

ABSTRACT
Montelukast is an anti-asthmatic medication, and has recently showed its inhibitory effects on the proliferation of cancers. The purpose of this study was to identify the cytotoxic effects of montelukast on multiple myeloma (MM) cells and the combination effects of montelukast and carfilzomib in the treatment of MM. Results revealed that montelukast induced a dose- and time-dependent cytotoxicity in MM cells lines and significantly suppressed the colony formation of myeloma cells. Furthermore, montelukast enhanced the cytotoxicity of carfilzomib in MM cell lines. This anti-tumor effect was associated with decreased c-Myc via the inhibition of mTOR signaling pathway. Moreover, the combination of montelukast and carfilzomib induced apoptosis of myeloma cells effectively, even in the presence of bone marrow stromal cells (BMSCs). It is more important to note that the co-treatment exhibited similar cytocidal effects in carfilzomib-resistant cell lines (U266R and 8226R). In addition, the combined effects were noted in two MM xenograft mice models and 7 cases of human CD138+ myeloma cells (4 newly diagnosed cases and 3 relapsed cases) with no cytotoxicity on peripheral blood mononuclear cells (PBMCs) from 5 healthy donors. Our data suggested that montelukast enhanced the cytotoxicity of carfilzomib in both carfilzomib-sensitive and carfilzomib-resistant MM cell lines. These findings may facilitate the development of therapeutic strategies and provide a promising therapeutic combination regimen for the treatment of refractory myeloma.
Keywords: Montelukast, Cytocidal effects, Carfilzomib, Multiple myeloma, mTOR pathway
Introduction
Multiple myeloma (MM) is characterized by clonal proliferation of malignant plasma cells in the systemic bone marrow microenvironment and associated with a poor prognosis. According to the latest statistics, the incidence of MM is followed by lymphoma and leukemia, ranking third in the hematological system malignant tumors.1 The application of new therapeutic regimens has prolonged the survival time of patients suffering from MM in the past decade.2 Recently, carfilzomib, a second-generation proteasome inhibitor (PI), is used for the treatment of patients suffering from relapsed/refractory MM, which showed a high selectivity for the proteasome and induced a lower rate of neurotoxicity compared to bortezomib.3,4 However, there is no distinct difference in the survival time of relapsed/refractory MM patients between those who received carfilzomib monotherapy and those who accepted full supportive therapies in the phase 3 study.5 For these reasons, the currently available therapies require further improvement.
According to Chng WJ et al, c-Myc activation may be a common transforming event in myeloma. This activation may be mediated by multiple mechanisms, and tumors with c-Myc activation have a more aggressive course associated with shorter survival.6 However, the c-Myc-targeted therapies have not been successfully developed for the treatment of tumors.
Montelukast, a cysteinyl-leukotriene receptor (CysLTR) antagonist, has long been used in the treatment of asthma. Usage of old drugs in an innovative way is attracting the attention of researchers currently in the development of anticancer drugs. Previous studies have reported that montelukast showed its activity against the growth of colon cancer and reversal of chemoresistance.7,8 In the present study, our study results have implicated cytocidal effects with the use of montelukast in MM cells lines, where c-Myc was decreased in a dose- and time-dependent manner.
The mammalian target of rapamycin (mTOR) signaling pathway controls cell growth and proliferation by regulating protein synthesis through phosphorylation of downstream effectors including ribosomal p70S6 kinase (p70S6K) and eukaryotic initiation factor 4E binding protein 1 (4EBP1).9–12 Activated mTOR signaling pathway is regarded as an essential pathway associated with disease progression13,14 and drug resistance of MM.15 Furthermore, it has been reported that the proteasome system regulates the intracellular pool of amino acids,16,17 and participates in the activation of mTOR.18,19 Previous studies have shown that PI3K inhibitor combined with carfilzomib reduced the translation of c-Myc via the inhibition of mTOR pathway.20
Herein, we showed that the combination of montelukast and carfilzomib inhibited the growth and caused apoptosis of both carfilzomib-sensitive and carfilzomib-resistant MM cell line. Furthermore, the combined cytocidal effects of montelukast and carfilzomib were detected in CD138+ plasma cells from primary and relapsed patients, and in xenograft mouse models. Our findings suggested that montelukast may act as an adjunctive therapy to enhance the cytotoxicity of carfilzomib in suppressing the growth and inducing apoptosis of MM cells.
Materials and methods
Cell lines, cell culture and reagents
Myeloma cells were cultured in a CO2 incubator at 5% CO2, suspended in RPMI 1640 medium (HyClone, Logan, UT), and then added 10% fetal bovine serum (Sigma-Aldrich, St.Louis, MO), 100 μg/ml streptomycin and 100 U/ml penicillin (Invitrogen). Carfilzomib-resistant cells were sieved and induced by ongoing treatment. The dose of carfilzomib was gradually enhanced to 50 nM, and was maintained up to 3 months.
The agents were purchased from Selleckchem (Houston, TX, USA). Antibodies including caspases 3, 8, and 9, PARP1, p-mTOR (Ser2448), mTOR, 4EBP1, p-4EBP1 (Ser 65), P70S6K, p-P70S6K (Thr389) and c-Myc were purchased from Cell Signaling Technology (Beverly, MA, USA). CysLTR antibody was purchased from abcam (Cambridge, UK).
CD138+primary MM cells, BMSCs, and PBMCs
Patients and healthy volunteers were informed to sign the informed consent forms before sample collection. The study was conducted in accordance with the Declaration of Helsinki protocol, and the study has been approved by the Ethics Committee of Affiliated Rui-Jin Hospital of Shanghai Jiao-Tong University School of Medicine.
Mononuclear cells (MNCs) were isolated from the bone marrow specimens using Ficoll-Hypaque density gradient sedimentation (Pharmacia, Piscataway, NJ). CD138+ myeloma cells of the active MM patients were obtained from the bone marrow samples using EasyStep CD138+ microbeads (Stem Cell Technologies, Vancouver, Canada). Bone marrow MNCs extracted from the bone marrow samples were initially suspended in mesenchymal stem cell growth medium (MSCGM, Cambrex, Verviers, Belgium). After 3 days, the adherent cells were considered as human BMSCs. For co-culturing of BMSC-MM cells, BMSCs were seeded when near to 70% confluence in 6-well plates. Then, the MM cells were added into the complete growth medium and co-cultured with BMSCs for 24 h. PBMCs obtained from healthy volunteers were separated according to the above mentioned methods.
Calculation of the synergy index
Cells were incubated in 96-well plate (2 × 104cells/100μL/well for 6 replicates). Then combinations of different concentrations of agents were added. After 24h, 20 μl of Cell Counting kit-8 solution (Dojindo, Kumamoto, Japan) was added into each well and then incubated for 2 hours. Finally, the absorbance of every each well was measured at 450 nm using a microplate reader.
Cell apoptosis assays and DNA content assay
Cell apoptosis was detected by Annexin V/propidium iodide (PI) assay apoptosis detection kit (BD PharmingenTM). Cell cycle distribution was determined by DNA content assay. Cells were treated with montelukast for 16 h, fixed in 75% ethanol at −20 °C for 24 h, and then stained using 20μg/mL PI at 37 °C for 30 min. The cell populations were analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA).
Western blot and quantitative reverse transcription real-time PCR (qRT-PCR)
Western blot was conducted as described in the previous study21. Trizol (Invitrogen) was used to extract the total RNA from the agent-treated cells. The cDNA reverse transcription kit (TransGen Biotech, Beijing, China) was used to obtain cDNA. qRT-PCR was performed on 7900HT real-time PCR instrument (Applied Biosystems, CA, USA) using the SYBR Green Mix (Applied Biosystems). The PCR primers were as follows: human β-actin: 5ʹCATCCTCACCCTGAAGTACCC3ʹ and 5ʹGGCTCCTGGCAAAAGGTCA3ʹ and c-Myc: 5ʹCTGCGTAGTTGTGCTGATGT3ʹ and 5ʹ ATCATTTCCATGACGGCCTGT 3ʹ.
RNA interference, MYC overexpression and transfection
Pairs of complementary oligonucleotides against CysLTR and non-target control shRNA (NC) were synthesized by Sangon Biotech (Shanghai, China) and then annealed and ligated to the pGIPZ vector (Clontech Laboratories, Inc., Palo Alto, CA, USA). The MYC open reading frame was subcloned into the MIGR1 plasmid (Clontech Laboratories, Inc., Palo Alto, CA, USA). The two restriction enzyme sites are NcoI and Sal I.
The retroviruses produced in 293T cells were used to infect RPMI 8226 and U266 cells.
Xenograft mouse model
All animal studies were performed with the approval from the Institutional Animal Care and Use Committee of Affiliated Rui-Jin Hospital of Shanghai Jiao-Tong University School of Medicine.
The xenograft mouse model experiments included two types of female mice, BALB-c nu/nu and NOD-SCID, aged approximately 4–6 weeks. Each BALB-c nu/nu mouse was subcutaneously implanted with 1 × 107 MM.1S cells into the right flank. Each NOD/SCID mice was implanted with RPMI 8226 cells (200μL cell suspension containing 1 × 107 cells and 100μL Matrigel (Corning, Bedford, MA, USA) in the right flank by hypodermic injection. When tumors reached the volume of 200-400mm3, mice were randomly divided into control, montelukast, carfilzomib or co-treatment groups.
Clonogenicity
Single-cell suspensions of RPMI 8226 cells were plated (1,0000 cells per well) in six-well plates with complete methylcellulose-based medium (Stem Cell Technologies, Vancouver, Canada) containing different concentrations of montelukast. The cells were grown in a humidified atmosphere of 95% air and 5% CO2 for 15 days.
Statistical analysis
Statistical differences between each group were calculated using one-way ANOVA followed by Tukey’s post hoc test. P ≤ 0.05 was considered to be statistically significant difference. Synergistic effects between montelukast and carfilzomib groups were calculated by CompuSyn software program (BioSoft, Milltown, NJ, USA) using isobologram analysis, and combination index (CI) <1.0 indicated that the two drugs has synergistic effects. All experiments were conducted thrice.
Results
Montelukast inhibits cell growth, induces cell cycle arrest, and causes apoptosis in human myeloma cells
To test the effect of montelukast on cell growth, RPMI 8226, U266, LP-1 and MM.1S cells were treated with different concentrations of montelukast for 48h. The results showed that montelukast strongly inhibited the viability of all myeloma cells in a dose- and time-dependent manner (Figure 1(a,b)). Also montelukast significantly inhibited the cell colony formation of RPMI 8226 cells (Figure 1(c)). Apoptosis often occurs as a consequence of cell cycle arrest,22 and montelukast could induce cell cycle arrest in G0/G1 phase both in U266 and RPMI 8226 cells (Figure 1(d)). To evaluate whether montelukast could induce apoptosis, RPMI 8226 and U266 cells were treated with montelukast as shown in Figure 1(e). The cleaved-PARP1 was increased, while the c-Myc was decreased in a dose- and time-dependent manner. We also determined the cytotoxicity of other CysLTR antagonists in MM cells. Results showed that neither the pranlukast nor the zafirlukast showed the effect on MM cell viability (Fig. S1A,B), while c-Myc level was not decreased in a dose-dependent manner (Fig. S1 C,D). Furthermore, knockdown studies have been conducted in RPMI 8226 and U266 cells. The proliferation of myeloma cells did not influence after knock down of CysLTR (Fig. S2A-D).
Figure 1.
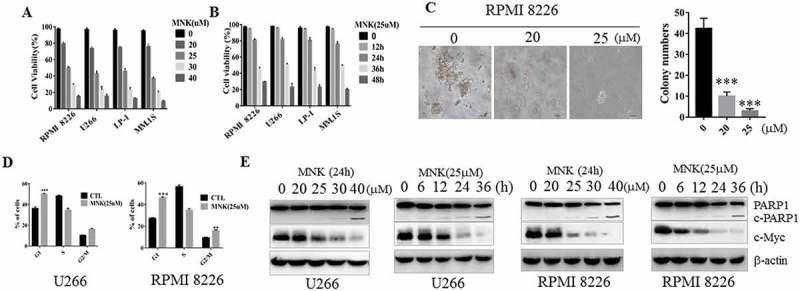
Montelukast induced cytotoxicity in human MM cell lines. (a) MM cell lines were treated with increasing concentrations of montelukast for 48 hours, or (b) montelukast (25μM) for 12, 24, 36, 48 hours, and the cell viability of MM cell lines were assessed by Beckman-Coulter cell counter. (c) Montelukast inhibited clonogenicity, as determined by colony formation assay. Colonies with diameters ≥0.1 mm were counted. 0 μM: 0.1% DMSO. (d) U266 and RPMI 8226 cells were incubated with montelukast (25μM) for 16 h and MM cells were arrested in the G0/G1 phase, RPMI 8226 was also arrested in the G2/M phase. The cell cycle distribution was determined by flow cytometry analysis of the DNA content. (e) U266 and RPMI 8226 were cultured with increasing doses of montelukast (15–40 μM) for 24 hours or montelukast (25μM) for 6, 12, 24, 36 hours and cell lysates were assessed by western blot for PARP1 and c-Myc. Data are presented as mean ± SD derived from 3 independent experiments. ***P ≤ 0.001 compared with control group. MNK: montelukast. c-PARP1: cleaved PARP1. CTL: control.
Montelukast potentiates cytotoxic effect induced by carfilzomib in myeloma cells lines and patient-derived myeloma cells
The CI values of montelukast with carfilzomib were <1, indicating that the synergy was presented between carfilzomib and montelukast (Tables 1 and 2). According to the CI values, RPMI 8226 and U266 cells were treated with montelukast alone or with carfilzomib in combination for 24h. As shown in Figure 2(a), co-treatment significantly increased Annexin V/PI+ cells when compared with montelukast or carfilzomib alone (P ≤ 0.001). Similar synergistic effects have been confirmed in LP-1 and MM.1S cells. Accordingly, the apoptotic-associated proteins, including the cleavage of PARP1, caspases-3, −8 and −9 were significantly activated in the combined treatment group in RPMI 8226 and U266 cells (Figure 2(b)). More importantly, we analyzed the tumoricidal effect of the combined treatment on newly diagnosed and relapsed myeloma samples. CD138+ plasma cells, including 4 newly diagnosed MM patients (Case 1, 2, IgG-λ subtype; Case 3, 4 IgA-λ subtype) and 3 relapsed MM patients (Case 5, 6, IgG-λ subtype; Case 7 IgA-λ subtype) were treated by montelukast and 10nM carfilzomib for 24h and the combination had significantly higher anti-tumor effects compared to each drug when treated alone (P ≤ 0.001) (Figure 2(c)). Meanwhile, no apoptotic evidence was found in PBMCs isolated from healthy volunteers (Figure 2(d)).
Table 1.
Montelukast has synergistic effect with carfizomib U266.
| MNK (μM) | CFZ (nM) | CI |
|---|---|---|
| 20.0 | 6.0 | 0.90689 + 0.10619 |
| 20.0 | 8.0 | 0.90363 + 0.15684 |
| 20.0 | 10.0 | 0.75439 + 0.14615 |
| 20.0 | 12.0 | 0.81819 + 0.12679 |
| 25.0 | 6.0 | 0.85210 + 0.10687 |
| 25.0 | 8.0 | 0.76484 + 0.14780 |
| 25.0 | 10.0 | 0.55163 + 0.11679 |
| 25.0 | 12.0 | 0.71626 + 0.13587 |
| 30.0 | 6.0 | 0.87361 + 0.10681 |
| 30.0 | 8.0 | 0.86799 + 0.13282 |
| 30.0 | 10.0 | 0.53618 + 0.12892 |
| 30.0 | 12.0 | 0.61214 + 0.11693 |
Table 2.
Montelukast has synergistic effect with carfizomib RPMI 8226.
| MNK (μM) | CFZ (nM) | CI |
|---|---|---|
| 20.0 | 6.0 | 0.84830 + 0.12610 |
| 20.0 | 8.0 | 0.80136 + 0.10416 |
| 20.0 | 10.0 | 0.70207 + 0.12519 |
| 20.0 | 12.0 | 0.82724 + 0.11617 |
| 25.0 | 6.0 | 0.75761 + 0.13167 |
| 25.0 | 8.0 | 0.59127 + 0.15611 |
| 25.0 | 10.0 | 0.64510 + 0.10613 |
| 25.0 | 12.0 | 0.69893 + 0.12519 |
| 30.0 | 6.0 | 0.74996 + 0.10716 |
| 30.0 | 8.0 | 0.59699 + 0.15418 |
| 30.0 | 10.0 | 0.66290 + 0.17613 |
| 30.0 | 12.0 | 0.64696 + 0.10601 |
Figure 2.
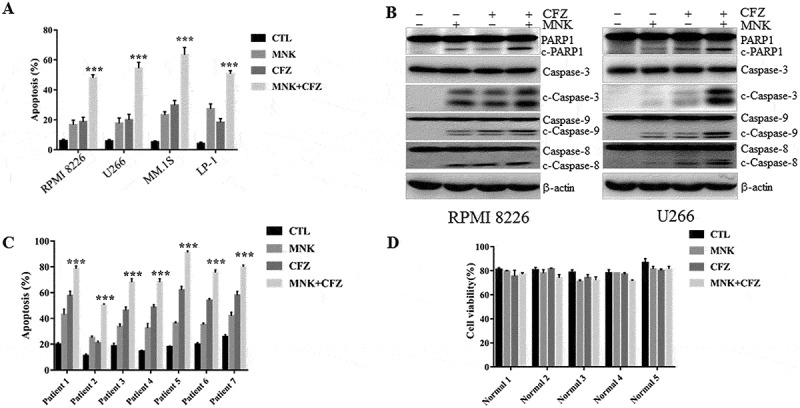
Montelukast potentiates cytotoxic effect induced by carfilzomib in myeloma cells lines and CD138+ plasma cells. (a) RPMI 8226, U266, MM.1S, and LP-1 were treated with montelukast (25 μM) or carfilzomib (10nM) separately or montelukast concurrently with carfilzomib for 24 h. (b) Cell lysates were assessed by western blot for PARP1, cleaved caspase 3, cleaved caspase 9, and cleaved caspase 8. (c) Patients’ mononuclear cells were separated by Ficoll-Hypaque density gradient sedimentation and CD138+ plasma cells were isolated and treated with 25μM of montelukast alone and/or 10nM of carfilzomib for 24 hours. (d) Freshly isolated PBMCs from 5 healthy donors were cultured with 25μM montelukast and/or 10 nM carfilzomib for 48 h. Cell apoptosis was assessed using flow cytometry by annexin V/PI. Data was presented as mean ± SD derived from 3 independent experiments. β-actin was used as a loading control. ***P ≤ 0.001 compared with other groups. PBMC: Peripheral Blood Mononuclear Cell. CTL: control. MNK: montelukast. CFZ: carfilzomib. c-PARP1: cleaved PARP1. c-caspases 3, 8, and 9: cleaved caspases 3, 8, and 9.
Montelukast in combination with carfilzomib has therapeutic efficacy in MM xenograft model
Next, we determined the tumoricidal ability of montelukast in combination with carfilzomib towards MM cells in vivo. BALB/c nu/nu female mice bearing subcutaneous xenografts of MM.1S cells were treated with montelukast (15mg/kg) daily and carfilzomib (2mg/kg) twice a week. The results showed significant reduction in tumor volumes in models with combined treatment, as compared with montelukast or carfilzomib treatment alone (P ≤ 0.01, Figure 3(a)). Furthermore, the mice did not exhibit weight loss (Figure 3(b)). As shown in Figure 3(c), immunostaining using anti-Ki67/anti-c-Myc Abs demonstrated that co-treatment could decrease proliferation in tumors, and increase the number of TUNEL-positive apoptotic tumor cells.
Figure 3.
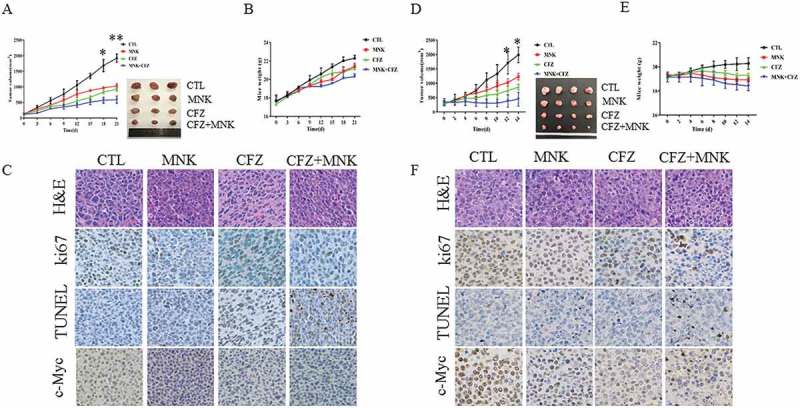
Montelukast and carfilzomib inhibited MM cell growth in vivo. BALB/c nu/nu mice bearing MM.1S tumors were treated with montelukast (15mg/kg; intraperitoneally) daily in the presence and/or absence of carfilzomib (2 mg/kg; intraperitoneally) for 21 days. Tumor volumes (a) and the weight of mice (b) were measured every 3 days. NOD/SCID mice bearing RPMI 8226 tumors were treated with montelukast (15 mg/kg; intraperitoneally) daily in the presence and/or absence of carfilzomib (2 mg/kg; intraperitoneally) for 14 days. Tumor volumes (d) and the weight of mice (e) were measured every 2 days. The tumor volumes were calculated using the following formula: V = (length × width2)/2. (c and f) Tumor sections from 4 groups were subjected to immunostaining using TUNEL and anti-Ki67/c-Myc Abs and demonstrated tumor cell apoptosis and proliferation. *P ≤ 0.05 compared with other groups. **P ≤ 0.001compared with other groups. MNK: montelukast. CFZ: carfilzomib. CTL: control.
The anti-tumor effects of the co-treatment were further demonstrated in RPMI 8226 cells xenograft model. As shown in Figure 3(d), the co-treatment further decreased the tumor volume compared with either single agent-treated animals (P ≤ 0.05). Furthermore, no significant differences were revealed in the weight of the mice among the four treatment groups (Figure 3(c)). Meanwhile, the expression of c-Myc, Ki67, and TUNEL detected by immunohistochemical staining also confirmed the aforementioned effects (Figure 3(f)). This suggested that the combined treatment significantly inhibited MM tumor proliferation and promoted apoptosis compared with monotherapy in vivo.
c-Myc is essential for the survival of MM, montelukast combined with carfilzomib decrease c-Myc expression via suppressing its protein synthesis
c-Myc remains to be the crucial regulator in cell proliferation, differentiation, cycle progression, and apoptosis23, 24. To determine whether the inhibition of c-Myc expression could induce apoptosis of myeloma cells, 10058-F4 (a c-Myc inhibitor) was used. Treatment with 10058-F4 induced apoptosis in MM cells (Fig. S2A). Overexpression c-Myc in RPMI 8226 cells promoted cell proliferation (Fig. S2B). Furthermore, we found that c-Myc was weakened in a time-dependent manner when RPMI 8226, U266 and CD138+ myeloma cells were treated with montelukast/carfilzomib alone or in combination (Figure 4(a-c)). Treatment with the two agents did not apparently reduce the c-Myc mRNA levels of MM cells lines (Figure 4(d)). Cycloheximide (CHX) was used to restrain the synthesis of new proteins in MM cells. However, the degradation rate of c-Myc was parallel either in the absence or presence of the two agents (Figure 4(e)). These data indicated that montelukast and carfilzomib more likely regulated c-Myc protein by inhibiting protein synthesis. Then, we subcloned the c-Myc open reading frame into MIGR1 plasmid, which has the Polio virus internal ribosome entry site (IRES). The results showed that the endogenous c-Myc in myeloma cells was reduced apparently, while the exogenous c-Myc was less influenced by montelukast and carfilzomib (Figure 4(f)). The apoptotic rate was decreased significantly in c-Myc overexpression samples compared with the empty vector samples after treatment with montelukast and carfilzomib for 24 h (Figure 4(g)).
Figure 4.
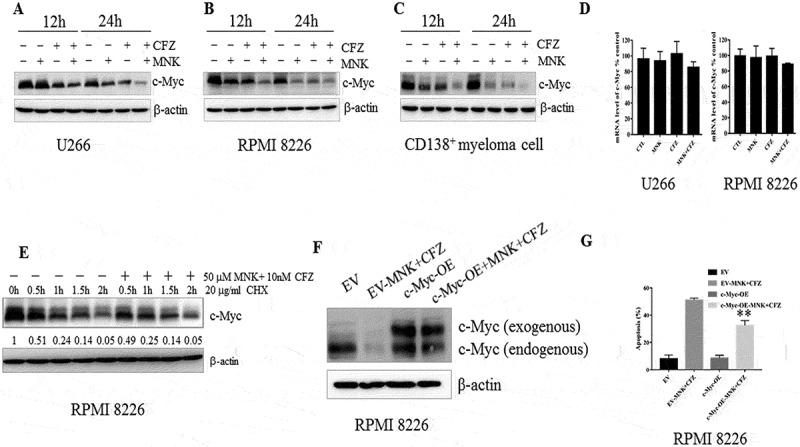
Montelukast and carfilzomib downregulated c-Myc protein levels. (a-c) U266, RPMI8226 and one case with CD138+ plasma cells were treated similarly as done in Figure 2A for 12 or 24 hours, and c-Myc protein levels were assessed by western blot. (d) U266, RPMI8226 cells were treated as described in Figure 2A for 24 hours, and c-Myc mRNA levels were assessed by q-RCR. Values represent the relative expression ratio of c-Myc to β-actin. (e) RPMI 8226 cells were pretreated with 10 μg/mL CHX for 0.5 hour, followed by DMSO or 25μM montelukast and 10 nM carfilzomib for 0.5, 1, 1.5, 2 hours, demonstrating a decrease in the protein level when the protein synthesis was blocked. (f) The exogenous c-Myc was less influenced after treatment with montelukast and carfilzomib for 24 hours. (g) RPMI 8226 with MYC+ plasmid was significantly more resistant than those cells with empty vector to the combination of montelukast and carfilzomib. CHX: cyclohexane. DMSO: dimethylsulfoxide. MNK: montelukast. CFZ: carfilzomib. CTL: control. EV: empty vector. OE: overexpression. **P ≤ 0.001compared with EV-MNK+ CFZ groups.
Montelukast potentiates carfilzomib cytotoxic effect in myeloma cells via suppression of mTOR pathway
As previously reported, mTOR signaling pathway plays a central role in the regulation of proliferation and growth in myeloma cells. Suppression of mTOR kinase, decreased the phosphorylation of mTOR, p70S6K and 4EBP1, leading to the suppression of c-Myc translation.23 To test this hypothesis, RPMI 8226 and U266 cells were treated with montelukast/carfilzomib alone or in combination for 24h, and the results revealed that p-mTOR (Ser2448), and its downstream signaling members namely, p-p70S6K (Thr489), p-4EBP1 (Ser 65) were obviously diminished (Figure 5(a,b)). Similar results were obtained in MM.1S cells (Fig. S3C).
Figure 5.
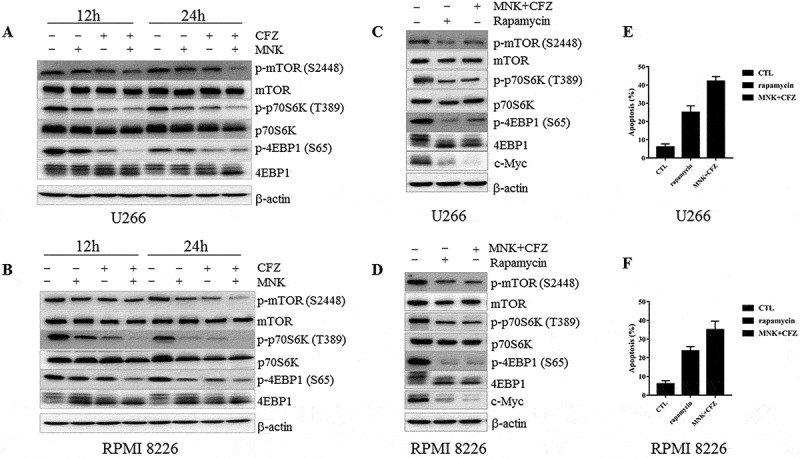
Montelukast potentiates the cytotoxic effect of carfilzomib in myeloma cells via suppression of mTOR pathway. (a-b) RPMI 8226 and U266 were treated as described in Figure 2A for 12 or 24 hours and with increased inhibition of mTOR pathway, and the total and phosphoprotein expression was evaluated using western blot. (c-d) RPMI 8226 and U266 were treated with rapamycin (20μM) or montelukast concurrently with carfilzomib, cell lysates were assessed by western blot in mTOR pathway after 12h, (e-f) and cell death was assessed using annexin V/PI staining after 24h. β-actin was used as a loading control. MNK: montelukast. CFZ: carfilzomib. CTL: control.
In order to further delineate the role of mTOR pathway, we treated the RPMI 8226 and U266 cells with montelukast concurrently and carfilzomib or rapamycin (mTOR inhibitor). Rapamycin inhibited the phosphorylation of mTOR, p70S6K and 4EBP1, leading to decreased c-Myc protein level and induced the apoptosis of myeloma cells, which was similar with the effects of the combination of montelukast and carfilzomib (Figure 5(c-f)). These data indicated that the addition of montelukast enhanced the pro-apoptotic effects of carfilzomib mostly via the inhibition of mTOR pathway in myeloma cells.
Apoptotic effects induced by montelukast and carfilzomib in carfilzomib-resistant cells and co-culture system of MM cells and BMSCs
To examine the cytocidal effects of montelukast and carfilzomib on carfilzomib-resistant cells, we used U266R and 8226R in our experiments (Figure 6(a)). U266R and 8226R cells were treated with montelukast and carfilzomib for 24h. Combined treatment significantly overcame the carfilzomib resistance (Figure 6(b)). Meanwhile, co-treatment increased the cleavage of PARP1, caspase-3, caspase-8 and caspase-9 (Figure 6(c,d)). We also analyzed the downstream proteins of mTOR pathway, and similar results were obtained with Figure 5(a,b) (Figure 6(e,f)). These findings suggested that the co-treatment overcame the carfilzomib resistance via suppression of mTOR signaling pathway.
Figure 6.
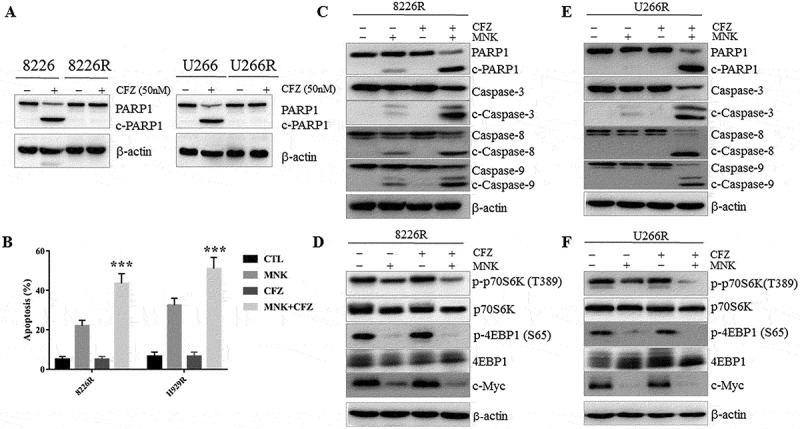
Montelukast and carfilzomib induced the pro-apoptotic effect in carfilzomib-resistant cells. (a) 8226R and U266R were treated with 50 nM carfilzomib for 24 hours and cell lysates were assessed for PARP1 by western blot. (b) Combined treatment significantly overcame carfilzomib resistance and increased apoptotic cells compared with montelukast alone treatment. (c-f) U266R and 8226R cells were treated with 30μM montelukast or 10nM carfilzomib or combination for 24 hours, and cell lysates were assessed by western blot for PARP1, cleaved caspase 3, cleaved caspase 9, cleaved caspase 8 and mTOR signaling pathway. MNK: montelukast. CFZ: carfilzomib. ***P ≤ 0.001 compared with other groups. CTL: control. c-caspases 3, 8, and 9: cleaved caspases 3, 8, and 9. c-PARP1: cleaved PARP1.
The contact between tumor cells and BMSCs could directly or indirectly activate the signaling pathways related with cell growth and upregulate anti-apoptotic proteins in the tumor cells.24 Our previous study has demonstrated that BMSCs could protect MM cells from bortezomib-induced apoptosis (Figure 7(a)). Therefore, we are interested to know whether the cytotoxicity of montelukast and carfilzomib on the myeloma cells was reduced in the presence of patient-derived BMSCs. The two agents demonstrated no cytotoxicity in BMSCs (Figure 7(b)). Notably, the apoptotic effect could be seen in the combination group even in the presence of BMSCs (Figure 7(c,d)). Furthermore, decreased p-4EBP1 and c-Myc were detected (Figure 7(e,f)). These results suggested that montelukast and carfilzomib could overcome the cytoprotective effects mediated by MM-BMSCs contact.
Figure 7.
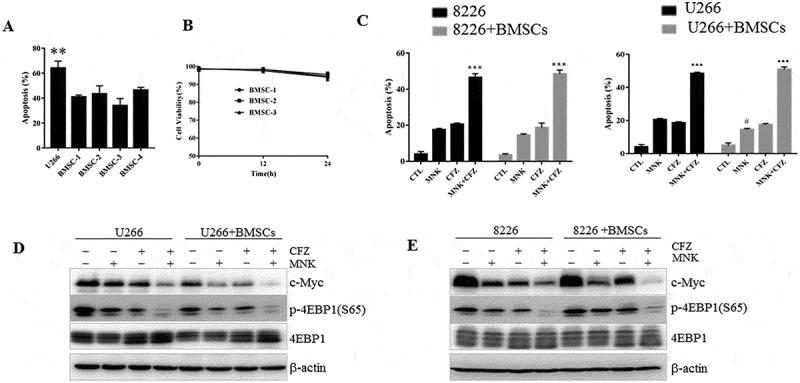
Cytotoxic effect induced by montelukast and carfilzomib in the co-culture system of MM cells and BMSCs. (a) U266 cultured with or without patient-derived BMSCs were exposed to bortezomib (2.5nM) for 24 hours and cell death was assessed using annexin V/PI staining. (b) Montelukast and carfilzomib had minimal effect on patient-derived BMSCs (BMSC 1–3) seeded at densities of 1 × 103 per 96-well plate as assessed by CCK-8 assay after treatment for 24 hours. (c-d) U266 and RPMI 8226 cells cultured with or without BMSCs were exposed to montelukast (25μM) or carfilzomib (10nM) separately or combination for 24 hours and cell death was assessed using annexin V/PI staining. (e-f) Cell lysates were assessed by western blot for p-4EBP1 and c-Myc. CCK-8: Cell Counting Kit-8. MNK: montelukast. CFZ: carfilzomib. **P ≤ 0.01 compared with other groups. ***P ≤ 0.001 compared with montelukast or carfilzomib mono-treatment groups. # P ≤ 0.05 compared with montelukast group without co-culture. CTL: control.
Discussion
Despite the advances in pathological mechanisms and therapeutic regimens, MM largely remains incurable with high mortality rate. PIs or immunomodulatory drugs (IMIDs) are the backbone of current MM treatment regimens. However, MM in patients is usually not curable and most patients suffer from palindromia after current treatment and have poor treatment outcomes.25,26 Currently, according to the statistics, the 10-year survival rate of MM patients is less than 20%.27–29 Consequently, new therapy strategies are urgently needed to boost the efficacy of therapies.
Previous research has shown that montelukast could inhibit the growth of colon cancer cells8 and overcome chemoresistance,7 and may be utilized as a potent anti-cancer drug in the clinic. Our study demonstrated that montelukast induced a dose- and time-dependent cytotoxicity in MM cells lines (RPMI 8226, U266, LP-1 and MM.1S) and significantly inhibited cell colony formation in RPMI 8226 cells. Recently, the second-generation PI, carfilzomib has superior clinical effects in the treatment of MM with a lower rate of neurotoxicity, compared with the first-generation PI bortezomib4,30 and is gradually merged into the MM first-line therapies and relapsed settings. In the current work, we demonstrated that montelukast enhanced the cytotoxicity of carfilzomib and overcame the carfilzomib-resistant in MM cells. The combined cytotoxicity effects were also obtained in CD138+ plasma cells separated from bone marrow of the primary and relapsed MM patients. Moreover, the co-culture of MM cells with BMSCs did not protect the MM cells from co-treatment-induced cell death. It is important to note that the combined treatment induced the tumor mass reduction in MM xenograft models, suggesting the combination of montelukast and carfilzomib as promising therapies.
Previous study has reported that montelukast reduced proliferation and induced apoptosis of tumor cells through the inhibition of CysLTR.8 However, the proliferation of myeloma cells was not influenced after knocking down CysLTR. We compared montelukast with the other two CysLTR antagonists (pranlukast and zafirlukast), and of the three drugs tested, montelukast was the most promising drug that could apply in the treatment of MM. In addition, our data demonstrated that the cytotoxic effect of montelukast occurred via suppression of mTOR pathway other than antagonizing CysLTR.
c-Myc is an essential regulator in cell proliferation, differentiation, cycle progression, and apoptosis.31,32 As known, c-Myc is aberrantly expressed in MM.33,34 Small molecule inhibitors targeting c-Myc showed a great potential in MM treatment.33,34 In the present study, we observed that the co-treatment of montelukast with carfilzomib produced a pro-apoptotic effect as demonstrated by the increase in the cleavage of PARP1, caspases-3, −8 and −9. Meanwhile, the co-treatment significantly downregulated the c-Myc expression and suppressed the mTOR pathway.
mTOR signaling pathway is an important modulator of MM cell tumor development, proliferation, and drug-resistance. Blocking of this signaling pathway could induce tumor regression and cell apoptosis, prolonging the survival time of MM patients.35 The function of p70S6K on mRNA translation is regulated by multiple elements.36 Moreover, 4EBP1 could negatively modulate the translation initiation factor eIF4E. It was released after the phosphorylation of 4EBP1, and consequently recruited to mRNA in the 40S ribosomal subunit.37 c-Myc expression is closely regulated by different links, such as mRNA transcription, protein translation and stability. In this study, the combination of carfilzomib and montelukast did not influence the transcription of c-Myc apparently. Meanwhile, the co-treatment did not accelerate the degradation of c-Myc after blocking the protein synthesis using CHX. To confirm that the c-Myc downregulation is a mechanistic target of the combinatorial treatment, we subcloned the c-Myc open reading frame into MIGR1 plasmid, which has the Polio virus IRES. Viral IRES-driven translation has a generally reduced the requirement for canonical translation initiation factors, particularly the members of the eIF4Fcomplex (initiation factors eIF4E and eIF4G).38 As a result, translation of exogenous c-Myc is expected to be less prone to inhibition by 4EBP1. According to the previous studies, 20 the c-Myc overexpression in myeloma cells could partly overcome the apoptosis induced by montelukast and carfilzomib. Our results suggested that montelukast and carfilzomib affected the protein synthesis of c-Myc via repressing the mTOR pathway.
In spite of greatly improved prognosis, MM remains incurable, and MM patients faced several difficulties of relapse and drug-resistant.39 Also the co-treatment showed similar anti-MM activity in 8226R and U266R cells. Then, we also analyzed the downstream proteins in mTOR signaling in carfilzomib-resistance cells, and obtained similar results as mentioned previously in the carfilzomib-sensitive MM cell lines. To examine whether these effects have clinical impact, after the co-treatment showed no toxic activity on PBMCs of healthy volunteers, we verified the pro-apoptotic effects of montelukast and carfilzomib in 4 newly diagnosed and 3 relapsed MM patients, indicating that the combined treatment could be an anti-MM regardless of carfilzomib sensitivity.
In the bone marrow microenvironment, BMSCs play a crucial role in supporting the growth and proliferation of abnormal cloned plasma cells. To determine whether the co-treatment of montelukast and carfilzomib could partly overcome the protective effects of the bone marrow microenvironment, MM cells were co-cultured with BMSCs directly and then treated with montelukast and carfilzomib alone or in combination. The results showed that the combination of montelukast and carfilzomib could overcome the cytoprotective effects mediated by the MM-BM microenvironment.
As a clinical medication, montelukast could be rapidly absorbed after oral administration. The mean peak plasma concentration (Cmax) was about 500–600 ng/ml, achieving within 3 to 4 h after the administration of 10mg montelukast. The mean area under the plasma concentration (AUC) and the half-life (t1/2) of montelukast were approximately 2.5 μg· h/mL, and 6–7 h, respectively.7,40,41 The Cmax achieved with the conventional dose was much lower than the drug concentration in our studies. However, previous clinic trials have demonstrated that oral administration of 900 mg montelukast for seven days showed no significant short-term toxicities in humans. Moreover, in our study, the additive effect of montelukast and carfilzomib was proved in two xenograft MM cells mice models and the weight of the mice did not change significantly after treatment with 15mg/kg montelukast for 21 days. No accumulation of toxicities was detected even with high doses, and meanwhile, with high AUC and long plasma t1/2, montelukast implicated its utility as an adjunctive therapy to anti-cancer clinically.
Conclusion
In conclusion, montelukast markedly enhanced the effect of carfilzomib in a synergistic or additive way. Indeed, treatment with montelukast and carfilzomib showed cooperative tumor cytotoxicity in both carfilzomib-sensitive and carfilzomib-resistant cell lines. Importantly, we demonstrated the therapeutic benefit of this novel combination both in vitro and in vivo. Taken together, these data supported the clinical development of montelukast as a potential therapy for myeloma and also suggested that co-treatment of montelukast and carfilzomib may provide significant clinical benefit for MM patients.
Funding Statement
This study was supported by the Shanghai Commission of Science and Technology (grant nos.16ZR1421400), the National Natural Science Foundation of China (grant nos. 8167010684).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the the publisher's website.
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Michels TC, Petersen KE. Multiple Myeloma: diagnosis and Treatment. Am Fam Physician. 2017;95:373–383. [PubMed] [Google Scholar]
- 3.Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, Habtemariam B, Bullock J, Bray JD, Hawes J, et al. U.s. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013;19:4559–4563. doi: 10.1158/1078-0432.CCR-13-0755. [DOI] [PubMed] [Google Scholar]
- 4.Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ, Muchamuel T, Bennett MK, Driessen C, Ball AJ, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res. 2011;17:2734–2743. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- 5.Hajek R, Bryce R, Ro S, Klencke B, Ludwig H. Design and rationale of FOCUS (PX-171-011): a randomized, open-label, phase 3 study of carfilzomib versus best supportive care regimen in patients with relapsed and refractory multiple myeloma (R/R MM). BMC Cancer. 2012;12:415. doi: 10.1186/1471-2407-12-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, Mulligan G, Chesi M, Bergsagel PL, Fonseca R. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25:1026–1035. doi: 10.1038/leu.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy U, Chakravarty G, Honer Zu Bentrup K, Mondal D. Montelukast is a potent and durable inhibitor of multidrug resistance protein 2-mediated efflux of taxol and saquinavir. Biol Pharm Bull. 2009;32:2002–2009. doi: 10.1248/bpb.32.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savari S, Liu M, Zhang Y, Sime W, Sjolander A. CysLT(1)R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS One. 2013;8:e73466. doi: 10.1371/journal.pone.0073466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem. 2007;282:24514–24524. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- 10.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 12.Fasolo A, Sessa C. mTOR inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2008;17:1717–1734. doi: 10.1517/13543784.17.11.1717. [DOI] [PubMed] [Google Scholar]
- 13.Maiso P, Liu Y, Morgan B, Azab AK, Ren P, Martin MB, Zhang Y, Liu Y, Sacco A, Ngo H, et al. Defining the role of TORC1/2 in multiple myeloma. Blood. 2011;118:6860–6870. doi: 10.1182/blood-2011-03-342394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci. 2015;36:124–135. doi: 10.1016/j.tips.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Tsubaki M, Takeda T, Ogawa N, Sakamoto K, Shimaoka H, Fujita A, Itoh T, Imano M, Ishizaka T, Satou T, et al. Overexpression of survivin via activation of ERK1/2, Akt, and NF-kappaB plays a central role in vincristine resistance in multiple myeloma cells. Leuk Res. 2015;39:445–452. doi: 10.1016/j.leukres.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science (80-). 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suraweera A, Munch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell. 2012;48:242–253. doi: 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Nicholatos J, Jr D, Sj R, Sb W, Gs H, Dj K, Bd M. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513:440–443. doi: 10.1038/nature13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng C, Lipstein MR, Scotto L, Jirau Serrano XO, Mangone MA, Li S, Vendome J, Hao Y, Xu X, Deng SX, et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kdelta and CK1epsilon in hematological malignancies. Blood. 2017;129:88–99. doi: 10.1182/blood-2016-08-731240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang RF, Wang Y, Zhang N, Xu WB, Cao Y, Tong J, Li JM, Wu YL, Yan H. MK2206 enhances the cytocidal effects of bufalin in multiple myeloma by inhibiting the AKT/mTOR pathway. Cell Death Dis. 2017;8:e2776. doi: 10.1038/cddis.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evan GI, Brown L, Whyte M, Harrington E. Apoptosis and the cell cycle. Curr Opin Cell Biol. 1995;7:825–834. [DOI] [PubMed] [Google Scholar]
- 23.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004;279:2737–2746. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 24.Can A, Balci D. Isolation, culture, and characterization of human umbilical cord stroma-derived mesenchymal stem cells. Methods Mol Biol. 2011;698:51–62. [DOI] [PubMed] [Google Scholar]
- 25.Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J, Haessler J, Feather J, Hoering A, Moreau P, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012;26:149–157. doi: 10.1038/leu.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nooka AK, Kastritis E, Dimopoulos MA, Lonial S. Treatment options for relapsed and refractory multiple myeloma. Blood. 2015;125:3085–3099. doi: 10.1182/blood-2014-11-568923. [DOI] [PubMed] [Google Scholar]
- 27.Bianchi G, Richardson PG, Anderson KC. Promising therapies in multiple myeloma. Blood. 2015;126:300–310. doi: 10.1182/blood-2015-03-575365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Avigan D, Rosenblatt J. Current treatment for multiple myeloma. N Engl J Med. 2014;371:961–962. doi: 10.1056/NEJMe1407442. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi G, Anderson KC. Understanding biology to tackle the disease: multiple myeloma from bench to bedside, and back. CA Cancer J Clin. 2014;64:422–444. doi: 10.3322/caac.21252. [DOI] [PubMed] [Google Scholar]
- 30.Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hajek R, Facon T, Ludwig H, Oriol A, Goldschmidt H, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17:27–38. doi: 10.1016/S1470-2045(15)00464-7. [DOI] [PubMed] [Google Scholar]
- 31.Zhou M, Gu L, Zhu N, Woods WG, Findley HW. Transfection of a dominant-negative mutant NF-kB inhibitor (IkBm) represses p53-dependent apoptosis in acute lymphoblastic leukemia cells: interaction of IkBm and p53. Oncogene. 2003;22:8137–8144. doi: 10.1038/sj.onc.1206911. [DOI] [PubMed] [Google Scholar]
- 32.Jacquel A, Colosetti P, Grosso S, Belhacene N, Puissant A, Marchetti S, Breittmayer JP, Auberger P. Apoptosis and erythroid differentiation triggered by Bcr-Abl inhibitors in CML cell lines are fully distinguishable processes that exhibit different sensitivity to caspase inhibition. Oncogene. 2007;26:2445–2458. doi: 10.1038/sj.onc.1210034. [DOI] [PubMed] [Google Scholar]
- 33.Holien T, Sundan A. Oncogene addiction to c-MYC in myeloma cells. Oncotarget. 2012;3:739–740. doi: 10.18632/oncotarget.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holien T, Vatsveen TK, Hella H, Waage A, Sundan A. Addiction to c-MYC in multiple myeloma. Blood. 2012;120:2450–2453. doi: 10.1182/blood-2011-08-371567. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Zhu J, Cao B, Mao X. The mTOR signaling pathway is an emerging therapeutic target in multiple myeloma. Curr Pharm Des. 2014;20:125–135. [DOI] [PubMed] [Google Scholar]
- 36.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 37.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komar AA, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle. 2011;10:229–240. doi: 10.4161/cc.10.2.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust JA, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng H, Leff JA, Amin R, Gertz BJ, De Smet M, Noonan N, Rogers JD, Malbecq W, Meisner D, Somers G. Pharmacokinetics, bioavailability, and safety of montelukast sodium (MK-0476) in healthy males and females. Pharm Res. 1996;13:445–448. [DOI] [PubMed] [Google Scholar]
- 41.Zhao JJ, Rogers JD, Holland SD, Larson P, Amin RD, Haesen R, Freeman A, Seiberling M, Merz M, Cheng H. Pharmacokinetics and bioavailability of montelukast sodium (MK-0476) in healthy young and elderly volunteers. Biopharm Drug Dispos. 1997;18:769–777. doi:. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.