products.
Reaction carried out for 40 h.
The reactivity of substituted benzo[b]thiophenes was then tested with 4-iodotoluene 2a as the coupling partner (Table 4). In general, 4- and 5-substituted benzo[b]thiophenes afforded high yields and regioselectivities of the corresponding α-arylated products (3ba–3fa). Consistently with the scope of the iodoarene coupling partner, the methodology showed compatibility with alcohols (3ea) among other functional groups. Substituents at C7 were also found to be compatible, albeit with decreased α:β regioselectivity (3ga). 3-Substituted benzo[b]thiophenes could be coupled with 4-iodotoluene 2a generating the α-arylated products in remarkably high yields (5aa and 5ca). Even compound 5cr was obtained in a noteworthy yield of 87% considering that both the bromine atom and the methyl group at the ortho-position of the iodoarene are considerably sterically hindered. Moreover, the methodology could be successfully applied to the synthesis of α,β-bisarylated benzo[b]thiophenes (5aa–5ab). Finally, the same protocol was applied to substituted thiophenes 6a–e obtaining the α-arylated products 7aa–7ea in moderate to good yields and selectivity (Table 4).20,21
Table 4. Direct C–H Arylation of Substituted Benzo[b]thiophene 1b–g or 4a–c with Iodoarene 2a and Substituted Thiophenes 6a–e with Iodoarene 2aa.
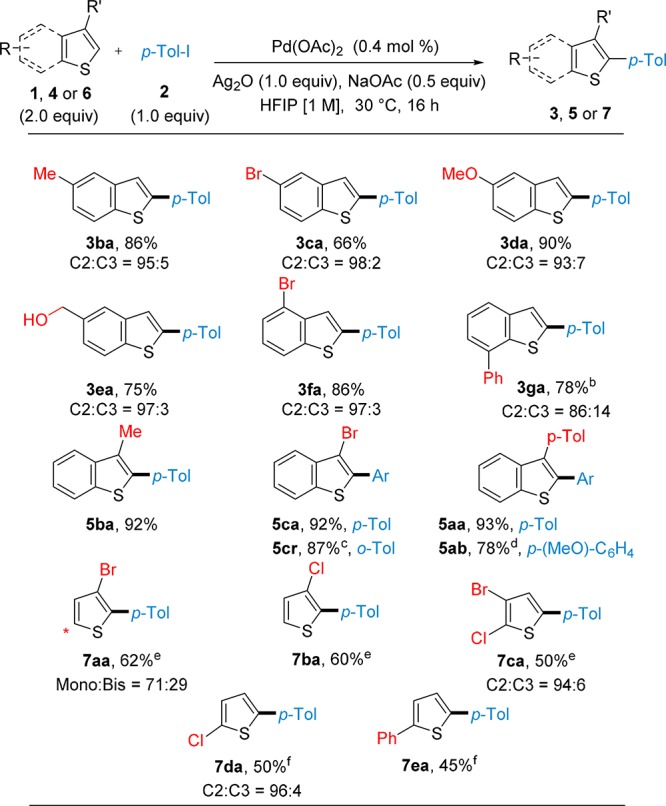
Reaction carried out on a scale of 0.75 mmol of 2. Yields are given as isolated. C3/C2 ratios were determined by GC-MS analysis of the crude reaction mixtures.
Isolated as an inseparable mixture 6:1 of C2/C3 arylated products.
2r instead of 2a.
2b instead of 2a.
Reaction carried out using Pd(OAc)2 (0.8 mol %) and performed at 50 °C for 16 h.
Reaction carried out using 1.0 equiv of 6 and 2.0 equiv of 2a with Pd(OAc)2 (0.8 mol %) and performed at 50 °C for 16 h.
2.2. Mechanistic Considerations
2.2.1. Competition Experiments
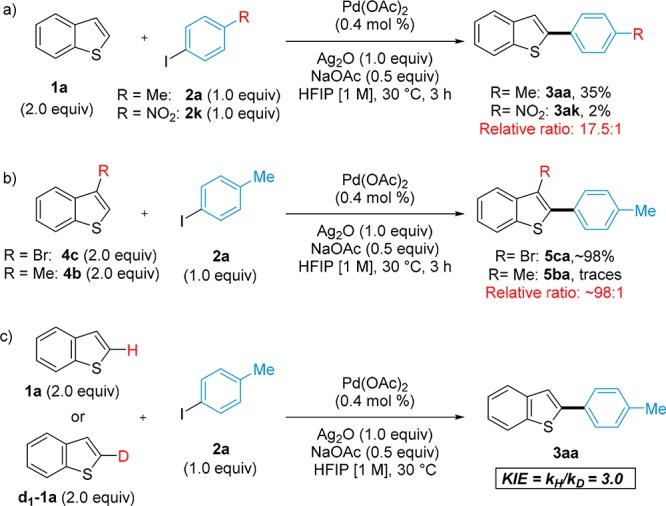
With the aim of gaining information on the mechanism, we carried out a competition experiment between 4-iodotoluene 2a and 1-iodo-4-nitrobenzene 2k (Scheme 5a). The higher reactivity of the more electron-rich iodoarene 2a suggests that the oxidative addition is reversible and happening before the rate-limiting step.9,22 A competition experiment between 3-bromobenzo[b]thiophene 4c and 3-methylbenzo[b]thiophene 4b was also tested (Scheme 5b). The similar steric size of CH3 and Br (van der Waals radii of 1.85 and 1.97 Å (average), respectively)22,23 allows extracting conclusions on the electronic effects of these substituents. Thus, the much higher reactivity of the more acidic 4c suggests that the C–H activation is rate limiting.24,25 Further evidence was obtained by measuring a H/D KIE of 3.0 for this system, suggesting a rate-limiting C–H activation that is likely proceeding via a concerted metalation deprotonation-like process (Scheme 5c).11,12,26 This is in contrast to the KIE of 1.0 measured for the C3-arylation protocol.9
Scheme 5. Competition Experiments.
2.2.2. Kinetic Studies
To gain further information on the mechanism of this reaction, we proceeded with the acquisition of kinetic data. We elected to use the experimental design of Blackmond’s reaction progress kinetic analysis (RPKA)27 and analyzed the data with the variable-time normalization analysis (VTNA) graphical methods recently developed by Burés (Scheme 6).28,29 In order to use these tools, catalyst deactivation and product inhibition cannot be present. Under the standard conditions, the same “excess” reaction analyzed with the time-adjusted method reveals that the reaction was not affected by these issues (Scheme 6a). We began the analysis with an investigation on the order of the Pd catalyst. Similarly to our previously reported discovery of Ag(I)-mediated C–H activation,11 an order of zero was obtained for Pd(OAc)2 at loadings between 0.4 and 0.8 mol %. Given that the Pd species are fully soluble in the reaction conditions, these results imply that a process external to the Pd-catalytic cycle is rate limiting (Scheme 6b),11,30 in agreement with our proposal of a Ag-mediated C–H activation of benzo[b]thiophene 1a (Scheme 3, path B). An order of 1 in 1a and an order 0 in iodoarene 2a (Scheme 6c,d) provide further support for this hypothesis. The order of 1 in benzo[b]thiophene together with the order of 0 in Pd catalyst suggests that Pd is not involved in the C–H activation step. An order of 0 was obtained for NaOAc, suggesting that this species is not involved in the rate-limiting step (Scheme 6e). We speculate that NaOAc helps the reaction by lowering the rate of insertion of the aryl–Pd complex III to the double bond of benzo[b]thiophene (to IV in Scheme 3) in the path to C3 formation, thus favoring the C2 pathway;31 on the other hand, the rate from III to VI remains controlled by the Ag(I)–C–H activation rate-limiting step and therefore unaffected by NaOAc. Demonstrating the involvement of Ag in the C–H activation step of the catalytic process proved to be nontrivial. Two practical issues were faced: (1) the low solubility of Ag2O in HFIP and (2) when lowering the rate of the C2-arylation process, C3-arylation becomes competitive again, producing a mixture of C2 and C3 which is extremely difficult to deconvolute into mechanistic information (Supporting Information, Figure S.7). These issues were overcome by changing the substrate to 3-bromobenzo[b]thiophene 4c. This allowed us to measure an order of 0.5 in Ag2O at concentrations between 0.6 and 0.4 M (Scheme 6f). This order in Ag is consistent with an inactive dimeric resting state of the type Ag2Xn in equilibrium with the active monomeric AgX species. We speculate that AgOCH(CF3)2 could form in situ in low concentrations by acid–base reaction of Ag2O with HFIP and could be responsible for the observed reactivity.32,33 Taken together, these kinetic data point to a mechanism involving a rate-limiting Ag-mediated C–H activation of 1a consistent with our proposal in Scheme 3 (path B).
Scheme 6. (a) Same “Excess” Experiment Where VTNA Enables the Determination of the Order in (b) Pd Catalyst, (c) Benzo[b]thiophene 1a, (d) ArI 2a, (e) NaOAc, and (f) Ag2O.
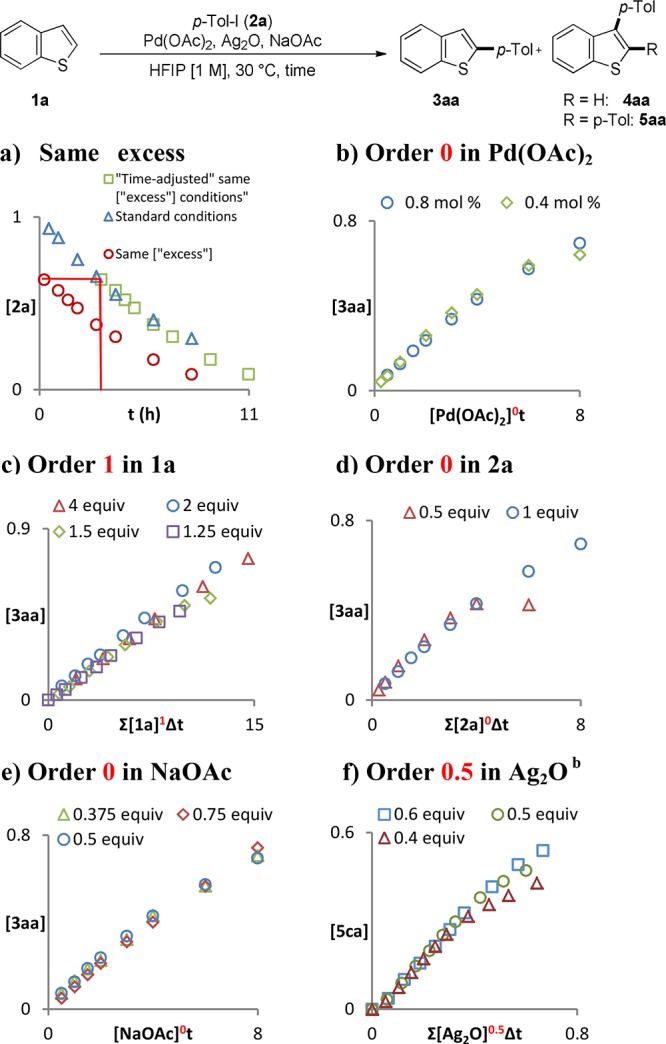
In the graphs, equiv are referred to the amounts of reactants whose orders are determined. For experimental details and unmodified temporal reaction profiles, see Supporting Information.
The kinetic run was performed with 4c instead of 1a, to give product 5ca.
3. Conclusion
In conclusion, we have developed the first protocol for the near-room-temperature α-arylation of benzo[b]thiophenes, which also found application to the α-arylation of substituted thiophenes. The excellent regioselectivity and mild conditions of this methodology are derived from a novel approach that utilizes Ag(I) to carry out C2-selective C–H activation, before transmetalation to Pd and subsequent C–C bond formation. The use of very low concentrations of the Pd catalyst is possible due to the key role played by Ag. D/H scrambling, competition experiments, KIE, and kinetic studies support a mechanism involving Ag(I)–C–H activation.
4. Experimental Section
General Procedure
Pd(OAc)2 (0.4 mol %), silver oxide (1.0 equiv), NaOAc (0.5 equiv), aryl iodide 2 (1.0 equiv), and (substituted)-benzo[b]thiophene 1 or 4, or (substituted)-thiophene 6 (2.0 equiv) were stirred in hexafluoro-2-propanol (1 M) at 30 °C for 16 h. After this time, the resultant mixture was diluted with EtOAc (5 mL) and filtered through a plug of silica. The silica plug was flushed with EtOAc (30 mL), and the filtrate was evaporated to dryness under reduced pressure. Purification via column chromatography afforded the desired arylated (benzo)thiophenes 3, 5, or 7.
Representative Example
2-(p-Tolyl)benzo[b]thiophene (3aa; 0.75 mmol Scale Reaction: Table 3)
Benzo[b]thiophene 1a (205 mg, 1.5 mmol, 2.0 equiv), 4-iodotoluene 2a (165 mg, 0.75 mmol, 1.0 equiv), Pd(OAc)2 (0.4 mol %), silver oxide (174 mg, 0.75 mmol, 1.0 equiv), and NaOAc (31 mg, 0.375 mmol, 0.5 equiv) were stirred in 0.75 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at 30 °C for 16 h. After this time, the resultant mixture was diluted with EtOAc (5 mL) and filtered through a plug of silica. The silica plug was flushed with EtOAc (30 mL), and the filtrate was evaporated to dryness under reduced pressure. Product 3aa was then isolated by column chromatography (hexane) as a white solid in 84% yield (141 g, 0.63 mmol). Rf (hexane): 0.47. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.82 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.51 (s, 1H), 7.37–7.27 (m, 2H), 7.24 (d, J = 8.0 Hz, 2H), 2.40 (s, 3H). 13C NMR (101 MHz, CDCl3): δ (ppm) 144.7, 141.1, 140.0, 138.6, 131.8, 130.0, 126.7, 124.8, 124.4, 123.7, 122.6, 119.2, 21.6. HRMS: calcd for C15H12S (M+), 224.0654; found, 224.0654. Mp: 166–168 °C.
Acknowledgments
We gratefully acknowledge the European Research Council for a Starting Grant (to I.L.), the Engineering and Physical Sciences Research Council (EP/L014017/2 and EP/K039547/1), and the Spanish MECD (grant FPU 13/05325, EST15/00774, to J. F.-C.). A.P. is funded by a scholarship from the Commonwealth Scholarship Commission.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05361.
Experimental procedures and characterization data (PDF)
Author Contributions
† C.C. and A.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected applications of arylated benzo[b]thiophenes and thiophenes, see:; a Lu R.-J.; Tucker J. A.; Pickens J.; Ma Y.-A.; Zinevitch T.; Kirichenko O.; Konoplev V.; Kuznetsova S.; Sviridov S.; Brahmachary E.; Khasanov A.; Mikel C.; Yang Y.; Liu V.; Wang J.; Freel S.; Fisher S.; Sullivan A.; Zhou J.; Stanfield-Oakley S.; Baker B.; Sailstad J.; Greenberg M.; Bolognesi D.; Bray B.; Koszalka B.; Jeffs P.; Jeffries C.; Chucholowski A.; Sexton C. J. Med. Chem. 2009, 52, 4481–4487. 10.1021/jm900330x. [DOI] [PubMed] [Google Scholar]; b Senthil Kumar N.; Arul Clement J.; Mohanakrishnan A. K. Tetrahedron 2009, 65, 822–830. 10.1016/j.tet.2008.11.044. [DOI] [Google Scholar]; c Bridges T. M.; Kennedy J. P.; Hopkins C. R.; Conn P. J.; Lindsley C. W. Bioorg. Med. Chem. Lett. 2010, 20, 5617–5622. 10.1016/j.bmcl.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Segawa Y.; Maekawa T.; Itami K. Angew. Chem., Int. Ed. 2015, 54, 66–81. 10.1002/anie.201403729. [DOI] [PubMed] [Google Scholar]; e Liger F.; Bouhours P.; Ganem-Elbaz C.; Jolivalt C.; Pellet-Rostaing S.; Popwycz F.; Paris J.; Lemaire M. ChemMedChem 2016, 11, 320–330. 10.1002/cmdc.201500463. [DOI] [PubMed] [Google Scholar]; f Rodríguez-Cantó P. J.; Martínez-Marco M.; Sánchez-Royo J. F.; Martínez-Pastor J. P.; Abargues R. Polymer 2017, 108, 413–422. 10.1016/j.polymer.2016.12.003. [DOI] [Google Scholar]
- a Hassan J.; Sévignon M.; Gozzi C.; Schulz E.; Lemaire M. Chem. Rev. 2002, 102, 1359–1469. 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]; b de Meijere A.; Diederich F.. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, 2004. [Google Scholar]; c Seechurn C. C. C. J.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]; d Colacot T. J.New Trends in Cross-Coupling: Theory and Applications; RSC: Cambridge, 2015. [Google Scholar]
- Ohta A.; Akita Y.; Ohkuwa T.; Chiba M.; Fukunaga R.; Miyafuji A.; Nakata T.; Tani N.; Aoyagi Y. Heterocycles 1990, 31, 1951–1958. 10.3987/COM-90-5467. [DOI] [Google Scholar]
- For [Pd] catalysis, see:; a Nandurkar N. S.; Bhanushali M. J.; Bhor M. D.; Bhanage B. M. Tetrahedron Lett. 2008, 49, 1045–1048. 10.1016/j.tetlet.2007.11.209. [DOI] [Google Scholar]; b Liégault B.; Lapointe D.; Caron L.; Vlassova A.; Fagnou K. J. Org. Chem. 2009, 74, 1826–1834. 10.1021/jo8026565. [DOI] [PubMed] [Google Scholar]; c Tamba S.; Okubo Y.; Tanaka S.; Monguchi D.; Mori A. J. Org. Chem. 2010, 75, 6998–7001. 10.1021/jo101433g. [DOI] [PubMed] [Google Scholar]; d René O.; Fagnou K. Adv. Synth. Catal. 2010, 352, 2116–2120. 10.1002/adsc.201000397. [DOI] [Google Scholar]; e Baghbanzadeh M.; Pilger C.; Kappe C. O. J. Org. Chem. 2011, 76, 8138–8142. 10.1021/jo201516v. [DOI] [PubMed] [Google Scholar]; f Ghosh D.; Lee H. M. Org. Lett. 2012, 14, 5534–5537. 10.1021/ol302635e. [DOI] [PubMed] [Google Scholar]; g Zhao L.; Bruneau C.; Doucet H. Tetrahedron 2013, 69, 7082–7089. 10.1016/j.tet.2013.06.037. [DOI] [Google Scholar]; h Dao-Huy T.; Haider M.; Glatz F.; Schnürch M.; Mihovilovic M. D. Eur. J. Org. Chem. 2014, 2014, 8119–8125. 10.1002/ejoc.201403125. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Li Y.; Wang J.; Huang M.; Wang Z.; Wu Y.; Wu Y. J. Org. Chem. 2014, 79, 2890–2897. 10.1021/jo402745b. [DOI] [PubMed] [Google Scholar]; j Luo B.-T.; Liu H.; Lin Z.-J.; Jiang J.; Shen D.-S.; Liu R.-Z.; Ke Z.; Liu F.-S. Organometallics 2015, 34, 4881–4894. 10.1021/acs.organomet.5b00181. [DOI] [Google Scholar]; k Jakab A.; Dalicsek Z.; Soós T. Eur. J. Org. Chem. 2015, 2015, 56–59. 10.1002/ejoc.201402586. [DOI] [Google Scholar]; l Korenaga T.; Sasaki R.; Shimada K. Dalton Trans. 2015, 44, 19642–19650. 10.1039/C5DT01991E. [DOI] [PubMed] [Google Scholar]; m Ouyang J.-S.; Li Y.-F.; Shen D.-S.; Ke Z.; Liu F.-S. Dalton Trans. 2016, 45, 14919–14927. 10.1039/C6DT02544G. [DOI] [PubMed] [Google Scholar]; n Ahmed J.; Sau S. C.; P S.; Hota P. K.; Vardhanapu P. K.; Vijaykumar G.; Mandal S. K. Eur. J. Org. Chem. 2017, 2017, 1004–1011. 10.1002/ejoc.201601218. [DOI] [Google Scholar]; o Chikhi S.; Djebbar S.; Soulé J.-F.; Doucet H. J. Organomet. Chem. 2017, 831, 55–63. 10.1016/j.jorganchem.2016.12.026. [DOI] [Google Scholar]; For a bimetallic system involving [Pd]/[Cu] complexes, see:; p Yin S.-C.; Zhou Q.; He Q.-W.; Li S.-W.; Qian P.-C.; Shao L.-X. Tetrahedron 2017, 73, 427–431. 10.1016/j.tet.2016.11.053. [DOI] [Google Scholar]
- Do H.-Q.; Kashif Khan R. M.; Daugulis O. J. Am. Chem. Soc. 2008, 130, 15185–15192. 10.1021/ja805688p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Join B.; Yamamoto T.; Itami K. Angew. Chem., Int. Ed. 2009, 48, 3644–3647. 10.1002/anie.200806358. [DOI] [PubMed] [Google Scholar]
- For recent reviews on C–H functionalization of α-/β-arylation of benzo[b]thiophenes and other heterocycles, see:; a Ackermann L.; Vicente R.; Kapdi A. R. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; b Bellina F.; Rossi R. Tetrahedron 2009, 65, 10269–10310. 10.1016/j.tet.2009.10.015. [DOI] [Google Scholar]; c Roger J.; Gottumukkala A. L.; Doucet H. ChemCatChem 2010, 2, 20–40. 10.1002/cctc.200900074. [DOI] [Google Scholar]; d Rossi R.; Bellina F.; Lessi M.; Manzini C. Adv. Synth. Catal. 2014, 356, 17–117. 10.1002/adsc.201300922. [DOI] [Google Scholar]; e Djakovitch L.; Felpin F.-X. ChemCatChem 2014, 6, 2175–2187. 10.1002/cctc.201402288. [DOI] [Google Scholar]; f El Kazzouli S. E.; Koubachi J.; El Brahmi N. E.; Guillaumet G. RSC Adv. 2015, 5, 15292–15327. 10.1039/C4RA15384G. [DOI] [Google Scholar]; g Rossi R.; Lessi M.; Manzini C.; Marianetti G.; Bellina F. Adv. Synth. Catal. 2015, 357, 3777–3814. 10.1002/adsc.201500799. [DOI] [Google Scholar]; h Bheeter C. B.; Chen L.; Soulé J. F.; Doucet H. Catal. Sci. Technol. 2016, 6, 2005–2049. 10.1039/C5CY02095F. [DOI] [Google Scholar]; i Badenock J. C.; Gribble G. W. In Adv. Heterocycl. Chem.; Scriven E. F. V., Ramsden C. A., Eds.; Elsevier: USA, 2016; pp 99–136. [Google Scholar]; j Maes J.; Maes B. U. W. In Adv. Heterocycl. Chem.; Scriven E. F. V., Ramsden C. A., Eds.; Elsevier: USA, 2016; pp 137–194. [Google Scholar]; k Théveau L.; Schneider C.; Fruit C.; Hoarau C. ChemCatChem 2016, 8, 3183–3194. 10.1002/cctc.201600489. [DOI] [Google Scholar]; l Yin S.; Zhou Q.; He Q.; Li S.; Qian P.; Shao L. Tetrahedron 2017, 73, 427–431. 10.1016/j.tet.2016.11.053. [DOI] [Google Scholar]; m Komiyama T.; Minami Y.; Furuya Y.; Hiyama T. Angew. Chem., Int. Ed. 2018, 57, 1987–1990. 10.1002/anie.201712081. [DOI] [PubMed] [Google Scholar]
- α-Arylation of benzo[b]thiophenes at room temperature has been previously claimed by:Xu Z.; Xu Y.; Lu H.; Yang T.; Lin X.; Shao L.; Ren F. Tetrahedron 2015, 71, 2616–2621. 10.1016/j.tet.2015.03.051. [DOI] [Google Scholar]; However, a careful analysis of 1H and 13C NMR spectra provided in the Supporting Information reveals that the regiochemistry was incorrectly assigned, and the β-arylated benzo[b]thiophenes were obtained instead. Indeed, when we attempted this protocol, low yields of the β-arylated product were obtained exclusively.
- Colletto C.; Islam S.; Juliá-Hernández F.; Larrosa I. J. Am. Chem. Soc. 2016, 138, 1677–1683. 10.1021/jacs.5b12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We have previously shown that this transformation is likely to proceed via a Heck-type Pd0/II pathway as that shown in Scheme 3a. See ref (9).
- Whitaker D.; Burés J.; Larrosa I. J. Am. Chem. Soc. 2016, 138, 8384–8387. 10.1021/jacs.6b04726. [DOI] [PubMed] [Google Scholar]
- Lotz M. D.; Carnasso N. M.; Canty A. J.; Sanford M. S. Organometallics 2017, 36, 165–171. 10.1021/acs.organomet.6b00437. [DOI] [Google Scholar]
- Yunmi Lee S.; Hartwig J. F. J. Am. Chem. Soc. 2016, 138, 15278–15284. 10.1021/jacs.6b10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- While it is tempting to assume from this experiment that NaOAc is accelerating the C–H activation step, this is not necessarily so. Subsequent experiments (see Mechanistic Section) suggest that NaOAc is only accelerating the H/D exchange step after the Ag(I)–C–H activation (see Supporting Information, section IV-1, for a detailed explanation).
- The requirement of stoichiometric Ag2O arises from the secondary role of the Ag as iodine scavenger (as depicted in Scheme 3).
- Trace amounts of C2/C3-bisarylated product, 5aa, were also detected. See Supporting Information for details.
- The coupling between benzo[b]thiophene 1a and 4-iodotoluene 2a was tested using the standard conditions in the presence of N,N-dimethylaniline (20 mol %) obtaining the desired product 3aa in only traces.
- The lower C2/C3 selectivity observed when using ortho-substituted aryl iodides could result from a more difficult transmetalation step between the benzo[b]thiophenyl–Ag complex and the Ar–Pd III (Scheme 3), thus favoring the formation of C3-arylated product. Nonetheless, no clear regioselectivity trend depending on the steric and electronic properties of the substituents has been observed.
- Racemization in the Stille coupling of a similar substrate has been observed at temperatures above 70 °C.
- For recent examples of arylation of thiophenes, see:; a Yin S.; Zhou Q.; He Q.; Li S.; Qian P.; Shao L. Tetrahedron 2017, 73, 427–431. 10.1016/j.tet.2016.11.053. [DOI] [Google Scholar]; b Natarajan P.; Bala A.; Mehta B. Tetrahedron 2016, 72, 2521–2526. 10.1016/j.tet.2016.03.087. [DOI] [Google Scholar]
- No relevant side products and complete recovery of the starting materials were observed.
- a Fors B. P.; Buchwald S. L. J. Am. Chem. Soc. 2009, 131, 12898–12899. 10.1021/ja905768k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vinogradova E. V.; Fors B. P.; Buchwald S. L. J. Am. Chem. Soc. 2012, 134, 11132–11135. 10.1021/ja305212v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charton M. J. Am. Chem. Soc. 1969, 91, 615–618. 10.1021/ja01031a016. [DOI] [Google Scholar]
- CH3 and Br present similar Taft’s ES steric parameters: −1.24 and −1.16, respectively.
- a Shen K.; Fu Y.; Li J.-N.; Liu L.; Guo Q.-X. Tetrahedron 2007, 63, 1568–1576. 10.1016/j.tet.2006.12.032. [DOI] [Google Scholar]; b Gorelsky S. I.; Lapointe D.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 10848–10849. 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]; c Liégault B.; Lapointe D.; Caron L.; Vlassova A.; Fagnou K. J. Org. Chem. 2009, 74, 1826–1834. 10.1021/jo8026565. [DOI] [PubMed] [Google Scholar]; d Ueda K.; Yanagisawa S.; Yamaguchi J.; Itami K. Angew. Chem., Int. Ed. 2010, 49, 8946–8949. 10.1002/anie.201005082. [DOI] [PubMed] [Google Scholar]
- Importantly, D/H exchange was not observed in the KIE experiment with d1-1a. This is consistent with the transmetalation from the benzo[b]thiophenyl–Ag complex to Pd-intermediate III (Scheme 3) being faster than the D/H exchange process and a rate-determining Ag–C–H activation.
- a Blackmond D. G. Angew. Chem., Int. Ed. 2005, 44, 4302–4320. 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]; b Mathew J. S.; Klussmann M.; Iwamura H.; Valera F.; Futran A.; Emanuelsson E. A. C.; Blackmond D. G. J. Org. Chem. 2006, 71, 4711–4722. 10.1021/jo052409i. [DOI] [PubMed] [Google Scholar]; c Baxter R. D.; Sale D.; Engle K. M.; Yu J.-Q.; Blackmond D. G. J. Am. Chem. Soc. 2012, 134, 4600–4606. 10.1021/ja207634t. [DOI] [PubMed] [Google Scholar]
- a Burés J. Angew. Chem., Int. Ed. 2016, 55, 2028–2031. 10.1002/anie.201508983. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Burés J. Angew. Chem., Int. Ed. 2016, 55, 16084–16087. 10.1002/anie.201609757. [DOI] [PubMed] [Google Scholar]
- The temporal reaction profiles have been analyzed considering either [3aa] alone or in combination with [5aa] obtaining identical results. For more details, see Supporting Information.
- a Pandey R. N.; Henry P. M. Can. J. Chem. 1975, 53, 2223–2231. 10.1139/v75-312. [DOI] [Google Scholar]; b Tanaka Y.; Takahara J. P.; Lempers H. E. B. Org. Process Res. Dev. 2009, 13, 548–554. 10.1021/op900001p. [DOI] [Google Scholar]; c Yayla H. G.; Peng F.; Mangion I. K.; McLaughlin M.; Campeau L.-C.; Davies I. W.; DiRocco D. A.; Knowles R. R. Chem. Sci. 2016, 7, 2066–2073. 10.1039/C5SC03350K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A plausible explanation for NaOAc having the effect of reducing C3-arylation could be associated to the formation of ArPdX species III containing OAc as X, instead of OCH(CF3)2. Indeed, we had previously shown (ref (9)) that “Ar-Pd-OCH(CF3)2”-type intermediates were essential for achieving C3 regioselectivity, while addition of any carboxylate species resulted in significantly reduced activity.
- Attempts at independently preparing AgOCH(CF3)2 by a variety of methods led to decomposition products.
- For a reaction with a dimerization off-cycle equilibrium the order on these species must be in the range 0.5 to 1. See ref (28)(a).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.