

ABSTRACT
Although a mountain of papers have showed that metformin plays a role in inhibiting cancers, but the mechanism underpinning this has not yet fully elucidated. Herein, we used AOM/DSS model, the clinicopathological features are similar to those found in humans, to investigate the effects of metformin as well as combination with 5-FU in the prevention of colitis and colitis associated cancer (CAC). Oral metformin significantly inhibited DSS-induced ulcerative colitis and AOM/DSS-induced CAC. Metformin also ameliorated 5-FU-induced colorectal gastrointestinal symptoms in mice. Metformin combination with 5-FU strongly inhibited colorectal cancer. Metformin reduced levels of the NFκB signaling components p-IKKα/β, p-NFκB, p-IκBα in colorectal mucosal cells. Transmission electron microscopy analysis suggested that the inhibition of metformin on colitis and CAC might associate with its biological activity of protecting mitochondrial structures of colorectal epithelial cells. Further analysis by Mito Tracker Red staining assay indicated that metformin prevented H2O2-induced mitochondrial fission correlated with a decrease of mitochondrial perimeter. In addition, metformin increased the level of NDUFA9, a Q-module subunit required for complex I assembly, in colorectal epithelial cells. These observations of metformin in the inhibition of colitis and CAC might associate with its activity of activating the LKB1/AMPK pathway in colorectal epithelial cells. In conclusion, metformin inhibited colitis and CAC through protecting the mitochondrial structures of colorectal epithelial cells.
Keywords: Colitis, colorectal cancer, metformin, Mitochondria, LKB1/AMPK pathway
Introduction
Colorectal cancer (CRC) is one of the most prevalent cancers worldwide and second leading cause of cancer-related death in Western countries.1 CRC includes hereditary, sporadic and colitis-associated cancer (CAC). Currently, the link of colitis with CAC has been well recognized, which laid the foundation of routine screening drugs for early treatment of CAC.2,3
Metformin is currently used extensively in the treatment of type 2 diabetes mellitus (T2DM), but there is potential for additional indications in many other diseases, such as inflammatory disorders and cancers.4–7 Epidemiological studies revealed an association between the use of metformin and decreased incidence of many types of cancers.8,9 Diabetic patients taking metformin showed a decreased incidence of pancreatic cancer. Patients taking metformin display approximately 30% reduced overall cancer incidence as well as cancer mortality.10 However, the mechanisms of metformin on cancers have still not fully understood. A number of studies suggest that the inhibition of metformin on cancer cells might associate with its activity of targeting cellular mitochondria.11,12 It is assumed that mitochondria are the major target of metformin, leading to the inhibition of mitochondrial respiration, AMPK activation and bioenergetic reprogramming.13 On the contrary, metformin also found to activate AMPK in cardiac myoblast H9c2 cells and many other type of normal cells like smooth muscle cells.14,15 The mechanisms of these pleiotropic effects of metformin have not yet understood. Furthermore, fairly amounts of clinical trials on its signal administration have not yet demonstrated a convincing efficacy. Recently, some reports suggested an indication of metformin combination with clinical commonly used drugs for increasing their biological activities.9,16 These therapeutic strategies might display some promising clinical benefits for patients with cancers.
We aimed to investigate the effect of metformin on colitis and colitis-associated cancer (CAC) in mice model. Our results demonstrated that metformin prevented the DSS-induced colitis. Metformin combination with 5-FU strongly inhibited colorectal cancer, resulting in the integrity of mucous membranes and normal structure of colorectal tissues. Our further study suggest that the mechanism of metformin action on colitis and cancer might associate with its activity of protecting the mitochondrial structures of colorectal epithelial cells. These results deepen the understanding of metformin in the inhibition of colitis and colorectal cancer.
Results
Metformin prevented dss-induced ulcerative colitis
Mice exposed to DSS were analyzed the severity of colitis. During colitis, diarrhea, rectal bleeding, hair bristling and body weight loss were observed in mice. Body weight loss was started at day 7 (Figure 1A). Bloody stools were observed from second DSS cycle. Disease severity scored by DAI reached highest level in third DSS cycle (Figure 1B). 5-FU is an important therapeutic drug for the treatment of colonic cancers. However, 5-FU-induced gastrointestinal symptoms might harm patients’ prognosis. Our results showed that metformin attenuated the severity of colitis, prevented bloody stools and body weight loss (Figure 1A, P < 0.01 vs. colitis model). Metformin combination with 5-FU showed an increase of body weight (Figure 1A, P < 0.01 vs. colitis model). A reduction of DAI was observed in metformin- (Figure 1B, P < 0.01 vs. colitis model) and metformin combination 5-FU-treated mice (Figure 1B, P < 0.001 vs. colitis model).
Figure 1.
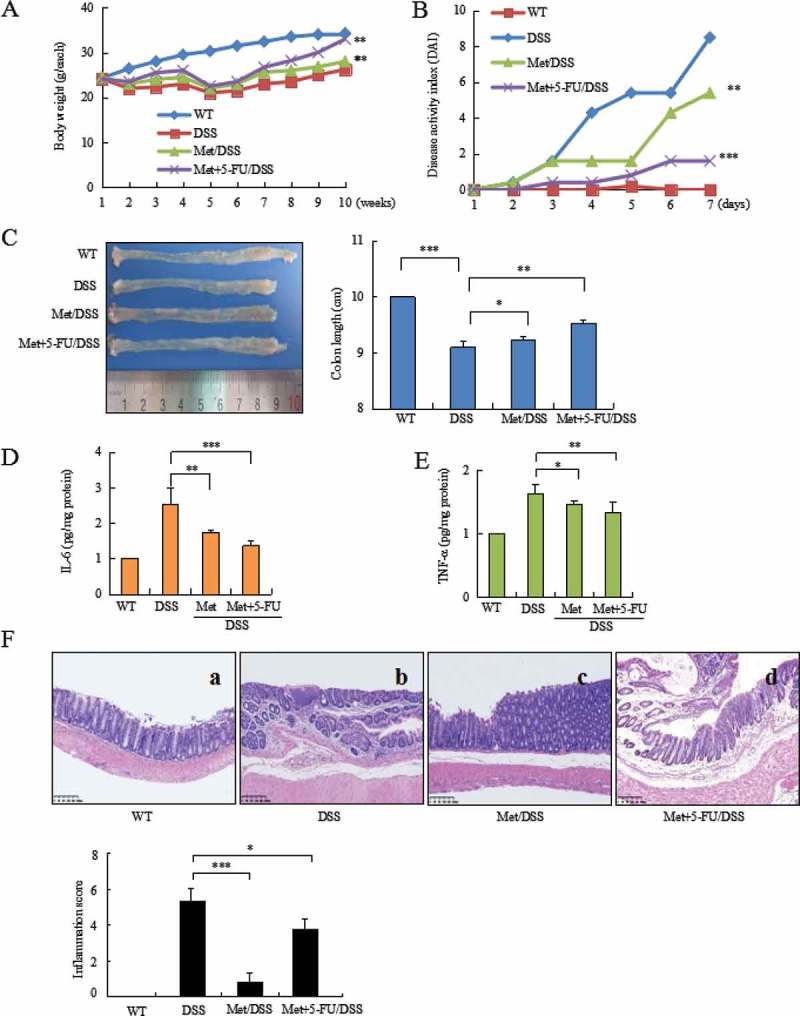
Metformin prevented DSS-induced colitis in mice. (A) DSS induced body weight loss. (B) Metformin inhibited DAI in colitis mice. (C) Metformin prevented shortening of colonic length. (D) ELISA assay analyzed IL-6 level in colorectal mucosa. (E) ELISA assay analyzed TNF-α level in colonic mucosa. (F) Histology analyzed the severity of colitis in DSS model (b), metformin-treated mice (c), and 5-FU combination with metformin-treated mice (d). Metformin reduced the score of inflammation; Metformin reduced DSS- and 5-FU-induced inflammation score. ***P < 0.001, *P < 0.05 between colitis model and metformin-treated mice or metformin combination with 5-FU-treated mice. Met = Metformin.
The inhibitory effect of metformin on colitis was further confirmed by a comparison of colorectal length in two groups of mice with or without metformin treatment. DSS induced shortening of colorectal length (Figure 1C, P < 0.001 vs. WT mice). In contrast, metformin prevented DSS-induced shortening of colorectal length (Figure 1C, P < 0.05 vs. colitis model).
IL-6 and TNF-α are critical inflammatory mediators involved in the stimulation of tumor microenvironment in colonic mucosa and tumors.17 ELISA analysis showed a strong inhibition of IL-6 (Figure 1D, P < 0.01 between colitis model and metformin-treated mice; P < 0.001 between colitis model and metformin + 5-FU-treated mice) and a significant decrease of TNF-α (Figure 1E, P < 0.05 between colitis model and metformin-treated mice; P < 0.01 between colitis model and metformin +5-FU-treated mice) in the colonic mucosa of metformin-treated mice.
Histopathologic analysis of colorectal tissues showed inflammatory lesions in 100% of colitis model mice but not in metformin-treated mice. Histological features of colorectal inflammation included different degrees of structural changes ranging from swelling and degeneration of villous epithelia to extensive denudation and collapse of villi, surface erosion with exuberant inflammatory exudate, patchy reepithelization, lamina propria fibrosis with acute and chronic inflammatory immunocytes infiltration and submucosa edema (Figure 1F-b). 5-FU also induced intestinal mucositis.18,19 In colitis model mice, 5-FU further exacerbate DSS-induced gastrointestinal symptoms, leading to colorectal ulcer. Metformin attenuated colorectal inflammatory severity. Metformin-treated mice demonstrated the integrity of mucous membranes and essential normal structure of colorectal tissues (Figure 1F-c, P < 0.001 vs. colitis model). Metformin attenuated the severity of DSS- and 5-FU-induced colorectal inflammation (Figure 1F-d, P < 0.05 vs. colitis model).
Metformin prevented aom/dss-induced colitis associated cancer (CAC)
Metformin prevented AOM/DSS-induced body weight loss (Figure 2A, P < 0.01 vs. CAC model) and reduced DAI score (Figure 2B, P < 0.01 vs. CAC model). AOM/DSS induced colonic tumor by 100% in model mice. Metformin prevented shortening of colorectal length (Figure 2C, P < 0.05, vs. CAC model), reduced the incidence of tumor by 50% (Figure 2D, P < 0.01 vs. CAC model), total colorectal tumor by 71% (Figure 2E, P < 0.01 vs. CAC model). CAC model mice developed greater numbers and larger sized tumors (> 5 mm), but not in metformin-treated mice (Figure 2F).
Figure 2.
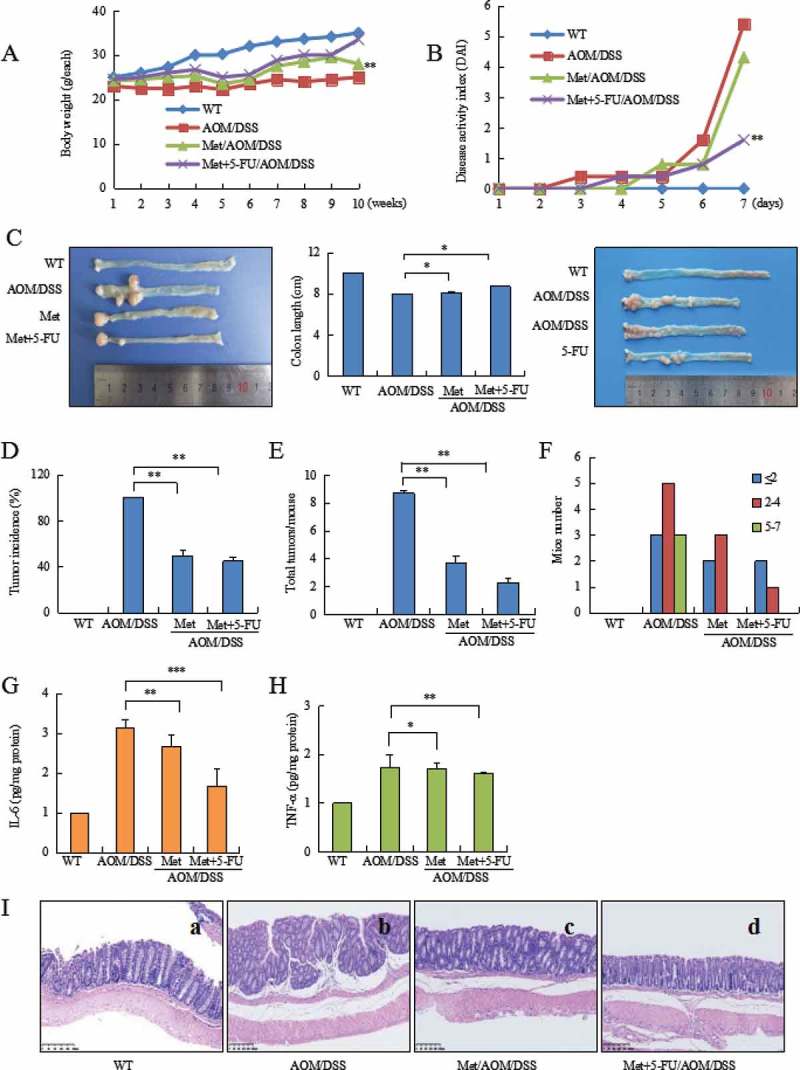
Metformin plus 5-FU inhibited AOM/DSS-induced CAC. (A) Body weight loss during CAC. (B) Metformin reduced DAI in AOM/DSS-treated mice. (C) Representative of image of colorectal cancer (left). Metformin prevented shortening of colonic length during CAC (middle). 5-FU moderately inhibited colorectal cancer growth in CAC model (right). (D) Tumor incidence in CAC model, metformin-treated and metformin plus 5-FU-treated mice. (E) Metformin plus 5-FU strongly reduced total tumor number. (F) Analysis the distribution of tumor size. Larger size of tumor (5–7 mm) did not found in metformin-treated mice. (G) ELISA assay analyzed IL-6 level in colorectal mucosa. (H) ELISA assay analyzed TNF-α level in colorectal mucosa. (I) Histology showed advanced colorectal tumor (b). Metformin prevented AOM/DSS-induced CAC (c). 5-FU plus metformin strongly inhibited CAC (d). ***P < 0.001, **P < 0.01, *P < 0.05 between CAC model and metformin-treated mice or metformin plus 5-FU-treated mice. Met = Metformin.
ELISA assay showed a decrease of IL-6 (Figure 2G, P < 0.01 between CAC model and metformin-treated mice; P < 0.001 between CAC model and metformin combination with 5-FU-treated mice) and TNF-α (Figure 2H, P < 0.05 between CAC model and metformin-treated mice; P < 0.01 between CAC model and metformin + 5-FU) in colorectal mucosa of metformin-treated mice.
Histopathology analysis of colorectal tissues indicated a strong inhibition of metformin combination with 5-FU on colorectal cancer. AOM/DSS-induced CAC showed advanced tubular adenoma with focal high grade dysplasia evidenced by marked nuclear pleomorphism, loss of nuclear polarity, frequent mitoses and architectural distortion. Tumors with gross ulceration were frequently observed in model mice (Figure 2I-b). Metformin significantly inhibited colorectal cancer (Figure 2I-c). Metformin combination with 5-FU strongly inhibited colorectal cancer, resulting in normal glands containing goblet cells in colorectal epithelium (Figure 2I-d).
We investigated the mechanism of metformin action on colorectal tumorigenesis. NFκB pathway plays a key role in colitis and colorectal tumorigenesis. Metformin significantly reduced DSS- or AOM/DSS-induced high levels of the NFκB signaling components IKKβ, IKKα, NFκB, IκBα (Figure 3A) and p-IKKα/β, p-NFκB, p-IκBα (Figure 3B) (**P < 0.01, ***P < 0.001, between colitis model and metformin- or 5-FU plus metformin-treated mice; #P < 0.05, ##P < 0.01, ###P < 0.001, between CAC model and metformin- or 5-FU plus metformin-treated mice).
Figure 3.
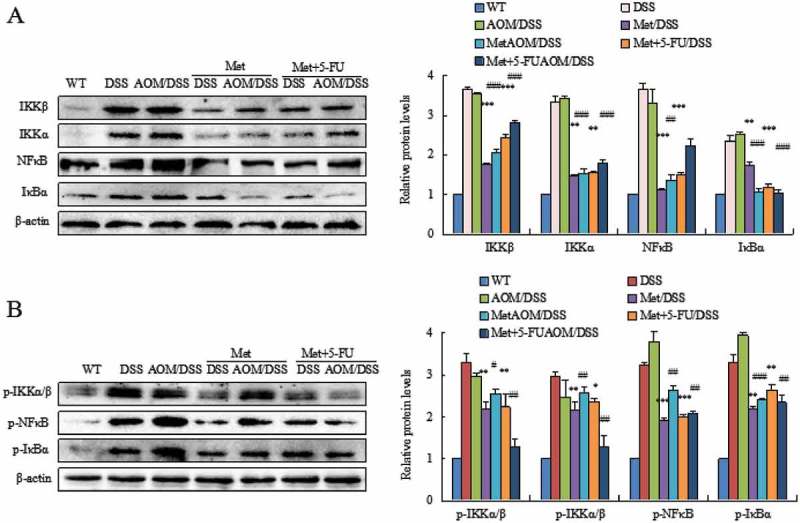
Metformin inhibited activation of the NF-κB pathway. DSS or AOM/DSS induced high level of the NFκB signaling pathway in colorectal epithelial cells. Metformin inhibited the NFκB signaling components IKKβ, IKKα, NFκB, IκBα (A) and p-IKKα/β, p-NFκB, p-IκBα (B). *P < 0.05, **P < 0.01, ***P < 0.001, between colitis model and metformin-treated mice or 5-FU plus metformin-treated mice; #P < 0.05, ##P < 0.01, ###P < 0.001, between CAC model and metformin-treated mice or 5-FU plus metformin-treated mice.
Metformin prevented DSS or aom/DSS-induced damage on mitochondria
We analyzed the mitochondrial structures of colorectal epithelial cells. Mitochondrial structures were damaged in DSS and AOM/DSS model mice, showing swollen mitochondria with cristae fracture, double-membrane decomposition and bound vacuoles with remnants. The counts of mitochondria were also decreased (Figure 4A-b, Figure 4B-b). Metformin prevented DSS or AOM/DSS-induced alteration of mitochondrial structures. In contrast, metformin-treated mice maintained essentially normal mitochondrial structures (Figure4A-c, Figure 4B-c). The counts of mitochondria were significantly increased compared to model mice. Administration of 5-FU might also damage mitochondrial dynamics and structures through inducing a reactive oxygen species (ROS)-mediated mitochondria-caspase-dependent apoptotic pathway and triggering mitochondrial dysfunction, therefore leading to gastrointestinal symptoms.20,21 Our results revealed that metformin could attenuate 5-FU-induced damage of mitochondrial structures (Figure 4A-d, Figure 4B-d).
Figure 4.
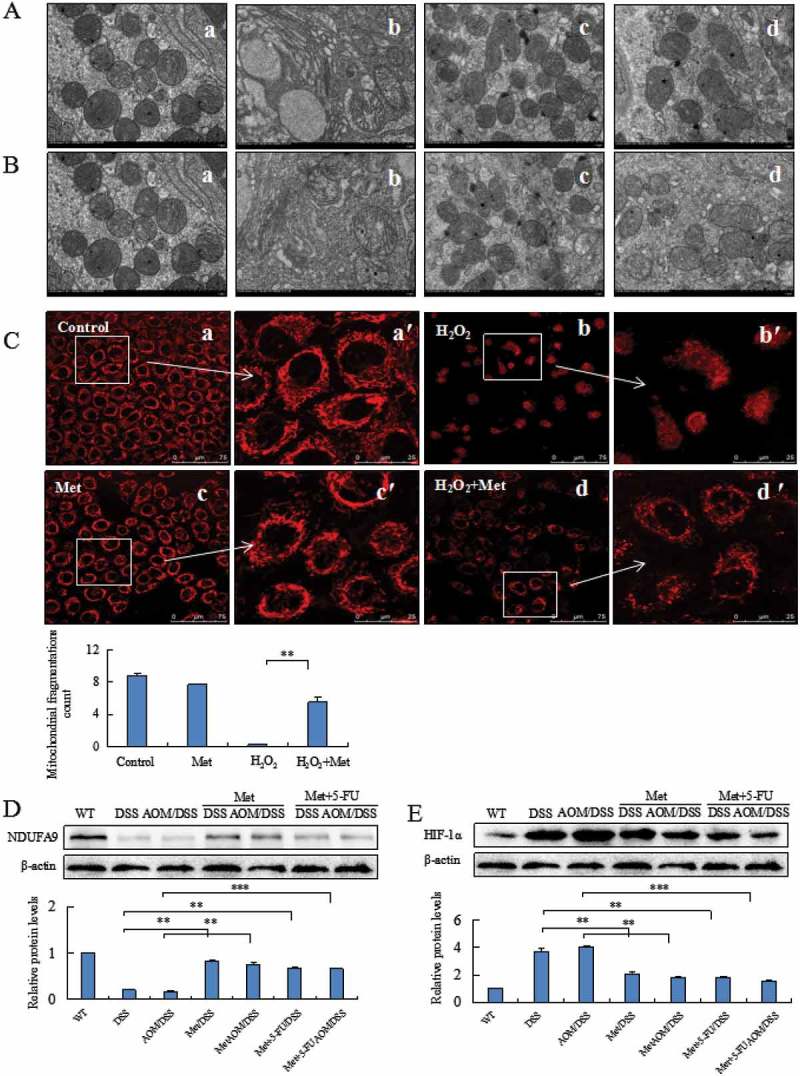
Metformin prevented alteration of mitochondrial structures damaged by DSS, AOM/DSS or H2O2. Metformin prevented DSS- (A) or AOM/DSS- (B) induced damage on mitochondrial structures in colorectal epithelial cells analyzed under a transmission electron microscopy. A-a and B-a: normal mitochondria counts and structures in colorectal mucosal cells. Mitochondria counts were decreased and mitochondrial structures were damaged by DSS (A-b) or AOM/DSS (B-b). A-c and B-c: Metformin prevented DSS- or AOM/DSS-induced damage on mitochondrial structures of colorectal epithelial cells. A-d and B-d: Metformin attenuated DSS- or AOM/DSS- and 5-FU-induced alteration of mitochondrial structures. The mitochondrial morphology of human normal colonic cells was analyzed by Mito Tracker red staining assay (C). Metformin prevented H2O2-induced-mitochondrial fission correlated with the decrease of mitochondrial perimeter (C, P < 0.01 vs. H2O2). Western blotting analysis of colorectal epithelial cells showed an increase of NDUFA9 (D) and a decrease of HIF-1α (E) in the metformin-treated mice. **P < 0.01, ***P < 0.001 vs. model mice.
MitoTracker Red staining analyzed mitochondria filamentous shape in human normal colonic cells exposed to H2O2 (Figure 4C-a). H2O2 induced mitochondrial fusion which is characterized by filamentous shape and the perimeter of fused mitochondria. H2O2 increased the fragmentation of mitochondria into granules shape and loss of staining with MitoTracker Red (Figure 4C-b). Metformin alone did not affect the filamentous shape (Figure 4C-c). Metformin prevented H2O2-induced mitochondrial fission correlated with a significant decrease of mitochondrial perimeter (Figure 4C-d, P < 0.01 vs. cells exposed to H2O2).
Western blotting analysis indicated that metformin prevented DSS- or AOM/DSS-induced low level of NDUFA9, a Q-module subunit required for complex I assembly, in colorectal epithelial cells (Figure 4D, **P < 0.01, ***P < 0.001 vs. model mice). Since hypoxia caused enlargement of mitochondria during colitis and tumorigenesis, we thus analyzed the inhibitory effect of metformin on HIF-1α, a marker of hypoxia, in colonic epithelial cells. Metformin suppressed DSS- or AOM/DSS-induced HIF-1α in colorectal epithelial cells (Figure 4E, **P < 0.01, ***P < 0.001 vs. model mice).
Metformin protected mitochondrial structures through activating the LKB1/AMPK pathway
DSS and AOM/DSS suppressed levels of liver kinase B1 (LKB1) and AMP-activated protein kinase (AMPK) in colorectal epithelial cells. Metformin prevented DSS- and AOM/DSS-induced low level of LKB1 and AMPK (Figure 5A, P < 0.01 between colitis model and metformin-treated mice; P < 0.01 between colitis model and metformin plus 5-FU-treated mice). Metformin also increased levels of LKB1 and AMPK in AOM/DSS-induced CAC model (Figure 5B, P < 0.01 between CAC model and metformin- or 5-FU plus metformin-treated mice). These results suggest that metformin protect the structure of mitochondria probable through activating the LKB1/AMPK pathway.
Figure 5.
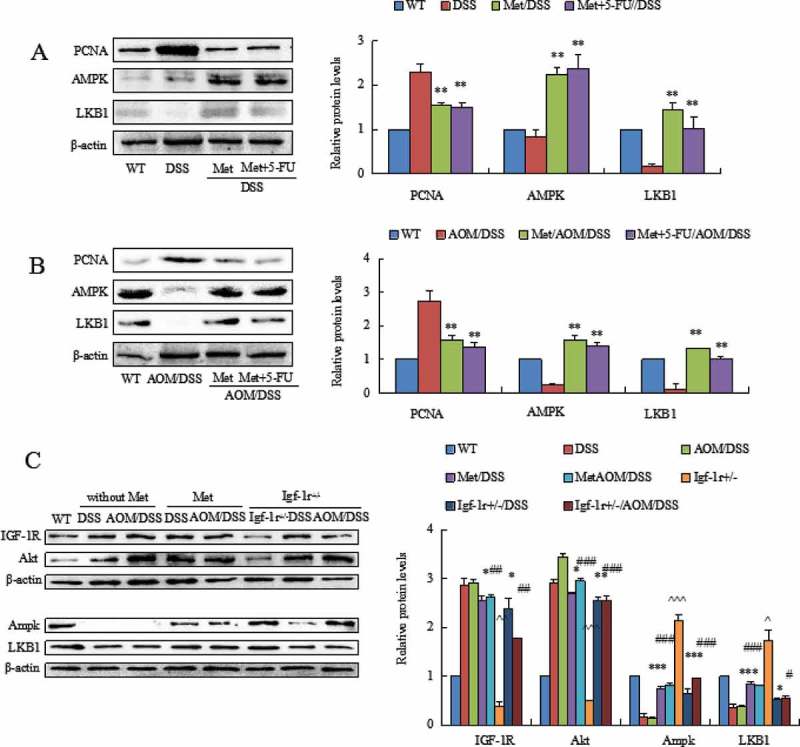
Western blot analyzed the levels of AMPK and LKB1 and PCNA in colorectal mucosal cells isolated from DSS- or AOM/DSS-treated mice. DSS or AOM/DSS decreased levels of AMPK and LKB1 and increased expression of PCNA in colorectal mucosal cells. Metformin increased the levels of AMPK and LKB1. Metformin reduced PCNA in colorectal mucosal cells. **P < 0.01 vs. model mice. (C) The comparison of LKB1 and AMPK levels in colorectal mucosa from WT mice and Igf1r± mice exposed to DSS and AOM/DSS. Both DSS and AOM/DSS induced high level of Akt. The levels of AMPK and LKB1 were consequently reduced. Metformin inhibited Akt and increased AMPK and LKB1 in colorectal mucosal cells. In Igf1r± mice, knockdown of IGF-1R prevented DSS- and AOM/DSS-induced high level of Akt and increased AMPK and LKB1 levels. *P < 0.05, **P < 0.01, ***P < 0.001, between colitis model and metformin-treated mice or Igf1r± mice; #P < 0.05, ##P < 0.01, ###P < 0.001, between CAC model and metformin-treated mice or Igf1r± mice. ^P < 0.05, ^^P < 0.01, ^^^P < 0.001, between Igf1r± mice and WT mice.
In addition, we surprisingly found that knockdown of insulin-like growth factor-1 receptor (IGF-1R) also activated the LKB1/AMPK pathway as metformin had. We compared the expression levels of LKB1 and AMPK in colonic mucosa from metformin-treated mice and Igf1r± mice. Knockdown of IGF-1R prevented DSS- and AOM/DSS-induced high level of Akt, a upstream of LKB1/AMPK pathway, in Igf1r± mice (Figure 5C). Our further results demonstrated an activation of LKB1 and AMPK in the colonic mucosa of Igf1r± mice (Figure 5C, *P < 0.05, **P < 0.01, ***P < 0.001, between colitis model and metformin-treated mice; and between colitis model and Igf1r± mice; #P < 0.05, ##P < 0.01, ###P < 0.001, between CAC model and metformin-treated mice; and between CAC model and Igf1r± mice; ^P < 0.05, ^^P < 0.01, ^^^P < 0.001, between Igf1r± mice and WT mice). These results further support an effect of metformin in the prevention of these deadly diseases.
Discussion
Metformin is a widely-used antidiabetic drug, and there is evidence among the diabetic patients that metformin is a chemopreventive agent against cancers. There are evidence in vitro and in vivo studies that metformin has been considered a cancer chemotherapeutic agent.4,5 Many clinical trials have been performed to investigate the inhibitory effects of metformin alone or in combination with other drugs on various cancers. Currently, two major pathways are recognized as the main ways in which metformin exerts its antitumor effect. Firstly, metformin might inhibit the activation of IGF-1/IGF1R pathway, thereby inactivating its downstream PI3K/Akt signaling to prevent tumor growth. Secondly, metformin could prevent inflamed colorectal epithelial cells and cancer cells through activating the AMPK signaling pathway.9 In this study, we used DSS- and AOM/DSS models, the widely-used mice models that resemble human colitis and CAC, to evaluate the inhibitory effects of metformin on colitis and colorectal cancer. Metformin strongly prevented DSS-induced ulcerative colitis. Metformin inhibit ulcerative colitis through suppressing the NFκB signaling pathway. Further, although 5-FU could treat intestinal tumors, 5-FU-induced intestinal mucositis has been considered as a major problem which lead to failure of treatment.22,23 In our study, we found that 5-FU combination with metformin strongly inhibited colitis and colorectal cancer. The mechanisms underpinning these effects of metformin might associate with its biological activity of inhibiting the NFκB signaling pathway.
It is well accepted that the connection between colitis and colorectal cancer is well established. Colitis is an important risk factor for the development of colorectal tumorigenesis. As compared with sporadic CRC, CRC arising in patients with colitis typically affects young individuals, tends to be multifocal, and has a higher proportion of mucinous and signet ring cell histology.24 Clinically, CAC is more aggressive, frequently presents at a more advanced stage as compared to sporadic CRC.3,24 Mechanically, both genetic and environmental factors might contribute to the progression of this deadly pathogenesis, including genetic instability; epigenetic alteration; immune response by mucosal inflammatory mediators; oxidative stress; and intestinal microbiota.25 Among these risk factors, one of the major causes is oxidative stress that is provoked by reactive oxygen species (ROS) and nitrogen species (RONS).26 Oxidative stress occurs as an imbalance of generation and elimination of ROS and RONS produced by inflammatory cells. Although in particular conditions ROS and RONS can even become part of several signaling pathways, their upregulation is frequently harmful to intestinal cells, which have evolved several antioxidant mechanisms to regulate ROS and RONS homeostasis.27 Further, NF-κB and cytokine signalling activation are crucial for the generation of colorectal tumorigenesis. The NF-κB pathway has been found amplification by the stimulation of ROS and RONS. 28 In AOM/DSS model, DSS-induced colitis is known to increase production of ROS and RONS that could cause DNA oxidation.29 Mechanically, ROS and RONS are able, in turn, to damage either directly or indirectly mitochondria, creating a positive feedback loop, thus resulting in mitochondrial dysfunction.30 It has long thought that mitochondria are less functional in inflamed intestinal epithelium and in tumor cells. Decreased mitochondrial function is considered to be tumorigenic, mainly because of enhanced ROS production. Our results suggest that metformin might prevent colitis and colorectal tumorigenesis through suppressing the NF-κB signaling pathway. Mitochondria are highly sensitive to various oxidative stimuli, which could cause substantial damage to the mitochondrial electron transport, leading to damage on mitochondria.31 It has become increasingly clear that the mitochondrial functions are involved in oxidative cell injury. The challenges of DSS or AOM/DSS could damage the mitochondrial structures, leading to swollen mitochondria with cristae fracture, double-membrane decomposition, and bound vacuoles with remnants. Our MitoTracker Red staining assay showed an increase of mitochondrial fusion and filamentous shape in human normal colonic cells induced by H2O2. We found that metformin could protect mitochondrial structure from H2O2-induced damage on mitochondria.
Mitochondrial dynamics proteins are critical for mitochondrial turnover and maintenance of mitochondrial health. NDUFA9 functions as an accessory subunit of multi-protein mitochondrial membrane respiratory chain NADH dehydrogenase complex. NDUFA9 defect might cause mitochondrial complex I deficiency, a mitochondrial disorder with wide symptoms ranging from cardiomyopathy, myopathy, liver disease, neurological disorders and tumorigenesis. Hypoxia-inducible factor 1 (HIF-1), a transcription factor regulating cellular response to hypoxia, plays a key role during colorectal tumorigenesis as well as invasion and metastasis.32 Importantly, the LKB1–AMPK–mammalian target of rapamycin (mTOR) pathway is a key moderator of HIF-1-targeted genes and HIF-1-mediated cellular metabolism.33 We therefore analyzed the biological activity of metformin in the inhibition of HIF-1 and the activation of AMPK and LKB1 pathway. Our results suggest that metformin significantly inhibited hypoxia-induced HIF-1α probable through activating the AMPK and LKB1 pathway. In addition, many reports showed that the inhibition of metformin on cancer cells might associate with its activity of inhibiting the respiratory complex I, leading to reduced oxidative phosphorylation and reduced ATP production, resulting in a reduction in cellular ATP and activation of AMPK.34 Metformin might trigger disorganization of cristae and inner mitochondrial membrane in several cancer cells. Mechanistically, these alterations might be due to calcium entry into the mitochondria in cancer cells. On the contrary, metformin was reported to activate AMPK in normal cells.35 These reports indicate that metformin has pleiotropic effects on modulating AMPK in cancer cells or normal cells. From a metabolic standpoint, AMPK is a metabolic sensor that helps maintaining cellular energy homeostasis. AMPK and LKB1 are both negative regulators of aerobic glycolysis under metabolic stress.36 During colitis, AMPK promotes ATP conservation under the condition of metabolic stress by activating the pathway of catabolic metabolism, thus AMPK improves intestinal epithelial differentiation and barrier function and maintains intestinal homeostasis and protects against colitis.37 In our study, we suggest that the activation of AMPK/LKB1 pathway might be an important strategy for the prevention of colitis and colorectal cancer. Metformin prevented DSS or AOM/DSS-induced damage on mitochondrial structures through activating AMPK and LKB1 pathway in colorectal mucosal cells, thus leading to the inhibition of colitis and colorectal cancer.
In summary, metformin could prevent colitis and CAC in mice. The mechanism underpinning these inhibitory effects of metformin might associate with its activity of prevention damage on mitochondrial structures in colorectal epithelial cells. Our results will hopefully contribute toward bridging the gap between preclinical research and the development of realistic novel therapeutic strategies to treat colitis and colorectal tumor.
Materials and methods
Animal care and diet
All experiments related to animal were approved by Animal Welfare Committee of Capital Medical University. C57BL/6 mice were purchased from Charles River Laboratories (Beijing, China). Knockdown of IGF-1 receptor (Igf1r±) mice were generated by Cyagen Biosciences Inc. (Guangzhou, China) as described.38 Mice were caged under a controlled room temperature, humidity and light (12/12 h light/dark cycle) and allowed unrestricted access to standard mouse chow containing 52% carbohydrate, 12% fat, 23% protein, 4% fiber, 6% ash, and 3% moisture and tap water.39
Animal model of colitis and CAC and pharmacological treatment protocol
Experimental procedures of colitis and CAC model were performed as described. Briefly, mice (6–8 weeks, male) were injected intraperitoneally with 10 mg/kg azoxymethane (AOM, Sigma). After 7 days, mice were received drinking water containing 2.0% dextran sulfate sodium (DSS, MP) for another 7 days. Mice were then given regular drinking water for 14 days, followed by two additional DSS cycles.40 Mice were randomly divided into following groups. Group 1: negative control (saline solution); Group 2: DSS-induced colitis model; Group 3: DSS-induced colitis model treated by metformin; Group 4: DSS-induced colitis model treated by 5-FU plus metformin; Group 5: AOM/DSS-induced CAC model; Group 6: AOM/DSS-induced CAC model treated by metformin; Group 7: AOM/DSS-induced CAC model treated by 5-FU plus metformin. The therapies of metformin and 5-FU were performed after first DSS or AOM/DSS cycle exposure. Metformin (Sigma) was added to drinking water (0.5 g in 50 ml, po.) once every two days.41 5-FU (15 mg/kg, i.p.) was intraperitoneal injected every 3 days.42 The therapies were performed for consecutive 12 weeks from second day after AOM injection. Mice were weighed twice a week and observed daily for any signs of illness. The pathological process of AOM/DSS-induced ulcerative colitis was scored as disease activity index (DAI) based on weight loss, stool consistency and bloody excreta as follows: weight loss score = 0: < 1%, 1: 1–5%, 2: 5–10%, 3:10–15%, 4: > 15%; stool consistency score = 0: normal, 2: loose, 4: diarrhea; blood in excreta score = 0: normal; 2: reddish, 4: bloody.43
Tissue processing and colonic cancer scoring
After 12 weeks’ treatment, mice were sacrificed. Whole colorectal tissues were removed and macroscopic tumors were counted and measured with digital calipers under a dissecting microscope based on size (diameter) into < 2 mm, 2–4 mm and 5–7 mm. Portions of colonic tissues were either fixed with 10% phosphate-buffered formalin or 2% paraformaldehyde and 2.5% glutaraldehyde in PBS.
Histological examination and colitis gradation
Histological examination was performed following the regular method. Colitis gradation was scored according to previous report.44 The presence of a normal amount of inflammatory cells in lamina propria was assigned a value of 0, increased numbers of inflammatory cells in the lamina propria a value 1, confluence of inflammatory cells extending into the submucosa was assigned a value of 2, and transmural extension of the infiltrate a value of 3. For tissue damage, no mucosal damage was scored as 0, discrete lymphoepithelial lesions were scored as 1, surface mucosal erosion or focal ulceration was scored as 2, and extensive mucosal damage and extension into deeper structures of bowel wall were scored as 3. The combined histological score ranged from 0 (no changes) to 6 (extensive cell infiltration and tissue damage).
Transmission electron microscopy
The excised sigmoid colonic tissues were fixed in 2% paraformaldehyde and 2.5% glutaraldehyde in PBS for 2 h at room temperature. After washing in PBS, tissues were postfixed in osmium tetroxide for 45 min at room temperature. Dehydration of the samples was accomplished by transferring the samples through a series of graded ethanol and then 100% propylene oxide. The tissue was then infiltrated by transferring the samples into increasing concentrations of Epon to propylene oxide solutions; 1:3, 1:1, and 3:1, then 100% Epon and finally embedded. Sections were made with a Leica EM UC7 μl tramicrotome (Leica), stained for 15 min with 7% (saturated) aqueous uranyl acetate, washed, stained with lead citrate. Obtained sections were stained and observed under a transmission electron microscope (HT7700, Hitachi, Japan).
Preparation of supernatant of colonic mucosa and determination of protein level
Fresh colonic mucosa was weighed and homogenised in PBS (pH 7.2) at 4°C. The supernatant was centrifuged and protein level was quantitatively determined by using BAC 100 protein determination kit (Sigma).
ELISA assay
The levels of TNF-α (CSB-E04741m), IL-6 (CSB-E04639m) in the supernatant of colorectal mucosa were quantified by using ELISA kits according to manufacturer’s protocol (Cusabio).
Human normal colon cell line, treatment
Human normal colon cell line NCM460 purchased from American Type Culture Collection was cultured in RPMI 1640 medium (Gibco, BRL, UK) supplemented with 10% fetal bovine serum (Gibco), 100 U/ml penicillin (Invitrogen, Carlsbad, CA), and 100 µg/ml streptomycin (Invitrogen) under humidified air with 5% CO2 at 37°C. NCM460 cells (1.0 × 106 cells per flask) were treated with 5 mM of metformin (Sigma), 50 µM H2O2 or 50 µM H2O2 after 5 mM of metformin for 1 h. After 48 h the mitochondrial structures were analyzed by Mito Tracker Red staining assay.45,46
Mitochondrial membrane potential staining assay
Mitochondrial membrane potential was detected by Mito Tracker Red staining (Beyotime, China). Mito Tracker staining was performed to observe mitochondrial morphology under a fluorescence microscope (Olympus, XSZ-D2, Japan). Continuous mitochondrial structures were counted, and the number was normalized to the total mitochondrial area to obtain the count of mitochondrial fragmentation. Cells with greater fragmentation exhibit a higher mitochondrial fragmentation. Lengths of mitochondria were measured by using NIS-Elements software and scored as follows: fragmented (globular, < 2 μm diameter); intermediate (2–4 μm long); and filamentous (> 4 μm long).
Western blotting analysis
Western blotting was performed routinely to analyze the expression of following proteins. Colonic tissues were dispersed mechanically in PBS and then the supernatants were collected and total protein was determined. The proteins were electrotransferred onto PVDF membranes and then the protein levels were estimated by using the primary antibodies with appropriate dilution. The primary antibodies included NFκB pathway sampler kit (9936), LKB1 (3050), AMPKα (2973), PCNA (2567) (Cell Signaling); NDUFA9 (ab14713), HIF-1α (ab1), IGF-1R (ab39398), AKT (ab8805). Densitometric analyses of bands were adjusted with β-actin (β-actin (Sigma).
Statistical analysis
Data are expressed as mean ± SD. P values were calculated by independent t test, one-way and two-way ANOVA. P < 0.05 was considered significant.
Funding Statement
This work was supported by the Beijing Natural Science Foundation Program 495 and Scientific Research Key Program of Beijing Municipal Commissionof Education (KZ201710025020, KZ201810025033).
Acknowledgments
The authors thank Prof. Yu Xin Feng in Capital Medical University for her intellectual inputs and edited the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors.
References
- 1.Abulí A, Lozano JJ, Rodríguez-Soler M, Jover R, Bessa X, Muñoz J, Esteban-Jurado C, Fernández-Rozadilla C, Carracedo A, Ruiz-Ponte C, et al. Genetic susceptibility variants associated with colorectal cancer prognosis. Carcinogenesis. 2013; 34(10): 2286–2291.doi: 10.1093/carcin/bgt179. [DOI] [PubMed] [Google Scholar]
- 2.Low D, Mino-Kenudson M, Mizoguchi E.. Recent advancement in understanding colitis-associated tumorigenesis. Inflamm Bowel Dis. 2014;20(11):2115–2123. doi: 10.1097/MIB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345(2):235–241. doi: 10.1016/j. [DOI] [PubMed] [Google Scholar]
- 4.Thent ZC, Zaidun NH, Azmi MF, Senin MI, Haslan H, Salehuddin R. Is metformin a therapeutic paradigm for colorectal cancer: insight into the molecular pathway?. Curr Drug Targets. 2017;18(6):734–750. doi: 10.2174/1389450118666161205125548. [DOI] [PubMed] [Google Scholar]
- 5.Koh SJ, Kim JM, Kim IK, Ko SH, Kim JS. Anti-inflammatory mechanism of metformin and its effects in intestinal inflammation and colitis-associated colon cancer. J Gastroenterol Hepatol. 2014;29(3):502–510. [DOI] [PubMed] [Google Scholar]
- 6.Cameron AR, Morrison VL, Levin D, Mohan M, Forteath C, Beall C, McNeilly AD, Balfour DJ, Savinko T, Wong AK, et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res. 2016; 119(5): 652–665.doi: 10.1161/CIRCRESAHA.116.308445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwin PJ. Obesity and breast cancer outcomes: how much evidence is needed to change practice?. J Clin Oncol. 2016;34(7):646–648. doi: 10.1200/JCO. [DOI] [PubMed] [Google Scholar]
- 8.Fowke JH, Motley SS. Statin use linked with a decrease in the conversion from high-grade prostatic intraepithelial neoplasia (HGPIN) to prostate cancer. Carcinogenesis. 2018;39(6):819–825. doi: 10.1093/carcin/bgy050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zi F, Zi H, Li Y, He J, Shi Q, Cai Z. Metformin and cancer: an existing drug for cancer prevention and therapy. Oncol Lett. 2018;15(1):683–690. doi: 10.3892/ol.2017.7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo YS, Kim YJ, Kim MS, Suh KS, Kim SB, Han CJ, Kim YJ, Jang WI, Kang SH, Tchoe HJ, et al. Association of metformin use with cancer-specific mortality in hepatocellular carcinoma after curative resection: a nationwide population-based study. Medicine (Baltimore). 2016; 95(17): e3527.doi: 10.1097/MD.0000000000003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, You YJ. Regulation of organelle function by metformin. IUBMB Life. 2017;69(7):459–469. doi: 10.1002/iub.1633. [DOI] [PubMed] [Google Scholar]
- 12.Kalyanaraman B, Cheng G, Hardy M, Ouari O, Sikora A, Zielonka J, Dwinell MB. Modified metformin as a more potent anticancer drug: mitochondrial inhibition, redox signaling, antiproliferative effects and future EPR studies. Cell Biochem Biophys. 2017;75(3–4):311–317. doi: 10.1007/s12013-017-0796-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardie DG. Keeping the home fires burning: AMP-activated protein kinase. J R Soc Interface. 2018;15:.138. doi: 10.1098/rsif.2017.0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Li X, Zhang W, He J, Xu B, Lei B, Wang Z, Cates C, Rousselle T, Li J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism. 2018;83:.256–270. doi: 10.1016/j.metabol.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan Y, Liu L, Li S, Wang K, Ke R, Shi W, Wang J, Yan X, Zhang Q, Wang Q, et al. Activation of AMPK inhibits TGF-β1-induced airway smooth muscle cells proliferation and its potential mechanisms. Sci Rep. 2018; 8(1): 3624.doi: 10.1038/s41598-018-21812-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung EJ, Do EJ, Kim SY, Cho EA, Kim DH, Pak S, Hwang SW, Lee HJ, Byeon JS, Ye BD, et al. Combination of metformin and VSL#3 additively suppresses western-style diet induced colon cancer in mice. Eur J Pharmacol. 2017;794:1–7. doi: 10.1016/j.ejphar.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 17.Vendramini-Costa DB, Francescone R, Posocco D, Hou V, Dmitrieva O, Hensley H, de Carvalho JE, Pilli RA, Grivennikov SI. Anti-inflammatory natural product goniothalamin reduces colitis-associated and sporadic colorectal tumorigenesis. Carcinogenesis. 2017;38(1):51–63. doi: 10.1093/carcin/bgw112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Justino PF, Melo LF, Nogueira AF, Costa JV, Silva LM, Santos CM, Mendes WO, Costa MR, Franco AX, Lima AA, et al. Treatment with Saccharomyces boulardii reduces the inflammation and dysfunction of the gastrointestinal tract in 5-fluorouracil-induced intestinal mucositis in mice. Br J Nutr. 2014; 111(9): 1611–1621.doi: 10.1017/S0007114513004248. [DOI] [PubMed] [Google Scholar]
- 19.Grivicich I, Regner A, Da Rocha AB, Grass LB, Alves PA, Kayser GB, Schwartsmann G, Henriques JA. Irinotecan/5-fluorouracil combination induces alterations in mitochondrial membrane potential and caspases on colon cancer cell lines. Oncol Res. 2005;15(7–8):385–392. [DOI] [PubMed] [Google Scholar]
- 20.Wu KY, Lai YH, Yang YC, Wu WC, Hong SJ. 5-Fluorouracil-induced apoptosis changes in cultured corneal epithelial cells. J Ocul Pharmacol Ther. 2016;32(3):155–162. doi: 10.1089/jop.2015.0109. [DOI] [PubMed] [Google Scholar]
- 21.Bessler H, Straussberg R, Alexandrova S, Beilin B, Djaldetti M, Hart J. Effect of oral chemotherapy on the mitochondrial size of mouse intestinal cells. Cancer Chemother Pharmacol. 1996;38(1):35–38. doi: 10.1007/s002800050444. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Bastian SE, Cheah KY, Lawrence A, Howarth GS. Escherichia coli Nissle 1917-derived factors reduce cell death and late apoptosis and increase transepithelial electrical resistance in a model of 5-fluorouracil-induced intestinal epithelial cell damage. Cancer Biol Ther. 2014;15(5):560–569. doi: 10.4161/cbt.28159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheah KY, Howarth GS, Yazbeck R, Wright TH, Whitford EJ, Payne C, Butler RN, Bastian SE. Grape seed extract protects IEC-6 cells from chemotherapy-induced cytotoxicity and improves parameters of small intestinal mucositis in rats with experimentally-induced mucositis. Cancer Biol Ther. 8;2009:382–390. [DOI] [PubMed] [Google Scholar]
- 24.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:.2101–2114. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Li S, Cao Y, Tian X, Zeng R, Liao DF, Cao D. Oxidative stress and carbonyl lesions in ulcerative colitis and associated colorectal cancer. Oxid Med Cell Longev. 2016;2016:.9875298. doi: 10.1155/2016/9875298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Mandal AK, Saito H, Pulliam JF, Lee EY, Ke ZJ, Lu J, Ding S, Li L, Shelton BJ, et al. Arsenic and chromium in drinking water promote tumorigenesis in a mouse colitis-associated colorectal cancer model and the potential mechanism is ROS-mediated Wnt/β-catenin signaling pathway. Toxicol Appl Pharmacol. 2012; 262(1): 11–21.doi: 10.1016/j.taap.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waldner MJ, Neurath MF. Mechanisms of immune signaling in colitis-associated cancer. Cell Mol Gastroenterol Hepatol. 2014;1(1):6–16. doi: 10.1016/j.jcmgh.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hibiya S, Tsuchiya K, Hayashi R, Fukushima K, Horita N, Watanabe S, Shirasaki T, Nishimura R, Kimura N, Nishimura T, et al. Long-term inflammation transforms intestinal epithelial cells of colonic organoids. J Crohns Colitis. 2017; 11(5): 621–630.doi: 10.1093/ecco-jcc/jjw186. [DOI] [PubMed] [Google Scholar]
- 29.Zhao B, Kang Q, Peng Y, Xie Y, Chen C, Li B, Wu Q. Effect of angelica sinensis root extract on cancer prevention in different stages of an AOM/DSS mouse model. Int J Mol Sci. 2017;18:.8. doi: 10.3390/ijms18081750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickrell AM, Fukui H, Wang X, Pinto M, Moraes CT. The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J Neurosci. 2011;31(27):9895–9904. doi: 10.1523/JNEUROSCI.6223-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KU, Kandela I, Wei C, Singhal S, Koblinski JE, Raje NS, Rosen ST, et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin Cancer Res. 2015; 21(5): 1161–1171.doi: 10.1158/1078-0432.CCR-14-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Zhu XD, Wang WQ, Shen Y, Qin Y, Ren ZG, Sun HC, Tang ZY. Activation of beta-catenin by hypoxia in hepatocellular carcinoma contributes to enhanced metastatic potential and poor prognosis. Clin Cancer Res. 2010;16(10):2740–2750. doi: 10.1158/1078-0432.CCR-09-2610. [DOI] [PubMed] [Google Scholar]
- 33.Shackelford DB, Vasquez DS, Corbeil J, Wu S, Leblanc M, Wu CL, Vera DR, Shaw RJ. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci USA. 2009;106(27):11137–11142. doi: 10.1073/pnas.0900465106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loubiere C, Clavel S, Gilleron J, Harisseh R, Fauconnier J, Ben-Sahra I, Kaminski L, Laurent K, Herkenne S, Lacas-Gervais S, et al. The energy disruptor metformin targets mitochondrial integrity via modification of calcium flux in cancer cells. Sci Rep. 2017; 7(1): 5040.doi: 10.1038/s41598-017-05052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001; 108(8): 1167–1174.doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shackelford BD, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumor suppression. Nat Rev Cancer. 2009;9(8):563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun X, Yang Q, Rogers CJ, Du M, Zhu MJ. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017;24(5):819–831. doi: 10.1038/cdd.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin H, Joëlle D, Bertrand D, Patricia L, Alain G, Patrick CE, Pascale C, Yves LB. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421(6919):182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 39.Meidenbauer JJ, Ta N, Seyfried TN. Influence of a ketogenic diet, fish-oil, and calorie restriction on plasma metabolites and lipids in C57BL/6J mice. Nutr Metab (Lond). 2014;11:.23. doi: 10.1186/1743-7075-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parang B, Barrett CW, Williams CS. AOM/DSS model of colitis-associated cancer. Methods Mol Biol. 2016;1422:.297–307. doi: 10.1007/978-1-4939-3603-8_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thangthaeng N, Rutledge M, Wong JM, Vann PH, Forster MJ, Sumien N. Metformin imparis spatial memory and visual acuity in old male mice. Aging Dis. 2017;8(1): 17–30. doi: 10.14336/AD.2016.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ke M, Wang H, Zhou Y, Li J, Liu Y, Zhang M, Dou J, Xi T, Shen B, Zhou C. SEP enhanced the antitumor activity of 5-fluorouracil by up-regulating NKG2D/MICA and reversed immune suppression via inhibiting ROS and caspase-3 in mice. Oncotarget. 2016;7(31):49509–49526. doi: 10.18632/oncotarget.10375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69(2):238–249. [PubMed] [Google Scholar]
- 44.Anzai H, Hata K, Kishikawa J, Ishii H, Yasuda K, Otani K, Nishikawa T, Tanaka T, Kiyomatsu T, Kawai K, et al. Appendiceal orifice inflammation is associated with proximal extension of disease in patients with ulcerative colitis. Colorectal Dis. 2016; 18(8): O278–82.doi: 10.1111/codi.13435. [DOI] [PubMed] [Google Scholar]
- 45.Moniruzzaman M, Ghosal I, Das D, Chakraborty SB. Melatonin ameliorates H2O2-induced oxidative stress through modulation of Erk/Akt/NFkB pathway. Biol Res. 2018;51(1):17. doi: 10.1186/s40659-018-0168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase–dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]