

Abstract
MicroRNAs (miRNAs) have been reported to play significant roles in the pathogenesis of various polyQ diseases. This study aims to investigate the regulation of ATXN3 gene expression by miRNA. We found that miR-25 reduced both wild-type and polyQ-expanded mutant ataxin-3 protein levels by interacting with the 3’UTR of ATXN3 mRNA. miR-25 also increased cell viability, decreased early apoptosis, and downregulated the accumulation of mutant ataxin-3 protein aggregates in SCA3/ MJD cells. These novel results shed light on the potential role of miR-25 in the pathogenesis of SCA3/MJD, and provide a possible therapeutic intervention for this disorder.
Keywords: Spinocerebellar ataxia type 3/Machado-Joseph disease, ATXN3, Cytotoxicity, Neuronal intranuclear inclusion, miR-25, Gene expression regulation
1. Introduction
Spinocerebellar ataxia type 3/Machado-Joseph disease (SCA3/ MJD), the most common type of inherited spinocerebellar ataxia worldwide, is a polyglutamine (polyQ) disease; these neurodegenerative illnesses are caused by glutamine-encoding CAG nucleotide expansions within endogenous human genes. In healthy individuals, the wild-type ataxin-3 protein (encoded by the wild-type ATXN3 gene) has between 12 and 40 CAG repeats, and results in no cellular toxicity. However, in SCA3/MJD, the polyQ-expanded ATXN3 gene encodes between 51 and 86 CAG repeats (http://neuromuscular.wustl.edu/index.html), resulting in an abnormal polyQ tract in the polyQ-expanded mutant ataxin-3 protein [1–6]. This protein, in turn, accumulates microscopically discernible aggrega tions—called neuronal intranuclear inclusions (NII)—in the nucleus and adjacent areas of the affected neurons, and can exacerbate cell death [2,7–9]. Downregulation of toxic ataxin-3 protein levels prevents the progression of SCA3/MJD [10–12].
MicroRNAs (miRNAs) are a group of endogenous non-coding RNAs that play a critical role in metabolism, neurodevelopment, neuroplasticity, apoptosis, and other fundamental neurobiological processes and diseases [13,14]. Recent studies have suggested that miRNAs are a significant player in mediating the pathogenesis of various polyQ diseases, including spinocerebellar ataxia, SBMA and Huntington’s disease [15–19]. For instance, in a human cell model of SCA1, miR-19, miR-101, and miR-130 were shown to directly target ATXN1 mRNA expression; in contrast, mutation of the target sites of miR-101 and miR-130 within the 3’UTR exacerbated ATXN1 cytotoxicity [20]. Moreover, miR-150 levels in SCA1 Purkinje neurons were found to modulate disease pathogenesis by targeting the 3’UTR of Rgs8 and Vegfa [21]. Another study found that miR-29a and miR-29b were downregulated in a cellular model of SCA17, and that miR-29a/b expression was inversely correlated with BACE1 expression [22]. In SCA3/MJD, the miRNA bantam was found to be a potent modulator of ataxin-3 cytotoxicity in Drosophila, and eliminating miRNAs via mutation in Dicer led to enhanced polyQ-mediated toxicity [23]. In a SCA3/MJD mouse model, anti- ATXN3 miRNA mimics effectively suppressed human ATXN3 expression and cleared the abnormal nuclear accumulation of mutant ataxin-3 throughout the cerebellum [24].
A previous study from our laboratory reported four dysregulated miRNAs in SCA3/MJD patients: miR-25, miR-125b, miR-29a, and miR-34b [25]. Building on this work, we hypothesized that miRNA-mediated regulation of ATXN3 might also modulate SCA3/ MJD neuropathology via previously unknown mechanisms. The present study investigated the role of miRNAs in the regulation of SCA3/MJD.
2. Materials and methods
2.1. Plasmid and siATXN3 construction
For SCA3/MJD model cells, the ATXN3 cDNA in the plasmid, which was a truncated fragment of 68 CAG repeats, was inserted into the pEGFP-C1 vector (Clontech, Palo Alto, CA, USA) and the pcDNA3.1/myc-His(−)B vector (Invitrogen, Carlsbad, CA, USA), respectively. For the pmirGLO-ATXN3 3’UTR-WT vector (referred to as ATXN3 3’UTR-WT), the 1531 bp region of the human ATXN3 gene (NM_004993.5) containing the miR-25 binding site (259–266) was cloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Madison, WI, USA). For the pmir-GLO-ATXN3 3’UTR-MUT vector (referred to as ATXN3 3’UTR-MUT), the GTGCAATA sequence was mutated to CTCGATAA. For positive control in luciferase assay, the perfect miR-25 binding sites (The sequence was TCAGACCGAGACAAGTGCAATG) were inserted into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (referred to as positive control). The following were used as positive controls for siATXN3 sequences: siATXN3-WT: AGCAGCAGGGG- GACCTATC and siATXN3-MUT: AGCAGCAGCGGGACCTATC.
2.2. Cell cultures
HEK 293T cells (National Key Laboratory of Medical Genetics, Changsha, China) were routinely cultured in high glucose DMEM supplemented with 10% heat-inactivated fetal bovine serum, 100U/ml penicillin, and 100U/ml streptomycin (Gibco, Carlsbad, CA, USA) at 37 °C in a 5% CO2 incubator. By using lipofectamine 2000 (Invitrogen), HEK 293T cells transfected with pcDNA3.1/myc-His(−)B-ATXN3–68Q pcDNA3.1/myc-His(−)B-ATXN3–68Q.−3’UTR- WT, pEGFP-C1-ATXN3–68Q or pEGFP-C1-ATXN3–68Q-3’UTR-WT (referred to as pcSCA3/MJDtr-68Q, pcSCA3/MJDtr-68Q-UTR, pSCA3/MJDtr-68Q or pSCA3/MJDtr-68Q-UTR, respectively), were used to establish SCA3/MJD model cells, and also routinely cultured using the same methods as for HEK 293T cells (Supplement material). SH-SY5Y human neuroblastoma cells (SH-SY5Ycells) were cultured in DMEM/F-12 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin, 1% MEM non-essential amino acids, and 4 mM L-glutamine (Gibco) at 37 °C in a 5% CO2 incubator.
2.3. Quantitative real-time polymerase chain reaction (RT-qPCR) for mRNA
HEK 293T cells, SH-SY5Y cells, and SCA3/MJD model cells (pSCA3/MJDtr-68Q-UTR) were plated in six-well plates and transfected with miRNA mimics or siATXN3 (The final concentration of miRNA mimics and siATXN3 was 50 nmol/L in the whole experiment). Forty-eight hours later, total RNA was extracted using Tri zol reagent (Invitrogen). Reverse transcription was carried out in a 20 il reaction volume using RevertAid First Strand cDNA Synthesis Kit (Fermentas, Glen Burnie, Maryland, USA). Quantitative PCR was carried out in a 25 μl reaction volume containing SYBR® Premix Ex Taq™ (TaKaRa, Dalian, China) and 0.2 μM primers (ATXN3-F: GGCTCACTTTGTGC TCAACATTG; ATXN3-R: TCTCATCCTCTCCTCCT- CATCCAG) according to the manufacturer’s instructions. A BioRad thermal cycler was programmed for an initial denaturation step (95 °C, 30 s) followed by 40 amplification cycles (95 °C, 5 s; 60 °C, 30 s). β-Actin was used as an internal reference. Differences between control and experimental samples were calculated using the 2-ΔΔCt method.
2.4. Western blotting
The same cell types and transfection steps were used as in the RT-qPCR method described above. Forty-eight hours later, cells were harvested and lysed with 100 μl radio-immunoprecipitation assay (RIPA) buffer containing 1 × protease inhibitor cocktail on ice for 15min. The cell lysate was centrifuged at 13000xg for 20 min, and the supernatant was mixed with an equal volume of sample buffer. Proteins were separated by 10% sodium dodecyl sulfate- polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). After blocking with 5% non-fat milk at room temperature for one hour, the membrane was washed three times with Tris-Buffered Saline and Tween 20 (TBST), then incubated with diluted primary antibodies overnight at 4 °C, or for 2 h at room temperature with anti-ATXN3 (Millipore) at 1:10000, anti- GFP (Clontech, Mountain View, CA, USA) at 1:10000, or anti-β-actin (Sigma, St. Louis, MO, USA) at 1:10000. After washing three times with TBST, the membrane was incubated with diluted secondary antibody for one hour at room temperature with antimouse antibody at 1:10000. Protein bands were visualized using enhanced chemiluminescence (ECL) reagents (Thermo Fisher Scientific, Rochford, IL, USA) as recommended after three washes. The film was analyzed using ImageJ software. The stable transfection of pcSCA3/MJDtr-68Q model cells was also verified by Western Blot.
2.5. Dual luciferase assay
HEK 293T cells were co-transfected with miRNAs and pmirGLO- ATXN3-3’UTR-WT vector (or pmirGLO-ATXN3-3’UTR MUT vector) in a 24-well plate. Twenty-four hours after transfection, cells were harvested and lysed in passive lysis buffer (PLB; Promega, Madison, WI, USA) followed by the addition of 20 μl of Dual-Glo® Reagent. Ten minutes later, firefly luminescence was measured in a Glomax luminometer (Turner Designs, Sunnyvale, CA, USA). A volume of Dual-Glo® Stop & Glo® Reagent equal to the Dual-Glo® Reagent was added to each well, and Renilla luminescence was measured 10 min later. Firefly luciferase activity was normalized by Renilla luciferase activity, and this ratio was then normalized to that of a control well transfected with miRNA negative mimics. Finally, the relative response ratios were calculated from the normalized ratios.
2.6. Cell viability analysis using 3-(4,5-dimethylthiazol-2-yl)-2,5- dipheyltetrazolium bromide (MTT) assay and clonogenic assay
For the MTT assay [4], SCA3/MJD model cells established using the HEK 293T cell line (pcSCA3/MJDtr-68Qor pcSCA3/MJDtr-68Q- UTR) were cultured in 96-well plates and transfected with miRNA mimics or siATXN3-MUT. Cells were harvested at 0, 24, and 48 h after transfection. Each well received 100 μl of MTT solution (5 g/l; Sigma) and the supernatant was carefully removed four hours later. MTT crystals were dissolved in l00 μl dimethyl sulfoxide (DMSO; Sigma), and optical density was measured at the optimal absorbance of 562 nm using an enzyme-labeling instrument (Thermo Fisher Scientific). The background absorbance of the medium was subtracted.
Clonogenic assays were performed to determine the effect of miR-25 on model SCA3/MJD cell growth. As MTT, we harvested cells at 24 h after miRNA mimics or siATXN3-MUT transfection. Then cells were seeded in 6-well plates with 500 cells/well. Each well contained 1.5 mL DMEM medium containing 10% FBS. Colonies were counted 15 days later. The colonies were stained with Giemsa and quantified. Cloning efficiency = number of colonies/ number of seeded cells * 100%. Untreated control group was assigned a value of 100% growth.
Each assay was performed in triplicate, and the mean for each experiment was calculated.
2.7. Early cell apoptosis rate analysis by Flow cytometry (FCM)
For the FCM assay [5], SCA3/MJD model cells (pcSCA3/MJDtr-68Qor pcSCA3/MJDtr-68Q-UTR) were plated in 6 cm tissue culture dishes and transfected with miRNA mimics or siATXN3-MUT. Forty-eight hours later, cells were harvested and apoptotic cells were quantified by double staining with fluorescein isothiocyanate (FITC)-conjugated Annexin V and propidium iodide (PI) using Annexin V/PI apoptosis kit (Invitrogen) to label dead or damaged cells. Briefly, cells were washed with PBS twice and stained with Annexin V and PI for 15 min at room temperature in the dark. The level of apoptosis was determined by measuring the fluores cence of the cells with an automatic flow cytometer (Beckman Coulter FC500, Fullerton, CA, USA). Samples were immediately measured for fluorescence emission at an excitation wavelength of 488 nm. The number of cells used by was approximately 20000. Four cellular subpopulations were evaluated: vital cells (annexin V-/PI-), early apoptotic cells (annexin V+/PI-), late apoptotic cells (annexin V+/PI+), and necrotic/damaged cells (annexin V-/PI+). Annexin V+/PI- cells were considered to be early apoptotic cells.
2.8. Immunofluorescence assays and laser scanning confocal detection
For immunofluorescence, SCA3/MJD model cells (pSCA3/MJDtr-68Qor pSCA3/MJDtr-68Q-UTR) were plated in 24-well plates and transfected with miRNA mimics or siATXN3-MUT [4,5]. Forty-eight hours after transfection, cells were counted in random fields from three different quadrants of the culture well using an inverted microscope (Olympus IX-71, Melville, NY, USA). Cells were counted in fields selected at random from five different quadrants of the culture well. Counting was performed by an investigator blind to the experimental conditions. Also, the immunofluorescence density of aggregates was measured by ImageJ software. For laser scanning confocal detection, cells were fixed with 4% paraformaldehyde at 37 °C for 30 min and permeabilized with 0.1% Triton-X100 in PBS at 37 °C for 30 min. After incubation with 4’- 6-diamidino-2-phenylindole (DAPI, 5 mg/ml) for 15 min, samples were mounted in fluoromount medium (Sigma) and examined with a laser scanning confocal system installed on a confocal laser microscope (Leica TCS SP5, Heidelberg, Germany). The images were analyzed using the Metaphor software package.
2.9. Statistical analysis
Results were drawn from at least three independent experiments. All data are expressed as mean ± S.E.M. Statistical analyses were performed using SPSS 18.0 by one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant.
3. Results
3.1. miR-25 regulates ATXN3 expression
Using miRCURY™ LNA Array followed by RT-qPCR analysis, our previous study found that four miRNAs were dysregulated in SCA3/ MJD patients (miR-25, miR-125b, miR-29a, and miR-34b). Among the four miRNAs, miR-34b expression was significantly elevated (The fold change was 2.35), whereas the levels of miR-29a, miR- 25, and miR-125b were decreased (The fold change was 0.79, 0.69 and 0.56, respectively.) [25]. Because these four miRNAs appear relevant to the pathophysiology of neurological, neurode generative, and neuropsychiatric diseases (Table 1), the present study further explored the mechanisms underlying their involvement in regulating SCA3/MJD pathogenesis.
Table 1.
Four candidate miRNAs and related central nervous system (CNS) diseases.
| miRNAs | Related diseases |
|---|---|
| miR-29a | Spinocerebellar ataxia 17 [22], Alzheimer’s disease [26–28] |
| miR-125b | Spinocerebellar ataxia 1 [21], Alzheimer’s disease [29], Huntington’s disease [30] |
| miR-25 | Schizophrenia [31], autism spectrum disorders [32] |
| miR-34b | Huntington’s disease [19], Parkinson’s disease [33,34] |
To test the effects of miR-25, miR-29a, miR-125b, and miR-34b on ATXN3 gene expression, we transfected each miRNA mimic into HEK 293T cells, then measured wild-type endogenous ATXN3 mRNA and ataxin-3 protein. We used HEK 293T cells because our previous studies have found that polyQ expanded ATXN3 contribute to SCA3 pathogenesis in this cell line [4,5], and more importantly, because molecular mechanisms that contribute to SCA3 pathogenesis in the cerebellum have been reproduced in this cell line in terms of the toxic polyQ expanded ataxin-3 protein [35]. None of these miRNA mimics affected ATXN3 mRNA levels. However, miR-25 significantly decreased wild-type endogenous ataxin-3 protein levels; the other three miRNAs had no such effect (Fig. 1A and B). As a result, all additional experiments were carried out only on miR-25.
Fig. 1.
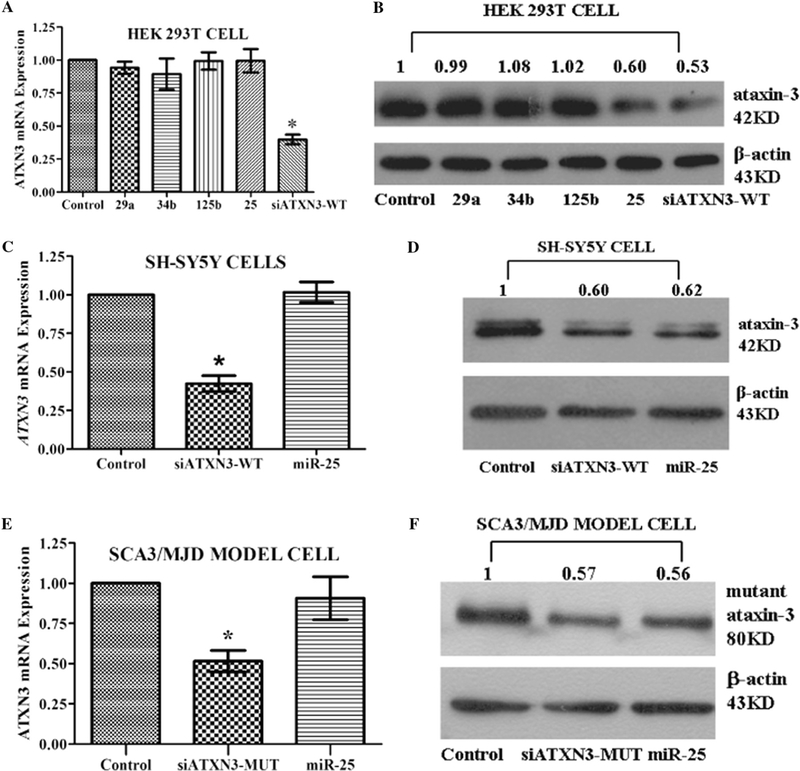
Regulation of ATXN3 gene expression by miR-25. (A, C, and E) Using RT-qPCR analysis, no difference was observed in levels of either wild-type or polyQ-expanded mutant ATXN3 mRNA expression in the miR-25 group. In contrast, siATXN3 acting as a positive control showed significant reductions in ATXN3 mRNA levels. (B, D, and F) miR-25 reduced levels of both wild-type and polyQ-expanded mutant ataxin-3 protein in three cell lines. Representative Western blot images and mean relative levels of ataxin-3 protein are shown (negative control = 1). Error bars represent S.E.M.*P < 0.05 compared to negative control. In Fig. 1A, control, 29a, 34b, 125b, 25, and siATXN3-WT refer to negative control, miR-29a mimics, miR-34b mimics, miR-125b mimics, miR-25 mimics, and siATXN3-WT mimics, respectively.
SH-SY5Y cells were used as a more physiologically relevant model because the ATXN3 gene is abundantly expressed in these neurons. Moreover, because SCA3/MJD is caused by the polyQ-expanded mutant ATXN3 gene rather than the wild-type ATXN3 gene, investigating the effect of miR-25 on the wild-type ATXN3 gene in SH- SY5Y cells and on the polyQ-expanded ATXN3 gene in SCA3/MJD model cells is of particular interest. As expected, no change in ATXN3 mRNA was observed. Ataxin-3 protein levels were markedly decreased in both SH-SY5Y(Fig. 1C and D) and SCA3/MJD model cells (Fig. 1E and F), which was consistent with the effects observed in HEK 293T cells.
3.2. miR-25 directly targets the 3’ UTR of ATXN3
miRNAs normally target the 3’UTR of mRNAs [36–39]. To understand the possible mechanisms of SCA3/MJD that miR-25 participated, we searched for potential gene targets of miR-25 using the bioinformatics algorithms, including miRBase, PicTar, TargetScan and miRanda. All of the algorithms identified ATXN3 as a target of miR-25, based on putative target sequences at position 259–266 of the ATXN3 3’ UTR (Fig. 2A). To verify the targets of miR-25 on the 3’ UTR of ATXN3, an ATXN3 3’UTR-WT (the 259–266 sequence was GTGCAAT) or an ATXN3 3’UTR-MUT (the 259–266 sequence was mutated to CTCGATA) was fused to the 3’ end of a luciferase gene (Fig. 2B). As shown in Fig. 2C, co-transfection of miR-25 mimics significantly suppressed the luciferase activity elicited by the luciferase-ATXN3 3’UTR-WT construct, which was similar to the positive control, but had no effect on the luciferase activity elicited by an ATXN3 3’UTR-MUT construct(Fig. 2C). These results suggest that miR-25 might regulate ATXN3 expression by targeting the specific 3’UTR of ATXN3 mRNA.
Fig. 2.
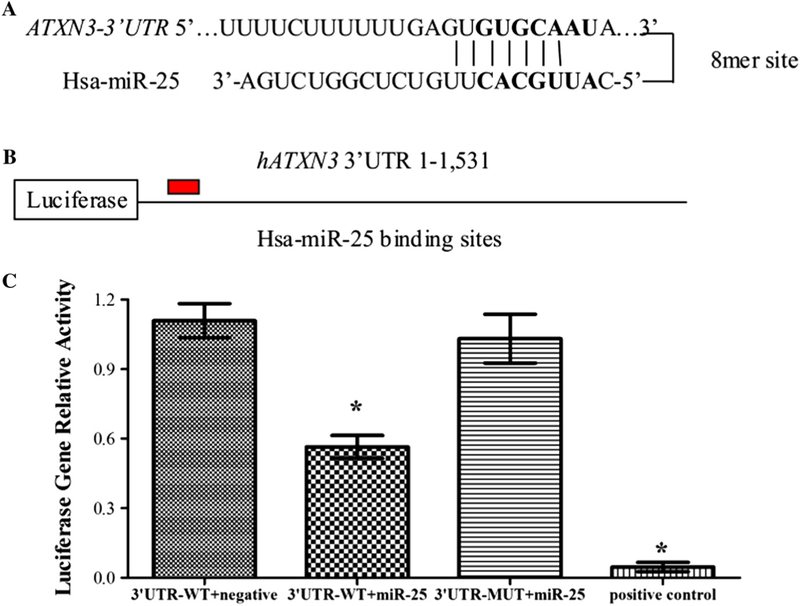
miR-25 directly targets the 3’ UTR of ATXN3. (A) The bioinformatics algorithms, including miRBase, PicTar, TargetScan and miRanda, identified the ATXN3 mRNA contains a miR-25 binding site at position 259–266 of the ATXN3 3’UTR. (B) The ATXN3 3’ UTR-WT (the 259–266 sequence was GTGCAATA) or ATXN3 3’ UTR-MUT (the 259–266 sequence was mutated to CTCGATAA) was fused to the 30 end of a luciferase gene. (C) Dual luciferase activity assays of co-transfections were performed using ATXN3 30 UTR-WT (with a partly miR-25 binding site), ATXN3 3’ UTR-MUT (with a mutated miR-25 binding site) or positive control (with a perfect miR-25 binding site) and miR-25 mimics. The results revealed that miR-25 mimics significantly reduced the expression of firefly luciferase when fused to the ATXN3 3’UTR-WT or positive control, but not the ATXN3 3’UTR-MUT. Error bars represent S.E.M.*P < 0.05 compared to the negative mimic control.
3.3. miR-25 increases cell viability in SCA3/MJD model cells
To examine whether miR-25 affects the cytotoxicity induced by polyQ-expanded mutant ataxin-3 protein, we transfected miR-25 mimics into SCA3/MJD model cells (pcSCA3/MJDtr-68Q-UTR), and measured cell viability via MTT and clonogenic assay. As shown in Fig. 3A, miR-25 significantly increased cell viability 48 h after transfection compared with the corresponding negative control; no significant effects were observed at 0 or 24 h. siATXN3-MUT transfection also significantly increased pcSCA3/MJDtr-68Q-UTR cell viability at 48 h. Further, miR-25 significantly increased colony formation of model SCA3/MJD cells compared with negative control group (Fig. 3B). Interestingly, a statistically significant increase in cell viability was observed between pcSCA3/MJDtr-68Q-UTR+ miR-25 and pcSCA3/MJDtr-68Q+miR-25, further confirming that the neuroprotective effects of miR-25 were caused by interactions between miR-25 and 3’UTR.
Fig. 3.
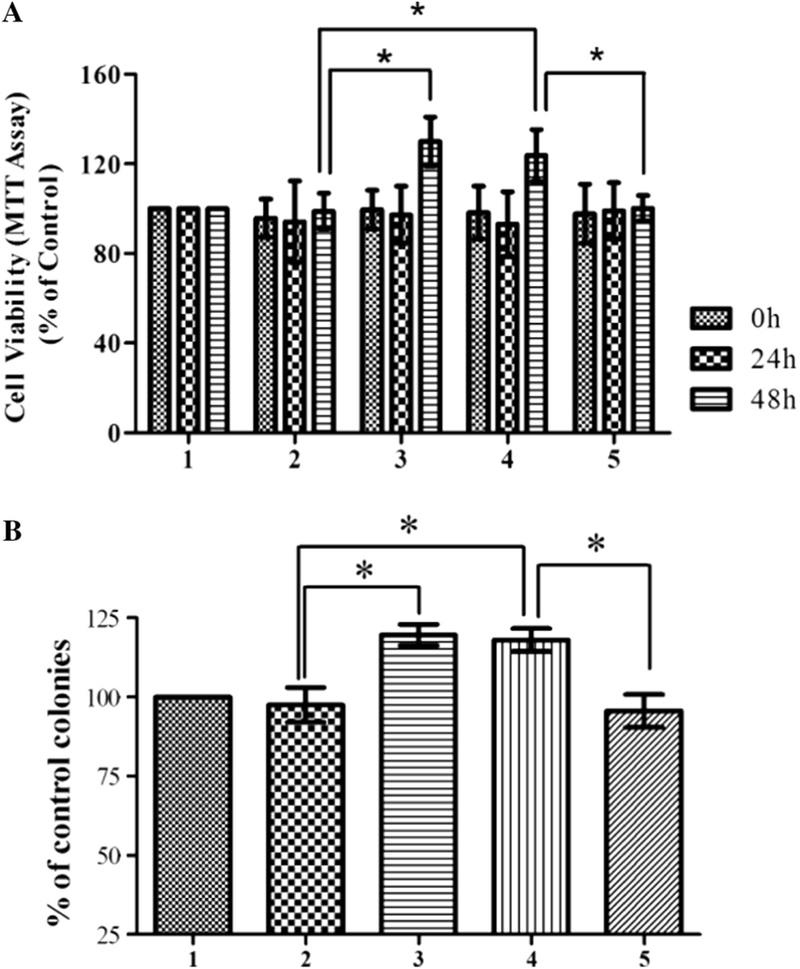
miR-25 increases cell viability in SCA3/MJD model cells. Statistical analysis of cell viability as measured by MTT assay of SCA3/MJD model cells untreated or treated with miR-25 mimics, siATXN3-MUT, and negative mimics at 0, 24, and 48 h.(A) miR-25 mimics significantly increased cell viability 48 h after transfection; no difference was detected at 0 or 24 h after transfection. As a positive control, siATXN3-MUT also significantly increased cell viability of SCA3/MJD model cells 48 h after transfection. (B) Significant increase in colony number was observed in miR-25 and siATXN3-MUT group. (A and B) Statistically higher cell viability wasalso observed in pSCA3tr-68Q-UTR+miR-25 versus pSCA3tr-68Q+miR-25. Error bars represent S.E.M. *P < 0.05 compared to the corresponding negative mimic control. Group 1: pcSCA3/MJDtr-68Q-UTR; group 2: pcSCA3/MJDtr-68Q-UTR+negative mimics; group 3: pcSCA3/MJDtr-68Q-UTR+siATXN3-MUT; group 4: pcSCA3/ MJDtr-68Q-UTR+miR-25 mimics; group 5: pcSCA3/MJDtr-68Q+miR-25 mimics.
3.4. miR-25 suppresses apoptosis in SCA3/MJD model cells
Although the molecular pathogenesis of SCA3/MJD remains unknown, considerable evidence suggests that apoptosis is at least partly involved in the neurodegenerative aspects of this disorder [40–42]. The stable transfection of pcSCA3/MJDtr-68Q model cells was verified by Western blot method (Fig. 4A). To assess the effects of miR-25 on apoptosis in SCA3/MJD model cells, flow cytometry assays were conducted at 24 h and 48 h after transfection with or without miR-25 mimics (Fig. 4B). As shown in Fig. 4C, transfection of miR-25 mimics significantly lowered early apoptotic rates in SCA3/MJD model cells at 48 h, but without 24 h. However, miR-25 failed to alter the early apoptotic rate in the stably transfected cells expressing the pcSCA3/MJDtr-68Q lacking the 3’UTR of ATXN3, again suggesting a specific role for 3’UTR of ATXN3 in mediating miR-25 function, Furthermore, late apoptotic rates both at 24 h and 48 h had no changes (Fig. 4D).
Fig. 4.
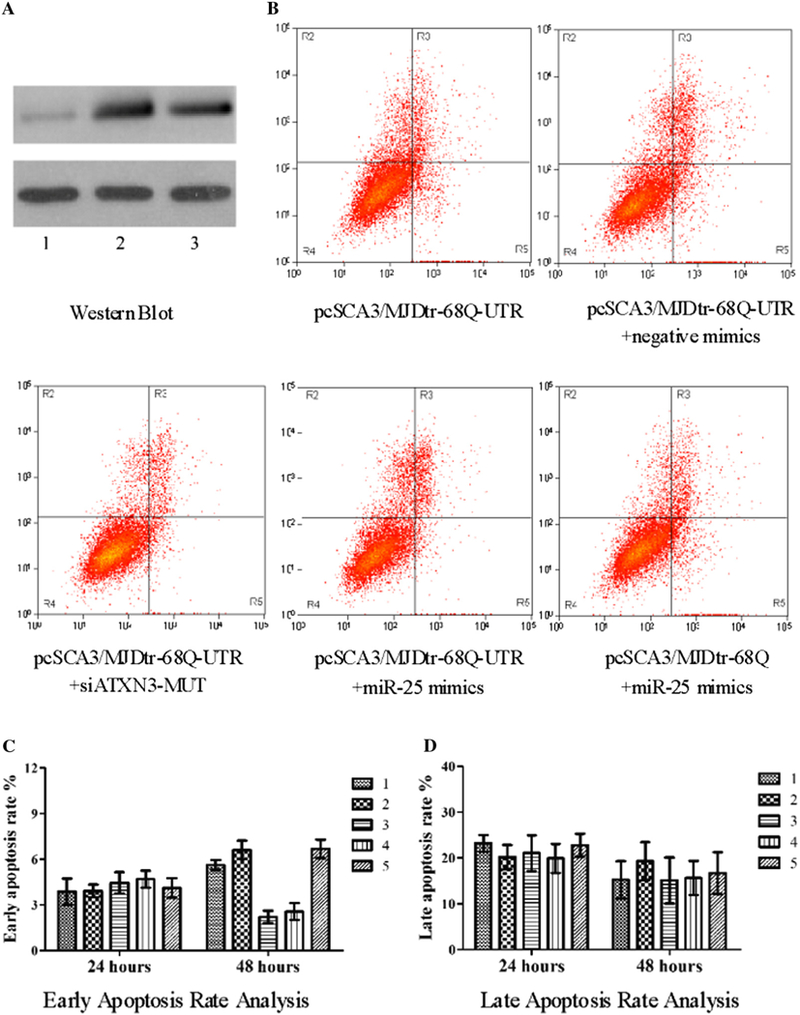
miR-25 suppresses apoptosis in SCA3/MJD model cells. (A) The stable transfection of pcSCA3/MJDtr-68Q model cells was verified by Western blot method (Fig. 4A). Group 1: pcDNA3.1/myc-His(−)B; group 2: pcSCA3/MJDtr-68Q-UTR; group 3: pcSCA3/MJDtr-68Q. (B) The upper right quadrant [Annexin V (+)/PI (+)] contained the late apoptotic and necrotic population, and the lower right quadrant [Annexin V (+)/PI (−)] contained the early apoptotic population in the flow cytometry assay. (C and D) Apoptosis was analyzed by flow cytometry analysis after staining with Annexin V and PI. miR-25 mimics significantly reduced the early apoptotic rate in SCA3/MJD model cells after 48 h, without affecting early apoptotic rate at 24 h and late apoptotic rate both at 24 h and 48 h. Data represent mean ± S.E.M.*P < 0.05 compared to the corresponding negative control. Group 1: pcSCA3/MJDtr-68Q-UTR; group 2: pcSCA3/MJDtr-68Q-UTR+negative mimics; group 3: pcSCA3/MJDtr-68Q-UTR+siATXN3-MUT; group 4: pcSCA3/MJDtr-68Q-UTR+miR-25 mimics; group 5: pcSCA3/MJDtr-68Q+miR-25 mimics.
3.5. miR-25 suppresses polyQ-expanded ataxin-3 protein aggregate formation
PolyQ-expanded mutant ataxin-3 protein has been found to be misfolded and aggregated in the nucleus and nearby loci, leading to accelerated cell death [4,7,8]. In order to expand our molecular analysis, GFP-tagged SCA3/MJD model cells (pSCA3/MJDtr-68Q or pSCA3/MJDtr-68Q-UTR) were used to detect mutant ataxin-3 protein aggregation. And we quantified aggregate formation cells and immunofluorescence density of aggregates by fluorescence imaging and imageJ computational analysis [4]. As predicted, miR-25 reduced the amount of mutant ataxin-3 protein aggregations in mutant ataxin-3-expressing cells (Fig. 5A). However, the immunofluorescence density of aggregates had no significant difference with or without miR-25 mimics (Fig. 5B). To locate the ataxin-3 protein aggregates, nuclei were stained by DAPI, and fluorescent microscopy was performed. Results showed the expression of ataxin-3–68Q-GFP protein (green) and DAPI stained nuclei (blue) in the same SCA3/MJD model cells. Diffused distribution of the mutant ataxin-3 protein was detected in the area adjacent to the nucleus (Fig. 5C).
Fig. 5.
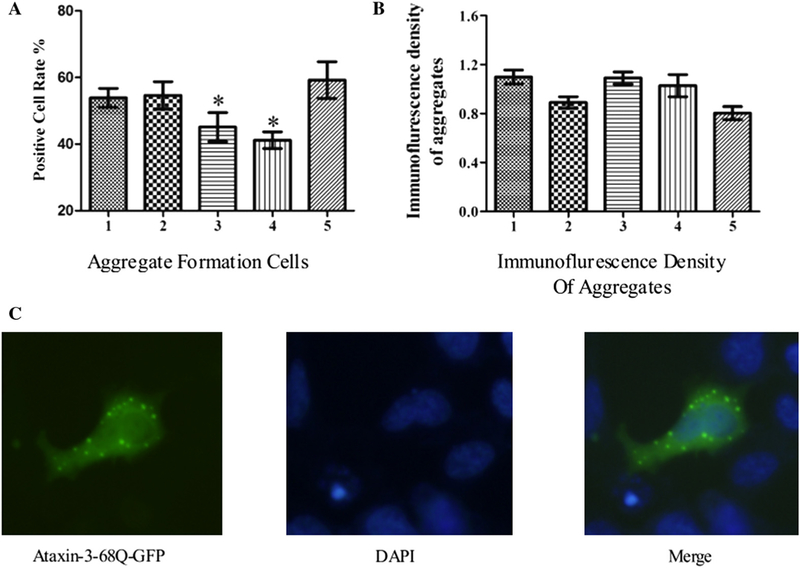
miR-25 suppresses aggregate formation of polyQ-expanded ataxin-3 protein. (A) Positive cell rate indicates the percentage of cells with NII in total cells. A statistically significant reduction in the amount of mutant ataxin-3 protein aggregates in siATXN3-MUT and miR-25 groups was observed. (B) miR-25 mimics did not affect the immunofluorescence density of aggregates. Error bars represent S.E.M. *P < 0.05. Group 1: pSCA3/MJDtr-68Q-UTR; group 2: pSCA3/MJDtr-68Q-UTR+negative mimics; group 3: pSCA3/MJDtr-68Q-UTR+siATXN3-MUT; group 4: pSCA3/MJDtr-68Q-UTR+miR-25 mimics; group 5: pSCA3/MJDtr-68Q+miR-25 mimics. (C) Laser scanning confocal detection showing the expression of ataxin-3–68Q-GFP protein (green) and DAPI stained nucleus (blue) in the same SCA3/MJD model cell. Significant diffuse staining of mutant ataxin-3 protein was observed in the perinuclear region.
4. Discussion
The current study investigated the role of miRNAs in the regulation of SCA3/MJD. We provide the first evidence that miR-25 overexpression led to reduced levels of endogenous wild-type ataxin-3 protein without affecting ATXN3 mRNA expression. The three other miRNAs investigated—miR-29a, miR-125b, miR-34b— all failed to affect endogenous wild-type ATXN3 expression. To further validate miR-25 function, we also transfected miR-25 mimics into SH-SY5Y cells, with similar results to those obtained in HEK 293T cells. These data suggest that miRNAs participate in the post-transcriptional regulation of gene expression, while siRNAs modulate gene expression through mRNA degradation. In SCA3/ MJD model cells, miR-25 decreased exogenous polyQ-expanded ataxin-3 protein expression without altering ATXN3 mRNA levels. Together, the data show that miR-25 levels correlated at least partially with ATXN3 gene expression.
No previous study has implicated miR-25 in polyQ diseases. Our results provide the first evidence that ATXN3 mRNA is a target for miR-25, which is consistent with the main regulatory mechanisms of miRNAs in other diseases such as SCA1 and Huntington’s Disease [20,43]. Using MTT and FCM assays in SCA3/MJD model cells, we also found that miR-25 substantially affected polyQ-mediated cytotoxicity, resulting in increased cell viability and decreased early apoptosis. miR-25 was found to reduce NII formation, which is mainly concentrated in the area adjacent to the nucleus (Fig. 5). These results echoed the effects of treatment with the mood stabilizers valproate and lithium in a Drosophila model of SCA3/MJD [5,12]. Taken together, the results implicate miR-25 as a possible therapeutic target candidate for SCA3/MJD, and suggest that miR-NA-mediated regulation of RNA metabolism has a potential role in treating SCA3/MJD and other polyQ diseases.
In summary, our study found that miR-25 reduced both endogenous wild-type and exogenous polyQ-expanded ataxin-3 protein expression levels in different cell types without affecting ATXN3 mRNA expression. We confirmed that miR-25 could directly target the 3’UTR of the ATXN3 gene and, further, found that miR-25 overexpression reduced polyQ-induced toxicity and NII formation in SCA3/MJD model cells. This identification of a common mechanism of miRNA-mediated neuroprotection or neurodegeneration not only enhances our understanding of the pathophysiology of SCA3/MJD, but also provides a promising prospect for its treatment. These novel results shed new light on the role of miR-25 in the pathogenesis of SCA3/MJD and suggest a possible new therapeutic strategy for treating this and other polyQ diseases.
Supplementary Material
Acknowledgements
The authors thank loline Henter and Peter Leeds of the NIMH, NIH, USA for excellent editorial assistance.
Role of funding source
This study was supported in part by the National Basic Research Program (973 Program) (Nos. 2012CB944601, 2012CB517902 and 2011CB510002 to Hong Jiang), the National Natural Science Foundation of China (Nos. 81410308019, 81471156, 81271260, 30971585 to Hong Jiang), Hunan Funds for Distinguished Young Scientists (No. 14JJ1008 to Hong Jiang), Xinjiang Natural Science Foundation (No. 201318101–4 to Hong Jiang), the Undergraduate Innovation Project of Central South University (No. YB13028 to Hong Jiang), High-level medical personnel of Hunan province “225” Project, and the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (IRP- NIMH-NIH). The funders had no further role in study design; in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016Zj.febslet.2014.11.013.
Footnotes
Conflict of interest
The authors have no competing personal or financial interests.
References
- [1].Lin X, Cummings CJ and Zoghbi HY (1999) Expanding our understanding of polyglutamine diseases through mouse models. Neuron 24 (3), 499–502. [DOI] [PubMed] [Google Scholar]
- [2].Bettencourt C and Lima M (2011) Machado-Joseph disease: from first descriptions to new perspectives. Orphanet J. Rare Dis, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, et al. (1997) Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19 (2), 333–344. [DOI] [PubMed] [Google Scholar]
- [4].Zhou YF, Liao SS, Luo YY,Tang JG, Wang JL, Lei LF, et al. (2013) SUMO- 1 modification on K166 of polyQ-expanded ataxin-3 strengthens its stability and increases its cytotoxicity. PLoS One 8 (1), e54214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yi J, Zhang L, Tang B, Han W, Zhou Y, Chen Z, et al. (2013) Sodium valproate alleviates neurodegeneration in SCA3/MJD via suppressing apoptosis and rescuing the hypoacetylation levels of histone H3 and H4. PLoS One 8 (1), e54792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Paulson H (2012) Machado-Joseph disease/spinocerebellar ataxia type 3. Handb. Clin. Neurol, 103437–103449. [Google Scholar]
- [7].Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, et al. (2007) A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 14 (4), 332–340. [DOI] [PubMed] [Google Scholar]
- [8].Bulone D, Masino L, Thomas DJ, San Biagio PL and Pastore A (2006) The interplay between PolyQ and protein context delays aggregation by forming a reservoir of protofibrils. PLoS One, 1e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Takahashi T, Katada S and Onodera O (2010) Polyglutamine diseases: where does toxicity come from? What is toxicity? Where are we going? J. Mol. Cell. Biol. 2 (4), 180–191. [DOI] [PubMed] [Google Scholar]
- [10].Boy J, Schmidt T, Wolburg H, Mack A, Nuber S, Bottcher M, et al. (2009) Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum. Mol. Genet. 18 (22), 4282–4295. [DOI] [PubMed] [Google Scholar]
- [11].Hu JX, Gagnon KT, Liu J, Watts JK, Syeda-Nawaz J, Bennett CF, et al. (2011) Allele-selective inhibition of ataxin-3 (ATX3) expression by antisense oligomers and duplex RNAs. Biol. Chem. 392 (4), 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jia DD, Zhang L, Chen Z, Wang CR, Huang FZ, Duan RH, et al. (2013) Lithium chloride alleviates neurodegeneration partly by inhibiting activity of GSK3beta in a SCA3 Drosophila model. Cerebellum 12 (6), 892–901. [DOI] [PubMed] [Google Scholar]
- [13].Aw S and Cohen SM (2012) Time is of the essence: microRNAs and age- associated neurodegeneration. Cell Res. 22 (8), 1218–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kosik KS and Krichevsky AM (2005) The elegance of the microRNAs: a neuronal perspective. Neuron 47 (6), 779–782. [DOI] [PubMed] [Google Scholar]
- [15].Persengiev S, Kondova I, Otting N, Koeppen AH and Bontrop RE (2011) Genome-wide analysis of miRNA expression reveals a potential role for miR- 144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol. Aging 32 (12), 2316.e17–2316.e27. [DOI] [PubMed] [Google Scholar]
- [16].Miyazaki Y, Adachi H, Katsuno M, Minamiyama M, Jiang YM, Huang Z, et al. (2012) Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat. Med. 18 (7), 1136–1141. [DOI] [PubMed] [Google Scholar]
- [17].Jovicic A, Zaldivar Jolissaint JF, Moser R, Silva Santos Mde F. and Luthi- Carter R. (2013) MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington’s disease-related mechanisms. PLoS One 8 (1), e54222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jin J, Cheng Y, Zhang Y, Wood W, Peng Q, Hutchison E, et al. (2012) Interrogation of brain miRNA and mRNA expression profiles reveals a molecular regulatory network that is perturbed by mutant huntingtin. J. Neurochem. 123 (4), 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gaughwin PM, Ciesla M, Lahiri N, Tabrizi SJ, Brundin P and Bjorkqvist M (2011) Hsa-miR-34b is a plasma-stable microRNA that is elevated in premanifest Huntington’s disease. Hum. Mol. Genet. 20 (11), 2225–2237. [DOI] [PubMed] [Google Scholar]
- [20].Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT and Zoghbi HY (2008) miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat. Neurosci. 11 (10), 1137–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rodriguez-Lebron E, Liu G, Keiser M, Behlke MA and Davidson BL (2013) Altered Purkinje cell miRNA expression and SCA1 pathogenesis. Neurobiol. Dis, 54456–54463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Roshan R, Ghosh T, Gadgil M and Pillai B (2012) Regulation of BACE1 by miR-29a/b in a cellular model of spinocerebellar ataxia 17. RNA Biol. 9 (6), 891–899. [DOI] [PubMed] [Google Scholar]
- [23].Bilen J, Liu N, Burnett BG, Pittman RN and Bonini NM (2006) MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol. Cell 24(1), 157–163. [DOI] [PubMed] [Google Scholar]
- [24].Rodriguez-Lebron E, do Carmo Costa M, Molina-Luna K, Peron TM, Fischer S, Boudreau RL, et al. (2013) Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shi Y, Huang F, Tang B, Li J, Wang J, Shen L, et al. (2014) MicroRNA profiling in the serums of SCA3/MJD patients. Int. J. Neurosci. 124 (2), 97–101. [DOI] [PubMed] [Google Scholar]
- [26].Shioya M, Obayashi S, Tabunoki H, Arima K, Saito Y, Ishida T, et al. (2010) Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 36 (4), 320–330. [DOI] [PubMed] [Google Scholar]
- [27].Bettens K, Brouwers N, Engelborghs S, Van Miegroet H, De Deyn PP, Theuns J, et al. (2009) APP and BACE1 miRNA genetic variability has no major role in risk for Alzheimer disease. Hum. Mutat. 30 (8), 1207–1213. [DOI] [PubMed] [Google Scholar]
- [28].Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. (2008) Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105 (17), 6415–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lukiw WJ and Alexandrov PN (2012) Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer’s disease (AD) brain. Mol. Neurobiol. 46(1), 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sinha M, Ghose J and Bhattarcharyya NP (2011) Micro RNA −214,−150,- 146a and-125b target Huntingtin gene. RNA Biol. 8 (6), 1005–1021. [DOI] [PubMed] [Google Scholar]
- [31].Santarelli DM, Beveridge NJ, Tooney PA and Cairns MJ (2011) Upregulation of dicer and microRNA expression in the dorsolateral prefrontal cortex Brodmann area 46 in schizophrenia. Biol. Psychiatry 69(2), 180–187. [DOI] [PubMed] [Google Scholar]
- [32].Sarachana T, Zhou R, Chen G, Manji HK and Hu VW (2010) Investigation of post-transcriptional gene regulatory networks associated with autism spectrum disorders by microRNA expression profiling of lymphoblastoid cell lines. Genome Med. 2 (4), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mouradian MM (2012) MicroRNAs in Parkinson’s disease. Neurobiol. Dis. 46(2), 279–284. [DOI] [PubMed] [Google Scholar]
- [34].Minones-Moyano E, Porta S, Escaramis G, Rabionet R, Iraola S, Kagerbauer B, et al. (2011) MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 20 (15), 3067–3078. [DOI] [PubMed] [Google Scholar]
- [35].Nguyen HP, Hubener J, Weber JJ, Grueninger S, Riess O and Weiss A (2013) Cerebellar soluble mutant ataxin-3 level decreases during disease progression in spinocerebellar ataxia type 3 mice. PLoS One 8 (4), e62043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136 (2), 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kosik KS (2006) The neuronal microRNA system. Nat. Rev. Neurosci. 7 (12), 911–920. [DOI] [PubMed] [Google Scholar]
- [38].Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 (2), 281–297. [DOI] [PubMed] [Google Scholar]
- [39].Agra Andrieu N, Motino O, Mayoral R, Llorente Izquierdo C, Fernandez- Alvarez A, Bosca L, et al. (2012) Cyclooxygenase-2 is a target of microRNA-16 in human hepatoma cells. PLoS One 7 (11), e50935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].U M., Miyashita T, Ohtsuka Y, Okamura-Oho Y, Shikama Y and Yamada M (2001) Extended polyglutamine selectively interacts with caspase-8 and - 10 in nuclear aggregates. Cell Death Differ. 8 (4), 377–386. [DOI] [PubMed] [Google Scholar]
- [41].Ikeda H, Yamaguchi M, Sugai S, Aze Y, Narumiya S and Kakizuka A (1996) Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 13 (2), 196–202. [DOI] [PubMed] [Google Scholar]
- [42].Berke SJ, Schmied FA, Brunt ER, Ellerby LM and Paulson HL (2004) Caspase-mediated proteolysis ofthe polyglutamine disease protein ataxin-3. J. Neurochem. 89 (4), 908–918. [DOI] [PubMed] [Google Scholar]
- [43].Kozlowska E, Krzyzosiak WJ and Koscianska E (2013) Regulation of huntingtin gene expression by miRNA-137, −214, −148a, and their respective isomiRs. Int. J. Mol. Sci 14 (8), 16999–17016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.