

Abstract
Next‐generation sequencing (NGS) has been instrumental in solving the genetic basis of rare inherited diseases, especially neurodevelopmental syndromes. However, functional workup is essential for precise phenotype definition and to understand the underlying disease mechanisms. Using whole exome (WES) and whole genome sequencing (WGS) in four independent families with hypotonia, neurodevelopmental delay, facial dysmorphism, loss of white matter, and thinning of the corpus callosum, we identified four previously unreported homozygous truncating PPP1R21 alleles: c.347delT p.(Ile116Lysfs*25), c.2170_2171insGGTA p.(Ile724Argfs*8), c.1607dupT p.(Leu536Phefs*7), c.2063delA p.(Lys688Serfs*26) and found that PPP1R21 was absent in fibroblasts of an affected individual, supporting the allele's loss of function effect. PPP1R21 function had not been studied except that a large scale affinity proteomics approach suggested an interaction with PIBF1 defective in Joubert syndrome. Our co‐immunoprecipitation studies did not confirm this but in contrast defined the localization of PPP1R21 to the early endosome. Consistent with the subcellular expression pattern and the clinical phenotype exhibiting features of storage diseases, we found patient fibroblasts exhibited a delay in clearance of transferrin‐488 while uptake was normal. In summary, we delineate a novel neurodevelopmental syndrome caused by biallelic PPP1R21 loss of function variants, and suggest a role of PPP1R21 within the endosomal sorting process or endosome maturation pathway.
Keywords: early endosome, endo‐lysosome, neurodevelopmental syndrome, PPP1R21, storage disease
1. BACKGROUND
Increased utilization of next‐generation sequencing (NGS), such as whole exome (WES) and whole genome sequencing (WGS) has enabled rapid expansion of our understanding of the genetic etiology of neurodevelopmental disease. Historically, diagnostic evaluation consisted of phenotype‐driven genetic testing; however, this approach has limited efficacy for common and genetically heterogeneous phenotypes like developmental delay. The increased utilization of NGS‐based testing, including exome analysis, has enabled a gene‐ and variant‐driven approach to diagnosis and has greatly accelerated new syndrome and new disease gene discovery (Boycott et al., 2017; Chong et al., 2015). Majority of the newly recognized disease genes have been identified in individuals with neurological disorders. The underlying molecular mechanisms for these disorders range from gene defects resulting in true brain malformations to causing degenerative disease. Neurological deficits are also frequently observed with metabolic disorders where defective lipid, mucopolysaccharide or glycoprotein processing results in a specific subgroup of disease entities referred to as (lysosomal) storage disorders. The common clinical features of lysosomal storage disorders include coarse facial features and skeletal abnormalities, enlargement of liver and spleen, neurodevelopmental delay (mainly manifesting as neurodegenerative phenotype), lung and heart disease as well as hearing and visual impairment. More than 50 disease entities have been described to date and although individually very rare, together they represent a significant disease burden affecting approximately 1:5,000 individuals (Fuller, Meikle, & Hopwood, 2006). Age of onset of symptoms varies from prenatally to adult age. Inheritance patterns are mostly autosomal‐recessive and rarely, X‐linked.
Lysosomes are the recycling centers of cells and degradation of macromolecules requires a number of enzyme‐driven steps within the endosomal–autophagic–lysosomal system. Mechanistically, functional deficits in degradation pathways cause accumulation of such material within lysosomes as well as within the cytosol with toxic effects, eventually resulting in cell death. Most defective genes encode for enzymes, however defects have also been identified in genes encoding for proteins involved in transport processes or transmembrane proteins (Bennett & Hofmann, 1999; Fuller et al., 2006; Parkinson‐Lawrence et al., 2010). Lysosomal degradation of cellular material can be accomplished via different pathways, including endosomal degradation where late endosomes fuse with lysosomes, including transient docking for content exchange (Platt, Boland, & van der Spoel, 2012). Endosome maturation into late endosomes occurs after approximately 5–10 min of fusion with novel endocytic vesicles (Maxfield, 2014). Early endosomes (EE) serve as a focal point of the endocytic pathway where internalized proteins and lipids are destined for recycling to the plasma membrane, for degradation in lysosomes or delivered to the trans‐Golgi network and functional deficits have been shown to play a role in a number of human diseases often demonstrating a neurological phenotype such as Alzheimer's disease, Down syndrome, Lowe syndrome, and Huntington's disease (Jovic, Sharma, Rahajeng, & Caplan, 2010).
Despite the progress made in recent years, substantial gaps remain in understanding the heterogeneity of clinical phenotypes of neurological disorders and the underlying molecular mechanisms. Here, we report on six patients from four unrelated consanguineous families with similar neurological phenotype. We detected homozygous truncating variants in PPP1R21 (Protein Phosphatase 1, regulatory subunit 21) in the affected individuals by WES or WGS. In parallel to our study, PPP1R21 loss of function alleles have been identified in three individuals with a similar syndromic neurodevelopmental phenotype, however no functional studies to understand the underlying patho‐mechanism and PPP1R21 protein function had been performed and PPP1R21 function had remained elusive. Here, we now suggest a role for PPP1R21 within the endo‐lysosomal compartment and propose PPP1R21 loss of function results in a mild disturbance of endosome function.
2. METHODS
2.1. Ethics statement and family ascertainment
The study was conducted in accordance with the protocol approved by the Institutional Review Board at the Baylor College of Medicine in Houston, Sidra Medicine in Qatar, University of Geneva in Switzerland, University of Netherlands, Royal Children's Hospital, Melbourne, Australia. Written informed consents were obtained from all live participating individuals and parents of the minors. Patients at Baylor Genetics laboratories in Baylor College of Medicine were referred by physicians for clinical exome sequencing. Patients in Nijmegen were included via the diagnostic route (innovative diagnostic program). Inclusion criteria used in the Nijmegen part of the study were patients with neurodevelopmental delay of likely genetic origin with exclusion of individuals in whom previously chromosomal aberrances were detected either by chromosome analysis or array‐CGH, proven intrauterine TORCH infections or perinatal asphyxia. Patients at University of Geneva were recruited during a research protocol that aimed to identify novel autosomal recessive genes in offspring of consanguineous families. Patients at the Royal Children's Hospital, Melbourne were recruited as part of the Australian Genomics Acute Care Flagship, which provides rapid genomic testing to infants and children admitted to intensive care units with suspected monogenic conditions.
2.2. Whole exome and whole genome sequencing and subsequent analyses
Peripheral blood samples were collected from available members of the four families and genomic DNA was extracted using standard protocol. WES was performed either at Baylor Genetics Laboratories using VCRome v3 in‐solution exome probes followed by sequencing on Illumina HiSeq 2500 instrument (Family 1, subject II‐6), using kit Agilent SureSelect Human All Exon v5 and sequencing on Illumina HiSeq 2500 at University of Geneva (Family 1, subject II‐5), using Agilent Sure select V6 with subsequent sequencing on a HiSeq machine at Novogene LTD Hong Kong for Nijmegen samples (Family 3, subject V:4) or using Agilent Sureselect QXT CREv1 kit, followed by sequencing on Illumina NextSeq500 at Victorian Clinical Genetics Services (Family 4). Further, OmniExpress 750K array was used in Family 2 for genome‐wide homozygosity mapping using DNA from six family members at Sidra Medical & Research Center in Qatar (Figure 1A) which showed three regions of homozygosity co‐segregating with the disease (Supp. Figure S1). Subsequently, DNA sample from individual II‐3 was subjected to WGS to a mean coverage of 30× on the Illumina HiSeq X platform and data was analyzed for potential pathogenic variants within the three regions of homozygosity.
Figure 1.
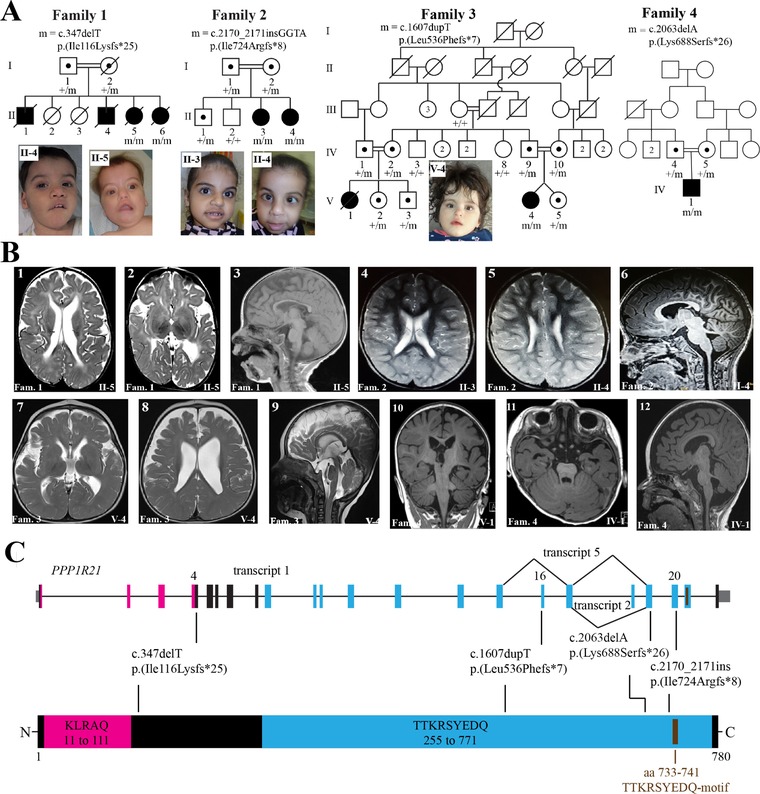
Biallelic pathogenic variants identified in the PPP1R21 gene. (A) Pedigrees and photographs of four families that co‐segregate homozygous truncating PPP1R21 variants with a neurodevelopmental phenotype. Filled squares and circles represent affected males and females, respectively. A diagonal line across a symbol means individual is deceased. Numbers within a symbol correspond to additional same‐sex siblings. PPP1R21 genotypes of available family members are shown under their symbols. m, mutant allele; +, reference allele. (B) MRI images of the brain of the subjects. Family numbers and individual IDs shown at the bottom of each image correspond to the pedigrees in panel A. Family 1: Image 1; axial T2‐w image showing an irregular outline to prominent bodies of both lateral ventricles with abnormal periventricular T2 hyperintensity returned from the deep white matter. Image 2; demonstrates generous extra‐axial CSF spaces with bilateral and symmetric underopercularization. Image 3; sagittal T1‐w image showing foreshortening and thinning of an otherwise intact corpus callosum. Family 2: Image 4; axial T2‐w image demonstrating slight prominence to the supratentorial ventricular system with increased T2 hyperintensity returned from the bilateral posterior centra semiovale. Image 5; demonstrates irregular outline to the bodies of both lateral ventricles in addition to the increased T2 hyperintensity returned from the periventricular white matter. Image 6; sagittal T1‐w midline image again demonstrating thinning and foreshortening to the body of the corpus callosum. Family 3: Image 7; axial T2‐w image demonstrates prominence to the bodies of the lateral ventricles with generous extra‐axial CSF spaces with bilateral and symmetric underopercularization. Image 8; axial T2‐w image shows a slightly irregular outline to the bodies of both slightly prominent lateral ventricles. Image 9; sagittal T2‐w midline image demonstrates thinning and foreshortening of the corpus callosum. Family 4: images 10–12 obtained from affected individual IV.1 showing abnormally deep and box‐like Sylvian fissures, diffuse reduction in cerebral volume, particularly the deep white matter signs of dysmyelination, diffuse reduction in the brainstem and vermian volume (molar tooth sign in image 11) and dysmorphic appearance of the corpus callosum and brainstem. (C) Schematic representation of the PPP1R21 gene, its predicted protein product, and locations of the pathogenic variants. Introns, exons, and untranslated regions of the PPP1R21 gene are shown by a horizontal line, vertical thick bars, and thin gray bars, respectively. Exon numbering and variant nomenclature is based on RefSeq accession number NM_ NM_001135629.2 for cDNA and NP_001129101.1 for protein
Sequencing data from each family was independently analyzed by respective diagnostic or research laboratories. Variants with >1% allele frequency in public SNP databases were removed from further analysis and the remaining variants were prioritized based on genetic evidence including variant rarity (e.g., novel variant), allele transmission (e.g., biallelic variants), predicted variant severity, and phenotype overlap between reported genetic disorders and the patient phenotypes. Possible causal variants in known disease genes that could explain patient's phenotype were not detected in any subject. To further explore the genetic basis of disease in these families, we investigated all rare deleterious variants that are in nondisease genes especially homozygous variants because of consanguinity in the families and apparently recessive mode of inheritance (Figure 1A). Follow up confirmation and co‐segregation analyses for putative pathogenic variants were performed using Sanger sequencing for families 1–3 following standard protocols. Primer sequences can be found in a supplemental table in the supplemental material. Identified PPP1R21 variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
2.3. Immunofluorescence and cell culture studies
Immunofluorescence and cell culture analyses were performed as previously described (Schmidts et al., 2015). Briefly, IMCD3, hTERT‐RPE, SH‐SY5Y cells and patient as well as control fibroblasts were cultured in DMEM‐F12 medium with 5% FBS under standard conditions. For ciliation experiments, cells were serum starved for 24 hr. Primary antibodies used: Golgi marker GM130 (mouse, ab169276, Abcam, USA), Golgi marker 58K (ab27043 mouse, Abcam, USA), early endosome marker EEA1 (1G11 ab70521, mouse monoclonal, Abcam), PPP1R21 antibody (rabbit, HHPA036792 (ab1) and HPA 036791 (ab2), Atlas antibodies, Sweden), acetylated tubulin (mouse monoclonal 6‐11‐B1, Sigma, USA). In brief, cells were grown on coverslips, washed with PBS and fixed 5 min in 5% PFA. After three washes, cells were permeabilized using 0.1% Triton for 3 min, washed three times and incubated in 5% BSA for 1 hr at RT to bock unspecific reactions. Incubation in primary antibodies was then performed for 1 hr (1:200 in 5% BSA), after five washes, cells were incubated in corresponding fluorescence tagged secondary 1:1,000 antibody dilutions for 30 min, washed five times in PBS and then mounted in Vectashield with DAPI. Images were taken with a Zeiss Apotome Axiovert 200 and processed with AxioVision 4.8 and Fiji software.
2.4. Transferrin assay
Patient fibroblasts and control fibroblasts from an age matched healthy individual were grown under standard conditions and serum starved for 3 hr before start of the experiment. Cells were incubated in medium containing transferrin‐Alexa‐Fluor‐488 conjugate 1:1,000 (Invitrogen/ThermoFisher, USA) and fixed in 4% PFA after 5, 10, and 30 min of incubation. Cells were then mounted in vectashield containing DAPI and images taken using a Zeiss Axiovert Apotome microscope. For washout/chase, cells were repetitively washed and re‐incubated in fresh medium containing unlabeled transferrin 1:200 every 20 min and fixed in 4% PFA after 1 and 2 hr washout/chase, mounted in vectashield containing DAPI and images taken using a Zeiss Axiovert Apotome microscope.
2.5. q‐PCR
PPP1R21 mRNA‐quantification in fibroblasts carrying PPP1R21 p.Leu536Phefs*7 in a homozygous manner versus fibroblasts from a healthy control individual was performed using total RNA and q‐PCR performed using SYBR‐Green (Qiagen, Germantown, USA) according to the manufacturer`s protocol. PPP1R21 levels were normalized to GAPDH levels. Statistical analysis was performed using Student's t‐test.
3. RESULTS
3.1. Clinical findings
Salient clinical features of six subjects from the four study families are provided in Table 1 and pedigrees and clinical images shown in Figure 1. Subject II‐6 from Family 1 was born at 33 weeks to a 33‐year‐old G6P6 mother and 44‐year‐old father. Her parents are from Syria and are first cousins. Prenatal ultrasound showed hyperechogenic bowel, hand, and skull anomalies and the neonatal period was complicated by respiratory distress at birth requiring surfactant application and 2 days of respiratory support. She had dysmorphic facial features, severe central hypotonia, poor suck, weak cry, minimal vision, and esotropia. She was noted to have feeding difficulties, constipation, distended abdomen, and small thoracic cage. Cardiac concerns included mild left pulmonary stenosis and secundum atrial septal defect (ASD). She had three affected siblings with similar clinical presentation who had died due to disease‐related complications. Brain MRI of subject II‐5 showed irregular outline to prominent bodies of both lateral ventricles with abnormal periventricular T2 hyperintensity returned from the deep white matter, generous extra‐axial CSF spaces with bilateral and symmetric underopercularization, and foreshortening and thinning of an otherwise intact corpus callosum (Figure 1B). Previous genetic testing included karyotyping for two affected siblings (II‐4 and II‐5) and comparative genomic hybridization (CGH) for sibling (II‐5) and were nondiagnostic.
Table 1.
Clinical features of patients segregating homozygous truncating variants in the PPP1R21
| Characteristic | Family 1a | Family 2 | Family 3 | Family 4 | 17DG0773b , Child 1c | Child 2c | Child 3c | ||
|---|---|---|---|---|---|---|---|---|---|
| Origin | Syria | Sultanate of Oman | Iran | Saudi Arabia | Not described | Not described | Not described | ||
| Consanguinity | First cousins | Second cousins | First cousins | First cousins | Consanguineous | Not described | Not described | ||
| Sibling ID | II‐5 | II‐6 | II‐3 | II‐4 | V‐4 | IV‐1 | – | – | – |
| Sex | Female | Female | Female | Female | Female | Male | Female | Male | Female |
| Age at last follow‐up | 1 Year | 7 Weeks | 7 Years | 4 Years | 2 Years | 16 Months | 3 Years | 2 Years | 11 Years |
| Current status | Deceased | Deceased | Alive | Alive | Alive | Alive | Not reported | ||
| PPP1R21 | c.347delT p.(Ile116Lysfs*25) | c.347delT p.(Ile116Lysfs*25) | c.2170_2171insGGTA p.(Ile724Argfs*8) | c.2170_2171insGGTA p.(Ile724Argfs*8) | c.1607dupT p.(Leu536Phefs*7) |
c.2063delA p.(Lys688Serfs*26) |
c.2089C>T p.(Arg697*) | c.427C>T (p.Arg143*) | c.87_88delAG (p.Gly30Cysfs*4) |
| Dysmorphic features | High forehead, high eye brows, high arch palate, tented mouth | Skull asymmetry, high forehead, flat orbital ridges, hypertelorism, low set ears, high‐arched palate, small chin, small thorax | High forehead, bitemporal narrowing, coarse features, telecanthus, blue sclerae, prominent nasal bridge, low set ears, long philtrum | High forehead, bitemporal narrowing, coarse features, telecanthus, blue sclerae, prominent nasal bridge, low set ears, long philtrum | Slightly coarse features, low set ears, increased facial hair | Plagiocephaly, slightly coarse features | Abnormal facial shape, wide nasal bridge, upslanted palpebral fissures, coarse facial features, generalized hirsutism, low‐set posteriorly rotated ears, thick lower lip vermilion, high, narrow palate | Thick eyebrows, hypertelorism, broad nasal bridge, short nose with upturned nasal tip and broad low‐hanging columella, thick lips, high arched palate, low‐set ears, coarse facies with excessive facial hair, and flat occiput | Thick eyebrows, hypertelorism, broad nasal bridge, short nose with upturned nasal tip and broad low‐hanging columella, thick lips, high arched palate, low‐set ears, coarse facies with excessive facial hair, and flat occiput |
| Severe global DD | Yes | Not applicabled | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Neurologic findings | Generalized hypotonia, weak cry, swallowing problems, absent DTRs | Severe central hypotonia, poor suck, weak cry. | Hypotonia, attention deficit, dysarthria, clumsy/ataxic gait | Hypotonia, attention deficit, dysarthria, clumsy/ataxic gait | Hypotonia, no sitting or walking | Hypotonia, not sitting or walking | Hypotonia, absent DTRs, generalized muscle weakness | Hypotonia, hyporeflexia, muscle weakness | Hypotonia, hyporeflexia, muscle weakness |
| Brain imaging | Irregular outline to prominent bodies of both lateral ventricles with abnormal periventricular T2 hyperintensity, generous extra‐axial CSF spaces with bilateral and symmetric underopercularization, foreshortening and thinning of corpus callosum | Not available | Slight prominence to the supratentorial ventricular system with increased T2 hyperintensity returned from the bilateral posterior centra semiovale, cavum septum pellucidum | Irregular outline to the bodies of both lateral ventricles in addition to the increased T2 hyperintensity returned from the periventricular white matter, thinning and foreshortening of the body of the corpus callosum, cavum septum pellucidum | Prominence to the bodies of the lateral ventricles with generous extra‐axial CSF spaces with bilateral and symmetric underopercularization, thinning and foreshortening of the corpus callosum | Reduction in the volume of deep white matter with abnormal signal; reduction in brainstem and vermian volume; dysmorphic corpus callosum and brainstem | Reduced brain volume with prominent CSF spaces, cerebellar vermis hypoplasia with absent inferior vermis, thin corpus callosum, delayed myelination, cavum septum pellucidum and mega cisterna magna | cerebellar vermis hypoplasia, ventricular dilatation and prominent CSF spaces, reduced white matter volume) | Cerebellar vermis hypoplasia, ventricular dilatation and prominent CSF spaces, reduced white matter volume |
| Cardiac findings | Hypertrophic cardiomyopathy | Mild left pulmonary stenosis, ASD secundum | ASD | – | – | – | Small PDA, small PFO, and mild septal hypertrophy | – | – |
| Respiratory findings | Difficulty breathing and choking at 1 month of age, recurrent respiratory infections, laryngomalacia | Admitted to NICU at 3 weeks of age for respiratory distress | – | – | Respiratory distress, recurrent respiratory infections | Admitted to PICU for respiratory support following episode of decreased consciousness and hypothermia | At birth admitted to NICU for respiratory distress, recurrent respiratory infections | Recurrent chest infections with respiratory distress | Recurrent chest infections with respiratory distress |
| Gastrointestinal findings | Feeding difficulties, constipation, distended abdomen, failure to thrive, hepatosplenomegaly | Feeding difficulties, constipation, distended abdomen | Feeding difficulties | Feeding difficulties | Hepatosplenomegaly | Feeding difficulties | Feeding difficulties, hepatomegaly | Feeding difficulties | Feeding difficulties |
| Skeletal findings | Delayed fontanel closure | Small thoracic cage, arched back, clenched hands, bilateral overlapping of fingers and toes | Delayed bone age, scoliosis, pectus carinatum | Delayed bone age, scoliosis, pectus carinatum | – | – | Not reported | – | – |
| Ophthalmologic findings | Optic atrophy, esotropia | Minimal vision, esotropia | Esotropia, telecanthus, blueish sclerae | Esotropia, telecanthus, blueish sclerae | Optic atrophy, reduced visus, strabismus | – | Myopia, rotatory nystagmus | Rotatory nystagmus | Rotatory nystagmus |
| Other | Strawberry hematoma of right knee | – | Dental caries | Dental caries, myoclonic epilepsy | Hypothyroidism | Undescended testes | – | – | – |
The oldest sibling (II‐1) from Family 1 was on ventilator for 2 years and had deceased at 3 years of age due to complications of disease. Sibling II‐4 deceased at 1 year of age due to complications of disease. Both siblings had clinical presentations that were similar to the other affected family members.
Patient reported in Anazi et al. (2017).
Patient reported by Suleiman et al. (2018).
Patient last seen by a physician at 7 weeks of age.
ASD, Atrial septal defect; CSF, cerebrospinal fluid; DD, developmental delay; DTRs, deep tendon reflexes; NICU, neonatal intensive care unit; PDA, patent ductus arteriosus; PFO, patent foramen ovale, PICU; Pediatric intensive care unit.
Family 2 is of Arab ancestry as well and consists of two affected and two unaffected siblings born to a consanguineous union among second cousins. The parents are healthy with an unremarkable family history. Both female subjects were born at full term without neonatal complications and were noted to have dysmorphic coarse facial features, persisting hypotonia, and global developmental delay. Neuro‐ophthalmologic phenotype includes ataxia, dysarthria, and esotropia. Additional common features among both siblings were feeding difficulties, delayed bone age, pectus carinatum, and mild scoliosis. The older subject (II‐3) was diagnosed with ASD by echocardiogram whereas the younger subject (II‐4) presented to the hospital at the age of 4 years with fever and opsoclonus/myoclonus which settled after steroids administration. MRI studies demonstrated irregular outline to the bodies of both lateral ventricles in addition to the increased T2 hyperintensity returned from the periventricular white matter, and thinning and foreshortening to the body of the corpus callosum. Karyotyping and array CGH studies performed in the older subject were normal.
The proband (V:4) from Family 3 is a female born as result of the first pregnancy to first cousin Iranian parents. Her dizygotic twin sister is healthy. The proband was referred for genetic testing at the age of 8 months due to severe global developmental delays with fronto‐temporal pronounced thin cerebral cortex with enlarged outer liquor spaces and enlarged lateral ventricles, thin corpus callosum, and hypoplasia/atrophy of the optic nerve on brain MRI. Additional clinical features included hypotonia, respiratory distress, slightly coarse facial features, and hepatosplenomegaly. Family history showed that her deceased first cousin (V:1) had a similar clinical presentation including developmental delay/intellectual disability (IQ below 60), and ophthalmic features (strabismus, decreased visual acuity, and was blind when she passed away at the age of 12 years). The proband as well as the deceased affected cousin were not able to sit or walk. Additionally, hypothyroidism was noted in both affected individuals as well as some healthy family members. Prior genetic testing was not performed in this family.
The proband from Family 4 is the first child born to consanguineous Arab parents following a normal pregnancy. He was admitted at the age of 16 months to pediatric intensive care with an episode of decreased consciousness and hypothermia, on a background of global developmental delay, hypotonia, and feeding difficulties. He had plagiocephaly, torticollis, slightly coarse facial features, and undescended testes. He was smiling responsively, but unable to roll. MRI of the brain demonstrated abnormally deep and box‐like Sylvian fissures, diffuse reduction in cerebral volume, particularly the deep white matter, diffuse reduction in the brainstem and vermian volume, and dysmorphic appearance of the corpus callosum and brainstem.
3.2. Identification of disease‐causing variants
In order to establish the underlying genetic cause, we performed whole exome and whole genome sequencing in these four families. Analyses of WES and WGS data showed the proband in each of the four families carried a homozygous truncating variant in the PPP1R21 gene, which encodes a protein of unexplored function. Variant nomenclature provided here is based on RefSeq accession number NM_001135629.2. In Family 1, we detected a deletion of one nucleotide c.347delT p.(Ile116Lysfs*25) in exon 4 of PPP1R21. Subjects II‐5 and II‐6 are homozygous for c.347delT whereas their parents are obligate carriers. Genotypes from additional family members cannot be obtained because their DNA samples were not preserved before their deaths due to disease (II‐1 and II‐4) or the accident (II‐2 and II‐3). In Family 2, we found a homozygous insertion of four nucleotides c.2170_2171insGGTA p.(Ile724Argfs*8) in exon 20 of PPP1R21 to co‐segregate with disease state in individuals II‐3 and II‐4. Both parents are obligate carriers of the c.2170_2171insGGTA allele whereas the two healthy siblings have at least one reference allele of PPP1R21. In Family 3, proband V‐4 is homozygous for duplication of one nucleotide, c.1607dupT p.(Leu536Phefs*7), in exon 16 of PPP1R21 whereas her healthy di‐zygotic twin as well as remaining family members for whom DNA is available are either heterozygous for c.1607dupT or are homozygous for the reference allele (Figure 1A). In Family 4, the proband is homozygous for deletion of one nucleotide, c. 2063delA p.(Lys688Serfs*26), while the parents are heterozygous. All four truncating variants are absent from ExAC (http://exac.broadinstitute.org) and gnomAD (http://gnomad.broadinstitute.org) as well as from our in‐house database of approximately 10,000 clinical exomes. Additionally, no PPP1R21 homozygous loss of function variant is reported in the ExAC or gnomAD databases suggesting that biallelic loss of function variants in PPP1R21 are likely to cause a clinically detectable phenotype. Clinical phenotypes of all affected individuals included in this study (n = 6) as well as of three individuals reported in parallel to our study by Suleiman et al. (2018) are displayed in Table 1. PPP1R21 variants identified to date are shown in Table 2.
Table 2.
Identified variants in the PPP1R21 gene
| Family | Genomic coordinates (hg19) | Variant nomenclature (NM_001135629.2) | Zygosity and variant type | Allele freq. in ExAC | Allele freq. in gnomAD |
|---|---|---|---|---|---|
| Family 1 | chr2:48,685,338 | c.347delT p.(Ile116Lysfs*25) | Homozygous frameshift | 0.00 | 0.00 |
| Family 2 | chr2:48,737,238‐48,737,239 | c.2170_2171insGGTA p.(Ile724Argfs*8) | Homozygous frameshift | 0.00 | 0.00 |
| Family 3 | chr2:48,722,825 | c.1607dupT p.(Leu536Phefs*7) | Homozygous frameshift | 0.00 | 0.00 |
| Family 4 | chr2:48,734,502 |
c.2063delA p.(Lys688Serfs*26) |
Homozygous frameshift | 0.00 | 0.00 |
| 17DG0773a , b | chr2:48,737,157 | c.2089C>T p.(Arg697*) | Homozygous stop‐gain | 0.00 | 0.00 |
| bChild 2 | chr2:48,686,944 | c.427C>T (p.Arg143*) | Homozygous stop‐gain | 0.00 | 0.000007 |
| bChild 3 | chr2:48,678,176_77 | c.87_88delAG (p.Gly30Cysfs*4) | Homozygous frameshift | 0.00 | 0.00 |
3.3. Gene structure of PPP1R21
PPP1R21 resides on the short arm of chromosome 2 (2p16.3) and consists of 22 protein coding exons. The NCBI reference sequence database (RefSeq) lists three protein coding transcripts of human PPP1R21 designated as transcript variant 1 (NM_001135629.2), 2 (NM_152994.4), and transcript variant 5 (NM_001193475.1). The three transcripts have identical sequence content up to exon 15 and differ in their sequence toward the 3′ end due to in‐frame alternative exon splicing (Figure 1C). Transcript variant 1 is the longest and codes for 780 amino acids whereas transcript 2 lacks exon 18 (31 residues) and transcript 5 lacks exons 16 and 18 (31+11 = 42 resides). All protein truncating variants of PPP1R21 identified in patients affect the three isoforms, except c.1607dupT p.(Leu536Phefs*7) that resides in alternatively spliced exon 16 and affects isoforms 1 and 2 (functional consequences of c.1607dupT in patient's fibroblasts are described later in the article).
3.4. Protein structure and expression pattern of PPP1R21
The PPP1R21 protein belongs to a family of predicted coiled‐coil domain containing proteins and its function has not been studied in depth to date and no functional domains have been described. We queried the NCBI database of nonredundant protein sequences (NCBI BLAST: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) using the longest isoform (NP_001129101.1) of PPP1R21 and found two putative conserved domains within PPP1R21; an N‐terminal KLRAQ domain, and a C‐terminal TTKRSYEDQ domain (Figure 1C). The predicted 100 amino acid long KLRAQ domain is encoded by residues 11–111 and contains a characteristic KLRAQ sequence‐motif. The 500 amino acids long C‐terminal TTKRSYEDQ domain is encoded by residues 255–771 and has a characteristic TTKRSYEDQ sequence‐motif (Figure 1C). Both domains are conserved from nematodes to humans but their molecular function(s) are yet to be discovered.
The Human Protein Atlas database (https://www.proteinatlas.org) lists that PPP1R21 mRNA is expressed in a variety of tissues including brain, endocrine, lungs, liver, heart, pancreas, spleen, and gastrointestinal tract. At the protein level, the strongest expression appears to be in cerebellum, esophagus, pancreas, thyroid and parathyroid glands, among others (Supp. Figure S2). We subsequently confirmed cortical expression of PPP1R21 during murine development by performing immunofluorescence analysis on mouse E16 frozen cortex sections using HPA036792, Atlas antibodies, Sweden) (Supp. Figure S3).
3.5. Subcellular localization of PPP1R21
Small cerebellum noted on the MRI and the phenotype observed in some individuals with biallelic PPP1R21 loss of function mutations shows partially overlapping features with Joubert syndrome including ataxic wide‐based gait; further large‐scale affinity‐MS proteomics analyses has previously suggested PPP1R21 to interact with a known Joubert syndrome gene, PIBF1 (Huttlin et al., 2015; Wheway et al., 2015b). This prompted us to next test the putative protein–protein interaction between PPP1R21 and PIBF1 by co‐immunoprecipitation. Our tests could not confirm the interaction however (Supp. Figure S4 and Supp. Figure S5) and subcellular immunofluorescence studies in several cell lines such as hTERT‐RPE (retinal pigment epithelium), IMCD3 (inner medullary collecting duct), and the neuronal cell line SH‐SY5Y did not show ciliary or basal body or centrosomal PPP1R21 localization (Supp. Figure S6, α‐acetylated tubulin (T7451, clone 6‐11‐B) Sigma 1:500, α‐PPP1R21). Rather, we observed a vesicular staining pattern within the cytosol of all tested cell lines which prompted us to further investigate the nature of these vesicles by performing co‐staining with antibodies for Golgi and endosomal marker proteins. We did not detect co‐localization of PPP1R21 with the two Golgi markers that we tested (α‐58K; ab27043 from Abcam, and α‐GM130; ab169276, Abcam). However, we found a nearly complete co‐localization of PPP1R21 with the early endosome marker EEA1 (ab70521, mouse monoclonal, Abcam) using two different PPP1R21 antibodies, HPA036791 and HPA036792 (rabbit polyclonal, Atlas antibodies, Sweden) (Figure 2A).
Figure 2.
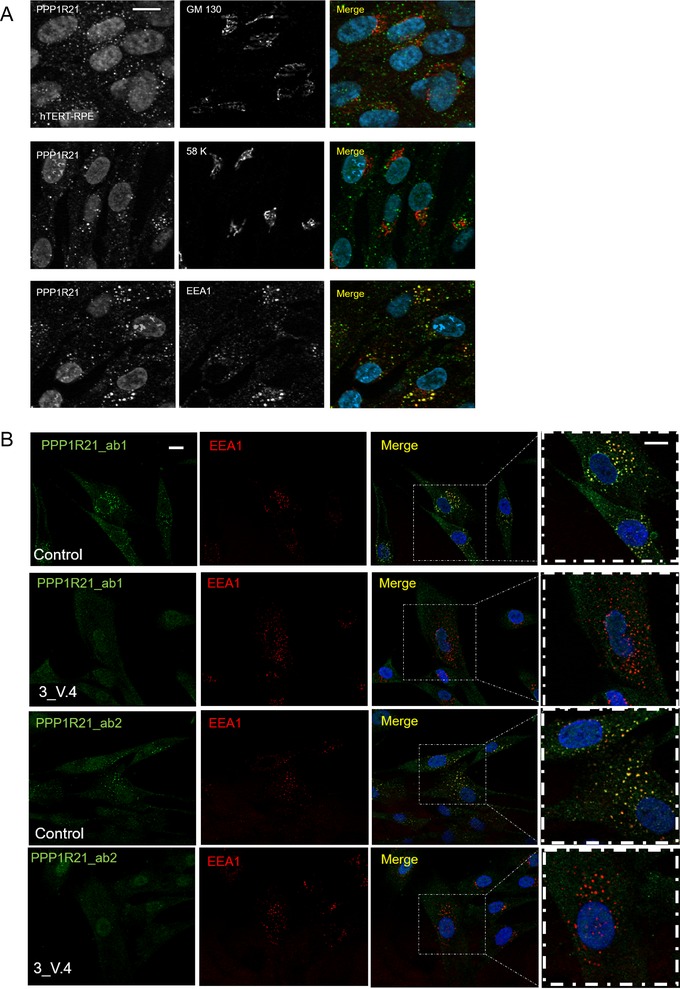
PPP1R21 co‐localizes with the main early endosome protein EEA1 but not Golgi proteins and PPP1R21 staining is absent in fibroblasts obtained from patient 3_V:4 who is homozygous for c.1607dupT p.(Leu536Phefs*7). (A) In order to determine the entity of PPP1R21 positive vesicles, we performed co‐localization studies using Golgi marker GM130 (mouse, ab169276, Abcam, USA) and 58K (ab27043 mouse, Abcam, USA) as well as EEA1 antibody to mark the early endosome. While no co‐localization with Golgi markers was observed (upper panels), PPP1R21 (rabbit, HPA036792, Atlas antibodies, Sweden) nearly completely co‐localized with EEA1 (lower panel). Scale bar 20 μm. (B) Using two different PPP1R21 antibodies, HPA036792 (ab1, antibody epitope is represented by aminoacid aa161‐256) and HPA 036791 (ab2, antibody epitope is represented by aminoacid aa572‐666), we found complete loss of the vesicular staining pattern in PPP1R21 mutant fibroblasts we previously observed in control cells while no difference regarding EEA1 staining was detected between PPP1R21 mutant and control fibroblasts. Scale bar: 20 μm
3.6. PPP1R21 expression is missing in patient fibroblasts
qPCR analyses revealed that PPP1R21 mRNA levels are greatly reduced in PPP1R21 deficient fibroblasts compared to control fibroblasts (Supp. Figure S7) but not completely absent. We, therefore, proceeded to perform immunofluorescence analysis to examine protein levels and localization in mutant cells versus control cell lines: as expected here we found that in 2 lines of human control fibroblasts, PPP1R21 consistently co‐localized with early endosome marker EEA1. However, this staining pattern was absent from fibroblasts of individual V:4 who is homozygous for the PPP1R21 c.1607dupT p.(Leu536Phefs*7) allele, proving specificity of both PPP1R21 antibodies and demonstrating the variant results in a true null allele (Figure 2B). EEA1 staining did not differ between PPP1R21 deficient and control cells, indicating PPP1R21 is not required for EEA1 localization. A weak nuclear staining pattern for PPP1R21 was still detected by immunofluorescence in PPP1R21 mutant cells, this could indicate that low levels of protein localizing to the nucleus is still present in patient cells but it is also possible the nuclear staining pattern observed in mutant and wildtype cells is nonspecific and truncated PP1R21 protein, if translated from the remaining low levels of RNA, is subjected to proteasomal degradation.
3.7. Uptake and clearance of transferrin‐488 in PPP1R21 mutant fibroblasts
Clinical features observed in patients with PPP1R21 variants, such as thin fronto‐temporal cortex with enlarged external liquor spaces in combination with coarse face, hepatosplenomegaly, and vision impairment, reminded us of the clinical picture observed in patients with inborn errors of metabolism such as Morbus Gaucher (Roshan Lal & Sidransky, 2017) or other neurometabolic/lysosomal (storage) diseases (Karimzadeh, 2015). This in combination with our subcellular localization studies suggesting PPP1R21 resides within the early endosome prompted us to next study uptake and clearance of fluorescence labelled transferrin (transferrin‐488) in control fibroblasts as well as fibroblasts from patient V:4 (Family 3). We found unchanged transferrin uptake in fibroblasts of an affected individual with definite PPP1R21 loss of function compared to control fibroblasts, indicating macro‐autophagy is not disturbed by PPP1R21 dysfunction and/or transferrin uptake may take place by receptor mediated endocytosis which is likewise intact. However, interestingly, transferrin‐488 clearance was mildly but statistically significantly delayed in the PPP1R21 mutant cells (p = 0.0015, Student's t‐Test) after 2 hr of repetitive washes with medium containing unlabeled transferrin (“chase”) (Figure 3 and Supp. Figure S8).
Figure 3.
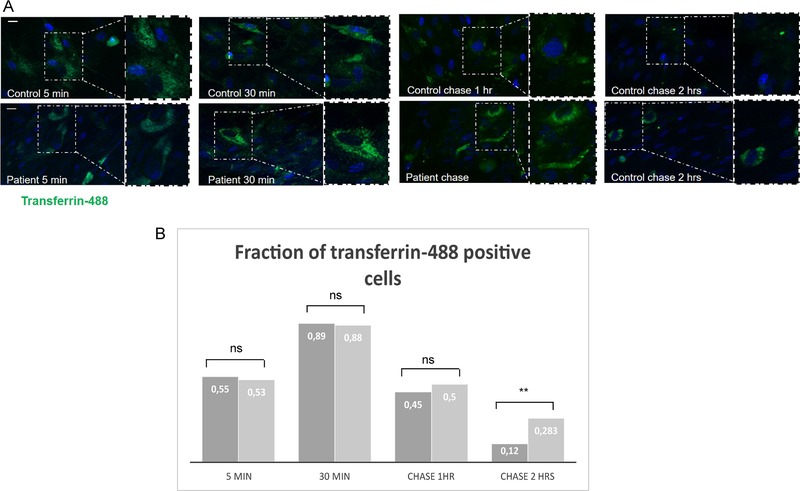
Transferrin‐488 uptake and clearance in PPP1R21 mutant fibroblasts and control cells. (A) Representative images of transferrin‐488 uptake and clearance in PPP1R21 mutant fibroblasts (lower panel) and control cells (upper panel) taken after 5 and 30 min of transferrin‐488 exposure and 1 and 2 hr of transferrin‐488 washout (chase). Scale bar = 30 μm. (B) Quantification of transferrin‐488 uptake 5 and 30 min after exposure to supplemented medium and 1 and 2 hr of washout (chase, exposure to medium where transferrin‐488 was replaced with unlabeled transferrin and renewal of the medium every 20 min). While the number of transferrin‐488 positive cells did not differ significantly between PPP1R21 mutant and control fibroblasts after 5 and 30 min or after 1 hr of chase, more cells were positive after 2 hr of chase, with a strong statistical significance (p = 0.0015, Student's t‐test). Control cells are shown in dark grey and mutant cells in light grey
3.8. Increased number of myelin figures in PPP1R21 deficient fibroblasts confirming mild endo‐lysosomal dysfunction
Our immunofluorescence data as well as the transferrin uptake and clearance assay suggested PPP1R21 locates to the early endosome and that patient fibroblasts exhibit mild endo‐lysosomal dysfunction. To assess if this defect results in abnormal storage of cellular material within cytoplasmic vesicles, we next performed electron microscopy (EM) analyses using PPP1R21 deficient fibroblasts as well as two control fibroblast lines. This revealed mildly increased number of myelin figures/myelin bodies in fibroblasts of patient V:4 carrying homozygous c.1607dupT p.(Leu536Phefs*7) variant in PPP1R21 while no myelin figures/myelin bodies were observed in the control lines (Figure 4).
Figure 4.
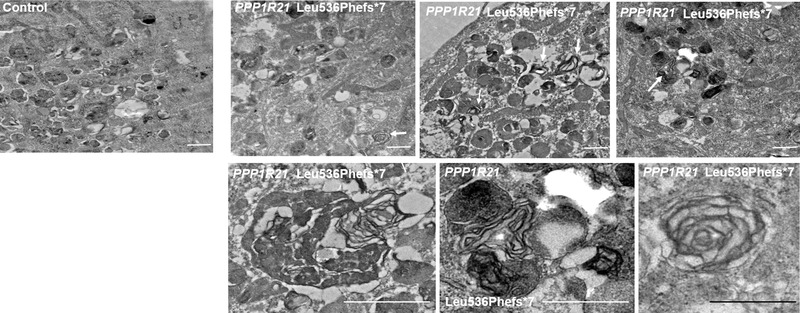
Increased number of myelin figures in PPP1R21 deficient fibroblasts confirming mild endo‐lysosomal dysfunction. Electron microscopy (EM) revealed mildly increased number of myelin figures/myelin bodies in fibroblasts of patient V:4 carrying homozygous c.1607dupT p.(Leu536Phefs*7) variant of PPP1R21. We observed 1–2 myelin bodies per vision field (arrows). These figures were not observed in two fibroblast control cell lines cultured in parallel. Scale bar: 500 nm
4. DISCUSSION
This study identified four previously unreported homozygous truncating alleles of PPP1R21 co‐segregating in four independent consanguineous families with affected individuals exhibiting overlapping clinical features. Consistent with our findings, a female subject homozygous for a PPP1R21 null allele was recently described within a larger cohort of developmental delay cases (Anazi et al., 2017). Her reported clinical presentation was very similar to what we observed in families 1–4 in our study including slightly coarsened dysmorphic facial features, severe global developmental delays, hypotonia, respiratory distress, feeding difficulties, hepatomegaly, and vision impairment. During preparation of this manuscript, two additional subjects with homozygous putative PPP1R21 null alleles have been described (Suleiman et al., 2018). We have summarized common clinical features observed among all nine subjects (six from this study and three previously reported (Anazi et al., 2017; Suleiman et al., 2018) in Table 1: most commonly observed features include hypotonia (9/9), severe global developmental delays (9/9), dysmorphic mildly coarsened facial features (9/9), vision impairment and/or nystagmus (8/9), respiratory problems (n = 7/9), feeding difficulties (n = 9/9), cardiac concerns (n = 4/9), and hepato/hepatosplenomegaly (n = 3/7; Table 1). Common findings on MRI of the brain include prominent and slightly dysmorphic ventricular system with irregular outline to the bodies of the lateral ventricles reminiscent of PVL (periventricular leukomalacia) and consistent with loss of white matter (Figure 1B and Table 1). There is some increased T2 hyperintensity returned from the deep white matter in all cases (where images were available). Further, generous extra‐axial CSF spaces with bilateral and symmetric underopercularization as well as slight foreshortening and thinning of the corpus callosum was noted as well as cerebellar hypoplasia in some but not all cases.
PPP1R21 is an evolutionarily conserved protein (Supp. Figure S9) with an undiscovered function and no investigations toward understanding the underlying patho‐mechanism had been performed. This prompted us to perform functional analysis to gain insight into the physiological role of PPP1R21 and to identify possible disease mechanism for the observed PPP1R21‐associated phenotype. Consistent with the severe neurodevelopmental phenotype observed, our immunofluorescence studies confirmed PPP1R21 expression within the developing mouse cortex (Supp. Figure S3). A large‐scale proteomics study had previously suggested PPP1R21 may interact with PIBF1, a protein found to be defective in some individuals with Joubert syndrome (Huttlin et al., 2015; Wheway et al., 2015a). However, our co‐immunoprecipitation analyses did not confirm the suggested interaction (Supp. Figure S4). Rather, using two different antibodies, we observed nearly complete co‐localization of PPP1R21 with the early endosome marker EEA1 (Figure 2A) and transferrin‐488 uptake and clearance assays (Figure 3) suggested a mild endo‐lysosomal functional defect. We found that although there was no difference of transferrin uptake between control and patient fibroblasts, its clearance was mildly but statistically significantly delayed in the PPP1R21 mutant cells after 2 hr of repetitive washes with medium containing unlabeled transferrin (“chase”). This could indicate a mild endosome maturation or fusion defect or could result from a trafficking defect back toward the plasma membrane. Such impaired proteolipid protein trafficking has been previously described to result from cathepsin D deficiency where phenotypically delayed central nervous system myelination was observed (Guo et al., 2018; Steinfeld et al., 2006) and hypomyelination was also noted in the brain MRI scans in Family 1 described here. However, PPP1R21 localization to early endosomes differs from cathepsin D localization to late endosomes/lysosomes, rendering such a retrograde trafficking defect as an unlikely major cause for the observed phenotype. Our EM studies confirmed mild endo‐lysosomal dysfunction in patient fibroblasts in comparison to control cell lines with a mildly increased number of myelin figures/myelin bodies observed in the PPP1R21 loss of function cell line (Figure 4).
The dynamics of the endocytic pathways are governed by several Rab family members with Rab 5 playing a major role in the early endosome traffic (Barbieri, Roberts, Mukhopadhyay, & Stahl, 1996; Murray, Panaretou, Stenmark, Miaczynska, & Backer, 2002). Some phenotypic features observed in the affected families may also directly result from Protein Phosphatase 1 (PP1) dysfunction. PP1 is a major serine/threonine phosphatase with an immense diversity of functions, including fundamental processes such as glycogen metabolism (Munro, Cuthbertson, Cunningham, Sales, & Cohen, 2002), cell cycle regulation, and regulation of centriole duplication (Peel et al., 2017), apoptosis (Garcia et al., 2003), and PP1 is expressed during development in murine brains (Collins & Sim, 1998). PP1 consists of a catalytic subunit (PPP1CA) and a high number of regulatory subunit determining the subcellular localization of PP1 and regulating its function. Future work will be required to further investigate this area.
Although previously published large scale proteomics data suggested an interaction with the Joubert protein PIBF1, patient phenotype did not match Joubert syndrome or related ciliopathies and our co‐immunoprecipitation studies and the subcellular localization to early endosomes in several cell lines points against a role within the Joubert syndrome protein network. Rather, our results point toward a defect within the endosomal‐lysosomal compartment, as it has been previously suggested for other neurodegenerative diseases in humans, resulting in an endocytic defects or impaired clearance of certain molecules from this compartment. For example, a phenotype somewhat resembling the phenotype we observe with PPP1R21 loss of function including coarse face, neurodevelopmental delay, and cerebellar atrophy was described to result from SNX14 (sorting nexin 14) loss of function mutations (Akizu et al., 2015; Thomas et al., 2014). SNX proteins are known to play a role in endosomal cargo sorting and SNX14 was shown to be essential for fusion of late endosome/autophagosomes with lysosomal vesicles in fibroblasts as well as neuronal precursor cells from individuals (Akizu et al., 2015).
In conclusion, we propose recessive PPP1R21 loss of function variants as a cause of a distinct neurodevelopmental syndrome with features of mild endosomal‐lysosomal dysfunction. All identified variants have protein‐truncating properties and studies in fibroblasts of one affected individual reveal complete absence of PPP1R21 protein. We further show that PPP1R21 co‐localizes with EEA1 in early endosomes and loss of PPP1R21 results in a mild transferrin clearance defect in PPP1R21 null‐fibroblasts, indicating PPP1R21 is essential for proper functioning of the early endosome compartment. Future studies including appropriate animal models are warranted to understand the precise molecular mechanism that leads to PPP1R21‐associated neurodevelopmental phenotype.
COMPETING INTERESTS
The Department of Molecular and Human Genetics at the Baylor College of Medicine derives revenue from molecular genetic testing offered at the Baylor Miraca Genetics Laboratories.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We thank the subjects and their families for making this study possible. We thank Prof. H. Omran, Munster, Germany, for evaluation of the MRI of Family 3 individual V‐4 and Thilo Bass, Freiburg, Germany for excellent technical assistance. We also thank Elizabeth A. Normand for her support.
PATIENT CONSENT
Obtained.
ETHICAL APPROVAL
The study was approved by respective ethics committees from Baylor College of Medicine in USA, Sidra Medicine in Qatar, University of Geneva in Switzerland, the Innovative diagnostics programme, Radboudumc Nijmegen, the Netherlands, and the Royal Children's Hospital, Melbourne.
Rehman AU, Najafi M, Kambouris M, et al. Biallelic loss of function variants in PPP1R21 cause a neurodevelopmental syndrome with impaired endocytic function. Human Mutation. 2019;40:267–280. 10.1002/humu.23694
Funding information
MS acknowledges funding from Radboudumc and RIMLS Nijmegen (Hypatia tenure track fellowship), the “Deutsche Forschungsgemeinschaft” (DFG CRC1140 KIDGEM), and the European Research Council (ERC StG TREATCilia, grant no. 716344). SEA is supported by grants from the Swiss National Science Foundation, and the European Research Council (AdG 249968). The Acute Care Flagship of the Australian Genomics Health Alliances is supported by grants from the RCH Foundation (2017‐906) and the National Health and Medical Research Council (GNT1113531).
Communicated by Garry R. Cutting
Contributor Information
Yaping Yang, Email: yapingy@bcm.edu.
Miriam Schmidts, Email: miriam.schmidts@uniklinik-freiburg.de, Email: miriam.schmidts@radboudumc.nl.
REFERENCES
- Akizu, N. , Cantagrel, V. , Zaki, M. S. , Al‐Gazali, L. , Wang, X. , Rosti, R. O. , … Gleeson, J. G. (2015). Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome‐autophagosome dysfunction. Nature Genetics, 47(5), 528–534. 10.1038/ng.3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anazi, S. , Maddirevula, S. , Salpietro, V. , Asi, Y. T. , Alsahli, S. , Alhashem, A. , … Alkuraya, F. S. (2017). Expanding the genetic heterogeneity of intellectual disability. Human Genetics, 136(11–12), 1419–1429. 10.1007/s00439-017-1843-2 [DOI] [PubMed] [Google Scholar]
- Barbieri, M. A. , Roberts, R. L. , Mukhopadhyay, A. , & Stahl, P. D. (1996). Rab5 regulates the dynamics of early endosome fusion. Biocell, 20(3), 331–338. [PubMed] [Google Scholar]
- Bennett, M. J. , & Hofmann, S. L. (1999). The neuronal ceroid‐lipofuscinoses (Batten disease): A new class of lysosomal storage diseases. Journal of Inherited Metabolic Disease, 22(4), 535–544. [DOI] [PubMed] [Google Scholar]
- Boycott, K. M. , Rath, A. , Chong, J. X. , Hartley, T. , Alkuraya, F. S. , Baynam, G. , … Lochmuller, H. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. American Journal of Human Genetics, 100(5), 695–705. 10.1016/j.ajhg.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, J. X. , Buckingham, K. J. , Jhangiani, S. N. , Boehm, C. , Sobreira, N. , Smith, J. D. , … Bamshad, M. J. (2015). The genetic basis of Mendelian phenotypes: Discoveries, challenges, and opportunities. American Journal of Human Genetics, 97(2), 199–215. 10.1016/j.ajhg.2015.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, E. , & Sim, A. T. (1998). Regulation of neuronal PP1 and PP2A during development. Methods in Molecular Biology, 93, 79–102. 10.1385/0-89603-468-2:79 [DOI] [PubMed] [Google Scholar]
- Fuller, M. , Meikle, P. J. , & Hopwood, J. J. (2006). Epidemiology of lysosomal storage diseases: An overview In Mehta A., Beck M., & Sunder‐Plassmann G. (Eds.), Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis. [PubMed] [Google Scholar]
- Garcia, A. , Cayla, X. , Guergnon, J. , Dessauge, F. , Hospital, V. , Rebollo, M. P. , … Rebollo, A. (2003). Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie, 85(8), 721–726. [DOI] [PubMed] [Google Scholar]
- Guo, D. Z. , Xiao, L. , Liu, Y. J. , Shen, C. , Lou, H. F. , Lv, Y. , & Pan, S. Y. (2018). Cathepsin D deficiency delays central nervous system myelination by inhibiting proteolipid protein trafficking from late endosome/lysosome to plasma membrane. Experimental Molecular Medicine, 50(3), e457 10.1038/emm.2017.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin, E. L. , Ting, L. , Bruckner, R. J. , Gebreab, F. , Gygi, M. P. , Szpyt, J. , … Gygi, S. P. (2015). The BioPlex network: A systematic exploration of the human interactome. Cell, 162(2), 425–440. 10.1016/j.cell.2015.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovic, M. , Sharma, M. , Rahajeng, J. , & Caplan, S. (2010). The early endosome: A busy sorting station for proteins at the crossroads. Histology and Histopathology, 25(1), 99–112. doi: 10.14670/HH-25.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimzadeh, P. (2015). Approach to neurometabolic diseases from a pediatric neurological point of view. Iranian Journal of Child Neurology, 9(1), 1–16. [PMC free article] [PubMed] [Google Scholar]
- Maxfield, F. R. (2014). Role of endosomes and lysosomes in human disease. Cold Spring Harbor Perspectives in Biology, 6(5), a016931 10.1101/cshperspect.a016931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro, S. , Cuthbertson, D. J. , Cunningham, J. , Sales, M. , & Cohen, P. T. (2002). Human skeletal muscle expresses a glycogen‐targeting subunit of PP1 that is identical to the insulin‐sensitive glycogen‐targeting subunit G(L) of liver. Diabetes, 51(3), 591–598. [DOI] [PubMed] [Google Scholar]
- Murray, J. T. , Panaretou, C. , Stenmark, H. , Miaczynska, M. , & Backer, J. M. (2002). Role of Rab5 in the recruitment of hVps34/p150 to the early endosome. Traffic (Copenhagen, Denmark), 3(6), 416–427. [DOI] [PubMed] [Google Scholar]
- Parkinson‐Lawrence, E. J. , Shandala, T. , Prodoehl, M. , Plew, R. , Borlace, G. N. , & Brooks, D. A. (2010). Lysosomal storage disease: Revealing lysosomal function and physiology. Physiology (Bethesda, MD), 25(2), 102–115. 10.1152/physiol.00041.2009 [DOI] [PubMed] [Google Scholar]
- Peel, N. , Iyer, J. , Naik, A. , Dougherty, M. P. , Decker, M. , & O'Connell, K. F. (2017). Protein phosphatase 1 down regulates ZYG‐1 levels to limit centriole duplication. PLoS Genetics, 13(1), e1006543 10.1371/journal.pgen.1006543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, F. M. , Boland, B. , & van der Spoel, A. C. (2012). The cell biology of disease: Lysosomal storage disorders–The cellular impact of lysosomal dysfunction. Journal of Cell Biology, 199(5), 723–734. 10.1083/jcb.201208152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshan Lal, T. , & Sidransky, E. (2017). The spectrum of neurological manifestations associated with Gaucher disease. Diseases, 5(1). 10.3390/diseases5010010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts, M. , Hou, Y. , Cortes, C. R. , Mans, D. A. , Huber, C. , Boldt, K. , … Witman, G. B. (2015). TCTEX1D2 mutations underlie Jeune asphyxiating thoracic dystrophy with impaired retrograde intraflagellar transport. Nature Communications, 6, 7074 https 10.1038/ncomms8074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinfeld, R. , Reinhardt, K. , Schreiber, K. , Hillebrand, M. , Kraetzner, R. , Bruck, W. , … Gartner, J. (2006). Cathepsin D deficiency is associated with a human neurodegenerative disorder. American Journal of Human Genetics, 78(6), 988–998. 10.1086/504159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleiman, J. , Al Hashem, A. M. , Tabarki, B. , Al‐Thihli, K. , Bi, W. , & El‐Hattab, A. W. (2018). PPP1R21 homozygous null variants associated with developmental delay, muscle weakness, distinctive facial features, and brain abnormalities. Clinical Genetics, 94(3–4), 351–355. 10.1111/cge.13387 [DOI] [PubMed] [Google Scholar]
- Thomas, A. C. , Williams, H. , Seto‐Salvia, N. , Bacchelli, C. , Jenkins, D. , O'Sullivan, M. , … Stanier, P. (2014). Mutations in SNX14 cause a distinctive autosomal‐recessive cerebellar ataxia and intellectual disability syndrome. American Journal of Human Genetics, 95(5), 611–621. 10.1016/j.ajhg.2014.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway, G. , Schmidts, M. , Mans, D. A. , Szymanska, K. , Nguyen, T. M. , Racher, H. , … Johnson, C. A. (2015a). An siRNA‐based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nature Cell Biology, 17(8), 1074–1087. 10.1038/ncb3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway, G. , Schmidts, M. , Mans, D. A. , Szymanska, K. , Nguyen, T. T. , Racher, H. , … Johnson, C. A. (2015b). An siRNA‐based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nature Cell Biology, 17(8), 1074–1087. 10.1038/ncb3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information