

Abstract
Dendritic arbors are crucial for nervous system assembly, but the intracellular mechanisms that govern their assembly remain incompletely understood. Here, we show that the dendrites of PVD neurons in Caenorhabditis elegans are patterned by distinct pathways downstream of the DMA-1 leucine-rich transmembrane (LRR-TM) receptor. DMA-1/LRR-TM interacts through a PDZ ligand motif with the guanine nucleotide exchange factor TIAM-1/GEF in a complex with act-4/Actin to pattern higher order 4° dendrite branches by localizing F-actin to the distal ends of developing dendrites. Surprisingly, TIAM-1/GEF appears to function independently of Rac1 guanine nucleotide exchange factor activity. A partially redundant pathway, dependent on HPO-30/Claudin, regulates formation of 2° and 3° branches, possibly by regulating membrane localization and trafficking of DMA-1/LRR-TM. Collectively, our experiments suggest that HPO-30/Claudin localizes the DMA-1/LRR-TM receptor on PVD dendrites, which in turn can control dendrite patterning by directly modulating F-actin dynamics through TIAM-1/GEF.
Research organism: C. elegans
Introduction
Neurons are highly polarized cells, which comprise a single axon and often elaborately sculpted dendritic arbors. Dendrites receive input from other neurons or the environment, whereas the single axon transmits information to other neurons. The nervous system is formed by a myriad of specific synaptic connections between neurons and the formation of these connections is influenced by the shape and complexity of dendritic arbors. Both genes that act within the developing neurons and in surrounding tissues are crucial to establish distinct dendritic structures during development (Jan and Jan, 2010; Dong et al., 2015; Lefebvre et al., 2015). Of note, defects in dendrite morphology have been found in various neurological disorders (Kaufmann and Moser, 2000; Kulkarni and Firestein, 2012).
Both dendritic and axonal morphology is driven by the cytoskeleton and regulators of the cytoskeleton have consequently important functions in neuronal development. Major components of the cytoskeleton include actin and tubulin, which form filamentous polymers named F-actin and microtubules, respectively (Brouhard, 2015; Pollard, 2016). F-actin exists in unbranched and branched forms, whereas microtubules are generally unbranched. These filament-like polymers are not static, but highly dynamic structures due to the constant association and dissociation of monomers at either end. A plethora of proteins bind to and modulate polymerization and depolymerization of both F-actin and microtubules in neurons (reviewed in (Dent et al., 2011; Kapitein and Hoogenraad, 2015; Konietzny et al., 2017). F-actin and microtubules are important for countless aspects of neuronal function and development in both axons and dendrites, including differentiation, migration and the elaboration of axonal and dendritic processes (Jan and Jan, 2010; Dent et al., 2011).
Regulation of the cytoskeleton is controlled by dedicated signaling pathways, which often originate with cell surface receptors. These receptors utilize regulatory proteins such as guanine nucleotide exchange factors (GEFs) or GTPase-activating proteins (GAPs), which, in turn, modulate the activity of small GTPases. For example, the RacGEF Tiam-1 regulates activity-dependent dendrite morphogenesis in vertebrates (Tolias et al., 2005), likely by activating the small GTPase Rac1. Activated GTPases such as Rac1 then bind the WASP family verprolin-homologous protein (WVE-1/WAVE) regulatory complex (WRC) of actin regulators to promote actin polymerization and branching (Chen et al., 2010).
The polymodal somatosensory neuron PVD in Caenorhabditis elegans has emerged as a paradigm to study dendrite development. The dendritic arbor of PVD neurons develops through successive orthogonal branching (Oren-Suissa et al., 2010; Smith et al., 2010; Albeg et al., 2011) (Figure 1A). During the late larval L2 stage primary (1°) branches first emerge both anteriorly and posteriorly of the cell body along the lateral nerve cord. In subsequent larval stages, secondary (2°) branches emanate orthogonally to bifurcate at the boundary between the lateral epidermis and muscle to form tertiary (3°) branches. These, in turn, form perpendicular quaternary (4°) branches to establish the candelabra-shaped dendritic arbors, which have also been called menorahs (Oren-Suissa et al., 2010). Previous studies have shown that an adhesion complex consisting of MNR-1/Menorin and SAX-7/L1CAM functions from the skin together with the muscle-derived chemokine LECT-2/Chondromodulin II to pattern PVD dendrites. This adhesion complex binds to and signals through the DMA-1/LRR-TM leucine rich transmembrane receptor expressed in PVD neurons (Liu and Shen, 2011; Dong et al., 2013; Salzberg et al., 2013; Díaz-Balzac et al., 2016; Zou et al., 2016). DMA-1/LRR-TM shows great similarity in domain architecture with the LRRTM family of leucine rich transmembrane receptors in humans (Laurén et al., 2003), but limited sequence homology (data not shown). The signaling mechanisms that operate downstream of the DMA-1/LRR-TM receptor in PVD dendrites have remained largely elusive.
Figure 1. The intracellular domain of DMA-1/LRR-TM is required for higher order branching of PVD somatosensory dendrites.
(A) Fluorescent images of PVD (left panels) and schematics (right panels) of wild-type control animals. PVD is visualized by the wdIs52 [F49H12.4p::GFP] transgene in all panels. 1°, 2°, 3°, 4°, and ectopic 3° (e3°) dendrites are indicated. Anterior is to the left and dorsal is up in all panels, scale bars indicate 20 µm. (B) Schematics of the DMA-1/LRR-TM protein and a variant used in transgenic rescue experiments (dma-1(ΔICD)). A PDZ-binding site (YFGI) at the extreme C-terminus of DMA-1/LRR-TM is indicated in lilac. The predicted deletion from the tm5159 deletion allele is shown. (C-F) Fluorescent images of PVD (left panels) and schematics (right panels) of the genotypes indicated. Scale bar indicates 20 µm. (G) Quantification of 2°, 3°, and 4° branch numbers per 100 µm anterior to the PVD cell body. Data for three and two independent transgenic lines for the dma-1 wild type cDNA or the dma-1(ΔICD), respectively, are shown next to the data for the dma-1(dz266[∆PDZ]) allele. The data for dma-1(tm5159) NTS are nontransgenic siblings of a representative transgenic line. For raw data see Figure 1—source data 1. Data are represented as mean ± SEM. Statistical comparisons were performed using one-sided ANOVA with Sidak’s correction. Statistical significance is indicated (ns, not significant; ****, p < 0.0001). n = 20 animals per genotype.

Figure 1—figure supplement 1. Genes functioning cell-autonomously in PVD somatosensory neurons.

Here, we show that DMA-1/LRR-TM forms a complex with the claudin-like molecule HPO-30 (Smith et al., 2013), which is required for surface localization and trafficking of the DMA-1/LLR-TM receptor in PVD dendrites. Consistent with these observations, DMA-1/LRR-TM functions genetically in the Menorin pathway together with hpo-30/Claudin, as well as the guanine nucleotide exchange factor (GEF) tiam-1/GEF and act-4/Actin. The signaling complex is required for both the correct localization of F-actin to, and the exclusion of microtubules from the distal endings of developing somatosensory dendrites. Intriguingly, TIAM-1/GEF appears to function independently of its Rac1 guanine nucleotide exchange factor activity. Biochemical experiments show that DMA-1/LRR-TM can form a complex with TIAM-1/GEF and ACT-4/Actin. Collectively, our experiments suggest that HPO-30/Claudin regulates surface expression of the DMA-1/LRR-TM receptor, which can modulate F-actin dynamics and localization through TIAM-1/GEF independently of GEF activity.
Results
Isolation of genes that function in PVD and FLP dendrite formation
From genetic screens for mutants with defects in PVD patterning (see Material and methods for details), we obtained recessive alleles of the leucine-rich repeat (LRR) single-pass transmembrane (TM) receptor dma-1/LRR-TM and claudin-like hpo-30 (Figure 2A–B, Figure 1—figure supplement 1A–D), both of which are expressed and function in PVD during patterning of the dendritic arbor (Liu and Shen, 2011; Smith et al., 2013). In addition, we isolated mutant alleles in tiam-1 (Figure 2C, Figure 1—figure supplement 1E), the C. elegans homolog (Demarco et al., 2012) of the multidomain vertebrate Rac1 guanine nucleotide exchange factor (GEF) Tiam1 (T-Lymphoma Invasion And Metastasis-Inducing Protein 1) (Habets et al., 1994). Transgenic expression of tiam-1/GEF with heterologous promoters in PVD neurons but not in other tissues efficiently rescued tiam-1/GEF mutant phenotypes, consistent with expression of a tiam-1/GEF reporter in PVD neurons (Demarco et al., 2012) (Figure 1—figure supplement 1F–H). Finally, we isolated a missense allele of act-4/Actin (Figure 2D, Figure 1—figure supplement 1I), one of five actins encoded in the C. elegans genome (Krause et al., 1989). This missense allele (dz222, G151E) is identical to the wy917 allele identified in a related screen by the Shen lab (Zou et al., 2018). The G151E mutation is analogous to a dominant allele in ACTA1 in a human patient with severe congenital myopathy where it resulted in abnormal actin aggregates (Ravenscroft et al., 2011). A reporter for act-4 is expressed in muscle ((Stone and Shaw, 1993), data not shown), but fluorescence in situ hybridization experiments also suggested neuronal expression (Birchall et al., 1995). We found that transgenic expression of act-4/Actin in PVD but not in muscle rescued act-4 mutant defects (Figure 1—figure supplement 1J–M). Additionally, expression of act-1, a paralog of act-4, rescued act-4 mutant phenotypes (Figure 1—figure supplement 1M). All isolated mutant alleles also affected patterning of FLP neurons, a related pair of neurons, which cover the head region of the animal with similar dendritic arbors (Figure 2—figure supplement 1A). We conclude that in addition to DMA-1/LRR-TM and HPO-30/Claudin, TIAM-1/GEF and ACT-4/Actin function cell-autonomously to pattern the dendritic arbor of PVD and, likely FLP neurons. Moreover, expression of any actin in PVD rather than a specific function of ACT-4/Actin is important for dendrite patterning in PVD neurons.
Figure 2. The hpo-30/Claudin, tiam-1/GEF, and act-4/Actin act genetically in the Menorin pathway.
(A – D) Fluorescent images of PVD (left panels, visualized by the wdIs52 [F49H12.4p::GFP] transgene) and schematics (right panels) of the genotypes indicated. The control image is identical to Figure 1A and shown for comparison only. Details on alleles of individual genes and images of other alleles are shown in Figure 1—figure supplement 1. Scale bar indicates 20 µm. (E – G) Quantification of 2°, 3°, and 4° branch numbers per 100 µm anterior to the PVD cell body. Data for additional single and double mutants of the Menorin pathway as well as the average length and aggregate length of secondary, tertiary, and quaternary branches are shown in Figure 2—figure supplement 2. All alleles used are molecular or genetic null alleles (Material and methods for details). Data for control, lect-2, sax-7, mnr-1 and dma-1 mutant animals is identical to data from Díaz-Balzac et al. (2016) and shown for comparison only. For raw data see Figure 1—source data 1. Data are represented as mean ± SEM. Statistical comparisons were performed using one-sided ANOVA with the Tukey correction. Statistical significance is indicated (ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001). n = 20 animals for all genotypes.
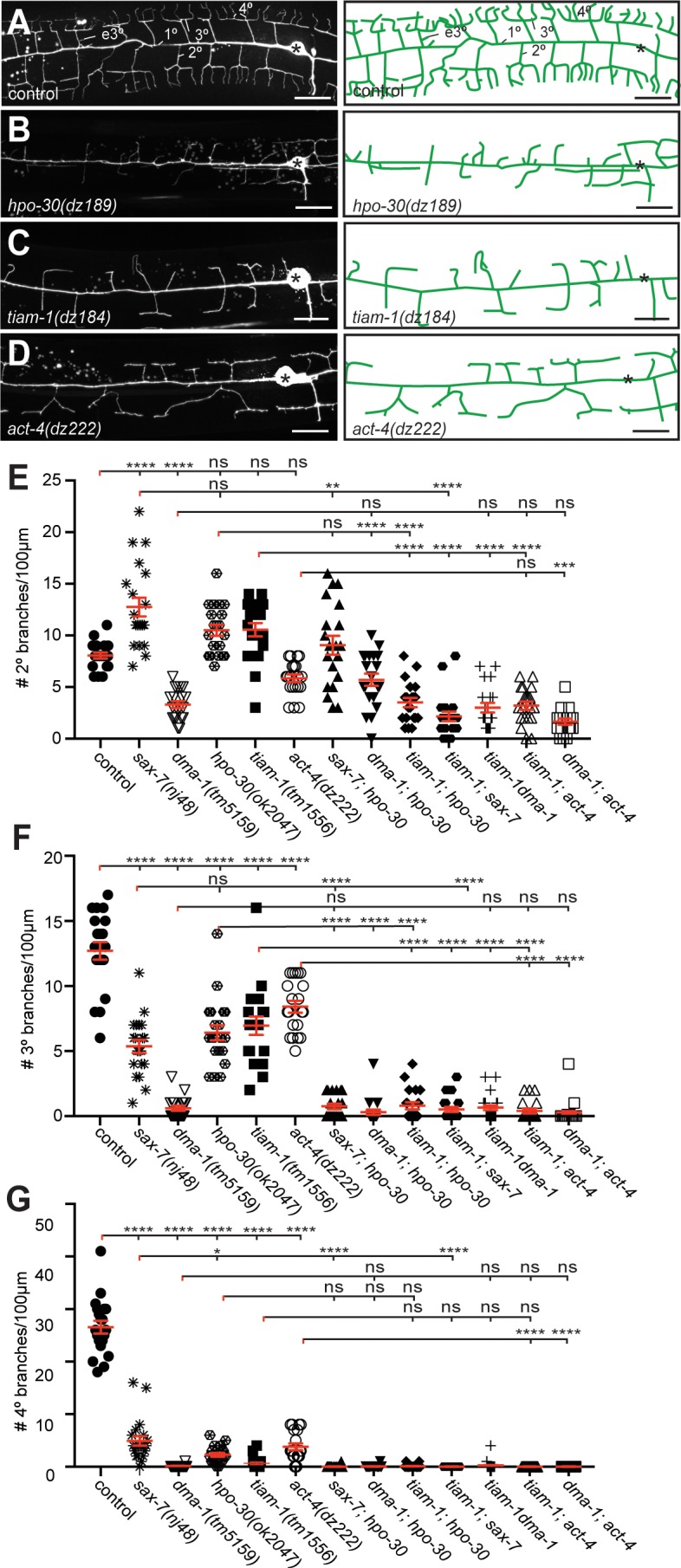
Figure 2—figure supplement 1. Effects of hpo-30/Claudin, tiam-1/GEF and act-4/Actin on localization of LECT-2:mNG, SAX-7::GFP and DMA-1::GFP.

Figure 2—figure supplement 2. The Genetics of hpo-30/Claudin, tiam-1/GEF, and act-4/Actin.

The PDZ-binding site of the DMA-1 leucine-rich transmembrane receptor (DMA-1/LRR-TM) is required for patterning of 4° branches but not 3° branches
Complete removal of dma-1/LRR-TM results in almost complete absence of 3° and 4° branches with additional effects on 2° branches (Liu and Shen, 2011; Dong et al., 2013; Salzberg et al., 2013) (Figure 1B,C, Figure 2—figure supplement 2). As previously shown (Liu and Shen, 2011), we found that the dma-1 mutant phenotype can be fully rescued by transgenic expression of a wild type DMA-1/LRR-TM cDNA (Figure 1C,D,G–I). In contrast, a mutant where the intracellular domain (ICD) of DMA-1 was replaced by mCherry (ΔICD) resulted in partial rescue, where the number of 3° branches, but not 4° branches were restored (Figures 1B,C,E,G–I). The C terminus of DMA-1/LRR-TM follows the consensus sequence of a PDZ-binding site (X- Φ –X–Φ). We therefore used CRISPR/Cas9 genome editing to generate mutant animals predicted to express a form of DMA-1 lacking the last four residues (referred to as ΔPDZ thereafter). The number of 2° and 3° branches in dma-1(ΔPDZ) mutant animals appear indistinguishable from wild-type animals, whereas the number of 4° branches is significantly decreased (Figure 1G–I). These results indicate (1) that the intracellular domain of DMA-1/LRR-TM is required for the formation of PVD higher order branches, and (2) that the PDZ-binding site at the extreme C-terminus of DMA-1/LRR-TM is important for formation of 4 ° branches.
The hpo-30/Claudin, tiam-1/GEF and act-4/Actin genes act in the menorin pathway
To genetically analyze the function of dma-1/LRR-TM, hpo-30/Claudin, tiam-1/GEF and act-4/Actin, we investigated the PVD mutant phenotype in single and double mutants using morphometric analyses. We determined the number and aggregate length for all classes of dendrites in a segment 100 µm anterior to the PVD cell body in different genetic backgrounds. We found that double mutants between sax-7/L1CAM and hpo-30/Claudin were not more severe than the more severe of the single mutants, indicating that hpo-30/Claudin functions in a pathway with sax-7/L1CAM for 2° branch patterning (Figure 2E). The dma-1/LRR-TM; hpo-30/Claudin and dma-1/LRR-TM; tiam-1/GEF were statistically indistinguishable from the dma-1/LRR-TM single mutant, suggesting that dma-1/LRR-TM is epistatic and required for most if not all functions during patterning of higher order branches in PVD. Interestingly, double mutants between tiam-1/GEF and mnr-1/Menorin, lect-2/Chondromodulin II, kpc-1/Furin, sax-7/L1CAM, hpo-30/Claudin, or act-4, respectively, appeared more severe than either of the single mutants alone, but indistinguishable from the dma-1/LRR-TM single mutant (Figure 2E, Figure 2—figure supplement 2). These findings suggest that tiam-1/GEF also serves in a genetic pathway that functions in parallel to mnr-1/sax-7/lect-2/hpo-30 and, possibly, act-4. Similar genetic relationships were observed regarding the number of 3° branches and the aggregate length of 2° and 3° branches with one notable exception. Double mutants between sax-7/L1CAM and hpo-30/Claudin displayed a phenotype for 3° branches that was statistically more severe than either of the single mutants, yet indistinguishable from the dma-1/LRR-TM mutant (Figure 2F, Figure 2—figure supplement 2), suggesting a parallel function for HPO-30/Claudin for higher order branching. Common to all single and double mutants was the near complete absence of 4° branches (Figure 2G, Figure 2—figure supplement 2). Together, our results suggest that (1) hpo-30/Claudin and tiam-1/GEF act in the Menorin pathway to pattern PVD dendritic arbors and, that (2) hpo-30/Claudin and tiam-1/GEF may also serve independent functions.
We next sought to place dma-1/LRR-TM, hpo-30/Claudin, tiam-1/GEF, and act-4/Actin within the genetic pathway using a combination of gain and loss of function approaches. Previous work showed that expression of the hypodermally derived cell adhesion molecule mnr-1/Menorin in muscle of wild-type animals results in the appearance of dendritic arbors that resembled African Baobab trees (Figure 3A) (Salzberg et al., 2013). We used this mnr-1/Menorin gain of function (gof) phenotype in combination with loss-of-function mutations in hpo-30/Claudin, tiam-1/GEF and act-4/Actin. We found that loss of hpo-30/Claudin, tiam-1/GEF or act-4/Actin suppressed the formation of baobab-like dendritic arbors (Figure 3A–E) to the same extent as removal of other genes in the Menorin pathway such as dma-1/LRR-TM, sax-7/L1CAM, or lect-2/Chondromodulin II (Salzberg et al., 2013; Díaz-Balzac et al., 2016). Moreover, protein localization of a LECT-2/Chondromodulin II or SAX-7/L1CAM reporter is not visibly affected in hpo-30/Claudin, tiam-1/GEF or act-4/Actin mutants (Figure 2—figure supplement 1B,C). These findings suggest that hpo-30/Claudin, tiam-1/GEF and act-4/Actin, just like dma-1/LRR-TM, sax-7/L1CAM, or lect-2/Chondromodulin II function genetically downstream of or in parallel to mnr-1/Menorin.
Figure 3. The tiam-1/GEF and act-4/Actin act downstream of the dma-1/LRR-TM receptor in PVD dendrites.
(A – D) Fluorescent images of animals in which mnr-1/Menorin was expressed ectopically in muscle (mnr-1(gof): dzIs43, [myo-3p::mnr-1] (Salzberg et al., 2013)) in different genetic backgrounds. A characteristic baobab-like tree is indicated by a white arrowhead in (A). PVD is visualized by the wdIs52 [F49H12.4p::GFP] transgene in all panels. Anterior is to the left, dorsal up, and scale bars indicate 20 µm in all panels. (E) Quantification of animals with baobab-like dendritic trees in the genotypes indicated. Data are represented as mean. For raw data see Figure 1— source data 1. Statistical comparisons were performed using the Z-test. Statistical significance is indicated (****p<0.0005). n = 20 animals for all samples. (F – G) Fluorescent images of PVD (left panels) and schematics (right panels) of wild type control and tiam-1(o/e) overexpressing animals (dzIs95 [ser-2p3::tiam-1]). The control image (F) is identical to Figure 1A and shown for comparison only. (H – J) Quantification of secondary (2°, (H), tertiary (3°, (I), and quaternary (4°, (J) branch numbers per 100 µm anterior to the PVD cell body in the genotypes indicated. Data for control, tiam-1(tm1556), and act-4(dz222) are identical to data in Figure 2 and shown for comparison only. For raw data see Figure 1—source data 1. Data are represented as mean ±SEM. Statistical comparisons were performed using one-sided ANOVA with the Tukey correction. Statistical significance is indicated (ns, not significant; *, p<0.05; ****p<0.0001). n = 20 for all samples. (K – L) Fluorescent images of PVD in dma-1/LRR-TM mutant animals alone (K) and in combination with a tiam-1(o/e) expression array (L). Anterior is to the left and ventral down. Scale bar indicates 20 µm. (M) Quantification of dendrite termini in a 100 µm section anterior to the PVD cell body in the genotype indicated. For raw data see Figure 1—source data 1. Data are represented as mean ±SEM. Statistical comparisons were performed using Student’s T-test. Statistical significance is indicated (****p<0.0001). n = 20 animals for all samples.

Figure 3—figure supplement 1. The tiam-1/GEF and act-4/Actin act downstream or in parallel of the dma-1/LRR-TM receptor in PVD dendrites.

We next asked in which order dma-1/LRR-TM, tiam-1/GEF and act-4/Actin function in PVD dendrites. Overexpression of tiam-1/GEF (tiam-1(o/e)) in an otherwise wild type background resulted in an increase of 2°, 3°, and 4° branches in PVD dendrites (Figure 3F,G). This excessive branching was completely suppressed by a mutation in act-4/Actin. Specifically, the tiam-1(o/e); act-4 double mutant was statistically indistinguishable from the act-4 single mutant, both with regard to the number and aggregate length of dendritic branches (Figure 3H–J, Figure 3—figure supplement 1A–C). These observations suggest that tiam-1/GEF function is dependent on ACT-4/Actin in PVD dendrites. We further hypothesized that tiam-1/GEF would function downstream of the dma-1/LRR-TM receptor. If that were the case, one would predict that loss of branching in dma-1/LRR-TM null mutants would be at least partially suppressed (i.e. reversed) by overexpression of tiam-1/GEF. Indeed, overexpression of tiam-1(o/e) in dma-1/LRR-TM null mutants significantly increased the number of branches in PVD dendrites (Figure 3K–M). Taken together, our experiments suggest a pathway, in which TIAM-1/GEF functions downstream of or in parallel to DMA-1/LRR-TM (and possibly hpo-30/Claudin) and in a manner that depends on ACT-4/Actin.
HPO-30/Claudin regulates DMA-1/LRR-TM surface expression and trafficking
Both DMA-1/LRR-TM and HPO-30/Claudin are predicted transmembrane proteins functioning in PVD dendrites. To determine the mechanistic relationship between these two factors, we asked where functional DMA-1::GFP and a HPO-30::tagBFP reporter fusions are localized in PVD neurons. As previously reported (Dong et al., 2016), the DMA::GFP reporter is localized in a punctate fashion in the cell body and along the dendritic tree as well as on the membrane, visible as diffuse membrane staining throughout the whole dendritic tree (Figure 4B). We found that an HPO-30::tagBFP fusion also exhibited localized punctate and diffuse membrane staining along the dendritic processes with less staining in the axon of PVD neurons (Figure 4A), much like other recently described HPO-30::GFP reporters (Smith et al., 2013; Zou et al., 2015). The DMA-1::GFP and HPO-30:: tagBFP puncta show significant but not complete colocalization in dendritic processes, suggesting that at least some fraction of these proteins are closely associated (Figure 4C–D).
Figure 4. HPO-30/Claudin forms a complex with, and, localizes DMA-1/LRR-TM to higher order branches of PVD somatosensory neurons.

(A – D) Images of an animal with PVD-specific expression of HPO-30::tagBFP (A, dzEx1837) and DMA-1::GFP (B, qyIs369) as well as a composite image (C), respectively. Both reporters show diffuse staining across the dendrite in addition to discreet punctate staining, with inset enlargement (D) where white arrows indicate puncta of HPO-30::tagBFP and DMA-1::GFP colocalization and a scale bar 10 µm. (E) Schematic showing the topography of the DMA-1/LRR-TM single pass transmembrane receptor and the four transmembrane, claudin-like, HPO-30 protein. Immuno tags (V5 and HA) used for co-immunoprecipitation experiments are shown. Not to scale. (F – G) Western blots of co-immunoprecipitation experiments. Transfected constructs are indicated above the panels. Antibodies used for immunopreciptation (IP) and western blotting (WB) are indicated. A molecular marker is denoted on the left. Note, that all cases the two lower panels are from a single western blot, which was developed repeatedly with two different antibodies after stripping. In G., DMA-1.HA ΔPDZ and DMA-1.HA ΔICD are lacking the PDZ-binding site or the intracellular domain, respectively. Similar results were obtained from at least three independent replicate experiments. (H – I) PVD-specific expression of DMA-1::GFP (qyIs369) imaged in the presence (upper panel) or absence (lower panel) of a transgene with PVD-specific overexpression of HPO-30::tagBFP (dzEx1837) using identical imaging parameters for comparison of fluorescent intensity. The intensity of DMA-1::GFP appears substantially dimmer on the 1° dendrites when HPO-30::tagBFP is overexpressed. The average fluorescent intensity of the 1° dendrites within 50 µm of the cell body is quantified and presented in I). ****, p<0.0001, Mann-Whitney test. (J – O). PVD-specific expression of DMA-1::GFP (qyIs369) imaged in a hpo-30(ok2047) null mutant or wild type control animal using identical imaging parameters for comparison of fluorescent intensity. Fluorescent intensity was quantified for 3° and 1° dendrites within 50 µm of the cell body in K and L, respectively. The number of puncta for 3° and 1° dendrites between 30 and 130 µm from the cell body was counted and presented in M and N, respectively; the ratio of the puncta between 3° and 1° dendrites is presented in O. For raw data see Figure 1— source data 1. Statistical significance is indicated as ns, not significant; **, p<0.01; ***, p<0.001; ****, p<0.0001; Mann-Whitney test. (P) – R. Images of selected time points in a time-lapse series of visualizing DMA-1::GFP (qyIs369) in wild-type control and hpo-30 null mutant animals, imaged in 2 min intervals for 30 min to observe the dynamics of DMA-1::GFP puncta. Puncta between 30 to 130 µm anterior to the cell body were identified at t = 0 min and classified as mobile or immobile. Puncta were deemed immobile if there was no perceived movement throughout the whole time-lapse assay (30 min), and mobile otherwise. The number of mobile and immobile puncta were quantified in R. For raw data see Figure 1— source data 1. Statistical significance is indicated as **, p<0.01, 2-way ANOVA.
To determine if DMA-1/LRR-TM and HPO-30/Claudin are part of the same biochemical complex, we conducted co-immunoprecipitation experiments by transfecting human embryonic kidney cells (HEK293T) with DMA-1 (tagged with HA) and HPO-30 (tagged with V5) (Figure 4E) alone and in combination. We found that DMA-1.HA efficiently co-precipitated HPO-30.V5 from a cellular lysate, suggesting that both proteins are part of the same biochemical complex (Figure 4F). This biochemical interaction was independent of the intracellular domain of DMA-1 (Figure 4G), suggesting that the proteins interact through either the transmembrane segments or the extracellular domains of DMA-1.
We noticed that overexpression of the HPO-30::tagBFP reporter in PVD resulted in significantly reduced fluorescent intensity of the DMA-1::GFP reporter in 1° dendrites compared to control animals (Figure 4H,I). Conversely, loss of hpo-30/Claudin resulted in an increase of diffuse dendrite staining of the DMA-1::GFP reporter in 3° but not in 1° dendrites (Figure J-L). In addition, we found the number of DMA-1::GFP puncta in 1° and 3° branches reduced in hpo-30 null mutants, although their relative distribution remained unchanged (Figure 4M–O). It has been suggested that the diffuse staining of DMA-1::GFP represents the surface membrane fraction while punctate staining represents the vesicular fraction (Taylor et al., 2015; Zou et al., 2015). Therefore, our results suggest that the normal function of HPO-30/Claudin is to negatively regulate the DMA-1/LRR-TM surface membrane fraction and to increase the vesicular fraction.
To further investigate this notion, we carried out time lapse imaging of the DMA-1::GFP reporter for 30 min in both wild type and hpo-30 mutant animals (Figure 4P–Q). We found that in both genotypes, DMA-1::GFP puncta fell into a mobile and a stationary fraction, respectively. The mobile fraction exhibited a wide range of moving speeds in both retrograde and anterograde directions. Intriguingly, the number of mobile DMA-1::GFP puncta was decreased in hpo-30 mutant animals compared to wild type animals, while the number of immobile DMA-1::GFP puncta remained unchanged (Figure 4R). The mobile DMA-1::GFP puncta may be vesicles that transport synthesized or internalized DMA-1::GFP, while immobile puncta may be vesicles or membrane fraction aggregates that serve signaling purposes. Collectively, we conclude that (1) HPO-30/Claudin exists in a complex with DMA-1/LRR-TM, that (2) HPO-30 negatively regulates DMA-1::GFP membrane levels and, that (3) HPO-30 serves a function in trafficking of DMA-1::GFP vesicles.
The TIAM-1/GEF guanine nucleotide exchange factor appears to function independently of its Rac1 GEF activity to pattern PVD dendrites
To investigate the functions downstream of DMA-1/HPO-30, we focused next on the function of the guanine nucleotide exchange factor TIAM-1/GEF. C. elegans TIAM-1/GEF is a multidomain protein (Figure 5A), which comprises, in order, a myristoylation signal, an EVH1 (Ena/Vasp homology) domain, a PDZ domain, and a DH/PH (Dbl homology domain/plekstrin homology domain) encoding the enzymatic guanine nucleotide exchange activity (Demarco et al., 2012). Consistent with work in flies and vertebrate neurons (Sone et al., 1997; Kunda et al., 2001; Tolias et al., 2005), C. elegans TIAM-1/GEF can shape neurons through mechanisms dependent on the small GTPases mig-2/RhoG and ced-10/Rac1 (Demarco et al., 2012). In addition to mig-2/RhoG and ced-10/Rac1, the C. elegans genome encodes one additional Rac1-like GTPase, rac-2 (Lundquist et al., 2001). Surprisingly, neither mutations in mig-2/RhoG and ced-10/Rac1 alone or in combination, nor a mutation in rac-2 resulted in obvious phenotypes in PVD dendritic arbor formation (Figure 5—figure supplement 1A–C), implying that TIAM-1/GEF could function independently of these Rac1-like GTPases, at least individually. To further investigate this notion, we conducted transgenic rescue experiments with deletions and point mutant variants of TIAM-1/GEF. We found that full length TIAM-1/GEF alone or as a C-terminal fusion with mCherry fully rescued the PVD dendrite patterning defects (Figure 5—figure supplement 1D). Surprisingly, a T548F point mutant of full length TIAM-1/GEF still rescued the PVD mutant phenotype (Figure 6B, Figure 5—figure supplement 1D). The fact that the very same mutation in the C. elegans TIAM-1 DH/PH domain (Demarco et al., 2012) as well as the analogous point mutation in the UNC-73/Trio GEF abolishes Rac1 GEF activity in vitro (Steven et al., 1998), lent further support to the idea that TIAM-1/GEF activity may not be required for PVD patterning (Figure 6B). It was conceivable, however, that mutant TIAM-1(T548F) retained some residual GEF activity that through high level transgenic expression could provide sufficient enzymatic GEF activity to rescue the PVD dendrite defects of tiam-1 mutants. To exclude this possibility, we used CRISPR/Cas9 genome editing to engineer the T548F missense mutation into the endogenous tiam-1/GEF locus. Two independently isolated alleles (dz264 and dz265) that harbored the T548F point mutation in the tiam-1/GEF locus appeared phenotypically normal in regard to PVD patterning (Figure 6C–D and data not shown). Thus, a point mutant form of TIAM-1 (T548F) devoid of measurable Rac1 GEF activity in vitro is sufficient for PVD patterning in vivo. In summary, our results suggest that TIAM-1/GEF can function independently of its GEF enzymatic activity although we can not formally exclude that the TIAM-1(T548F) shows enzymatic activity towards small GTPases other than Rac1 that may be involved in PVD patterning.
Figure 5. TIAM-1/GEF interacts with DMA-1/LRR-TM through PDZ motif/domain interaction to shape 4° dendrite branches.
(A – B) Fluorescent image with schematics of tiam-1 mutant animals carrying either a full length or deletion construct (GEF-only) of the tiam-1 cDNA under control of the PVD-specific ser-2p3 promoter. (B) (C – E) Quantification of 2°, 3°, and 4° branch numbers per 100 µm anterior to the PVD cell body in the genotypes indicated. For raw data see Figure 1 — source data 1. Statistical significance is indicated as Data are represented as mean ±SEM. Statistical comparisons were performed using one-sided ANOVA with the Tukey correction. Statistical significance is indicated (****p<0.0005). n = 10 animals per genotype. (F – H) Western Blots of co-immunoprecipitation experiments with corresponding schematic showing the topography of the DMA-1/LRR-TM single pass transmembrane receptor and the TIAM-1/GEF multidomain protein. Schematics not to scale. Immuno tags (V5 and HA) used for co-immunoprecipitation experiments are shown. Transfected constructs are indicated above the panels. Antibodies used for immunoprecipitation (IP) and western blotting (WB) are indicated. A molecular marker is on the left. Note, that the two lower panels are from a single western blot, which was developed repeatedly with two different antibodies after stripping in all panels. Panel F investigates the interaction between TIAM-1 and DMA-1, and panels G and H are structure function analyses to delineate the domains required for the TIAM-1/DMA-1 interaction in TIAM-1 and DMA-1, respectively. Similar results were obtained from at least three independent replicate experiments. (G) (H) (I) Fluorescent images of TIAM-1::GFP reporter expressed in PVD in wild type or dma-1(Δ|DZ) mutant animals. PVD is visualized by dzIs53 ([F49H12.4p::mCherry]). TIAM-1::GFP (wyIs1139) staining on 4° dendrite branches in wild-type animals is lost in dma-1(ΔPDZ) mutant animals.

Figure 5—figure supplement 1. The TIAM-1/GEF acts independently of Rac1 GEF activity.

Figure 5—figure supplement 2. Supplementary co-immunoprecipitation experiments (related to Figure 5 (A) Figure 7 (B)).

Figure 6. The TIAM-1/GEF functions independently of its guanine nucleotide activity.

(A) Genomic environs of the tiam-1 locus in linkage group I (LGI) with exons and introns indicated. The location of the two CRISPR/Cas9 engineered dz264 and dz265 alleles, encoding a missense mutation that causes the T548F mutation is shown. (B) Fluorescent images with schematics of tiam-1 mutant animals carrying a T548F point mutant form of the tiam-1 cDNA under control of the PVD-specific ser-2p3 promoter. The T548F point mutant is analogous to the S1216F mutation in UNC-73 (Steven et al., 1998) and the T516F mutation in a previously described TIAM-1 cDNA (Demarco et al., 2012). Importantly, the point mutations in both proteins have been shown to abrogate GEF activity toward Rac in vitro (Demarco et al., 2012). (C – D) Fluorescent images of PVD (left panels) and schematics (right panels) of wild-type control (C) and tiam-1(dz265) mutant animals (D). PVD is visualized by the wdIs52 [F49H12.4p::GFP] transgene in all panels.
The TIAM-1/GEF guanine nucleotide exchange factor interacts with DMA-1/LRR-TM through a PDZ ligand motif/domain to promote 4° branching
Since the PDZ ligand motif of DMA-1/LLR-TM is important for the formation of 4° branches (Figure 1), we tested if the PDZ domain of TIAM-1/GEF may mediate this function. We found that truncated TIAM-1, lacking the EV and PDZ domains (referred to as GEF-only), only partially rescued the PVD mutant phenotype (Figure 5B, Figure 5—figure supplement 1). Specifically, the GEF-only construct rescued formation of PVD 2° and 3° defects in the same manner as the full-length TIAM-1 construct, but failed to rescue defects in the number of 4° branches (Figure 5C–E). Intriguingly, overexpression of full length TIAM-1 resulted in more 4° branches, suggesting that TIAM-1 function is dosage dependent (Figure 5E). Thus, the DH/PH domain of TIAM-1 is sufficient for 2° and 3° branch formation, but not for 4° branch formation.
The remarkable similarity of phenotypes, namely a reduction of 4° branches, in the DMA-1(ΔPDZ) allele (Figure 1) and TIAM-1(GEF-only) rescued animals (Figure 5) suggested that DMA-1/LRR-TM and TIAM-1/GEF may function through a PDZ interaction for the formation of 4° branches. To test this hypothesis, we transfected DMA-1 (tagged with V5, DMA-1.V5) and TIAM-1 (tagged with HA, TIAM-1.HA) in human embryonic kidney cells (HEK293T) and conducted co-immunoprecipitation experiments from lysates of transiently transfected cells. We found that DMA-1.V5 efficiently co-immunoprecipitated TIAM-1.HA from lysates (Figure 5F). This interaction was dependent on the DMA-1 intracellular domain (ICD) and, specifically on the PDZ binding site, as the interaction was strongly reduced if either was removed (Figure 5G). Moreover, the interaction between TIAM-1/GEF and DMA-1/LRR-TM appeared specific, because replacing the DMA-1 PDZ binding site (YFGI) with the heterologous PDZ motif (EFYA) of the heparan sulfate proteoglycan SDN-1/Syndecan reduced the interaction between TIAM-1 and DMA-1 to the same extent as removing the PDZ binding site entirely (Figure 5—figure supplement 2A). Residual binding activity may be contributed by the EVH1 domain, as removing the EVH1 domain from TIAM-1/GEF also compromised the interaction with DMA-1 (Figure 6H). On the other hand, removing the PDZ domain from TIAM-1 (ΔPDZ) completely abrogated the interaction between DMA-1/LRR-TM and TIAM-1/GEF (Figure 6H). Collectively, our result showed (1) that TIAM-1/GEF interacts with DMA-1/LRR-TM through a PDZ domain-motif interaction with a possible contribution of the EVH1 domain in TIAM-1, and (2) that loss of PDZ domain or motif from either protein functionally result in a specific loss of 4° dendritic branches, suggesting that DMA-1/LRR-TM localizes TIAM-1/GEF to direct 4° dendrite growth. Consistent with this hypothesis, the localization of a TIAM-1::GFP reporter to 4° dendrite branches (but not lower branches) was lost in the dma-1 mutant in which the PDZ motif of DMA-1 was deleted (dma-1(dz266)[ΔPDZ]) (Figure 5I).
F-Actin is localized to the leading edge of dendrites
The filamentous F-actin and microtubule polymers are part of the cytoskeleton that provide stability and force during growth and development of neuronal processes (Dent et al., 2011; Kapitein and Hoogenraad, 2015). Having identified a function for act-4/Actin in PVD dendrite patterning, we sought to visualize F-actin in PVD neurons of live animals. To this end, we used the calponin homology domain of utrophin (UtrCH) fused to tagRFP (tagRFP::UtrCH), which has been previously shown to faithfully visualize F-actin without effects on actin dynamics (Burkel et al., 2007; Chia et al., 2014). Transgenic animals expressing tagRFP::UtrCH in PVD neurons showed the reporter localized to defined subcellular compartments during development of the dendritic arbor. During the L2 larval stage tagRFP::UtrCH was primarily localized to the cell body, the 1°, 2°, and budding 3° branches (Figure 7A). During the subsequent larval stages, tagRFP::UtrCH remained strongly expressed in the cell body, but otherwise became successively more localized to distal (i.e. developing) branches, rather than proximal branches. For example, at the young adult stage, tagRFP::UtrCH was primarily localized to 4° branches, and less to 3° and 2° branches (Figure 7A). These results indicate that tagRFP::UtrCH, and by inference F-actin, localizes to extending branches. Conversely, an α-tubulin fusion (tagRFP::TBA-1) fusion that labels microtubules, specifically expressed in PVD neurons is primarily localized to axons and 1° dendrites of PVD neurons (Figure 7—figure supplement 1), as previously described (Maniar et al., 2011). Thus, microtubules and F-actin occupy non-overlapping compartments in dendrites. Interestingly, this largely mutually exclusive localization of F-actin and microtubules is lost in mutants of the Menorin pathway. For example, mutations in dma-1/LRR-TM, hpo-30/Claudin, sax-7/L1CAM, or tiam-1/GEF result in a relocalization of the UtrCH reporter, and by inference F-actin, to more proximal dendrites (Figure 7B) with more dendritic endings lacking F-actin in tiam-1/GEF mutants (Figure 7C). Conversely, the tagRFP::TBA-1 fusion can be found in more distal dendrites in mutants of the Menorin pathway (Figure 7—figure supplement 1). These results suggest that the Menorin pathway establishes polarity and may function to localize F-actin to growing dendrites.
Figure 7. F-actin is localized to the leading edges of developing dendrites and requires the Menorin pathway for polarized localization.
(A) Fluorescent images of animals at different developmental stages carrying a F-actin reporter (ser-2p3::tagRFP::UtrCH [dzEx1564], left panels), a PVD cytoplasmic GFP reporter (wdIs52 [F49H12.4p::GFP], second panels), merged images (third panel), and corresponding schematics (right panels). L2: second larval stage, L3: third larval stage, L4 fourth larval stage. Scale bars indicate 10 µm. (B) Fluorescent images of animals in different genetic backgrounds carrying a F-actin reporter (ser-2p3::tagRFP::UtrCH [dzEx1564], left panels), a PVD cytoplasmic GFP reporter (wdIs52 [F49H12.4p::GFP], second panels), merged images (third panel), and corresponding schematics (right panels). Genotypes are indicated on the left. Scale bars indicate 10 µm. (C) Quantification of dendrite termini with F-actin rich staining in wild-type control (n = 10) and tiam-1(tm1556) mutant animals (n = 13). For details on quantification see Materials and methods. For raw data see Figure 1— source data 1. Statistical significance is indicated as Data are represented as mean ±SEM. Statistical comparisons were performed using the Mann-Whitney test. Statistical significance is indicated (***, p<0.005). (D – F) Western blots of co-immunoprecipitation experiments analyzing the interaction between TIAM-1 and wild type ACT-4 or mutant ACT-4(G151E). Transfected constructs are indicated above the panels. Antibodies used for immunopreciptation (IP) and western blotting (WB) are indicated. A molecular marker is on the left. Note, that panels in F are from a single western blot, which was developed repeatedly with two different antibodies after stripping. Similar results were obtained from at least three independent replicate experiments.

Figure 7—figure supplement 1. Localization of TBA-1::tagRFP in different genetic backgrounds.

TIAM-1/GEF may link DMA-1/LRR-TM to ACT-4/Actin
Based on our genetic findings, we next investigated the biochemical relationship of DMA-1/LRR-TM and TIAM-1/GEF with ACT-4/Actin to form a biochemical complex. We found that TIAM-1 co-immunoprecipitated ACT-4 from lysates of cells transiently transfected with tagged constructs of TIAM-1 and ACT-4 (Figure 7D). Intriguingly, we found that the ACT-4 G151E missense mutation encoded by the dz222 missense allele from our genetic screen, dramatically increased the strength of the biochemical interaction (Figure 7E). Deletion of individual domains of TIAM-1/GEF failed to abrogate the interaction with either wild type or G151E mutant ACT-4/Actin (Figure 5—figure supplement 2B), suggesting that more than one domain in TIAM-1/GEF may act in a partially redundant fashion to bind ACT-4/Actin. Alternatively, a multiprotein complex could account for this observation. To test this notion directly, we added DMA-1.V5 to the transfection mix of TIAM-1.HA and ACT-4.FLAG. Surprisingly, we found that ACT-4.FLAG could also be precipitated with an antibody against DMA-1.V5, with the interaction possibly stronger in the presence of TIAM-1/GEF (Figure 7F). Collectively, we conclude that TIAM-1/GEF is part of a complex containing both the DMA-1/LRR-TM receptor and ACT-4/Actin, potentially directly linking the receptor to the cytoskeleton.
Discussion
HPO-30/Claudin regulates DMA-1/LRR-TM surface expression and trafficking
We found that both membrane surface levels as well as trafficking of a DMA-1/LRR-TM reporter were affected by the amount of HPO-30/Claudin expressed in PVD. Loss of hpo-30/Claudin increased, whereas overexpression of hpo-30/Claudin decreased membrane surface staining of the DMA-1/LRR-TM reporter, respectively. In addition, loss of hpo-30/Claudin also reduced the fraction of mobile vesicles containing the DMA-1::GFP reporter within the dendrite. Together, these findings suggest that HPO-30/Claudin promotes internalization and trafficking of the cell surface receptor DMA-1/LRR-TM. This conclusion is also consistent with a recent study demonstrating that HPO-30/Claudin negatively regulates the level of the synaptic cell adhesion protein NLG-1/Neuroligin at the cholinergic NMJ (Sharma et al., 2018). There are several, not mutually exclusive, explanations for the functional significance of DMA-1/LRR-TM regulation by HPO-30/Claudin. First, the surface expression of DMA-1/LRR-TM may have to be tightly regulated, because overexpression of surface DMA-1/LRR-TM could result in too strong or sustained binding to its cognate binding partners, the hypodermally expressed cell adhesion molecules SAX-7/L1CAM and MNR-1/Menorin. Secondly, recycling of DMA-1 may be required for DMA-1 to remain functional. For example, cell adhesion molecules, such as cadherins are constantly recycled and, blocking endocytosis results in dysfunctional cell adhesion phenotypes (Cadwell et al., 2016). Alternatively, recycling of DMA-1/LRR-TM may be necessary for transporting DMA-1/LRR-TM to locations of particular demand, such as the tip of growing dendrites. Thirdly, the internalization of DMA-1/LRR-TM maybe part of the signal transduction itself, as internalization of surface receptors can be required to trigger signal transduction pathways occurring at a distance from the cell surface (Sorkin and von Zastrow, 2009).
DMA-1/LRR-TM regulates 3° and 4° branch formation through distinct mechanisms
Several lines of evidence suggest that signaling downstream of the DMA-1/LRR-TM receptor diverges into at least two molecularly distinct, partially redundant pathways to establish 2° and 3° branches and 4° branches, respectively. First, removing the PDZ ligand motif of DMA-1/LRR-TM results in a decrease in 4° branches but not 2° or 3° branches. Conversely, a GEF-only construct of TIAM-1/GEF, which lacks the PDZ domain, rescues the 3° branching defects, but not 4° branching defects of tiam-1 mutant animals. Thus, compromising the PDZ-mediated interaction between TIAM-1/GEF and DMA-1/LRR-TM results in specific loss of 4° branching. Furthermore, immunoprecipitation assays strongly indicate that TIAM-1/GEF and DMA-1/LRR-TM can interact directly through the PDZ motif at the C-terminus of DMA-1 and the PDZ-binding domain of TIAM-1. Since tiam-1 overexpression resulted in excessive branching, TIAM-1/GEF may promote branching in a dose-dependent manner. We propose that the PDZ interaction with DMA-1 recruits TIAM-1/GEF to specific subcellular locations, where a locally higher concentration of TIAM-1/GEF induces dendrite branching. Indeed, this is supported by the observation that TIAM-1::GFP fails to localize to 4° branches in DMA-1(ΔPDZ) animals. These findings are also consistent with a recent study, which also suggested that a TIAM-1::GFP reporter expressed in PVD is recruited to higher order dendritic branches by DMA-1/LRR-TM (Zou et al., 2018). Collectively, these findings suggest that the PDZ-mediated interaction of TIAM-1/GEF and DMA-1/LRR-TM interaction is required for normal 4° dendrite formation.
An intriguing finding is that transgenic expression of a TIAM-1(GEF-only) construct lacking the PDZ interaction domain can rescue both the formation of 2° and 3° PVD branches. Conversely, DMA-1/LRR-TM lacking the cytoplasmic domain was still capable of rescuing 2° and 3° branching number (but not extension), suggesting that protein interactions of the DMA-1 transmembrane or the extracellular domain, possibly in conjunction with TIAM-1/GEF may mediate an alternative parallel pathway. Indeed, imaging experiments and biochemical assays suggest that (1) DMA-1/LRR-TM colocalizes with the HPO-30/Claudin and that (2) both proteins can form a biochemical complex through an interaction independent of the cytoplasmic domain of DMA-1. Moreover, genetic double mutant analyses show that hpo-30 double mutants with tiam-1 or sax-7/L1CAM display enhanced phenotypes consistent with functions in parallel pathways. Previous studies have shown that mutating components of the WASP family verprolin-homologous protein (WVE-1/WAVE) regulatory complex (WRC) of actin regulators result in defects in PVD patterning (Aguirre-Chen et al., 2011; Zou et al., 2018). Moreover, HPO-30 can physically interact with the WRC and, compromising the molecular interactions between both DMA-1 and TIAM-1 and, between HPO-30 and the WRC at the same time result in synergistic defects consistent with the notion of parallel pathways downstream of the DMA-1 receptor (Zou et al., 2018). Zou et al. proposed that the DMA-1/LRR-TM receptor, in conjunction with HPO-30/Claudin and TIAM-1/GEF recruit actin regulators to the membrane to form dendritic branches. Alternatively, and consistent with our observation that HPO-30 negatively regulates the surface localization of the DMA-1/LRR-TM reporter, recruitment of the WRC by HPO-30 may also serve a role in endocytosis of DMA-1::GFP, as the WRC is known to play a role in clathrin-mediated endocytosis (Mooren et al., 2012). In possible support of such a function, the act-4(dz222) allele results in more distal localization of DMA-1::GFP, including at the tip of growing dendrites (Figure 2—figure supplement 1D,E).
The DMA-1/LRR-TM may shape 4° dendrites by directly regulating F-actin
F-actin clearly plays an important role in the formation of PVD dendritic branches. First, depolymerizing F-actin in vivo results in severe defects in PVD patterning (Harterink et al., 2017). Second, F-actin is localized to the distal tips of developing PVD dendrites and appears mutually exclusive with microtubules. Third, we find that F-actin localization to distal dendrites depends on the presence of transmembrane receptors on PVD dendrites and their ligands. Lastly, RNAi-mediated gene knock down of other genes with possible or established roles as regulators of F-actin in developing axons such as unc-44/Ankyrin (Otsuka et al., 1995), unc-115/Ablim (Lundquist et al., 1998; Struckhoff and Lundquist, 2003), or unc-70/β-spectrin (Hammarlund et al., 2000), display similar defects in PVD dendrite patterning as act-4/Actin and tiam-1/GEF mutants (Aguirre-Chen et al., 2011).
Our experiments show that the DMA-1/LRR-TM receptor is part of a biochemical complex with the TIAM-1/GEF and ACT-4/Actin. How could this complex mediate 4° dendrite growth or patterning? One possibility is that the DMA-1/TIAM-1/ACT-4 complex is part of a molecular clutch mechanism, first proposed as a means to explain actin-dependent cellular movements by Mitchison and Kirschner (1988). These authors suggested that the force generated by polymerizing F-actin is transmitted through engagement of F-actin filaments with the membrane through transmembrane receptors. For example, transmembrane receptors of the Ig domain superfamily have been shown to couple extracellular interactions to cytoskeletal actin dynamics (reviewed in Giannone et al. (2009)). In analogy, binding of the DMA-1/LRR-TM receptor by the extracellular SAX-7/MNR-1/LECT-2 ligand complex could result in recruitment of TIAM-1/GEF, ACT-4/Actin and DMA-1/LRR-TM in a complex, thereby directly linking the receptor to the cytoskeleton. The interaction between TIAM-1/GEF and ACT-4/Actin is likely specific, because animals carrying the act-4(dz222) missense allele are viable without other obvious phenotypes. In support of the specificity of the dz222 allele, it is worth noting that the exact same molecular change was identified by Zou et al. (2018) in their screen for genes involved in PVD patterning. The phenotype of act-4(dz222) is in stark contrast to the embryonic lethality observed upon continuous RNAi-mediated knockdown of act-4 (Gottschalk et al., 2005, data not shown). Moreover, knock down of ACT-4/Actin by RNAi during larval stages on the one hand and, the G151E missense mutation in ACT-4/Actin encoded by the act-4(dz222) allele on the other hand result in similar phenotypes in PVD dendrites. In other words, loss of ACT-4 is as detrimental for dendrite patterning as the stronger biochemical interaction between TIAM-1 and ACT-4(G151E). This suggests that the interaction between TIAM-1 and ACT-4, and possibly DMA-1/LRR-TM has to be transient or dynamic. Alternatively, the strong interaction between ACT-4(G151E) and TIAM-1/GEF could sequester actin from the polymerizing pool. These findings are reminiscent of a point mutation in β-spectrin, which by increasing the affinity of β-spectrin with actin one thousand fold, results in defects in Drosophila dendrite arborization neuron (Avery et al., 2017). Collectively, our studies and the findings by Zou et al. (Zou et al., 2018) suggest that at least two partially redundant pathways act downstream of the DMA-1/LRR-TM receptor. In one pathway, DMA-1 interacts via HPO-30/Claudin with the WRC during patterning of lower order branches, whereas the interaction with TIAM-1/GEF may directly link the receptor complex with the actin cytoskeleton during 4° dendrite formation. Further experiments will be required to determine how these two pathways are spatially and temporally regulated.
TIAM-1/GEF functions independently of its Rac1 GEF activity
Guanine nucleotide exchange factors such as TIAM-1 activate small GTPases by promoting the exchange of GDP for GTP. Tiam1 in vertebrates has been shown to serve important functions during nervous system patterning (Yoo et al., 2012). Knock down and overexpression experiments further established functions for Tiam1 in axon growth cone formation and, activity-dependent remodeling of dendritic arbors in vitro and in vivo (Kunda et al., 2001; Tolias et al., 2005; Tolias et al., 2007). Such functions may be conserved, since the drosophila homolog still life/Tiam1 and C. elegans TIAM-1/GEF function during synaptic and, axonal patterning downstream of the netrin receptor UNC-40/DCC (Deleted in Colorectal Cancer), respectively (Sone et al., 1997; Demarco et al., 2012). All known functions of Tiam1 appear dependent on the canonical enzymatic activity of exchanging GDP for GTP in small GTPases of the Rac1 type.
Unexpectedly, our findings imply that TIAM-1/GEF can function independently of its Rac1 GEF activity in shaping PVD dendrites. This conclusion is supported by three arguments. First, transgenic expression of a TIAM-1 (T548F) fully rescued the tiam-1 mutant phenotype. Second, engineering the T548F point mutation into the tiam-1 locus resulted in animals with PVD dendritic arbors that were indistinguishable from wild type animals. The T548F mutation is analogous to the S1216F mutation in UNC-73/Trio, which lacks Rac1 activity, but displays neuronal patterning defects (Steven et al., 1998). Moreover, the very mutation in C. elegans TIAM-1/GEF also shows no Rac1 GEF activity in vitro (Demarco et al., 2012). Lastly, single and double mutants of the small GTPases mig-2/RhoG and ced-10/Rac1, previously established as acting downstream of TIAM-1/GEF during axonal patterning in C. elegans (Demarco et al., 2012), displayed no defects PVD morphology. Similarly, a third Rac-like gene (rac-2/3) encoded in the C. elegans genome serves no function individually in PVD patterning. Taken together, these findings suggest that TIAM-1/GEF can function independently of Rac1 activity during PVD patterning. To our knowledge, there is only one other known example of GEF-independent functions of a guanine nucleotide exchange factor. The DOCK 180 family member DOCK7 functions during nuclear migration by antagonizing TACC3, a protein known to coordinate microtubule polymerization (Yang et al., 2012). Thus, GEF-independent functions that directly modulate the cytoskeleton may be an additional and, previously underappreciated property of guanine nucleotide exchange factors.
Materials and methods
C. elegans strains
All strains were maintained using standard conditions (unless stated otherwise). For details see Supplementary file 1.
Imaging and morphometric analyses
All strains were maintained using standard methods (Brenner, 1974). All experiments were performed at 20°C, and animals were scored as 1-day-old adults unless otherwise specified. The strains and mutant alleles used in this study are listed in the Supplementary file 1. Fluorescent images were captured in live C. elegans using a Plan-Apochromat 40×/1.4 or 63x/1.4 objective on a Zeiss Axioimager Z1 Apotome. Worms were immobilized using 5 mM levamisole and Z stacks were collected. Maximum intensity projections were used for further analysis.
For quantification of branching, synchronized starved L1 larvae were allowed to grow for 48 hr (Figure 1) or 50 hr (Figure 2) or, directly picked as L4 animals and mounted onto an agar pad for microscopy (Figure 5). Morphometric data was either obtained directly on the microscope (Figure 1) or, fluorescent images of immobilized animals (1–5 mM levamisol, Sigma) were captured using a Zeiss Axioimager equipped with an ApoTome. Z stacks were collected and maximum projections were used for counting dendritic branches manually (Figure 5) or by tracing dendrites as described (Salzberg et al., 2013) (Figure 2). In any case, all branches within 100 µm of the primary branch anterior to the cell body were counted and classified into primary, secondary, tertiary, and quaternary. All protrusions from the 1° dendrite were considered 2° branches.
Statistical comparisons were conducted using one-sided ANOVA with the Sidak or Tukey correction, the Kruskal-Wallis test, the Student t-test or the Mann-Whitney test as applicable using the (Prism 7 [GraphPad]) software suite.
Quantification of fluorescence reporters
For quantification of UtrCH::tagRFP staining, images were acquired at a setting where maximal intensity of UtrCH::tagRFP staining was within the dynamic range of the camera detector. All end branches were then manually examined and classified as displaying staining or no staining if no discernible differences between the dendritic branch and surrounding background signals was detected, that is that no structure was visible in the UtrCH channel, even with maximum gain and contrast.
For quantification of DMA-1::GFP reporter membrane staining, animals were imaged with identical parameters to allow for intensity comparisons. The average fluorescence was then quantified using ImageJ. Briefly, the freehand line tool was used to trace the branches to create an ROI with a width of 10 pixels (=1.6 µm). For quantification on the primary dendrite, ROI lengths of 30, 40, 50 and 60 µm from the cell body were defined and the mean of the average fluorescent intensity of these 4 ROIs was calculated for each animal. For quantification on the tertiary branches, 4 ROIs were traced through the whole length of 4 separate branches, and the mean of the average fluorescence intensity of these 4 ROIs was calculated for each animal. As background control, four additional ROIs of the same length were defined in an area inside the worm but outside of branches, and means of the average fluorescent intensity of these ROI was calculated as the background for each animal and subtracted from the mean fluorescent intensity for either primary or tertiary branches, respectively. Statistical comparisons were conducted using one-sided ANOVA with the Tukey correction, the Kruskal-Wallis test, the Mann-Whitney test as applicable using the (Prism 7 [GraphPad]) software suite.
Cloning of mutant alleles
Alleles of different genes were isolated during genetic screens for mutants with defects in PVD patterning. In several F1 clonal genetic screens for mutants with defects in PVD dendrite arborization, we isolated one allele of dma-1 (dz181), two alleles of hpo-30 (dz178 and dz189), and two alleles of tiam-1 (dz184 and dz206). One allele of act-4 (dz222) was isolated in an enhancer screen of the hypomorphic kpc-1(gk333538) allele. In addition, we obtained putative null deletion alleles for dma-1(tm5156), hpo-30(ok2047), and tiam-1(tm1556) and tiam-1(ok772). Details for cloning of individual point mutations are provided below.
tiam-1(dz184)
A SNP-mapping-WGS approach (Doitsidou et al., 2010) was used to map tiam-1(dz184) between 3 Mb and 6 Mb of chromosome I. Using SNP mapping (Davis et al., 2005), the mutation was further mapped to a physical interval between 4 Mb and 5.48 Mb. This region contained only one nonsense mutation. The mutation was confirmed by Sanger sequencing of the original isolate, confirming a non-sense C to T mutation in the tiam-1(dz184) allele. Additionally, the tiam-1(dz184) mutant showed non-complementation with all other loss-of-function alleles and its phenotype was not different from the dz206, tm1556 and ok772 deletion alleles (see List of complementation tests below). Therefore, our genetic and molecular evidence suggests that all tiam-1 alleles used in this study are likely complete loss-of-function alleles.
tiam-1(dz206)
Complementation tests with a deletion allele of tiam-1 showed non-complementation between tiam-1(tm1556) and tiam-1(dz206). The mutation was further confirmed by Sanger sequencing of the original isolate, identifying a non-sense mutation C to T mutation in dz206 in the tiam-1 locus (see List of tiam-1 mutant alleles below).
hpo-30(dz178)
A SNP-mapping-WGS approach (Doitsidou et al., 2010) was used to map hpo-30(dz178) between 3 Mb and 5 Mb of chromosome V. This region contained only one candidate mutation. The mutation was confirmed by Sanger sequencing of the original isolate, confirming a missense C to T mutation in hpo-30(dz178). Additionally, the hpo-30(dz178) phenotype was not different from the dz178 and ok2047 deletion allele. Therefore, our genetic and molecular evidence suggests that all hpo-30 alleles used in this study are likely complete loss-of-function alleles.
hpo-30(dz189)
Complementation tests with the other loss-of-function allele of hpo-30 showed non-complementation between hpo-30(dz178) and hpo-30(dz189). The mutation was further confirmed by Sanger sequencing of the original isolate, identifying a non-sense mutation C to T mutation in hpo-30(dz189) (see List of hpo-30 mutant alleles below).
act-4(dz222)
The dz222 allele was identified in a genetic screen as a recessive enhancer of the kpc-1(gk333538) hypomorphic allele. A SNP-mapping-WGS approach (Doitsidou et al., 2010) was used to map act-4(dz222) between 4 Mb and 6 Mb of chromosome X. This region contained five candidate mutations, including non-sense, missense, frameshift and splice site mutations. The mutant defect in dz222 was phenocopied by RNAi-mediated gene knock down of act-4 and, rescued by transgenic expression of act-4 under control of heterologous promoters. Sequencing of the original isolate identified a G to A missense mutation in act-4(dz222), resulting in glycine to glutamic acid change at position 151 in ACT-4. Finally, when the act-4(dz222) allele was placed over a deficiency the phenotype was not enhanced. Therefore, our genetic and molecular evidence suggests that the act-4(dz222) allele used in this study is likely complete loss of function allele of act-4 in the context of PVD dendrite morphogenesis.
List of mutant dma-1 alleles
| Allele | Molecular lesion | Defect in PVD | Physical Location† |
|---|---|---|---|
| dz181 | splice acceptor | Yes | 8,283,604, G - > A |
| tm5159 | deletion (671nt) | Yes | 8,282,281–8,282,951 |
| dz266 | Y600Amber (ΔPDZ) | Yes | 8,284,283, A - > T‡ |
†Nucleotide change according to nucleotide positions in Wormbase release WS260.
‡Note that additional synonymous changes were introduced as a result of the CRISPR procedure. See Repair oligos below for details.
List of mutant hpo-30 alleles
| Allele | Molecular lesion | Defect in PVD | Physical location† |
|---|---|---|---|
| dz178 | S155F | Yes | 12,309,368 |
| dz189 | Q168X | Yes | 12,309,330 |
| ok2047 | Deletion (1,294 bp) | Yes | 12,309,724–12,311,017 |
†Nucleotide change according to nucleotide positions in Wormbase release WS252.
List of mutant tiam-1 alleles
| Allele | Molecular lesion | Defect in PVD | Physical location† |
|---|---|---|---|
| dz184 | Q303X | Yes | 4,422,065 |
| dz206 | Q543X | Yes | 4,424,113 |
| dz264 | T548F | No | 4,424,128–4,424,129 |
| dz265 | T548F | No | 4,424,128–4,424,129 |
| tm1556 | Deletion (851 bp) | Yes | 4,423,487–4,424,337 |
| ok772 | Deletion (838 bp) and insertion (18 bp) | Yes | 4,424,394–4,425,231 |
†Nucleotide change according to nucleotide positions in Wormbase release WS252.
List of mutant act-4 alleles
| Allele | Molecular lesion | Defect in PVD | Physical location† |
|---|---|---|---|
| dz222 | G151E (M03F4.2a) | Yes | 4,963,194 |
| gk279371 | L9H (M03F4.2a) | No | 4,960,687 |
| gk279385 | G246R (M03F4.2a) | No | 4,963,478 |
| gk473333 | A30T (M03F4.2a) | No | 4,962,830 |
| gk785720 | E4K (M03F4.2c) | No | 4,962,048 |
†Nucleotide change according to nucleotide positions in Wormbase release WS252.
List of complementation tests for tiam-1
| Genotypes | % of defective PVDs | N | Results |
|---|---|---|---|
| +/+ (wdIs52 (control)) | 0 | 100 | Complementation |
| dz184/tm1556 | 100 | 100 | Non-complementation |
| dz184/ok772 | 100 | 100 | Non-complementation |
| dz184/+ | 0 | 100 | Complementation |
| dz206/tm1556 | 100 | 40 | Non-complementation |
| dz206/+ | 0 | 40 | Complementation |
Molecular biology and transgenesis
To establish DNA constructs used for rescue experiments or immunoprecipitation experiments, the respective cDNAs were cloned under control of either cell-specific promoters or promoters to drive expression in cell culture. Details for plasmid construction can be found in Supplementary file 1.
Transgenesis
A complete list of all transgenic strains created for this study is shown in the Supplementary file 1.
PVD-specific rescue of dma-1 PVD dendrite branching defect
Complete rescue of the dma-1 mutant phenotype was achieved in transgenic animals carrying a plasmid where the dma-1 cDNA was driven under control of a short version of the previously reported ser-2prom3 (=ser-2p3) promoter (Tsalik et al., 2003). The new promoter, which we term ser-2p3s encompasses 1825 bp upstream of the predicted translational start site of the C02D4.2d isoform.
For the deletion of the intracellular domain in dma-1 (ΔICD), a mCherry fluorescent tag was inserted in frame after E532 leaving a protein devoid of the last 71 residues. Both constructs were injected individually at 5 ng/µl into dma-1(tm5159) I; wdIs52 II, together with the myo-3p::tagRFP marker (at 50 ng/µl) for the injection mix containing the entire dma-1 cDNA or myo-2p::mCherry (at 50 ng/µl) for the dma-1 (ΔICD) version. The final injection mix was brought to 100 ng/µl of DNA concentration with pBluescript.
Heterologous rescue of tiam-1 PVD dendrite branching defect
The tiam-1 cDNA was cloned under control of heterologous promoters: hypodermal dpy-7p (Gilleard et al., 1997), body wall muscle myo-3p (Okkema et al., 1993), pan-neuronal rab-3p (Nonet et al., 1997), and a PVD-specific ser-2p3 promoter (Altun-Gultekin et al., 2001). All constructs were injected at 5 ng/µl into tiam-1(tm1556) I; wdIs52 II, together with the myo-3p::mCherry marker at 50 ng/µl and pBluescript at 50 ng/µl.
Heterologous rescue of act-4 PVD dendrite branching defect
The act-4 (or act-1) genomic DNA was cloned under control of heterologous promoters: hypodermal body wall muscle myo-3p (Okkema et al., 1993) and PVD-specific ser-2p3 (Altun-Gultekin et al., 2001). All constructs were injected at 5 ng/µl into wdIs52 II; act-4(dz222) X, together with the elt-2p::gfp marker at 5 ng/µl and pBluescript at 90 ng/µl.
Transcriptional reporter of act-4
The act-4 transcriptional reporter promoter was constructed by cloning 2.6 kb upstream of the predicted act-4 translational start site (based on Figure 1—figure supplement 1) into pPD95.75-NLS::mCherry, and injected at 5 ng/µl into wdIs52 II animals, together with the myo-2p::mNG marker at 5 ng/µl and pBluescript at 90 ng/µl.
Translational reporter of act-4
The act-4 cDNA was tagged at the C-terminus with tagRFP and expressed under the Pser-2p3 promoter (Okkema et al., 1993; Tsalik et al., 2003). It was injected at 5 ng/µl into wdIs52 II; act-4(dz222) animals together with the myo-2p::mNG marker at 10 ng/ µl and pBluescript at 85 ng/µl.
tagRFP::UtrCH and tba-1::tagRFP reporters
UtrCH and tba-1 were tagged with tagRFP and expressed under ser-2p3 promoter. All constructs were injected at 5 ng/µl into wdIs52 II, together with the myo-3p::tagBFP marker at 10 ng/µl and pBluescript at 85 ng/µl.
Generation of genome-edited strains through CRISPR/Cas9
For introducing mutations into endogenous loci of dma-1(Y600Amber) and tiam-1(T548F), the CRISPR co-conversion method as described in Arribere et al. (2014) was utilized. Briefly, two appropriate sgRNA sequences were identified for each gene and introduced into Cas9-sgRNA expression vector pDD162, while repair oligos were ordered from IDT. Both Cas9-sgRNA constructs and repair oligo were injected into N2 animals along with Cas9-sgRNA and a repair oligo for dpy-10(cn64) conversion. Rollers (dpy-10(cn64)/+animals) from the F1 generation were selected and genotyped using restriction sites engineered in the repair oligo. Wildtype F2 animals were then selected from positive F1 animals to segregate the intended edit from dpy-10(cn64).
sgRNA sequences
| dma-1(∆PDZ [Y600Amber]) | tiam-1 (T1548F) |
|---|---|
| ATGCCAAAATAGGATGATCC | AAGAATCATAAGAAGTAGTG |
| ATGCCAAAATAGGATGATCC | GATGGCTCTGCAAGAATTGT |
Repair oligo sequences
dma-1(∆PDZ[Y600Amber]):
TGCAAATAATCACACGGATTTAGATTTTTGAAACATTTGAAACTCTCAAAAGGATATTTATTATCTAGACTAAGAGGAACCTGGCTTCGGAGGTGCTGGTGGAATCAATGGAGGAGTGGCTTTAAATGTTTCAGTACTAACCAAGAATGG
tiam-1(T1548F):
TGTCAATAAGAAAACGGGCTGTGCAAAGTTGGCGATGGCTCTGCAAGAATTGTTAGTCTTTGAGAAGAAATATGTCAGCGATCTTCGAGAGGTAAGAGATCCCAAAAAGTTATAGAATTGAATAATTTACGATTTCAGATGA
Cell culture, transient transfection and co-immunoprecipitation
HEK293T human embryonic kidney cells were maintained with a standard protocol in DMEM supplemented with 10%FBS. Cells were not validated because they were merely used for heterologous protein expression. The identity of heterologously expressed proteins was verified in western blots via unique immunotags. Transient transfection was performed using Lipofectamine 3000 (Thermo-fisher) according to the manufacturer’s instruction. For each experimental permutation, 105 cells/well were transfected in duplicate in six-well plates with 1 μg of each construct.
For co-immunoprecipitation assays, the duplicate wells of 105 transfected cells were lysed in 500 μl of lysis buffer and combined (25 mM Hepes, 150 NaCl, 1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS, protease inhibitor cocktail (Thermo-Fisher)). The lysate was subsequently incubated at 4°C with agitation for an hour before centrifugation at 20,000 rcf for 10 min. The resulting lysate was combined with 20 µl of pre-equilibrated Protein A/G agarose (Thermo-fisher) and 1 µl of the appropriate antibody for pulldown and, incubated at 4°C with agitation overnight. The agarose was then washed 5 x with 500 µl of lysis buffer before SDS loading buffer was added. SDS-PAGE and western blot were then performed using standard protocols. All primary antibodies were used at 1:5000 dilutions and secondary antibodies were used at 1:10,000 dilutions. Supersignal West Femto ECL reagent (Thermo Fisher) was used for detection with X-ray films. For details on antibodies used see Supplementary file 1.
Acknowledgements
We thank T Boulin, S Cook, A Meléndez, R Townley and members of the Bülow laboratory for comments on the manuscript and for discussion during the course of this work. We thank the Caenorhabditis Genome Center for strains (funded by NIH Office of Research Infrastructure Programs, P40OD010440); O Hobert and K Shen for reagents and discussions. This work was funded in part through the NIH (R01NS096772 to HEB; T32GM007288 and F31HD066967 to CADB; F31NS100370 to MR; T32GM07491 to MILP; P30HD071593 & P30CA013330 to Albert Einstein College of Medicine), a Human Genome Pilot Project from Albert Einstein College of Medicine and the Binational Science Foundation (#2013188 to HEB). NJRS is the recipient of a Fulbright-Colciencias fellowship. HEB is an Irma T. Hirschl/Monique Weill-Caulier research fellow.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Hannes E Bülow, Email: hannes.buelow@einstein.yu.edu.
Shai Shaham, The Rockefeller University, United States.
Didier Y Stainier, Max Planck Institute for Heart and Lung Research, Germany.
Funding Information
This paper was supported by the following grants:
National Institute of General Medical Sciences T32GM007288 to Carlos A Diaz-Balzac.
Eunice Kennedy Shriver National Institute of Child Health and Human Development F31HD066967 to Carlos A Diaz-Balzac.
National Institute of Neurological Disorders and Stroke F31NS100370 to Maisha Rahman.
Fulbright-Colciencias to Nelson J Ramirez-Suarez.
National Institute of General Medical Sciences T32GM07491 to Maria I Lázaro-Peña.
Irma T Hirschl/Monique Weill-Caulier to Hannes E Bülow.
National Institute of Neurological Disorders and Stroke R01NS096672 to Hannes E Bülow.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Visualization, Methodology, Writing—original draft, Writing—review and editing.
Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Visualization, Methodology, Writing—original draft, Writing—review and editing.
Data curation, Formal analysis, Investigation, Visualization, Writing—review and editing.
Data curation, Formal analysis, Investigation, Visualization, Writing—review and editing.
Data curation, Formal analysis, Investigation, Visualization.
Investigation.
Conceptualization, Resources, Data curation, Formal analysis, Supervision, Funding acquisition, Validation, Visualization, Project administration, Writing—review and editing.
Additional files
In addition, Supplementary file 1 lists all transgenic strains created during the course of these studies.
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files.
References
- Aguirre-Chen C, Bülow HE, Kaprielian Z. C. elegans bicd-1, homolog of the Drosophila dynein accessory factor bicaudal D, regulates the branching of PVD sensory neuron dendrites. Development. 2011;138:507–518. doi: 10.1242/dev.060939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albeg A, Smith CJ, Chatzigeorgiou M, Feitelson DG, Hall DH, Schafer WR, Miller DM, Treinin M. C. elegans multi-dendritic sensory neurons: morphology and function. Molecular and Cellular Neuroscience. 2011;46:308–317. doi: 10.1016/j.mcn.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun-Gultekin Z, Andachi Y, Tsalik EL, Pilgrim D, Kohara Y, Hobert O. A regulatory cascade of three homeobox genes, ceh-10, ttx-3 and ceh-23, controls cell fate specification of a defined interneuron class in C. elegans. Development. 2001;128:1951–1969. doi: 10.1242/dev.128.11.1951. [DOI] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis Elegans. Genetics. 2014;198:837–846. doi: 10.1534/genetics.114.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery AW, Thomas DD, Hays TS. β-III-spectrin spinocerebellar ataxia type 5 mutation reveals a dominant cytoskeletal mechanism that underlies dendritic arborization. PNAS. 2017;114:E9376–E9385. doi: 10.1073/pnas.1707108114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchall PS, Fishpool RM, Albertson DG. Expression patterns of predicted genes from the C. elegans genome sequence visualized by FISH in whole organisms. Nature Genetics. 1995;11:314–320. doi: 10.1038/ng1195-314. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouhard GJ. Dynamic instability 30 years later: complexities in microtubule growth and catastrophe. Molecular Biology of the Cell. 2015;26:1207–1210. doi: 10.1091/mbc.E13-10-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel BM, von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motility and the Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell CM, Su W, Kowalczyk AP. Cadherin tales: regulation of cadherin function by endocytic membrane trafficking. Traffic. 2016;17:1262–1271. doi: 10.1111/tra.12448. [DOI] [PubMed] [Google Scholar]
- Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, Rosen MK. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia PH, Chen B, Li P, Rosen MK, Shen K. Local F-actin network links synapse formation and axon branching. Cell. 2014;156:208–220. doi: 10.1016/j.cell.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics. 2005;6:118. doi: 10.1186/1471-2164-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarco RS, Struckhoff EC, Lundquist EA. The rac GTP exchange factor TIAM-1 acts with CDC-42 and the guidance receptor UNC-40/DCC in neuronal protrusion and axon guidance. PLoS Genetics. 2012;8:e1002665. doi: 10.1371/journal.pgen.1002665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Gupton SL, Gertler FB. The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harbor Perspectives in Biology. 2011;3:a001800. doi: 10.1101/cshperspect.a001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Balzac CA, Rahman M, Lázaro-Peña MI, Martin Hernandez LA, Salzberg Y, Aguirre-Chen C, Kaprielian Z, Bülow HE. Muscle- and Skin-Derived cues jointly orchestrate patterning of somatosensory dendrites. Current Biology. 2016;26:2379–2387. doi: 10.1016/j.cub.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Pani AM, Heppert JK, Higgins CD, Goldstein B. Streamlined genome engineering with a Self-Excising drug selection cassette. Genetics. 2015;200:1035–1049. doi: 10.1534/genetics.115.178335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsidou M, Poole RJ, Sarin S, Bigelow H, Hobert O. C. Elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS ONE. 2010;5:e15435. doi: 10.1371/journal.pone.0015435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Liu OW, Howell AS, Shen K. An extracellular adhesion molecule complex patterns dendritic branching and morphogenesis. Cell. 2013;155:296–307. doi: 10.1016/j.cell.2013.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Shen K, Bülow HE. Intrinsic and extrinsic mechanisms of dendritic morphogenesis. Annual Review of Physiology. 2015;77:271–300. doi: 10.1146/annurev-physiol-021014-071746. [DOI] [PubMed] [Google Scholar]
- Dong X, Chiu H, Park YJ, Zou W, Zou Y, Özkan E, Chang C, Shen K. Precise regulation of the guidance receptor DMA-1 by KPC-1/Furin instructs dendritic branching decisions. eLife. 2016;5:e11008. doi: 10.7554/eLife.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G, Mège RM, Thoumine O. Multi-level molecular clutches in motile cell processes. Trends in Cell Biology. 2009;19:475–486. doi: 10.1016/j.tcb.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Gilleard JS, Barry JD, Johnstone IL. Cis regulatory requirements for hypodermal cell-specific expression of the caenorhabditis elegans cuticle collagen gene dpy-7. Molecular and Cellular Biology. 1997;17:2301–2311. doi: 10.1128/MCB.17.4.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk A, Almedom RB, Schedletzky T, Anderson SD, Yates JR, Schafer WR. Identification and characterization of novel nicotinic receptor-associated proteins in Caenorhabditis Elegans. The EMBO Journal. 2005;24:2566–2578. doi: 10.1038/sj.emboj.7600741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets GG, Scholtes EH, Zuydgeest D, van der Kammen RA, Stam JC, Berns A, Collard JG. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994;77:537–549. doi: 10.1016/0092-8674(94)90216-X. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Davis WS, Jorgensen EM. Mutations in beta-spectrin disrupt axon outgrowth and sarcomere structure. The Journal of Cell Biology. 2000;149:931–942. doi: 10.1083/jcb.149.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harterink M, da Silva ME, Will L, Turan J, Ibrahim A, Lang AE, van Battum EY, Pasterkamp RJ, Kapitein LC, Kudryashov D, Barres BA, Hoogenraad CC, Zuchero JB. DeActs: genetically encoded tools for perturbing the actin cytoskeleton in single cells. Nature Methods. 2017;14:479–482. doi: 10.1038/nmeth.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Branching out: mechanisms of dendritic arborization. Nature Reviews Neuroscience. 2010;11:316–328. doi: 10.1038/nrn2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Hoogenraad CC. Building the neuronal microtubule cytoskeleton. Neuron. 2015;87:492–506. doi: 10.1016/j.neuron.2015.05.046. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cerebral Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Konietzny A, Bär J, Mikhaylova M. Dendritic actin cytoskeleton: structure, functions, and regulations. Frontiers in Cellular Neuroscience. 2017;11:147. doi: 10.3389/fncel.2017.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Wild M, Rosenzweig B, Hirsh D. Wild-type and mutant actin genes in caenorhabditis elegans. Journal of Molecular Biology. 1989;208:381–392. doi: 10.1016/0022-2836(89)90503-2. [DOI] [PubMed] [Google Scholar]
- Kulkarni VA, Firestein BL. The dendritic tree and brain disorders. Molecular and Cellular Neuroscience. 2012;50:10–20. doi: 10.1016/j.mcn.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Kunda P, Paglini G, Quiroga S, Kosik K, Caceres A. Evidence for the involvement of Tiam1 in axon formation. The Journal of Neuroscience. 2001;21:2361–2372. doi: 10.1523/JNEUROSCI.21-07-02361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Airaksinen MS, Saarma M, Timmusk T. A novel gene family encoding leucine-rich repeat transmembrane proteins differentially expressed in the nervous system. Genomics. 2003;81:411–421. doi: 10.1016/s0888-7543(03)00030-2. [DOI] [PubMed] [Google Scholar]
- Lefebvre JL, Sanes JR, Kay JN. Development of dendritic form and function. Annual Review of Cell and Developmental Biology. 2015;31:741–777. doi: 10.1146/annurev-cellbio-100913-013020. [DOI] [PubMed] [Google Scholar]
- Liu OW, Shen K. The transmembrane LRR protein DMA-1 promotes dendrite branching and growth in C. elegans. Nature Neuroscience. 2011;15:57–63. doi: 10.1038/nn.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist EA, Herman RK, Shaw JE, Bargmann CI. UNC-115, a conserved protein with predicted LIM and actin-binding domains, mediates axon guidance in C. elegans. Neuron. 1998;21:385–392. doi: 10.1016/S0896-6273(00)80547-4. [DOI] [PubMed] [Google Scholar]
- Lundquist EA, Reddien PW, Hartwieg E, Horvitz HR, Bargmann CI. Three C. elegans rac proteins and several alternative rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development. 2001;128:4475–4488. doi: 10.1242/dev.128.22.4475. [DOI] [PubMed] [Google Scholar]
- Maniar TA, Kaplan M, Wang GJ, Shen K, Wei L, Shaw JE, Koushika SP, Bargmann CI. UNC-33 (CRMP) and ankyrin organize microtubules and localize kinesin to polarize axon-dendrite sorting. Nature Neuroscience. 2011;15:48–56. doi: 10.1038/nn.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Cytoskeletal dynamics and nerve growth. Neuron. 1988;1:761–772. doi: 10.1016/0896-6273(88)90124-9. [DOI] [PubMed] [Google Scholar]
- Mooren OL, Galletta BJ, Cooper JA. Roles for actin assembly in endocytosis. Annual Review of Biochemistry. 2012;81:661–686. doi: 10.1146/annurev-biochem-060910-094416. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis Elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. The Journal of Neuroscience. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Sequence requirements for myosin gene expression and regulation in caenorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren-Suissa M, Hall DH, Treinin M, Shemer G, Podbilewicz B. The fusogen EFF-1 controls sculpting of mechanosensory dendrites. Science. 2010;328:1285–1288. doi: 10.1126/science.1189095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka AJ, Franco R, Yang B, Shim KH, Tang LZ, Zhang YY, Boontrakulpoontawee P, Jeyaprakash A, Hedgecock E, Wheaton VI. An ankyrin-related gene (unc-44) is necessary for proper axonal guidance in Caenorhabditis Elegans. The Journal of Cell Biology. 1995;129:1081–1092. doi: 10.1083/jcb.129.4.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Actin and Actin-Binding proteins. Cold Spring Harbor Perspectives in Biology. 2016;8:a018226. doi: 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft G, Wilmshurst JM, Pillay K, Sivadorai P, Wallefeld W, Nowak KJ, Laing NG. A novel ACTA1 mutation resulting in a severe congenital myopathy with nemaline bodies, intranuclear rods and type I fibre predominance. Neuromuscular Disorders. 2011;21:31–36. doi: 10.1016/j.nmd.2010.08.005. [DOI] [PubMed] [Google Scholar]
- Salzberg Y, Díaz-Balzac CA, Ramirez-Suarez NJ, Attreed M, Tecle E, Desbois M, Kaprielian Z, Bülow HE. Skin-derived cues control arborization of sensory dendrites in caenorhabditis elegans. Cell. 2013;155:308–320. doi: 10.1016/j.cell.2013.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Li L, Liu H, Tikiyani V, Hu Z, Babu K. The Claudin-like protein HPO-30 is required to maintain LAChRs at the C. elegans Neuromuscular Junction. The Journal of Neuroscience. 2018;38:7072–7087. doi: 10.1523/JNEUROSCI.3487-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Watson JD, Spencer WC, O'Brien T, Cha B, Albeg A, Treinin M, Miller DM. Time-lapse imaging and cell-specific expression profiling reveal dynamic branching and molecular determinants of a multi-dendritic nociceptor in C. elegans. Developmental Biology. 2010;345:18–33. doi: 10.1016/j.ydbio.2010.05.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, O'Brien T, Chatzigeorgiou M, Spencer WC, Feingold-Link E, Husson SJ, Hori S, Mitani S, Gottschalk A, Schafer WR, Miller DM. Sensory neuron fates are distinguished by a transcriptional switch that regulates dendrite branch stabilization. Neuron. 2013;79:266–280. doi: 10.1016/j.neuron.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone M, Hoshino M, Suzuki E, Kuroda S, Kaibuchi K, Nakagoshi H, Saigo K, Nabeshima Y, Hama C. Still life, a protein in synaptic terminals of Drosophila homologous to GDP-GTP exchangers. Science. 1997;275:543–547. doi: 10.1126/science.275.5299.543. [DOI] [PubMed] [Google Scholar]
- Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nature Reviews Molecular Cell Biology. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven R, Kubiseski TJ, Zheng H, Kulkarni S, Mancillas J, Ruiz Morales A, Hogue CW, Pawson T, Culotti J. UNC-73 activates the rac GTPase and is required for cell and growth cone migrations in C. elegans. Cell. 1998;92:785–795. doi: 10.1016/S0092-8674(00)81406-3. [DOI] [PubMed] [Google Scholar]
- Stone S, Shaw JE. A caenorhabditis elegans act-4::lacZ fusion: use as a transformation marker and analysis of tissue-specific expression. Gene. 1993;131:167–173. doi: 10.1016/0378-1119(93)90290-J. [DOI] [PubMed] [Google Scholar]
- Struckhoff EC, Lundquist EA. The actin-binding protein UNC-115 is an effector of rac signaling during axon pathfinding in C. elegans. Development. 2003;130:693–704. doi: 10.1242/dev.00300. [DOI] [PubMed] [Google Scholar]
- Taylor CA, Yan J, Howell AS, Dong X, Shen K. RAB-10 regulates dendritic branching by balancing dendritic transport. PLOS Genetics. 2015;11:e1005695. doi: 10.1371/journal.pgen.1005695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Kane CG, Tolias CS, Hu L, Greenberg ME. The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptor-dependent dendritic spine development. PNAS. 2007;104:7265–7270. doi: 10.1073/pnas.0702044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalik EL, Niacaris T, Wenick AS, Pau K, Avery L, Hobert O. LIM homeobox gene-dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Developmental Biology. 2003;263:81–102. doi: 10.1016/S0012-1606(03)00447-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YT, Wang CL, Van Aelst L. DOCK7 interacts with TACC3 to regulate interkinetic nuclear migration and cortical neurogenesis. Nature Neuroscience. 2012;15:1201–1210. doi: 10.1038/nn.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Kim Y, Lee H, Park S, Park S. A gene trap knockout of the Tiam-1 protein results in malformation of the early embryonic brain. Molecules and Cells. 2012;34:103–108. doi: 10.1007/s10059-012-0119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Yadav S, DeVault L, Nung Jan Y, Sherwood DR. RAB-10-Dependent membrane transport is required for dendrite arborization. PLOS Genetics. 2015;11:e1005484. doi: 10.1371/journal.pgen.1005484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Shen A, Dong X, Tugizova M, Xiang YK, Shen K. A multi-protein receptor-ligand complex underlies combinatorial dendrite guidance choices in C. elegans. eLife. 2016;5:e18345. doi: 10.7554/eLife.18345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Dong X, Broederdorf TR, Shen A, Kramer DA, Shi R, Liang X, Miller DM, Xiang YK, Yasuda R, Chen B, Shen K. A dendritic guidance receptor complex brings together distinct actin regulators to drive efficient F-Actin assembly and branching. Developmental Cell. 2018;45:362–375. doi: 10.1016/j.devcel.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]