

ABSTRACT
Tumor markers of bladder cancer (BC) have been investigated for many years, but the clinical treatment based on these biomarkers is still unsatisfactory. STK32C, a member of the serine/threonine protein kinase of AGC superfamily, was first found to be highly expressed in brain tissues; however, the role of STK32C in malignant disease has not been determined. Data from TCGA database showed that the STK32C gene is overexpressed in BC and a number of other human tumors. In the current study, immunohistochemistry revealed that high expression of STK32C protein in tumor tissues was significantly associated with poor clinico pathologic features and a short relapse-free survival (RFS) in patients with BC. Slicing of STK32C inhibited tumor cell proliferation, migration and invasion in vitro. In vivo animal experiments demonstrated that knocking-down of STK32C restricted the growth of tumor cells in mice. Finally, microarray analysis revealed that silencing of STK32C inhibited the activity of the HMGB1 pathway and regulated the expression of key genes in this pathway. In conclusion, our study showed novel promoting roles for STK32C in human tumors, which may provide a new therapeutic target for the patients with BC.
Keywords: STK32C, protein kinase, bladder cancer, progression, TCGA
1. Introduction
Bladder cancer (BC) is the second most common malignancy of the genitourinary tract with an estimated 79030 newly diagnosed cases and 16870 deaths in USA, 2017.1 Most of BC cells are derived from epithelial cells. Approximately 25% of BCs are muscle invasive (MIBC) and approximately 75% of patients have non-muscle invasive (NMIBC) or metastatic BC. With the high rates of recurrence and the short time to progression and death in patients with NMIBC and MIBC, monitoring patient survival is extremely important and leads to a giant economic burden worldwide.2 Currently, cystoscopy and voided urine cytology are the basic standard diagnostic methods for BC.3 Although the treatment of BC has changed, the high recurrence rate and unsatisfactory 5-year overall survival rate are still in need of improvement.4 Tumor markers have been investigated for many years, and many conventional tumor markers and molecular pathways involved in BC have been reported to play important roles,5 yet clinical treatment based on these biomarkers is still unsatisfactory. Thus, novel molecular biomarkers for clinical outcome prediction and targeted treatment of BC need to be identified.
The human protein kinase family has been found to contain more than 500 members, which mediate most signal transductions in eukaryotic cells and control a number of cellular processes including metabolism, transcription, cell cycle, movement, apoptosis and differentiation by modification of substrate activity.6 Mutation and dysregulation of protein kinases are associated with many human diseases. Antagonists and agonists of these protein kinases provide possible therapeutic strategies for tumors.7,8 Serine/threonine kinases belong to the AGC protein kinase superfamily. Previous studies have shown that some serine/threonine protein kinases are associated with progression of malignant diseases. Specifically, STK33 promotes cancer cell viability and can be considered a target for treatment of mutant KRAS-driven cancers.9,10 Inactivation of STK11 leads to expansion of a tumor subpopulation in melanoma.11 In addition, the AGC superfamily also includes the PKA, PKC, and PKG families, most of which are also linked to tumor progression.12
Serine/threonine kinase 32C (STK32C) is a member of the serine/threonine protein kinase of AGC superfamily, but the specific functions of STK32C are unclear. It has been shown that STK32C is overexpressed in brain tissues and associated with adolescent depression based on DNA methylation differences that occur in monozygotic twins.13 Data mining from different studies have revealed that STK32C gene expression changes are correlated with CD4+Treg-cell function and lung inflammation.14,15 Tumor-related gene expression profile have shown that STK32C elevation may occur in some malignant diseases, such as prostate cancer.16 Bioinformatics analyses from the TCGA database in our study showed that STK32C gene is abnormally expressed in many tumors and associated with the prognosis of patients, which implies that STK32C might play a role in tumor progression, especially BC. Therefore, in this study we systematically explored the biological mechanism of STK32C in the progression of BC in vivo and in vitro. According to these, we expected to determine the functions of STK32C and find efficient novel therapeutic targets for BC.
2. Materials and methods
2.1. Bioinformatics
TCGA, the cancer genome atlas (https://cancergenome.nih.gov/), is an online bioinformatics database, which provides grand biological information. We downloaded the clinical data and mRNA expression data (normalized mRNASeq-FPKM.txt) of different cancers from TCGA. Then, the R.3.41 project was applied to organize data and analyze the expression of STK32C in these carcinomas. In addition, to confirm the reliability of the results, the preliminarily collated data from the UCSC Cancer Genomics Browser (http://xena.ucsc.edu) (an online analytical tool of TCGA) were also extracted and analyzed. The results were consistent with the data from TCGA. The data involving the mutation, amplification, and deletion of the STK32C gene in TCGA patients were obtained from cBioPortal (http://cbioportal.org).
2.2. Patients and samples
Ninety-six patients pathologically diagnosed with BC in the second hospital of Tianjin Medical University (Tianjin, China) from January 2010 to December 2014 were included. The tumor tissues of these patients were collected after surgical treatment. None of the patients had a history of preoperative radiotherapy and/or chemotherapy and neoadjuvant chemotherapy. The patients’ clinical information was recorded (Supplementary table S1). Patients were classified according to the 2009 UICC TNM staging, as well as in compliance with the 2004 WHO/ISUP classification.17 Sixty-four patients treated by transurethral resection of the bladder tumor (TURBT) were followed. The final follow-up date was 1 October 2016. The study was approved by the Human Ethics Committee of Tianjin Medical University (Tianjin, China).
2.3. Immunohistochemistry (IHC)
Tumors and matched normal adjacent tissues collected from the Second Hospital of Tianjin Medical University were fixed in 10% neutral-buffered formalin and embedded in paraffin. Then, 4-μm sections were cut from the paraffin blocks. Tissue sections were performed with citrate buffer (PH = 6.0) and 1.5% hydrogen peroxide in methanol. Slides were incubated with anti-STK32C (1:200, from rabbit, sigma) overnight at 4°C. Then, the second antibody was added to incubate at room temperature for 1 hour and 3, 3ʹ-diaminobenzidine was added for visualization. A semi-quantitative scoring system (H-score approach) was used to assess the level of STK32C expression.18 The proportion of positively stained cells was assigned a score from 0–4 and was termed as category A: 0 (0%); 1 (1–25%); 2 (26–50%); 3 (51–75%); and 4 (76–100%). Intensity of staining was assigned a score from 0–3 and was termed as category B: 0 (negative); 1 (weak); 2 (moderate); and 3 (strong). The final score, Z, was calculated by multiplying A by B (Z = A × B). We divided samples into two groups based on the final scores, as follows: STK32C over-expression group (Z > 3) and low-expression group (Z ≤ 3). The critical value Z was determined according to the distribution of the final data.
2.4. Cell culture
Human bladder cancer T24 and 5637cells were obtained from the ATCC. All cell lines were authenticated using short tandem repeat profiling analysis, and routinely tested for mycoplasma contamination within the last 6 months by Hoechst staining and PCR. Cells at passage numbers <8 after reception or thawing in our laboratory were used and cultured in RPMI-1640 medium plus 1% penicillin-streptomycin and 10 % fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2.
2.5. Plasmid construction and lentiviral transfection
A STK32C short hairpin RNA oligonucleotide sequence (shRNA) was inserted into a lentiviral vector (GV115) to construct a STK32C-RNAi plasmid. Three different STK32C-RNAi lentiviral vectors and an ordinary lentiviral vector as a control were incubated separately with BC cells (2 × 105/ml) (Supplementary Table S2). Real-time quantity PCR and Western blot were used to verify the final efficacy of knocking-down in both cell lines. Qualified cells were selected for later experiments. The biological behaviors of shSTK32C (cells transfected by the STK32C-RNAi lentiviral vector) and shCtrl (cells transfected by the control lentiviral vector) were compared, as described below. In addition, to certify the specificity of STK32C-RNAi lentiviral vector transfection and exclude an off-target effect on cell biological functions, a self-rescue experiment was performed.
2.6. Real-time quantity PCR
Total mRNA was extracted using the TRIzol reagent (Thermo, USA) according to the manufacturer’s instructions. RNA was reverse transcribed to cDNA using a cDNA Reverse Transcription Kit (Thermo, USA), which included 4μL of dNTP-mix, 2μL of primer-mix, 4μL of 5× PrimeScript buffer, 2μL of DTT, and DEPC water for a total volume of 20μL. Quantitative PCR was performed using SGExcel FastSYBR Mixture with High ROX) Plus (Thermo, USA). The primers of target genes were described (Supplementary Table S2).
2.7. Western blot
Proteins from different cells were extracted using lysis buffer and measured with the BCA protein assay (Thermo, USA). 20μg of proteins were denatured in sample buffer, then electrophoresed on 10% SDSPAGE. After electrophoresis, transfer, and blockage, the PVDF membranes (Thermo, USA) were incubated with primary antibodies overnight at 4°C. Then, secondary antibodies were added at room temperature for 1h and the immunoblots were marked using ECL (Thermo, USA). The details of antibodies were shown (Supplementary Table S2).
2.8. Cell proliferation assay
The transfected T24 cells were seeded into 24-well plates with a density of 2 × 103 cells/well and 5637 cells were seeded with a density of 1 × 103 cells/well. The quantity of cells contained in different groups was calculated using the Celigo image cytometry system every 24h. MTT assay was also used to measure the proliferation of both cell lines. The number of cells was measured every day. After 5 days, the proliferation curve was described to analyze the difference between groups. The assays were performed in triplicate.
2.9. Colony formation assay
Transfected T24 cells (300cells/well) and 5637 cells (400cells/well) were seeded into 6-well plates and incubated 9 days for colony formation assay. The cells were washed with PBS and fixed with 4% paraformaldehyde, then stained 10–30 min with Giemsa. Finally, the colony formation rates were observed with the unaided eye or under a microscope (low magnification). The colonies in which the number of cells exceeded 50 were counted. The assays were performed in triplicate.
2.10. Flow cytometry analysis of cell apoptosis and cycle
Cells in the shSTK32C and shCtrl groups were harvested for analysis of cell apoptosis and cell-cycle distribution respectively. Cell apoptosis was determined using an Annexin V-FITC apoptosis kit (Sigma-Company, USA). The experimental procedure was performed according to the manufacturer’s instructions. The levels of fluorescence were measured by flow cytometry (BD Biosciences, USA). The Annexin V-positive cells were defined as apoptotic cells. The cell growth cycle was also analyzed by flow cytometry. The assays were performed in triplicate.
2.11. Migration and invasion array
Transwell membrane with 8μm pore was used to determine the migration of cells. The shSTK32C and shCtrl cells (1 05/well) were suspended in 100μL cell suspensions and seeded into the upper chamber in 24-well plates. 600μL of medium with 30% FBS were added to the lower chamber. After a 24h incubation at 37°C, the cells that migrated into the lower membrane surface were stained with Giemsa and counted in 5 randomly selected photographs under a light microscope at × 200 magnification. For the cell invasion assay, the Transwell membrane with Matrigel substrate (BD, USA) was used and the processes were as described above. A wound and healing assay was also used to analyze the migration of cells. Transfected cells (3 × 104 cells/well) were seeded into 96-well plates, incubated, and harvested every 8 h. Photographs were obtained with fluorescence microscopy. Then, the distance between the edges of both sides was measured. All experiments were performed in triplicate.
2.12. Animal study
Animal studies were conducted with the approval of the Animal Care and Use Committee of Tianjin Medical University. (Permit Number: TMUaMEC2015008) Twenty 4-week-old female BALB/C nude mice (ten per group) were purchased from Ling Chang Biotechnology Company (Shanghai, China). T24 cells transfected with STK32C-RNAi lentiviral vector (KD) and control lentiviral vector (NC) were subcutaneously injected into the right axillae (2 × 107 cells per animal). 28 days after implantation, fluorescence imaging was performed on the live mice. The experimental animals were euthanized with a lethal dose of pentobarbital sodium and the tumors were harvested.
2.13. Gene microarrays
Three human gene expression profile chips with STK32C knocked down cells (KD) and three normal controls (NC) were performed on an Affymetrix platform. Total RNA was extracted from the samples using the Trizol method. The quality of total RNA extracted from the samples was tested on a NanoDrop 2000 (Thermo, USA) and Bioanalyzer 2100 (Agilent, USA) Qualified samples were used in the chip experiments. Then, hybridization, washing, and staining were completed using a GeneChip Hybridization Wash and Stain Kit according to the manufacturer’s instructions. Quality control of raw data from Genechip was performed. Subsequent function analysis was performed by IPA (Ingenuity Pathway Analysis).
2.14. Statistic analysis
SPSS 20.0 software was used for statistical analysis. Quantitative data were assessed by the mean ± SD. Parametric and non-parametric testing were used for data with and without variance heterogeneity, respectively. The associations between STK32C and clinico pathologic features were analyzed using a chi-square test. Kaplan-Meier survival analysis was applied to analyze the association between STK32C and recurrence-free survival (RFS). A P < 0.05 for the difference was significant.
3. Results
3.1. Bioinformation of the STK32C gene from TCGA
Data downloaded from the TCGA database were applied to analyze the gene expression of BC patients. Expression profiles of STK32C mRNA in 414 bladder tumor samples and 19 adjacent normal controls were included in our research. By comparing the expression of 414 tumor samples with 19 normal controls, a significant increase in STK32C mRNA expression was demonstrated in the tumor group (foldchange = 2.531; P value< 0.001; Figure 1(a)). By comparing gene expression in 19 of paired samples, we showed that the level of STK32C mRNA expression in the tumor group was dramatically increased (foldchange = 2.635; P value< 0.001; Figure 1(b)). Each of the 19 paired samples were analyzed individually; 12 samples were up-regulated, 1 sample was down-regulated, and 6 samples did not changed significantly (Figure1 (c-d)). Based on these results, we suspected that the STK32C gene possessed a higher level of expression in bladder tumors than adjacent normal tissues, which may play a role in tumor progression. After screening the state of the STK32C gene in different patients of the TCGA database, we found few patients with STK32C mutations or amplifications. The site of the mutation was located on R97W. Amplification of the STK32C gene was not correlated with expression according to the data from TCGA (Figure 1(e)).
Figure 1.
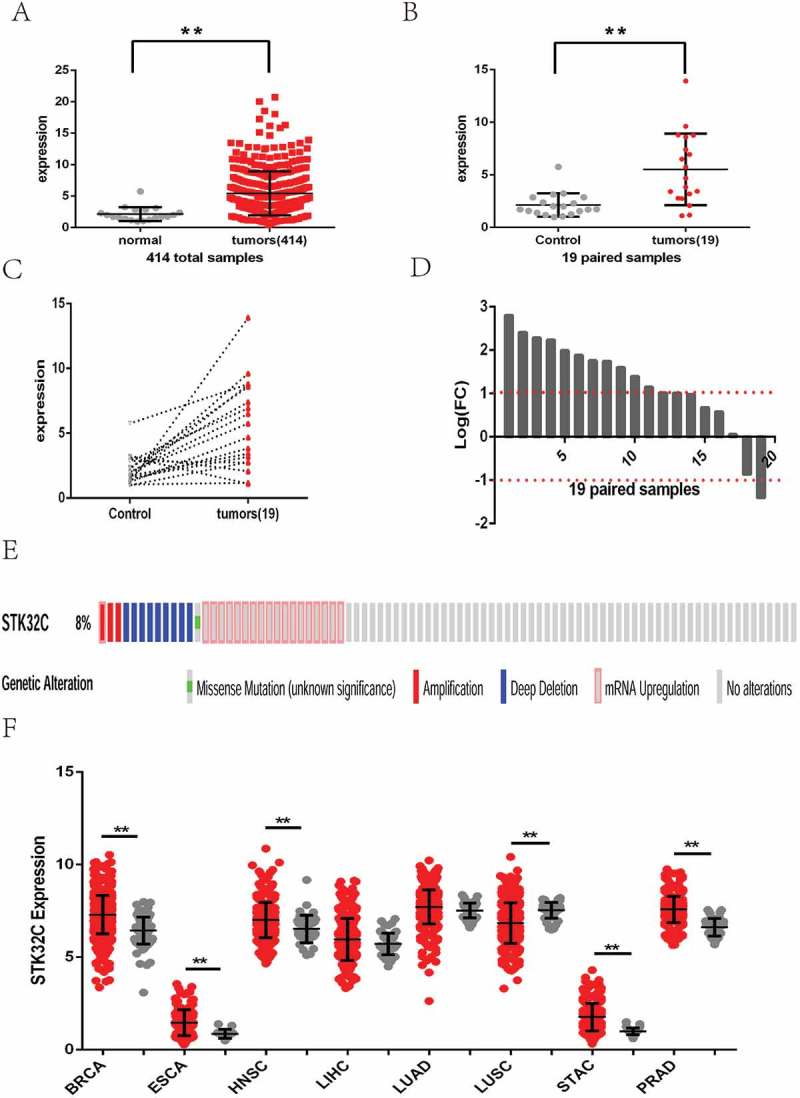
Expression of STK32C in bladder cancer and other cancers from TCGA. (a) Gene expression matrix of 414 of bladder cancer was downloaded from the TCGA database. Expression of the STK32C gene in overall bladder tumor samples was higher than normal controls. (P < 0.001) (b) 19 patients had tumors and adjacent normal control samples. Expression of the STK32C gene in 19 paired samples changed significantly. (P < 0.001) (c-d) Of 19 of paired samples, 12 samples were up-regulated, 1 was down-regulated, and 6 did not change insignificantly. (e)The mutation, amplification, deep deletion and mRNA upregulation of the STK32C gene in BC patients from TCGA. (f) Expression of STK32C was over-expressed in BRCA, ESCA, HNSC, STAC and PRAD, down-expressed in LUSC, and non-significant in LIHC and LUAD.
We also searched the level of STK32C gene expression in other common cancers from TCGA, including breast invasive carcinoma (BRCA), esophageal carcinoma (ESCA), head and neck squamous cell carcinoma (HNSC), liver hepatocellular carcinoma (LIHC), lung squamous cell carcinoma (LUSC), adenocarcinoma (LUAD), stomach adenocarcinoma (STAD), and prostate adenocarcinoma (PRAD). The results found that STK32C gene was still over-expressed in most of these tumors but down-regulated in LUSC, which also implied that STK32C is an active factor in cancers (Figure 1(f)).
3.2. Distribution of STK32C and associations with clinico pathologic features of BC patients from IHC
To determine if STK32C expression is associated with tumor progression of BC, tumor tissues from 96 samples were collected from the second hospital of Tianjin Medical University for IHC. The expression of STK32C was higher in tumors than adjacent normal tissues. More invasive BC tissues (T ≥ 2) showed a stronger dyeing intensity than non-invasive tumors, which indicated that elevated STK32C is correlated with high tumor pathologic T stage (Figure 2(a)). Interestingly, we showed that both the nucleus and cytoplasm of tumor cells could be dyed, and higher pathologic T stage of BC tended to be more easily stained in the nucleus (Figure 2(b)). The results implied that STK32C possibly plays a role in tumor cells by acting on the nucleus.
Figure 2.
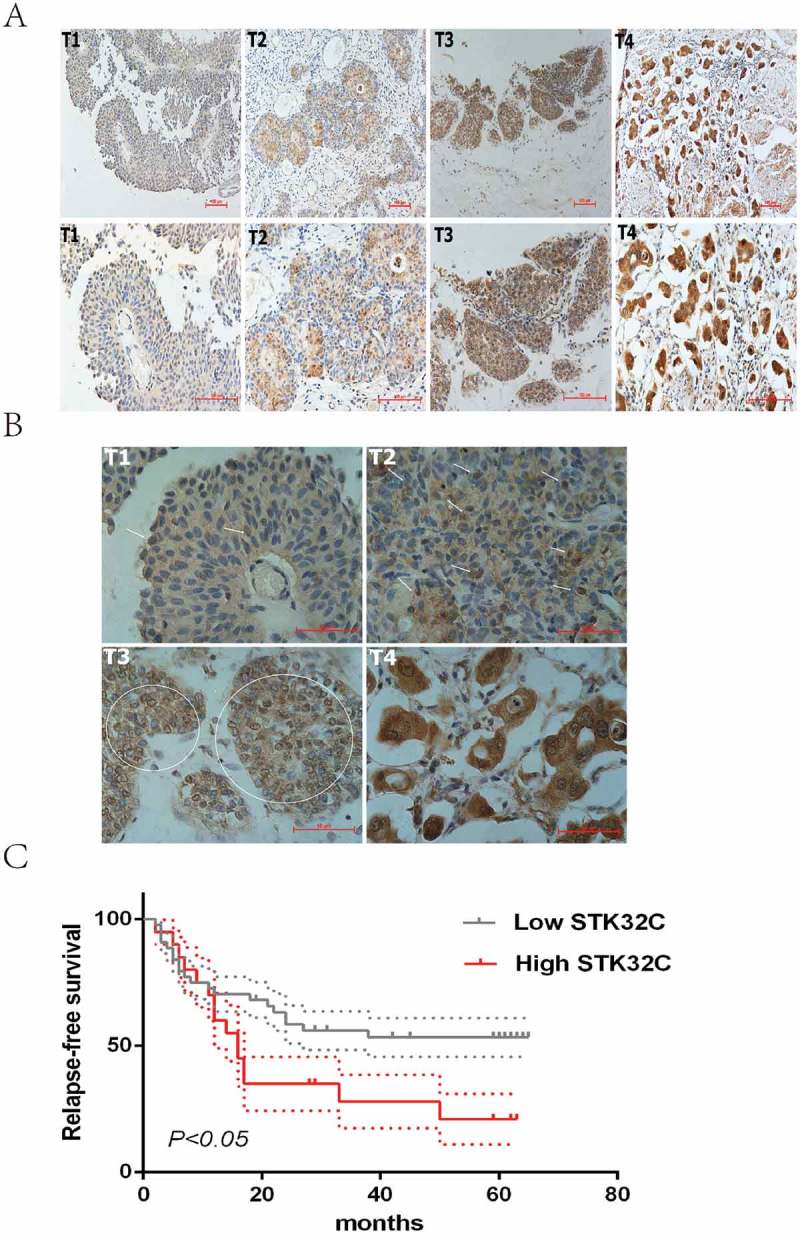
Immunohistochemistry. (a) Typical staining of STK32C in different stages of BC, a stronger density of staining existed in more invasive BC. (b) Nucleus and cytoplasm of tumor cells was dyed. The arrows and circles referred to the positively stained nucleus. Higher pathologic T stage of BC tended to be more easily stained in the nucleus. (c) Kaplan-Meier analysis was used to analyze the relapse-free survival (RFS) of patients treated by TURBT. High expression of STK32C was associated with a poor RFS with a P < 0.05.
According to IHC scoring, we divided samples into two groups (high and low expression groups). The expression of STK32C was significantly associated with clinico pathologic characters including age, gender, tumor size, tumor number, pTNM stage and recurrence. Males of an older age were more likely to present with rich STK32C. Pathologically, abundance of STK32C tended to be associated with large, multiple and invasive tumors; however, no significant associations were found between STK32C and smoking status and pathologic grade (Table 1). The correlation between STK32C and RFS in patients treated by TURBT was determined. A high level of STK32C was significantly associated with poor survival (P < 0.05; Figure 2(c)). In conclusion, STK32C possibly acted as an unfavorable factor, which was associated with poor clinico pathologic characteristics and short RFS in patients with BC.
Table 1.
An association between STK32C and clinical features.
| Clinical-pathological Features | STK32C |
P value | |||
|---|---|---|---|---|---|
| High | Low | Total | |||
| Age | ≥ 70 | 24 | 18 | 42 | 0.001* |
| < 70 | 13 | 41 | 54 | ||
| Sex | Male | 26 | 53 | 79 | 0.015* |
| Female | 11 | 6 | 17 | ||
| Smoking | YES | 19 | 23 | 42 | 0.234 |
| NO | 18 | 36 | 54 | ||
| Tumor number | 1 | 14 | 36 | 50 | 0.027* |
| ≥ 2 | 23 | 23 | 46 | ||
| Tumor size | < 2 | 7 | 26 | 33 | 0.012* |
| ≥ 2 | 30 | 33 | 63 | ||
| Grade | Low | 11 | 20 | 31 | 0.671 |
| High | 26 | 39 | 65 | ||
| pT stage | Ta, T1 | 14 | 38 | 52 | 0.011* |
| T2, T3, T4 | 23 | 21 | 44 | ||
| Recurrence† | YES | 15 | 20 | 35 | 0.028* |
| NO | 5 | 24 | 29 | ||
“*”, represents a significant association; P < 0.05 for the difference was significant.
“†”, Recurrence of bladder cancer patients only included patients treated by TURBT.
3.3. STK32C was over-expressed in bladder tumor cell lines, and knockdown of STK32C inhibited cell proliferation and promoted cell apoptosis
The STK32C gene was highly expressed in T24 and 5637 BC cell lines. To elucidate the role of STK32C, we stably knocked down the expression of STK32C using lentiviral shRNA transfection into T24 and 5637 cell lines. The transfection efficiency rates in both groups were significant (> 80%; Figure 3(a)). The cells with the highest knockdown were confirmed by qPCR as well as by Western blotting and selected for use (Figure 3(b)). Taken together, we confirmed that STK32C is overexpressed in BC cells, and we have effectively constructed a STK32C knock down model for both cell lines.
Figure 3.
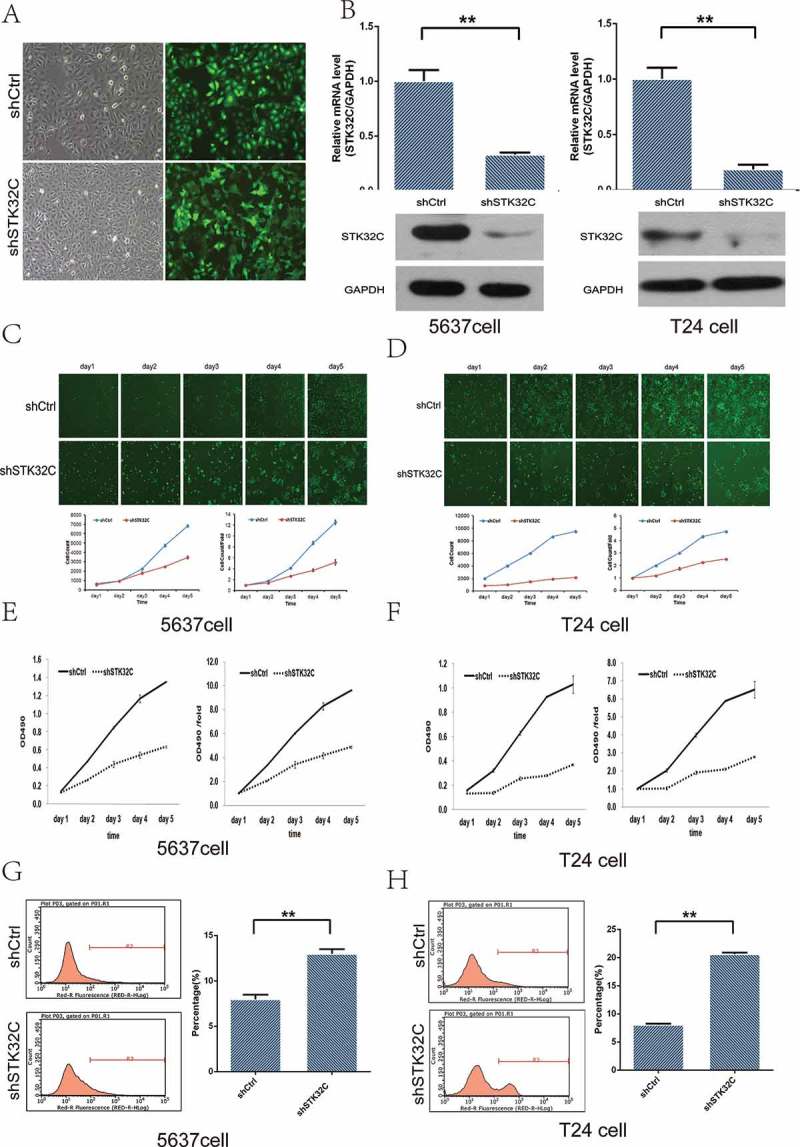
Influence of STK32C expression on proliferation of bladder tumor cell lines. (a) The efficiency of transfection was monitored by fluorescence microscopy. Greater than 80% cells were transfected successfully. (b) Expression of STK32C in shCtrl and shSTK32C groups of T24 and 5637 cells was measured by qPCR and Western blot. STK32C was successfully knocked down. (c-d) The proliferation of 5637 cells and T24 cells was significantly inhibited in the shSTK32C group compared with controls after incubation for 5 days. (P < 0.001) (e-f) MTT array showed that the cell proliferation in shSTK32C was inhibited in both cell lines. (g-h) Flow cytometry analysis found that the percentage of apoptotic cells was higher in the shSTK32C group than shCtrl in both cell lines. (P < 0.001).
The proliferation of T24 and 5637 cells in the shSTK32C and shCtrl groups were observed by two methods. The fluorescence measured with the Celigo instrument showed that the proliferation rates of T24 and 5637 cells in the shSTK32C group were significantly inhibited, which suggested that STK32C was significantly associated with the proliferation of T24 and 5637 cells (Figure 3(c-d)). The MTT arrays were in agreement with the above findings and showed that a lower level of STK32C gene expression could suppress the proliferation of cells dramatically (Figure 3(e-f)). In addition, the apoptotic cells dramatically increased in the shSTK32C group of both cell lines (Figure 3(g-h)). Taken together, the results clarified that knockdown of STK32C inhibited cell proliferation and promoted cell apoptosis in BC.
3.4. STK32C promoted cell colony formation, migration, and invasion in both cell lines
Colony formation assay was performed to investigate the effect of STK32C on the colony formation ability of BC cells. Compared with controls, the number of colonies in the shSTK32C group were markedly reduced (Figure 4(a-b)). Transwell and wound healing assays were performed to explore the migration and invasion of tumor cells. Silencing of STK32C expression notably inhibited migration in both T24 and 5637 cell lines (Figure 4(c-f)). The invasion of T24 cells was dramatically inhibited in the shSTK32C group, while 5637 cells changed insignificantly because of the inherently low-invasive ability (Figure 4(g)). In brief, STK32C was significantly associated with cell motility and knock down of STK32C expression significantly slowed down the migration and invasion in BC cells.
Figure 4.
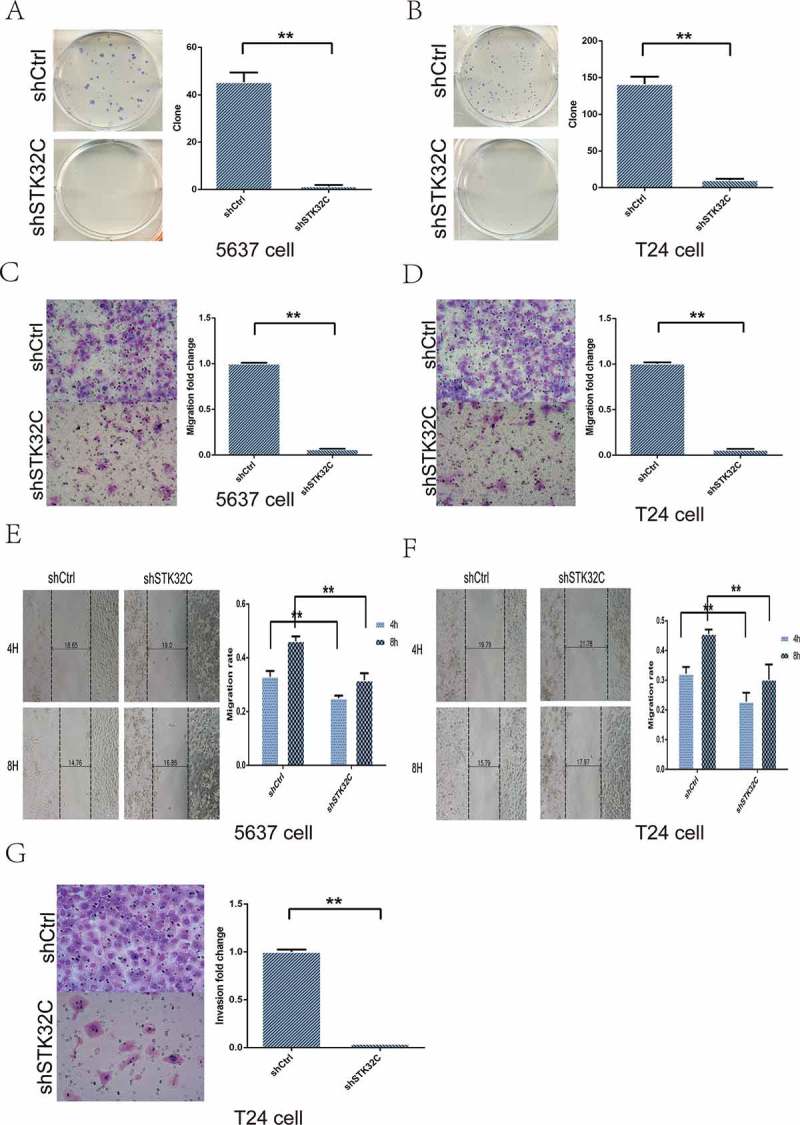
Influence of STK32C expression on colony formation, invasion, and migration of bladder tumor cell lines. (a-b) The colony formation assay was used to measure the colony formation ability of tumor cells. Colonies in shSTK32C groups were dramatically decreased compared with shCtrl. (P < 0.001) (c-d) The transwell assay showed that cells migrated into the lower chamber were decreased in the shSTK32C group of both cell lines. (P < 0.001) (e-f) In the wound and healing assay, the migration rates in the shSTK32C group were markedly inhibited at 4 h and 8 h in both cell lines. (P < 0.001) (g) The invasion of T24 cells was significantly inhibited in the shSTK32C group. (P < 0.001).
3.5. Knocking down STK32C blocked the growth of tumors in mice
To study the effect of STK32C on tumor growth, 1 × 107/ml of shSTK32C and shCtrl cells were separately injected into the right axillae of BALB/C nude mice. After 28 days of breeding, fluorescence imaging was performed on the living mice in vivo. All mice were alive and had no significant difference in body weight before final observation (Figure 5(a)). The results showed that fluorescence intensity in the KD group mice was stronger than the controls (Figure 5(b)). The weights of tumors in the KD group were significantly decreased compared to the controls (Figure 5(c)). The results were consistent with the in vitro assays, indicating that knock down of the STK32C gene expression inhibited tumor growth and the cells with low expression of STK32C had poor survivability.
Figure 5.
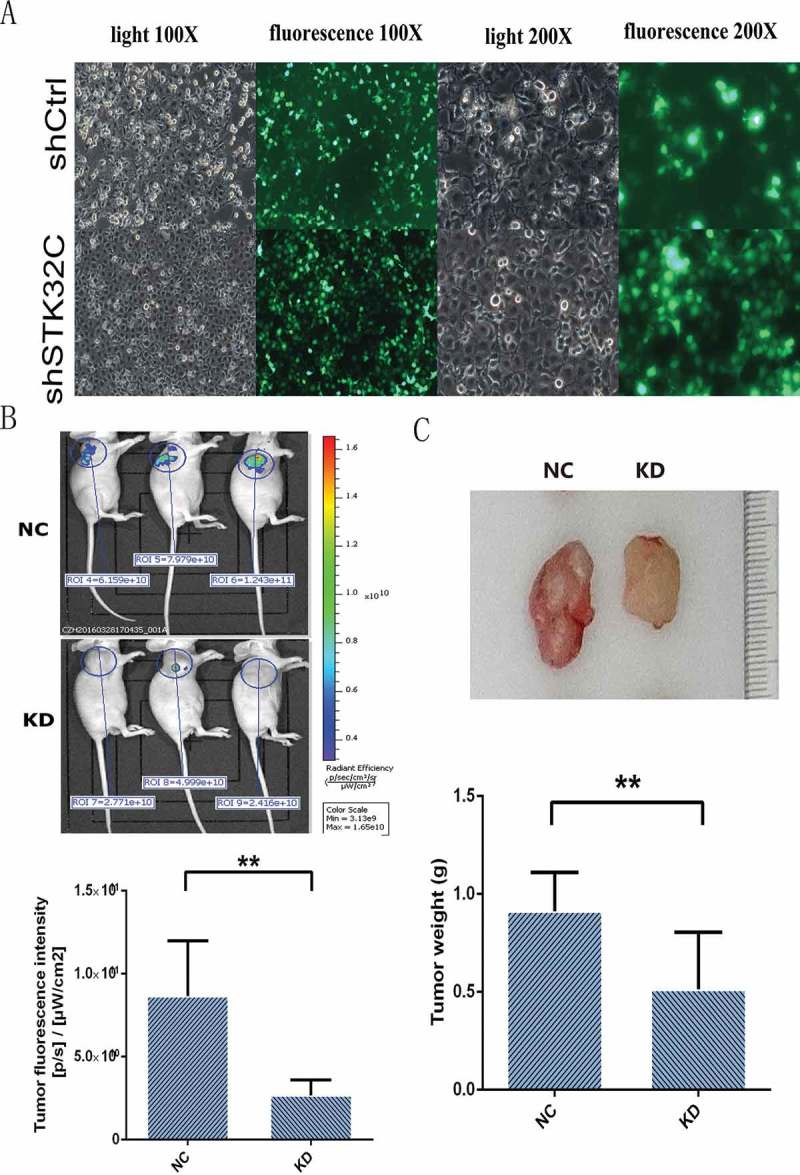
In vivo animal experiments. (a) T24 cells used for animal tumor formation experiments were observed by fluorescence microscopy, and most of the injected cells (> 90%) were transfected effectively. (b) The fluorescence imaging was observed from the live mice. The results found that tumor fluorescence intensity was decreased significantly in the KD groups. (c) After 28 days, tumors were harvested and mice were euthanized, tumor weights in the KD groups were heavier than the controls. (P < 0.001).
3.6. Microarray analysis of the gene expression after knocking down the STK32C gene
After comparing the normalized gene expression levels in KD with NC cells by human gene expression profile chips, a total of 519 genes were identified that significantly changed with 183 up-regulaed and 336 down-regulated (Foldchange> 1.5; P < 0.05; Figure 6(a)). IPA was applied to identify and predict the biological functions and pathways of target genes. First, pathway enrichment analysis of the changed genes showed that the most significant pathway was the HMGB1 signaling pathway, which was dramatically repressed by knocking-down STK32C (Figure 6(b)). Disease and functional analysis exhibited the enrichment of changed genes in the category of disease and function. The top term of increased activity was cell apoptosis. Conversely, the top terms of decreased activity were maturation of antigen presenting cells and phagocytes (Figure 6(c-d)). The functions of invasion and migration of tumor cell lines were also inhibited significantly. Regulation effect analysis showed the changes in genes involved in maturation of antigen presenting cells and phagocytes.
Figure 6.
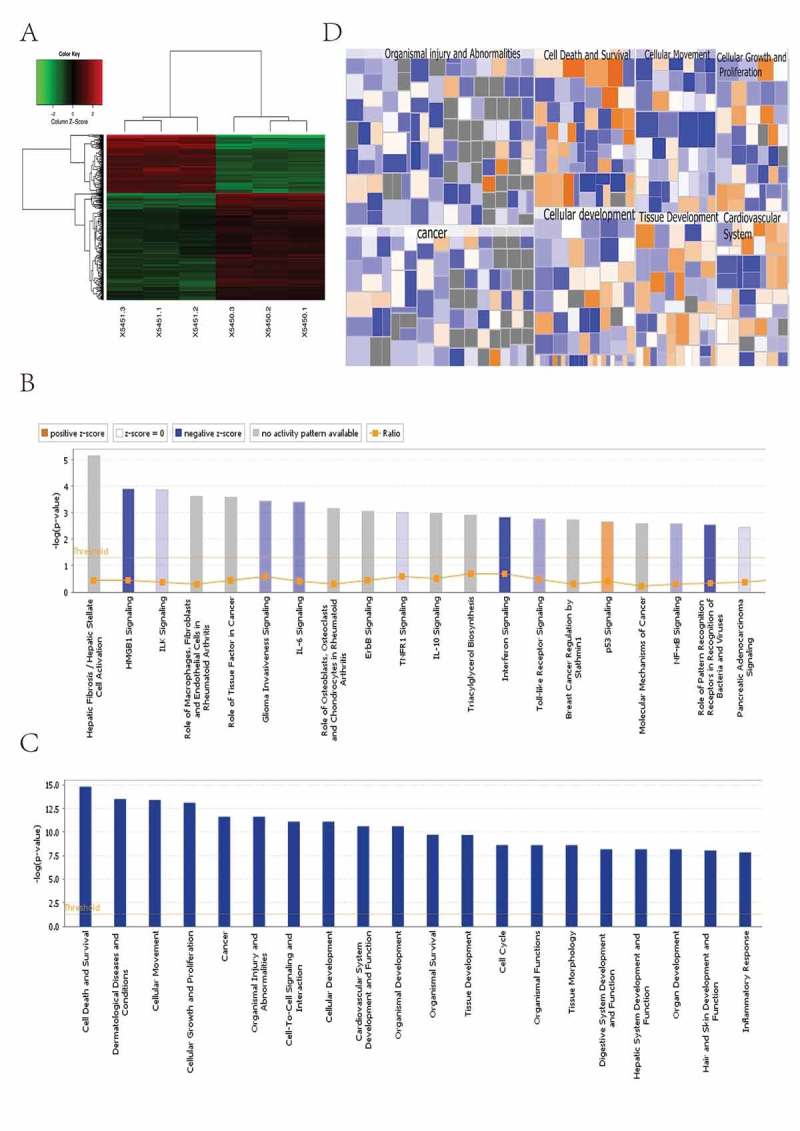
Microarray analysis of gene expression after knocking down the STK32C gene. All signal pathways, disease and functions were ordered using log (P-value); the area marked orange represented the Z-score> 0 and the area marked blue was the Z-score < 0; color intensity was positively associated with the Z-score. A Z-score> 2 represented a significant activation and a Z-score <-2 represented a significant inhibition. (a) The clustering heat map showed the aggregation of all samples and differential genes at the expression level. 183 genes were up-regulaed and 336 genes were down-regulated. (b) Pathway enrichment analysis (C-D) Disease and functional analysis.
3.7. Silencing of STK32C inhibited the HMGB1 signaling pathway and regulated expression of key factors in this pathway
According to enrichment pathway analysis of microarrays in our experiments, we found that the HMGB1 signaling pathway was significantly inhibited by knocking-down STK32C expression (Figure 7(a)). Then, to further explore the association between STK32C and the HMGB1 signaling pathway, five key genes in the HMGB1 signaling pathway were identified, including DDX58, PAK1, IL1, PIK3R1 and CDK2. Transcription and the protein levels of these genes were detected by qPCR and Western blot. When STK32C was inhibited, DDX58, PAK1, IL1 and PIK3R1 were significantly down-regulated, whereas CDK2 protein was up-regulated, which was consistent with the microarray results (Figure 7(b-c)). In conclusion, when we knocked down the expression of the STK32C gene, the activity of the HMGB1 signaling pathway was inhibited, and most key genes in the pathway were influenced with respect to transcription and protein levels. Therefore, we predicted that STK32C plays an essential role in epithelial bladder tumor progression through the HMGB1 signaling pathway.
Figure 7.
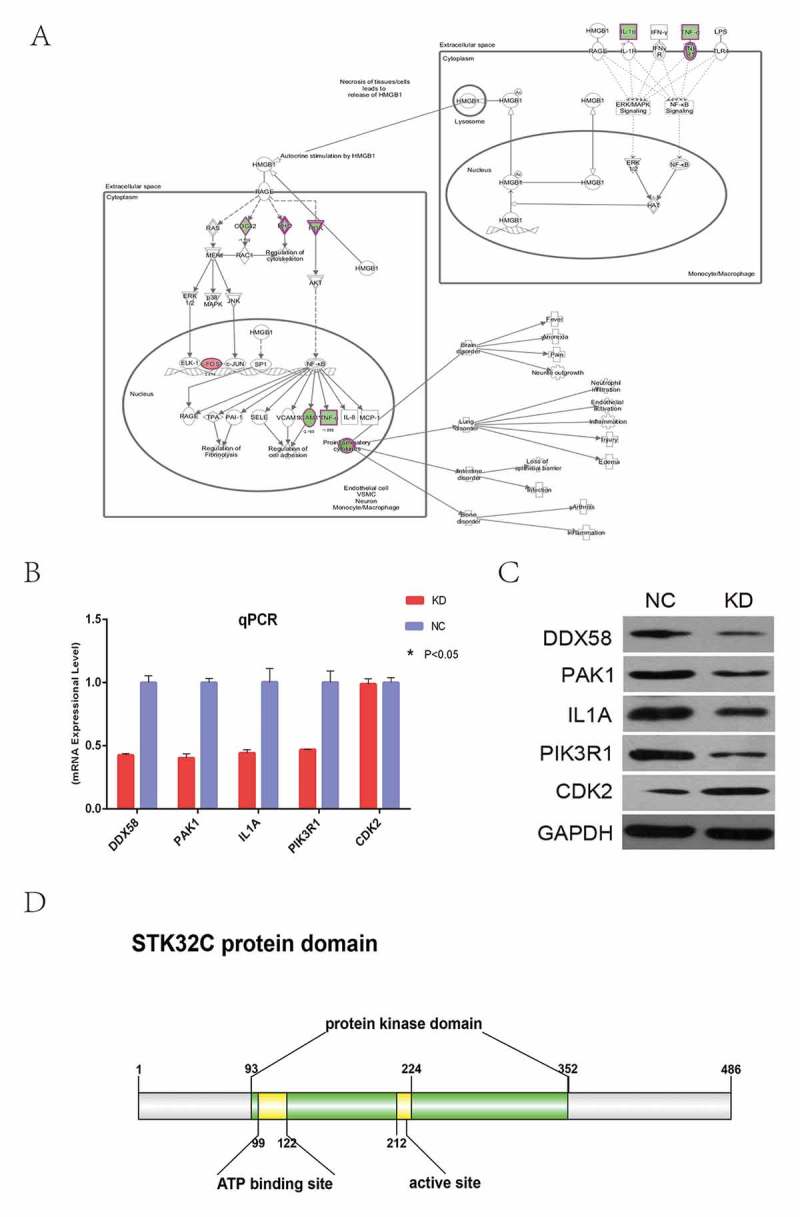
Knocking down STK32C influenced key genes in the HMGB1 pathway. (a) The important genes of the HMGB1 pathway map were shown. “Green” represented inhibited and “red” represented increased. (b) Transcriptional level of 13 genes on the HMGB1 pathway were detected by qPCR after knocking-down STK32C; all these genes except CDK2 were significantly regulated.(P < 0.001). (c) The protein levels of 5 genes verified by Western blot were consistent with the results of qPCR. (P < 0.001) (d) The protein domain of STK32C was predicted by public databases.
4. Discussion
Up to now, there was no research involving the relationship between STK32C and tumors. Our research was the precedent experiment that, for the first time, systematically studied the roles of STK32C in BC. Indeed, BC is a common malignant disease, although primary tumors can be eliminated by surgery, chemotherapy, or radiotherapy. Tumors recur frequently and may progress to muscle-invasive forms.19 Standard therapies could restrict the growth and development of a tumor; however, could not prevent patients from recurrence and drug-resistance.20 At present, many conventional tumor markers and molecular pathways involved in BC treatment and clinical outcomes have been investigated.21 TP53, ErbB2, and the phosphatidylinositol-3-OH kinase/AKT/mTOR pathways are the major genomic alternations in BC and other human malignancies.22 The status of many pathway genes are changed and used to predict the prognosis of BC patients. Representative known genes such as Tp53, FGFR3, HER2, VEGF, and EGFR, as targets for treatment of BC, have been reported to play a pivotal role in tumor recurrence and progression.23,24,25,26,27 Presently, although many associated genes involved in BC have been investigated, most of these targets are lacking in high specificity for diagnosis and treatment efficacy. Therefore, a standard biomarker for the prediction of clinical outcomes is still lacking, and the detection of novel effective therapeutic targets is needed.
STK32C, also known as PKE and YANK3, has low expression in normal bladder epithelial cells and high expression in human brain.28 The sequence of the STK32C gene in many species is highly conserved, such as chimpanzees, dogs, cattle and large mice. Two hundred organisms have orthologs of the human STK32C gene.6,29 The gene is located on human chromosome 10q26.3 and contains 18 introns and 6 transcripts (splice variants).30 STK32C encoding protein belongs to the AGC superfamily silk/threonine kinase, and many members of this superfamily have been reported to be associated with tumor progression.12 The protein domain of STK32C was shown by IBS,31(Figure 7(d)). It was predicted from NCBI () and Ensembl (http://www.ensembl.org/) that the functions of this protein consist of binding to nucleic acid, ATP, and metal ions, phosphorylation, activity of silk/threonine kinase, and possibly also included transferase activity to attune intracellular signaling. Phosphorylation of protein kinases is a fundamental process of cell signaling,32,33,34, the function or expression of which may be associated with pathogenesis.35,36 Protein phosphorylation increases or decreases enzymatic activity and alters other biological activities, such as transcription and translation.37,38 It has been shown that protein kinases, such as serine/threonine kinases, could promote tumor progression.10,11 The inhibition of protein kinase potentially provided a new strategy for treatment of many diseases, including cancer.39,40
In our study, we found that STK32C, which had low expression in normal human bladder epithelial cells, but was abundantly expressed in human bladder urothelial carcinoma, played an essential role in cell proliferation, migration, and invasion. Knocking down the expression of the STK32C gene reduced the ability of these tumor behaviors significantly. In vivo, suppression of STK32C gene expression resulted in quick death of tumor cells in mice subcutaneously implanted with xenograft tumor cells. In addition, we found that the level of STK32C expression was associated with the clinical features from immunohistochemistry analysis of clinical patients. Overall, these findings confirmed that STK32C plays an essential role in BC progression.
Microarray analysis showed that STK32C functioned probably via the HMGB1 signaling pathway. Some key genes in HMGBA signaling pathway were identified and validated. Most of them were shown to be associated with tumor progression. For example, CCNE/CDK2, which has long been considered an essential and master regulator of progression through the G1 phase of the cell cycle, plays an important role in tumoigenesis.41 PAK1 is associated with tumor cell growth, survival, motility, invasion, and cytoskeleton remodeling via its direct relevant substrates.42 By knocking down STK32C, the expression of these genes was dramatically regulated, which implied these were potential targets of STK32C. In summary, silencing of STK32C could inhibit the activity of HMGB1 pathway and regulate the expression of many key genes in this pathway, which shed a new light on the target treatment of bladder cancer. In the future, we plan to conduct comprehensive research on the exact targets and mechanisms of STK32C in BC.
5. Conclusion
In summary, our results demonstrated that STK32C was overexpressed in bladder tumors and associated with poor clinical features. Knocking down the expression of STK32C inhibited tumor progression in vitro and in vivo. Microarray analyses further revealed that STK32C inhibited the activity of the HMGB1 pathway and influenced the expression of many key genes in this pathway. Taken together, our study revealed that STK32C possesses the essential ability to promote BC progression.
Abbreviations
- STK32C
Serine/threonine kinase 32C;
- BC
Bladder cancer;
- NMIBC
Non-muscle invasive bladder cancer;
- MIBC
Muscle invasive bladder cancer;
- RFS
Relapse-free survival;
- OS
Overall survival;
- TCGA
The cancer genome atlas;
- IPA
Ingenuity pathway analysis;
- DDX58
DExD/H-box helicase 58;
- PAK1
p21 activated kinase 1;
- IL1
Interleukin-1;
- IL6
Interleukin-6;
- PIK3R1
Phosphoinositide-3-kinase regulatory subunit 1;
- CDK2
Cyclin dependent kinase 2
Authors’ contributions
The authors (ES and KL) contributed equally to this work. ES and KL were responsible for the coordination of the project and contributed to study design. The writing team consisted of ES and KL. ES, KL, KZ and LW performed most of in vitro and in vivo experiments. KL, KZ and LW performed immunohistochemistry and clinical data analysis. All authors read and approved the final manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s websit.
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmstrom PU, Choi W, Guo C C., Lotan Y., Kassouf W.. Bladder cancer. Lancet. 2016;388:2796–2810. doi: 10.1016/S0140-6736(16)30512-8. [DOI] [PubMed] [Google Scholar]
- 3.Jacobs BL, Lee CT, Montie JE.. Bladder cancer in 2010: how far have we come?. CA Cancer J Clin. 2010;60:244–272. doi: 10.3322/caac.20077. [DOI] [PubMed] [Google Scholar]
- 4.Jin X, Yun SJ, Jeong P, Kim IY, Kim WJ, Park S. Diagnosis of bladder cancer and prediction of survival by urinary metabolomics. Oncotarget. 2014;5:1635–1645. doi: 10.18632/oncotarget.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roupret M, Babjuk M, Comperat E, Zigeuner R, Sylvester RJ, Burger M, Cowan N C., Böhle A., Van Rhijn Bas W G., Kaasinen E., Palou J., Shariat S F.. European Association of Urology Guidelines on Upper Urinary Tract Urothelial Cell Carcinoma: 2015 Update. Eur Urol. 2015;68:868–879. doi: 10.1016/j.eururo.2015.06.044. [DOI] [PubMed] [Google Scholar]
- 6.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Sci. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 7.Fleuren ED, Zhang L, Wu J, Daly RJ. The kinome ‘at large’ in cancer. Nat Rev Cancer. 2016;16:83–98. doi: 10.1038/nrc.2015.18. [DOI] [PubMed] [Google Scholar]
- 8.Dancey J, Sausville EA. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov. 2003;2:296–313. doi: 10.1038/nrd1066. [DOI] [PubMed] [Google Scholar]
- 9.Kong F, Kong X, Du Y, Chen Y, Deng X, Zhu J, Du J., Li L., Jia Z., Xie D., Li Z., Xie K.. STK33 promotes growth and progression of pancreatic cancer as a critical downstream mediator of HIF-1alpha. Cancer Res. 2017;77:6851–6862. doi: 10.1158/0008-5472.CAN-17-0067. [DOI] [PubMed] [Google Scholar]
- 10.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver S J., Tamayo P., Wadlow R C., Ramaswamy S., Döhner K., Bullinger L., Sandy P., Boehm J S., Root D E., Jacks T., Hahn W C., Gilliland D G.. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 11.Liu W, Monahan KB, Pfefferle AD, Shimamura T, Sorrentino J, Chan KT, Roadcap D W., Ollila D W., Thomas N E., Castrillon D H., Miller C R., Perou C M., Wong K-K., Bear J E., Sharpless N E.. LKB1/STK11 inactivation leads to expansion of a prometastatic tumor subpopulation in melanoma. Cancer Cell. 2012;21:751–764. doi: 10.1016/j.ccr.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roskoski R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1–23. doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Dempster EL, Wong CC, Lester KJ, Burrage J, Gregory AM, Mill J, Eley T C.. Genome-wide methylomic analysis of monozygotic twins discordant for adolescent depression. Biol Psychiatry. 2014;76:977–983. doi: 10.1016/j.biopsych.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin H, Wang Z, Du W, Lee WH, Wu X, Riggs AD, Liu C-P.. Killer cell Ig-like receptor (KIR) 3DL1 down-regulation enhances inhibition of type 1 diabetes by autoantigen-specific regulatory T cells. Proc Natl Acad Sci U S A. 2011;108:2016–2021. doi: 10.1073/pnas.1019082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang DD, Lin Y, Moreno JR, Randall TD, Khader SA. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS One. 2011;6:e16161. doi: 10.1371/journal.pone.0016161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beaver LM, Buchanan A, Sokolowski EI, Riscoe AN, Wong CP, Chang JH, Löhr C V., Williams D E., Dashwood R H., Ho E.. Transcriptome analysis reveals a dynamic and differential transcriptional response to sulforaphane in normal and prostate cancer cells and suggests a role for Sp1 in chemoprevention. Mol Nutr Food Res. 2014;58:2001–2013. doi: 10.1002/mnfr.201400269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webber C, Gospodarowicz M, Sobin LH, Wittekind C, Greene FL, Mason MD, Compton C., Brierley J., Groome P A.. Improving the TNM classification: findings from a 10-year continuous literature review. Int J Cancer. 2014;135:371–378. [DOI] [PubMed] [Google Scholar]
- 18.Lykkegaard Andersen N, Brugmann A, Lelkaitis G, Nielsen S, Friis Lippert M, Vyberg M. Virtual double staining: a digital approach to immunohistochemical quantification of estrogen receptor protein in breast carcinoma specimens. Appl Immunohistochem Mol Morphol: AIMM. 2017. doi: 10.1097/PAI.0000000000000502. [DOI] [PubMed] [Google Scholar]
- 19.Bryan RT, Collins SI, Daykin MC, Zeegers MP, Cheng KK, Wallace DM, Sole G M.. Mechanisms of recurrence of Ta/T1 bladder cancer. Ann R Coll Surg Engl. 2010;92:519–524. doi: 10.1308/003588410X12664192076935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tada Y, Wada M, Migita T, Nagayama J, Hinoshita E, Mochida Y, Maehara Y., Tsuneyoshi M., Kuwano M., Naito S.. Increased expression of multidrug resistance-associated proteins in bladder cancer during clinical course and drug resistance to doxorubicin. Int J Cancer. 2002;98:630–635. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh M, Brancato SJ, Agarwal PK, Apolo AB. Targeted therapies in urothelial carcinoma. Curr Opin Oncol. 2014;26:305–320. doi: 10.1097/CCO.0000000000000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research N Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CH, Liu YM, Pan SL, Liu YR, Liou JP, Yen Y. Trichlorobenzene-substituted azaaryl compounds as novel FGFR inhibitors exhibiting potent antitumor activity in bladder cancer cells in vitro and in vivo. Oncotarget. 2016;7:26374–26387. doi: 10.18632/oncotarget.8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruthi RS, Nielsen M, Heathcote S, Wallen EM, Rathmell WK, Godley P, Whang Y., Fielding J., Schultz H., Grigson G., Smith A., Kim W.. A phase II trial of neoadjuvant erlotinib in patients with muscle-invasive bladder cancer undergoing radical cystectomy: clinical and pathological results. BJU Int. 2010;106:349–354. doi: 10.1111/j.1464-410X.2009.09101.x. [DOI] [PubMed] [Google Scholar]
- 25.Symanowski JT, Kim ES. Gene expression and prognosis in bladder cancer–real progress? Editorial on ‘S100A9 and EGFR gene signatures predict disease progression in muscle invasive bladder cancer patients after chemotherapy’. Ann Oncol. 2014;25:919–920. doi: 10.1093/annonc/mdu113. [DOI] [PubMed] [Google Scholar]
- 26.Ciccarese C, Massari F, Blanca A, Tortora G, Montironi R, Cheng L, Scarpelli M., Raspollini M R., Vau N., Fonseca J., Lopez-Beltran A.. Tp53 and its potential therapeutic role as a target in bladder cancer. Expert Opin Ther Targets. 2017;21:401–414. doi: 10.1080/14728222.2017.1297798. [DOI] [PubMed] [Google Scholar]
- 27.Lae M, Couturier J, Oudard S, Radvanyi F, Beuzeboc P, Vieillefond A. Assessing HER2 gene amplification as a potential target for therapy in invasive urothelial bladder cancer with a standardized methodology: results in 1005 patients. Ann Oncol. 2010;21:815–819. doi: 10.1093/annonc/mdp488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A., Horn-Saban S., Safran M., Domany E., Lancet D., Shmueli O.. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- 29.Chidambaram M, Radha V, Mohan V. Replication of recently described type 2 diabetes gene variants in a South Indian population. Metabolism. 2010;59:1760–1766. doi: 10.1016/j.metabol.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 30.Deloukas P, Earthrowl ME, Grafham DV, Rubenfield M, French L, Steward CA, et al. The DNA sequence and comparative analysis of human chromosome 10. Nature. 2004;429:375–381. doi: 10.1038/nature02462. [DOI] [PubMed] [Google Scholar]
- 31.Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, Lahrmann U., Zhao Qi, Zheng Y., Zhao Y., Xue Yu, Ren J.. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31:3359–3361. doi: 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang X, McGann JC, Liu BY, Hannoush RN, Lill JR, Pham V, Newton K., Kakunda M., Liu J., Yu C., Hymowitz S G., Hongo J-A., Wynshaw-Boris A., Polakis P., Harland R M., Dixit V M.. Phosphorylation of Dishevelled by protein kinase RIPK4 regulates Wnt signaling. Science (80-). 2013;339:1441–1445. doi: 10.1126/science.1232253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SH, Kim HS, Bahk S, An J, Yoo Y, Kim JY, Chung WS. Phosphorylation of the transcriptional repressor MYB15 by mitogen-activated protein kinase 6 is required for freezing tolerance in Arabidopsis. Nucleic Acids Res. 2017. doi: 10.1093/nar/gkx417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez D S., Joshi A., Gwinn D M., Taylor R., Asara J M., Fitzpatrick J., Dillin A., Viollet B., Kundu M., Hansen M., Shaw R J.. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (80-). 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter E W., Gallegos L L., Miller C J., Furnari F B., Hunter T., Brognard J., Newton A C.. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian X, Li X, Lu Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy. 2017;1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee TY, Bo-Kai Hsu J, Chang WC, Huang HD. RegPhos: a system to explore the protein kinase-substrate phosphorylation network in humans. Nucleic Acids Res. 2011;39:D777–87. doi: 10.1093/nar/gkq970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steffen M, Petti A, Aach J, D’Haeseleer P, Church G. Automated modelling of signal transduction networks. BMC Bioinformatics. 2002;3:34. doi: 10.1186/1471-2105-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buschbeck M. Strategies to overcome resistance to targeted protein kinase inhibitors in the treatment of cancer. Drugs R D. 2006;7:73–86. [DOI] [PubMed] [Google Scholar]
- 40.Cohen P. Protein kinases–the major drug targets of the twenty-first century?. Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 41.Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- 42.Kumar R, Li DQ. PAKs in human cancer progression: from inception to cancer therapeutic to future oncobiology. Adv Cancer Res. 2016;130:137–209. doi: 10.1016/bs.acr.2016.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.