

Abstract
Estrogen binding to estrogen receptors (ESR) triggers signaling cascades within cells. Historically, a major emphasis has been characterizing estrogen-induced genomic actions resulting from binding to nuclear estrogen receptor 1 (nESR1). However, recent evidence indicates the first receptors estrogens encounter as they enter a cell, membrane ESR1 (mESR1), also play crucial roles. Membrane and nuclear ESR are derived from the same transcripts but the former are directed to the membrane via palmitoylation. Binding and activation of mESR1 leads to rapid fluctuations in cAMP and Ca+2 and stimulation of protein kinase pathways. Endocrine disrupting chemicals (EDC) that mimic 17β-estradiol can signal through mESR1 and elicit non-genomic effects. Most current EDC studies have focused on genomic actions via nESR1. However, increasing number of studies have begun to examine potential EDC effects mediated through mESR1, and some EDC might have higher potency for signaling through mESR1 than nESR1. The notion that such chemicals might also affect mESR1 signaling via palmitoylation and depalmitoylation pathways has also begun to gain currency. Recent development of transgenic mice that lack either mESR1 or nESR1, while retaining functional ESR1 in the other compartment, will allow more precise in vivo approaches to determine EDC effects through nESR1 and/or mESR1. It is increasingly becoming apparent in this quickly evolving field that EDC directly affect mESR and estrogen signaling, but such chemicals can also affect proportion of ESR reaching the membrane.Future E D C studies should be designed to consider the full range of effects through mESR alone and in combination with nESR.
Keywords: Xenoestrogen, Calcium signaling, MAPK, ERK, Non-genomic actions, NOER
1. Introduction
Normal male and female reproduction is dependent upon estrogen signaling, and exposure to insufficient or increased amounts of endogenous estrogen or to xenoestrogens can cause reproductive and other disorders, leading Watson to describe estrogens as “triple-edged swords” (Watson et al. 2011). Circulating concentrations of endogenous and exogenous estrogens are one critical factor regulating estrogenic responses, but the differing location and function of estrogen receptors (ESR) that estrogens encounter as they enter a cell are also involved in ultimate cellular effects. Like a wave coming ashore, estrogens first encounter membrane estrogen receptors (mESR) as they enter a cell, and then as they continue further into the cellular interior, they bind and signal through nuclear estrogen receptors (nESR). Peripheral events, such as menopause, gonadal dysfunction, ovarian cysts, testicular tumors, etc., can result in a deficit or surfeit of estrogen delivery and corresponding health effects, including reduced fecundity, postmenopausal symptoms, osteoporosis, and estrogen-dependent cancer (Watson et al. 2011).
Moreover, humans have inadvertently impacted this complex system through the introduction of industrial and other endocrine disrupting chemicals (EDC) into the environment that are carried into target cells during the wave-like entry of these chemicals into cells (Kwiatkowski et al. 2016). Due to their chemical mimicry, such compounds signal through ESR at both the membrane and nuclear levels, just like endogenous ligands.
Before reaching membrane and nuclear ESR, circulating E2 and xenoestrogens can be free or bound to blood carrier proteins such as albumin or sex hormone binding globulins (SHBG), also termed estradiol-binding protein (E2BP), and binding affinity of E2 to SHBG approximates that of its binding to ESR (Iqbal et al. 1983; Mean et al. 1977; Saito et al. 1989). These circulating proteins that bind sex steroids play important roles in overall distribution of steroids to target tissue and subsequent actions, but how binding of steroids, such as estrogen, to such proteins may affect signaling through mESR remains to be determined. As described later, one method used in the initial characterization of these receptors and still is in use to study steroid activation exclusively through mESR was to link the steroid to a large carrier protein that allowed it to bind and signal through mESR but prevented it from entering a cell and inducing classic genomic actions through nESR. Typically, these protein-bound steroid molecules could still induce potent rapid actions through mESR. These findings at least suggest that possibility that E2 or other steroids bound to large carrier proteins in the circulation would be capable of binding and signaling through mESR. This possibility bears further investigation.
Most studies to date have examined EDC actions through nESR, where they induce genomic effects through DNA binding and subsequently alter gene transcription and translation (De Coster and van Larebeke 2012). In general, EDC bind with lower affinity to nESR compared to the endogenous ligand, 17β-estradiol (E2) (Kuiper et al. 1998; Laws et al. 2000). Less understood are potential interactions of EDC on the first line receptors, mESR, associated with non-genomic effects.
One hallmark of genomic changes induced by E2 is that these effects take hours to days. In sharp contrast, rapid effects are triggered by E2 binding to mESR (Watson and Gametchu 2003; Watson et al. 2010), with some data indicating that uterine responses to estrogen in the rat can be seen within seconds of intravenous administration of estrogen (Rambo and Szego 1983). In the process, concentrations of ions and cyclic nucleotides are altered and kinases and phosphatases are stimulated, with the ultimate effect that the phosphorylation state of key protein kinases (PK) is altered. Initial changes are induced swiftly, but lasting secondary cellular impacts, namely epigenetic alterations, occur in the aftermath of such physiological changes. While EDC might only bind classic nESR weakly, such chemicals, even at low concentrations (nM to pM), might potently stimulate non-nuclear, membrane-driven mechanisms, as detailed below (Alonso-Magdalena et al. 2005; Belcher et al. 2012; Quesada et al. 2002; Wozniak et al. 2005).
The mESR forms include estrogen receptor 1 (ESR1; also known as ERα), ESR2 (ERβ), and G protein-coupled estrogen receptor 1 (GPER, previously termed G protein-coupled receptor 30, or GPR30) (Soltysik and Czekaj 2013). Splice variants of ESR1 might also contribute to nongenomic estrogenic actions (Lin et al. 2013). In contrast to ESR1 and ESR2, which are located both in the nucleus and cell membrane and show extensive orthology in sequence and structure and can dimerize with each other, GPER is in a separate class of membrane receptor that is not found in the nucleus and is related to other seven-transmembrane G protein-coupled receptors (Thomas et al. 2005). While EDC can act through GPER (Huff et al. 2016; Xu et al. 2017), the focus of the current review will be on EDC actions through the membrane fraction of ESR1 and ESR2, with an emphasis on mESR1, because the majority of data in this area relates to this receptor.
Shuttling of ESR1 to the membrane following synthesis is dependent upon palmitoylation of the receptor at a cysteine residue (Cys447 for human ESR1) by palmitoyl-acyltransferase (PAT) proteins, namely DHHC-7 and -21, which facilitates association of these receptors with caveolin-1 in the membrane (Fig. 1) (Acconcia et al. 2003; Marino and Ascenzi 2006; Pedram et al. 2007; Pedram et al. 2012). Recent data suggest that palmitoylation of this key cysteine residue in human ESR1 evolutionary demarcates it from estrogen-related receptors (Lathe and Houston 2018). In a temporal and dose-dependent manner, E2 seemingly suppresses ESR 1 palmitoylation and consequent interaction of this receptor with caveolin-1 (CAV1) (Acconcia et al. 2004). How such mESR were first discovered and their specific cellular effects are intriguing and are explored next.
Fig. 1. Pathways for the palmitoylation or depalmitoylation of ESR1.
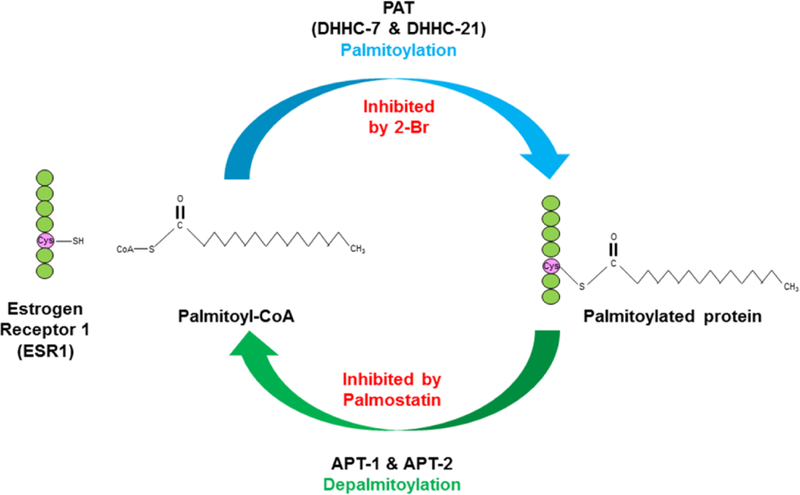
Palmitoylation of estrogen receptor 1 (ESR1) is stimulated by palmitoyl-acyltransferase(s) (DHHC-7 and -21), and this results in palmitoylated ESR1 becoming associated with the plasma membrane; this reaction can be inhibited with 2-bromohexadecanoic acid (2-Br). Conversely, acyl protein thioesterase(s) (APT-1 and -2) induce depalmitoylation of ESR1, and this reaction can be inhibited by with palmostatin.
2. Methods to study membrane-mediated estrogen effects and identification of mESR1
The demonstration by Elwood Jensen and colleagues that radioactively labeled estrogen preferentially localized to nuclei of cells in such organs as the uterus (Jensen et al. 1967) was a landmark discovery that paved the way for subsequent work showing that ligand-bound steroid receptors acted in the nucleus to induce gene transcription (O’Malley and McGuire 1968). Understanding genomic effects of steroid hormones became the principal focus over the next few decades for those seeking to elucidate the mechanism of estrogen’s actions.
Contemporaneous findings by Clara Szego that estrogen had rapid effects on uterine parameters such as cAMP (Szego and Davis 1967) were hard to reconcile with increasing evidence supporting the primacy of nuclear actions in estrogen signaling. In these studies, E2 was administered to ovariectomized female Sprague-Dawley rats, and within minutes of hormone administration, an increase in uterine cAMP levels and eosinophilic infiltration was observed, an effect that could not be accounted for by the nascent model of genomic signaling by hormones (Szego and Davis 1967). Subsequent work by Szego and collaborators provided evidence that estrogen could produce rapid shifts in uterine Ca+2 ion. These effects again were too rapid to be mediated through genomic pathways and initial evidence was obtained for estrogen binding at the membrane level (Pietras and Szego 1975 1977) Study of non-genomic effects of estrogen continued to progress in succeeding decades, but the escalating amount of data related to genomic effects of estrogen overshadowed those discoveries.
Studies in succeeding decades, mainly conducted in the central nervous system, provided additional evidence of rapid membrane effects of estrogens. E2 quickly suppressed spontaneous firing of medial preoptic neurons (Kelly et al. 1977) but sped up the firing rate of pituitary cells (Dufy et al. 1979). Administration of estrogen to guinea pigs rapidly suppressed hyperpolarization of hypothalamic arcuate neurons (Kelly et al. 1992). Potassium-stimulated dopamine release in the rat nucleus accumbens was directly potentiated by E2 (Thompson and Moss 1994), and Ca+2 currents in rat neonstriatal neurons were rapidly reduced following E2 (Mermelstein et al. 1996).
Rapid physiological alterations (generally within minutes) after E2 treatment are generally ascribed to membrane ESR when there would be insufficient time for transcriptional responses through nESR. However, experimental methods were needed that more clearly allowed membrane estrogen effects to be teased out from genomic effects. Initial studies seeking to answer this question tested E2 conjugated with albumin or other heavy molecular weight proteins or tethered the hormone to a substrate to prevent it from being able to penetrate through the cell membrane and enter the cytosol (Ke and Ramirez 1987; Pietras and Szego 1977). While such approaches advanced our understanding of non-genomic and genomic estrogenic effects, individually and in combination, there were limitations with this method, especially in vivo. Hormone administration to rodents in this type of study generally required direct injection into the site of interest, especially when examining neural roles of mESR (Sa et al. 2014). The conjugated protein might detach from E2, as suggested by Stevis et al (Stevis et al. 1999), thereby allowing E2 to enter the cell and induce both non-genomic and genomic effects. Other potential limitations exist with in vitro studies testing E2-BSA. The increased size and structure of E2-BSA may require more time and energy to bind to mESR1 or affect binding properties relative to free E2 (Taguchi et al. 2004; Temple and Wray 2005) These studies also assume that albumin does not affect cellular function, which might be incorrect. Pathological changes attributed to albumin include oxidative stress in renal proximal convoluted tubule cells (Lee et al. 2009), apoptosis (Huang et al. 2009), and extraction of molecules such as arachidonic acid from the plasma membrane and consequent effects on membrane fluidity and signaling (Beck et al. 1998).
To address potential concerns over utilizing E2-BSA as a means of stimulating mESR1 signaling without nESR1-mediated effects, several studies have begun to test an estrogen (17α-ethinyl estradiol; EE2) dendrimer conjugate that because of its charge and size is excluded from the nucleus and therefore only signals through mESR (Harrington et al. 2006). Besides distinguishing extragenomic from genomic effects of estrogens, such compounds might have important therapeutic properties resulting from their activation of mESR, but not nESR. For example, mice treated with estrogen dendrimer conjugate show reduced incidence of thromboembolism, suggestive that such conjugates can affect mESR in megakaryocytes or platelet derivatives (Valera et al. 2015). Other beneficial effects attributed to estrogen dendrimer conjugate include improvement of hepatic steatosis (Chambliss et al. 2016), reduction in cardiac ischemia-reperfusion injury (Menazza et al. 2017), and other cardioprotective effects (Chambliss et al. 2010).
Despite steps forward in understanding non-genomic effects of estrogens, slow progress towards identifying the factor responsible for estrogen binding in the membrane restrained interest in this area. The revelation decades after the initial demonstration of rapid uterine estrogen effects that a fraction of newly synthesized ESR1 is targeted to the cell membrane by palmitoylation finally answered the question as to the identity of the mESR1 and how membrane effects occurred (Pedram et al. 2006; Pedram et al. 2007). In essence, both nuclear and membrane forms are encoded by the same transcripts, and palmitoylation of 5–10% of the synthesized ESR1 directs that fraction to the membrane.
The realization that palmitoylation directs ESR1 to the membrane led to a number of initially in vitro approaches to exploit this in order to selectively interfere with mESR1 actions. Palmitoylacyltransferases (PAT) mediate the palmitoylation process (Acconcia et al. 2003; Marino and Ascenzi 2006; Pedram et al. 2007; Pedram et al. 2012), which can be reversed by acyl protein thioesterases (APT) 1 and 2 (Lin and Conibear 2015; Tabatadze et al. 2013; Won et al. 2018). Predictably, the PA , DHHC-7 and -21, and APT are co-expressed in several ESR positive cells (Meitzen et al. 2013; Pedram et al. 2012; Tabatadze et al. 2013). Thus, palmitoylation and depalmitoylation inhibitors (2-bromohexadecanoic acid [2-Br] and palmostatin, respectively) can be used to increase or decrease mESR1 (Fig. 1). For example, deficiency in palmitoylation results in ESR1 being vulnerable to E2-induced degradation, suppresses ESR1 S118 phosphorylation, and prevents estrogen response element (ERE) promoter occupancy by E2-stimulated ESR1, suggesting the non-genomic affects induced by mESR1 are required for nESR1 transcriptional responses (La Rosa et al. 2012). In vitro mutatgenesis of the cysteine residue at position 447 to alanine in human ESR blocks such forms from being pamitoylated and trafficked to the membrane, effectively creating mESR-deficient cells. HeLa cells transfected with wild-type human ESR1 and then treated with 2-Br have reduced E2-induced transactivation of an ERE construct probe (Acconcia et al. 2004). Comparable responses are obtained with HeLa cells transfected with ESR1 where the Cys447 residue is mutated to Ala, which predictably impaired ESR1 palmitoylation and E2-induced rapid ERK phosphorylation (Acconcia et al. 2004). While in vitro approaches helped decipher potential roles of mESR, subsequent development of transgenic mice with selective ablation of mESR or nESR have allowed the cumulative effects of loss of ESR1 in one cell compartment to be determined.
3. Development of mice lacking mESR1
The separate ESR1 populations in the cell membrane and nucleus raise obvious questions as to the physiological roles of each. Over the past 30 years, transgenic technology in various animal models has permitted inferences to be made on individual gene function. However, mESR1 and nESR1 populations are merely different pools of the same molecule in different locations, as described above. Thus, typical global gene knockout techniques could not be utilized to specifically delete mESR1 or nESR1. However, alternate approaches were developed that allowed elimination of either mESR1 or nESR1, while retaining ESR1 in the other cell compartment. These transgenic mice have played important roles in establishing relative functions of mESR1 and nESR1 and can provide critical information on EDC’s effects and potency through nESR1 and mESR1.
The discovery that ESR1 localization to the cell membrane depended on palmitoylation of a specific cysteine in ESR1 led to generation of a mutated form of ESR1 in which this cysteine was changed to an alanine, an amino acid that cannot be palmitoylated. This altered ESR1 molecule was used initially in in vitro studies to determine the effects of blocking ESR1 localization to the membrane, as described above (Acconcia et al.2004).This altered ESR1 molecule was eventually used to develop transgenic mice with ESR1 that could not be palmitoylated and thus did not migrate to the cell membrane, resulting in deficiencies of mESR1 but normal nESR1 expression/function. These nuclear-only ESR1 mice (NOER) were developed simultaneously by two laboratories, one in Europe and one in the U.S. (Adlanmerini et al. 2014; Pedram et al. 2014).
Despite the similar methodology used to develop these mice, which in both cases involved mutation of cysteine to alanine at position 451 in mouse ESR1, resultant mice demonstrated notable differences. One version of these transgenic mice appeared to have complete ablation of mESR1 (Pedram et al. 2014), while the other only showed reduced amounts of mESR1 (Adlanmerini et al. 2014). Despite normal amounts of functional nESR1, both NOER versions were similar in that they had extensive reproductive abnormalities and both males and females became sterile as adults (Adlanmerini et al. 2014; Nanjappa et al. 2016; Pedram et al. 2014). These results clearly indicate that mESR1 is essential for normal estrogen signaling. The NOER mice, along with other recently developed transgenic mouse lines, provide powerful tools for establishing effects of environmental and other estrogens on mESR1 and nESR1 signaling pathways.
4. Development of mice lacking nESR1
Early in vitro work by Levin and colleagues indicated that the E-domain of human ESR1 alone may be sufficient to mediate mESR1 action (Razandi et al. 2003). This group transfected cells lacking ESR1 with a plasmid encoding a fusion protein (called Emem) consisting of the human ESR1 E-domain, a N-terminal fragment of the CNS protein neuromodulin, and a cyan fluorescent variant of green fluorescent protein. The neuromodulin fragment contains a signal for posttranslational palmitoylation of two cysteines in this peptide, and many investigators have shown that this results in membrane targeting of all protein produced by the Emem transgene. The E-domain of human ESR1 contained in Emem retains the ligand-binding domain and also allows ESR1 dimerization, but lacks the DNA binding domain and thus does not bind DNA or alter transcription. Expression of Emem, driven by a cytomegalovirus promoter, allowed cells to produce mitogen-activated protein kinase (MAPK) and phosphoinositol 3-kinase (PI3K) normally in response to E2, while other ESR1 regions were unnecessary for full MAPK/PI3K responses (Razandi et al. 2003).
Levin and colleagues then developed membrane-only ESR1 (MOER) mice (Pedram et al. 2009), which have Emem knocked in to Esr1KO mice. Despite E2-induced MAPK and PI3K/AKT pathway activation in MOER mice, they were infertile, with uterine hypoplasia and ovaries containing hemorrhagic ovarian follicles similar to Esr1KO mice. Thus, the E-domain of human mESR1 alone without nESR1 could not mediate major E2 effects, confirming that nESR1 was essential for all major actions of E2. This MOER mouse has been used in studies of mESR1 function in liver, adipose and other tissues (Pedram et al. 2013; Pedram et al. 2016).
However, the Emem gene product in MOER mice differs significantly from endogenous mESR1 in that it only has the E-domain of the human ESR1 attached to the neuromodulin fragment and cyan fluorescent variant of green fluorescent protein described above, and the Emem construct is driven constitutively by a cytomegalovirus promoter. Thus, it is unclear whether this Emem transgene in MOER mice can fully substitute for endogenous mESR1 in normal E2 responses in vivo, although it mediates some typical non-genomic E2 effects.
A second transgenic mouse in which mESR1, but not nESR1, signaling is present was recently developed. In a series of reports by Korach and colleagues (Burns et al. 2011; Burns et al. 2014; Stefkovich et al. 2018) a new transgenic H2NES mouse was described and characterized that retains non-genomic ESR1 signaling but lacks genomic ES 1 signaling. The H2NES mouse contains mutations in the nuclear localization sequence in the hinge region (H2) of the ESR1 molecule and also has a nuclear export signal placed at the nuclear localization sequence site for forced exclusion of ESR1 from the nucleus. Critically, reporter assays indicated that ESR1 with the H2NES mutation could not stimulate signaling through EREs in estrogen target genes (Burns et al. 2011). Cells transfected with the H2NES knock-in construct exhibit E2-initiated rapid induction of MAPK in response to estrogen, indicating that rapid membrane E2 actions are maintained and functional in cells expressing H2NES ESR1 (Burns et al. 2011). Female H2NES mice are phenotypically similar to Esr1KO mice (Stefkovich et al. 2018), confirming an essential role of nESR1 in most E2 actions, as was seen with MOER mice. It remains to be determined whether these H2NES mice retain all aspects of normal mESR1-mediated signaling, but these mice provide a potential system to evaluate effects of E2 and other EDC through mESR1. Fig. 2 illustrates the different transgenic mice created to study mESR1 and/or nESR1 actions. We will now consider current evidence that specific EDC can act through mESR1 and in some cases alter the proportion of ESR shuttled to the membrane and thus affect mESR signaling cascades.
Fig. 2. Estrogen receptor 1 signaling in wild-type mice and in various transgenic mouse models used to study actions of ESR1.
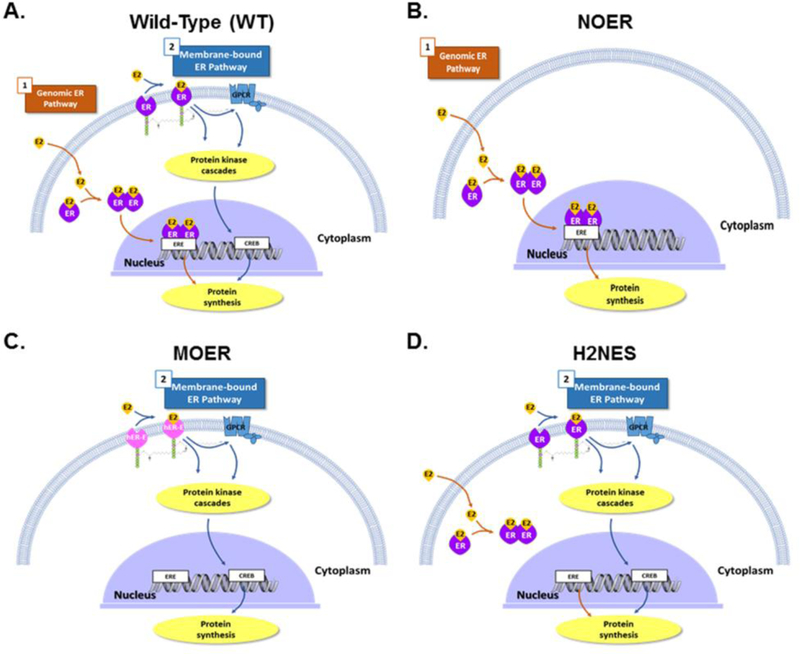
A) In wild-type (WT) mice, genomic estrogen receptor 1 (ESR1,ER) signaling occurs when estrogens bind to cytoplasmic/nuclear receptors, dimerize and then bind to estrogen response element (ERE) within target gene promoter regions and generally induce gene transcription that ultimately leads to translation and increase expression of specific proteins. In the second (non-genomic) pathway for estrogen receptor 1 responses, estrogens bind to membrane estrogen receptor (mESR1,ER), where they can stimulate several intracellular protein kinase cascades and/or G-protein coupled receptors (GPCR), which can result in enhanced cAMP response element-binding protein (CREB)-induced protein transcription, as well as activation of the phosphoinositol 3-kinase/ protein kinase B (PI3K/AKT) and mitogen-activated protein kinase (MAPK) pathways. In addition, 17β-estradiol (E2) or other estrogens could also bind and signal through membrane G protein-coupled estrogen receptor (GPER), which also results in increases in cAMP as well as increased release of epidermal growth factor (EGF) family ligands. In panel A, as well as all the other panels, nuclear and membrane ESR2, as well as GPER, would also be present, but these are not shown in the interest of simplicity. B) Nuclear-only ESR1 (NOER) mice lack membrane ESR1 due to a point mutation in the palmitolyation site (C451A, equivalent site in hESR1 would be Cys447) on this molecule, but the nESR1 pathway remains functional. C) Membrane-only ESR1 (MOER) mice lack both the nESR1 and mESR1 pathways, but a transgene consisting of the E-domain of human ESR1 (hER-E) and another protein fragment that directs all of the protein encoded by this transgene to the membrane has been knocked into these mice, and this mouse has the E-domain of human ESR1 expressed in the plasma membrane. D) Transgenic mice that retain non-genomic ESR1 signaling but lack genomic ESR1 signaling due to mutation in the hinge region of ESR1 (H2NES) possess mutations in the estrogen receptor 1 D-domain, which prevents retention of estrogen-bound ESR1 in the nucleus and thus blocks the classic nuclear pathway involving ligand-bound ESR1 associating with ERE in target genes. Signaling through mESR1 should be unaffected in this transgenic mouse model.
5. Diethylstilbestrol (DES), EE2 and Phytoestrogens
The potent synthetic estrogen DES was administered to pregnant women from 1940–1971 in a misguided and ineffectual attempt to prevent miscarriages (McLachlan 2016). Resultant prenatal DES exposure of female fetuses was later shown to induce adult clear cell vaginal adenocarcinoma and other reproductive abnormalities (Herbst et al. 1971). Prenatal DES exposure in men also produced adult reproductive problems such as undescended testicles, cryptorchidism, hypospadias and epididymal cysts, and some evidence suggests it could increase prostate cancer (reviewed in Palmer et al. 2009). Rodent models developed by McLachlan and colleagues (McLachlan 2016) and others were used extensively to understand and predict consequences of early and even transgenerational DES exposure, and literature on reproductive effects of DES is more extensive than for any other EDC. It is increasingly becoming apparent that pathological findings identified in rodents mirror those identified in humans directly and even transgenerationally exposed to this chemical (Newbold et al. 2006; Tournaire et al. 2018).
DES, as well as other natural and environmental estrogens, can act through mESR1 to increase signaling through the PI3K/AKT pathway and ultimately alter histone methylation in neonatal rodent uteri (Bredfeldt et al. 2010; Greathouse et al. 2012; Wong and Walker 2013).DES was the first xenoestrogen shown to be capable of producting epigenetic modifications in target organs, such as the uterus (Li et al. 1997). These epigenetic effects of DES have been described in males as well, where early DES administration leads to epigenetic changes in seminal vesicle genes (Li et al. 2014; Li et al. 2018a).
EE2, used extensively in human birth control pills, is a potent synthetic derivative of the endogenous estrogen E2. Up to 2 million women per year in the U.S. and Europe continue use of oral contraceptives during undetected early pregnancy (Smithells 1981), and this may continue to the 4th month of gestation (Kallen et al. 1991). Thus, developing fetal reproductive tracts may be exposed to exogenous estrogens during critical developmental periods. In addition, EE2 is also found in drinking water in the U.S. and elsewhere, although the consequences of this are unclear (Caldwell et al. 2010).
Developmental EE2 exposure in rodentsMANUSCRIPTresultsinabnormalitiesinestrogen-responsive organs such as the prostate (Timms et al. 2005). Rapid effects on MAPK expression are induced by EE2 acting through mESR1 in cells that express ESR (Prifti et al. 2003). Estrogens can produce epigenetic changes, and exposure to EE2 produces epigenetic changes that do not mirror those obtained with other environmental contaminants such as BPA (Bhandari et al. 2015; Cheong et al. 2018).
Genistein and daidzein are phytoestrogens present in high quantities in soy and soy-based food products. Soy-based formula for infants was first marketed in 1929 (Hill and Stuart 1929) and currently about 25% of formula-fed infants in the U.S. are given soy-based formula. Total isoflavone consumption for a soy formula-fed infant is 6–9 mg/kg bodyweight/day at 4 months of age (Setchell et al. 1997), and median genistein concentrations of 3.6 μM have been reported in these infants (Cao et al. 2009). Although ultimate health consequences remain to be determined, soy infant formula consumption has been associated with heavier adult menstrual bleeding and larger sizes of uterine fibroid tumors in adulthood (Upson et al, 2016a,b), and some countries actively discourage elective soy formula use due to concerns over consequences of estrogenic exposure of human infants (Westmark 2017).
Genistein induces uterine PI3K/AKT signaling through mESR1 (Greathouse et al. 2012). This results in phosphorylation and inactivation of the histone methyltransferase enhancer of zeste homolog 2 (EZH2) and a reduction in the histone mark H3K27me3. The H3K27me3 mark normally represses transcription of target genes, and decreases in H3K27me3 result in a more open or euchromatin conformation that makes target genes more accessible to the transcriptional apparatus, ultimately causing increased responsiveness of estrogen target genes. Furthermore, developmental exposure to genistein promotes uterine leiomyoma formation (Greathouse et al. 2008), although these effects likely involve signaling through both nESR1 and mESR1. Genistein increases MAPK activation of histone H3 phosphorylation in an immortalized human uterine leiomyoma cell line (Yu et al. 2016), consistent with the possibility of human health effects from consumption of this phytoestrogen.
The large human cohort numbering in the millions that have been fed soy-based infant formula provides a unique resource to determine effects of neonatal genistein exposure in young girls. A recent study of vaginal epithelial cells from infant girls that consumed soy-based infant formulas reported methylation changes associated with decreased expression of the estrogen-responsive gene proline rich 5 like (PRR5L ), with differences in soy formula-fed infants becoming more pronounced with age (Harlid et al. 2017). Mice exposed to genistein at doses that produced serum concentrations similar to those reported in infants fed soy formula showed similar changes in Prr51 expression as the soy formula-fed human infants (Harlid et al. 2017).
Although interest in phytoestrogens typically focused on genistein due to its greater abundance in soy products and greater estrogenic potency, work by Yanagihara et al (2008) showed that daidzein at low concentrations achievable in vivo (0.01 to 1 μM) can signal through mESR to increase extracellular signal-regulated kinase (ERK) 1/2 in bovine adrenomedullary cells. Recent work indicates that equol, a daidzein metabolite, can induce MAPK and thus may be capable of inducing rapid effects through the MAPK/ERK pathway that is typically regulated by mESR1 (Subedi et al. 2017).
Concern over potential epigenetic changes in response to phytoestrogens typically focused on females, and the assumption was that these changes would be undesirable, but some data indicate that genistein could potentially have beneficial effects on prostate cancer through such mechanisms. Genistein increases ESR2 expression in prostate cancer by reducing promoter methylation, and this effect involved downregulation of DNA methyl transferases in prostate cancer cell lines, potentially as a result of epigenetic effects of mESR1 signaling (Mahmoud et al. 2015). ESR2 has a tumor suppressor role in prostate cancer (Mahmoud et al. 2015), and genistein may reduce methylation of the ESR2 promoter, which may underlie preventative actions of genistein on prostatic cancer.
6. Phenolic Chemicals
Bisphenol A (BPA) is one of the most ubiquitous EDC (Schug et al. 2011). Approximately 15 billion lbs/year of this chemical were produced in 2013, with no evidence of any subsequent falloff (GrandViewResearch 2014). BPA is detectable in urine of 93% of the U.S. population, indicating that the vast majority of people are exposed to this chemical (Calafat et al. 2008). This EDC is also detected in fetal plasma and can be transmitted from mother to offspring via the placenta and breast milk (Vandenberg et al. 2007a; vom Saal et al. 2007a). Its environmental stability and pervasiveness has ensured continued human and animal exposures in coming decades (Environment Canada 2008; Vandenberg et al. 2009). Due to consumer concerns regarding BPA, select manufacturers have opted to use BPA substitutes such as bisphenol S (BPS), but such analogs can target similar pathways as BPA and induce comparable disorders (Rosenfeld 2017; Wu et al. 2018). The primary route of exposure to both phenolic chemicals is dietary (Galloway et al. 2010; Sieli et al. 2011), although other routes, including inhalation and transdermal, have been reported (Hines et al. 2018; Xue et al. 2016). A conference at the National Institute of Environmental Health Sciences in 2007 suggested that internal BPA exposure in humans likely exceeded 35 mg/day (~500 μg/kg/day) (Vandenberg et al. 2007b; vom Saal et al. 2007b). This estimate has been increased to reflect increasing production rates and concentrations in U.S. adults were 1.24 μg/L and 0.37 μg/L, respectively (Lehmler et al. 2018).
BPA and BPS act primarily by binding to ESR1 and ESR2 (Grignard et al. 2012; Molina-Molina et al. 2013), where they can induce agonistic (Li et al. 2018b; Sekar et al. 2016) or antagonistic (Molina-Molina et al. 2013; Vigezzi et al. 2016) effects. While most studies have focused on transcriptional effects induced through nESR binding, increasing evidence indicates that these chemicals can induce rapid effects through binding and activation of mESR.
The mESR effects of BPA have been validated in various cell lines and tissue explants. Exposure of human seminoma cells to low BPA concentrations (10−9 to 10−12 ), approximating those identified in human fluids, leads to increased cell proliferation, rapid activation of cAMP-dependent and cGMP-dependent PK pathways and phosphorylation of cAMP response element-binding protein (CREB) and retinoblastoma protein (Bouskine et al. 2009). Co-administration of an anti-estrogen fails to abolish such effects, and treatment with E2 or DES instead suppresses these pathways; activation was achieved only with administration of BSA-bound E2. The above effects of BPA might thus be due to binding to a non-classical ESR that is a G protein-coupled receptor (GPCR).
Treatment of female rat myocytes with E2 or BPA (10−6 to 10−12 M) stimulated a rapid (within minutes) increase in contraction (suggestive of Ca+2 influx) and arrhythmogenic effects in an inverted U-shape dose-dependent manner with 10−9 M eliciting the most pronounced effects (Belcher et al. 2012). Similar effects were not observed in male myocytes or those from ovariectomized mice, suggestive that endogenous estrogens are needed. Treatment with protein conjugated-E2 provided support that mESR mediates the above rapid actions. Further work with ESR inhibitors point to BPA and E2 promoting rapid contraction in female myocytes through mESR2.
Rapid effects of BPA through mESR might also influence neurophysiological responses. To test this possibility, long-term potentiation, which is a persistent strengthening of synapses due to enhanced and recent activity, was assessed in rat hippocampal slices (Chen et al. 2017). Contrasting dose results were obtained, with a low BPA dose (100 nM) enhancing long-term potentiation, whereas high BPA (1000 nM) suppressed LTP. apid effects induced by the low dose of BPA involve mESR activation of the ERK signaling pathway, which was suppressed by co-treatment with E2.
Similarly, the BPA substitute BPS stimulates rapid effects through mESR. In a non-monotonic dose-dependent manner, BPS phosphoactivated ERK in a rat pituitary cell line (CH3/B6/F10), and this reaction occurred within minutes after exposure to this chemical and in a non-monotonic dose-dependent manner at concentrations between 10−5 to 10−7 M (Vinas and Watson 2013b). Concurrent exposure of this cell line to BPS concentrations in the fM to nM range and E2 resulted in enhanced E2-induced c-jun-N-terminal kinase (JNK) activity, reduced cell numbers, and activated apoptosis-associated caspases. Lastly, BPS treatment inhibited the rapid E2 stimulation of prolactin release. A follow-up study by this research group demonstrated swift non-monotonic dose-responsive effects of BPS (fM to nM) on phosphoactivation of ERK,enhanced JNK activity and BPS-induced cellular proliferation when added alone, but BPS and E2 co-treatment resulted in increased apoptosis (Vinas and Watson 2013a).
7. Organochlorine Pesticides
Organochlorine pesticides (OCP) such as1,1,1-trichloro-2,2-bis[4-chlorophenyl]ethane (DDT), methoxychlor [1,1,1-trichloro-2,2-bis(p-methoxyphenyl)ethane], endosulfan, and dieldrin have been and are still widely used for agricultural and industrial applications in the U.S. and globally. Their environmental stability and ability to be concentrated in animals higher up the food chain has ensured wide-spread contamination and exposure for decades to come (Chopra et al. 2011). Despite longstanding health concerns and a ban in the .S., DDT is currently used in places such as Mexico and Africa to control mosquito-borne diseases (Thompson et al. 2017). Exposure at environmentally relevant concentrations can result in endocrine disruption via ESR and other steroid pathways (Leon-Olea et al. 2014). Similar to other xenoestrogens, OCP effects involve both mESR and nESR.
In the rat prolactinoma cell line GH3/B6/F10, multiple xenoestrogens, including OCP and metabolites such as endosulfan, diledrin, and DDE induce rapid (3–30 min post-treatment) and concentration-dependent (10−14 to 10−8M) ERK 1/2 phosphorylation, albeit with unique signature activation patterns (Bulayeva and Watson 2004). Inhibitors of ESR, epidermal growth factor receptors, a+2 signaling, and Src and PI3 kinases affect this stimulation. A later study by this group revealed that low concentrations in the pM to nM range of both E2 and xenoestrogens stimulated a surge in intracellular Ca+2 and time- and concentration-dependent prolactin secretion (Wozniak et al. 2005). Co-administration of the calcium-channel blocker nifedipine prevented xenoestrogen-induced increases in intracellular Ca+2 and prolactin secretion, suggesting Ca+2 influx is a prerequisite for this hormone release.
8. Flame Retardants
Polybrominated diphenyl ethers (PBDEs) are flame retardants commonly used in various consumer products, such as sofas, and are also abundant in the environment. Exposure to this class of chemicals, typically through contaminated dust or food products (Bramwell et al. 2016; Pietron and Malagocki 2017), has been linked with various human diseases (Malliari and Kalantzi 2017; Vuong et al. 2018).
Meerts et al (Meerts et al. 2001) were the first to report estrogenic activity of PBDEs in human T47D breast cancer cells. Within this group of chemicals, it is now apparent that several are ESR1 and ESR2 agonists (Li et al. 2013; Meerts et al. 2001; Mercado-Feliciano and Bigsby 2008). However, some forms, such as 6-OH-BD47, BDE-99, BDE-153, 4’-OH-BDE-17, and 4-OH-BDE-42, are ESR antagonists (Kojima et al. 2009; Mercado-Feliciano and Bigsby 2008).However, these studies did not selectively investigate effects of PBDEs on mESR.
One study testing effects of penta-bromodiphenyl (DE-71) and octa-bromodiphenyl (DE-79) ethers on proinflammatory responses in cultured human bronchial epithelial cells provides hints that such chemicals can bind and activate mESR. Both DE-71 and DE-79 induce increased production of pro-inflammatory cytokines (Koike et al. 2014). Co-treatment with inhibitors of EGFR-selective tyrosine kinase and MAPK attenuates PBDE-induction of IL-6 and IL-8. Such responses might occur via PBDE binding and activation of mESR, although agonistic effects through other receptor pathways cannot be excluded.
9. Heavy metal ions can function as estrogens and signal through mESR1
The EDC initially described to have estrogenic or anti-estrogenic activity were organic compounds that bore some structural resemblance to E2. Decades after the first environmental estrogens were described, it was reported that heavy metal ions could have estrogenic effects. Initial studies focused on cadmium, which was shown to produce estrogenic responses through ESR1 signaling (Garcia-Morales et al. 1994; Stoica et al. 2000). Subsequent work (Johnson et al. 2003) established that classical estrogenic effects, such as increased uterine epithelial height, could be mimicked by cadmium, and anti-estrogens blocked the cadmium effect, clearly indicating these actions occur through ESR. Cadmium also stimulated expression of classical estrogen target genes and promoted mammary gland growth. Perhaps of greatest concern, these effects were seen at environmentally relevant concentrations similar to those in human populations, especially cigarette smokers, who possess high cadmium body burdens. In addition to cadmium, a number of other metals, such as copper, cobalt, nickel, lead, mercury, tin, and chromium also have estrogenic effects (Martin et al. 2003); these estrogenic actions resulted from interactions of the metals with the ESR1 hormone-binding domain, which trigger downstream estrogenic actions though classical genomic pathways.
The amount of work on estrogenic effects of metal ions is far less than on some better-characterized environmental estrogens. Howevever, heavy metals, like other environmental estrogens, also appear to signal through mESR, and this must be taken into account in understanding their overall estrogenic effects. For example, cadmium induces phosphorylation and activation of ERK 1/2 and AKT in various human breast cancer cells lines (Liu et al. 2008; Zang et al. 2009). These effects appear to involve both mESR1 and GPER (Liu et al. 2008; Zang et al. 2009). Later work suggested that proliferation of these breast cancer tumor cell lines by cadmium could be mediated by GPER (Yu et al. 2010) and might also involve cadmium interaction with EGF receptor (Wei et al. 2015). Recent work concluded that proliferative effects of cadmium on human breast cancer cell lines could also involve ESR2 (Huff et al. 2016), although this is not consistent with the initial report of mitogenic effects of cadmium on human breast cancer cell lines which did not identify a role for ESR2 in cadmium-induced estrogenic effects on ERK1/2 and AKT (Liu et al. 2008).
10. Novel mechanisms by which EDCs can affect mESR1 signaling:
While many of the above studies show that EDC binding and activation of mESR induce similar non-genomic effects as E2, other potential mechanisms by which such chemicals can affect mESR function may exist. Palmitoylation directs trafficking of steroid receptors to the membrane (Fig. 1). PAT proteins, specifically DHHC-7 and -21, are required for shuttling of ESR and other steroid receptors to the membrane (Pedram et al. 2012). Depalmitoylation by APT 1 and 2 removes the thioester-linked long chain fatty acids from cysteine residues, including membrane steroid receptors (Tabatadze et al. 2013; Won et al. 2018). The fate of such steroid receptors following this process is uncertain, but presumably they are subjected to lysosomal protein degradation. Membrane ESR1, but perhaps not mESR2 (Pastore et al. 2018), also need to associate with caveolins, specifically CAV-1 and -3, to stimulate downstream pathways. Thus, disruption in expression or function of DHHC-7 and -21, APT, or caveolins will alter mESR function (Fig. 3).
Fig. 3. Mechanisms by which endocrine disrupting chemicals (EDC) can disrupt membrane estrogen receptor signaling.
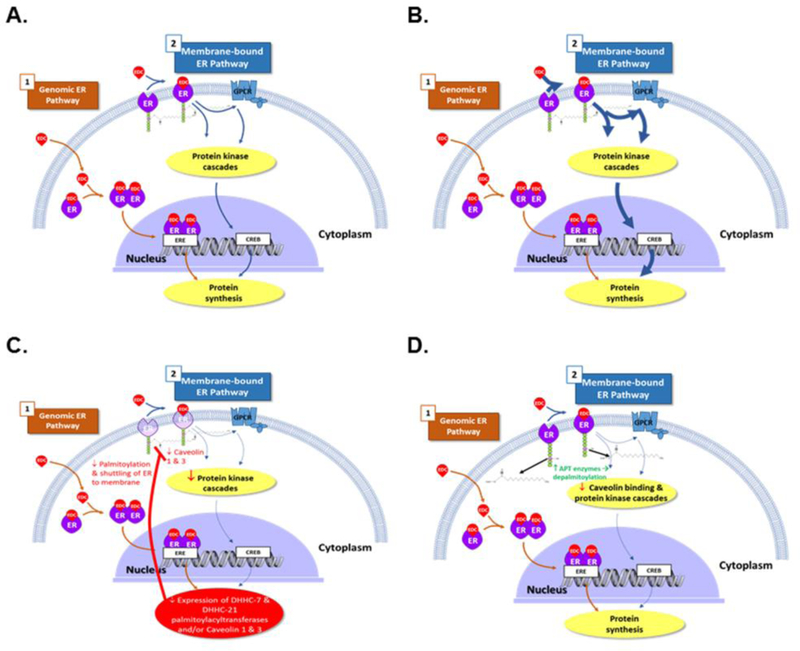
A) In this model, EDC bind with equal affinity to estrogen receptor 1 (ESR1, ER) located in the plasma membrane and cytoplasm/nucleus, and can also act through the G protein-coupled estrogen receptor (GPER) pathway. The EDC can also bind and signal through all of these receptors and disrupt estrogen signaling in this manner. B) Conversely, EDC may preferentially bind and activate membrane ESR1 (mESR1) relative to those located in the cytoplasm/nucleus, resulting in the EDC have more potent non-genomic effects in terms of disrupting mESR1 rather than nuclear ESR1 (nESR1) signaling. C) By inhibiting the DHHC-7 and -21 palmitoylacyltransferases, some EDC might reduce palmitoylation and shuttling of ESRs to the plasma membrane, which would result in decreased mESR1 signaling compared to nESR1 signaling. EDCs might also disrupt membrane ESR1 action by reducing expression of caveolin 1 3, which are essential for mESR1 action. D) EDC can potentially increase the transcription and translation of the depalmitoylating enzymes, acyl protein thioesterase(s) (APT1 and APT2), leading to depalmitoylation of membrane ESR1, decreased caveolin binding, and reduced signaling through the mESR1 pathway.
Although there is limited evidence, some reports suggest EDC can disrupt palmitoylation/depalmitoylation. In the rat hippocampus, intact females express reduced levels of Dhhc7 and Cav1 compared to males, suggestive that these genes are estrogen-sensitive (Meitzen et al. 2017). The flavonone naringenin induces anti-estrogenic effects in HepG2 and HeLa cells by stimulating ESR1 depalmitoylation more rapidly than E2 and consequent dissociation of the mESR from CAV1 (Galluzzo et al. 2008).
Additional studies suggest that estrogen/ESR regulate caveolin expression. In the neuronal cell line SK-N-MC, ESR1 signaling results in ligand-independent transcriptional suppression of CAV1 and 2, presumably due to epigenetic modifications (Zschocke et al. 2002). Ovariectomized rats show increased numbers of caveolae and elevated expression of CAV1 and 2 in uterine smooth muscle cells (Turi et al. 2001); Conversely, E2 treatment of ovariectomized rats diminishes the number of caveolae and protein expression of membrane-associated CAV1 and 2, and these efects on CAV expression are reversisble and even augmented by co-administration of an ESR antagonist. Microarray analyses reveal that treatment of ovariectomized mice with o,p′-DDT and EE2 reduces uterine expression of Cav1 and Cav3 (Kwekel et al. 2005; Kwekel et al. 2013)
11. Potential selective binding of EDC to membrane versus nuclear estrogen receptor 1
Net effects of EDCs are influenced by their binding affinity to mESR1 and nESR1. In many cases, it is presumed that such compounds bind with equal affinity to mESR and nESR. However, certain DC might have higher binding affinity for mESR that approximates that of E2 (Fig. 3). Moreover, splice variants exists for both mESR and nESR that can affect binding affinity to E2 and various EDC.
Most EDC bind with weaker affinity than E2 to classical nESR1 and 2 (Kuiper et al. 1998; Laws et al. 2000). Conversely, low dose (1 nM) BPA or E2 treatment of primary cultures of mouse pancreatic islets induces rapid (within five minutes) phosphoactivation of CREB and both chemicals show equal potency (Quesada et al. 2002). Similar responses are observed with membrane-impermeable E2 conjugated to horseradish peroxidase, but an ESR antagonist fails to suppress this response, implicating other non-classical mESR. Comparably, nM concentrations of BPA, DES, and E2-HRP rapidly suppress low-glucose-stimulation of intracellular Ca+2 oscillations in mouse α-pancreatic islet cells, which in turn activate glucagon secretion (Alonso-Magdalena et al. 2005). Competition studies with E2 conjugated to horseradish peroxidase indicate all three share a common membrane-binding site that is independent of classic ESR.
Other studies suggest that low doses of BPA and other EDC induce equivalent mESR responses as E2. For instance, both low doses of BPA and E2 (10−9 to 10−12 M) stimulate rapid increases in myocyte contraction and arrhythmogenic effects in females but not males (Belcher et al. 2012). Several xenoestrogens, including OCP, nonylphenol, BPA, coumestrol and DES at pM to nM concentrations elicit membrane ESR-induced Ca+2 fluxes and prolactin release by rat GH3/B6 pituitary tumor cells (Wozniak et al. 2005).
Using cell-free prokaryotic and eukaryotic expression systems with select mESR1 forms and based on saturation binding, 3H-E2 binds differentially to ESR isoforms mESR1 66 and mESR1 46, with Kd values of ~69 and 61 pM, respectively, but within the test range, no binding to human mESR1 36 is detected (Lin et al. 2013). Inhibition of palmitoylation by 2-Br decreases E2 binding affinity to mESR1 66 and mESR1 46 to Kd values of 185 pM and 337.5 pM, respectively. The isoforms mESR1 66 and mESR1 46 bind differentially to phytoestrogens (genistein, daidzein, and kaempferol), ESR agonists (PPT, DPN, and G1), and ESR antagonists (tamoxifen, raloxifene, and ICI 182,780). Further studies are needed to examine whether EDC, such as BPA, OCP, heavy metals, and PBDE differentially bind isoforms of mESR1 and mESR2. If so, remediation strategies might involve therapies designed to target specific mESR isoforms, such as with xenoestrogen dendrimer conjugates (Harrington et al. 2006).
12. Epigenetic effects induced by environmental endocrine disruptors
In recent years, there has been intense interest in epigenetic changes induced by estrogens. These epigenetic effects can increase tumor susceptibility or cause other reproductive pathologies. Epigenetic changes can heritably alter gene function, but do so without causing DNA changes. Although estrogen induced epigenetic modifications induced were reported 20 years ago (Li et al. 1997), the mechanism of these effects remains unclear. Early DES exposure stimulates target gene activity and produces short-term changes in uterine epithelial proliferation, secretory protein production and other classical estrogen effects. In addition, early exposure to DES or other EDC alters methylation of target genes and histones (Bromer et al. 2010; Doherty et al. 2010). This can result in long-term alteration of target gene expression that increases susceptibility to adult pathologies without changing the DNAsequence.
Evidence from various in vitro and in vivo studies indicates that epigenetic effects of estrogens may involve mESR1. Membrane estrogen receptors activate PI3K/AKT and MAPK and increase intracellular cAMP and Ca+2 (Adlanmerini et al. 2014; Pedram et al. 2014; Pietras and Szego 1975; Szego and Davis 1967). Estrogen binding causes mESR1 to interact with the p85α regulatory subunit of PI3K, leading to AKT activation (Cosentino et al. 2007). Estrogens, including environmental estrogens, can act through mESR1 to increase signaling through the PI3K/AKT pathway and ultimately alter histone methylation in neonatal rodent uteri (Bredfeldt et al. 2010; Greathouse et al. 2012; Wong and Walker 2013).
The polycomb repressive complex 2 (PRC2) enzyme complex is the most critical regulator of epigenetic changes such as histone methylation. This PRC2 is a histone methyltransferase that can silence gene activity through histone methylation. The PRC2 functions by adding methyl groups at lysine-27 of histone H3 (H3K27) to form trimethylated histone H3 (H3K27me3). Enhancer of Zeste homolog 2 (EZH2) is the catalytic subunit of the PRC2 complex that provides methyltransferase activity, and thus PRC2 activity is regulated primarily by expression of EZH2.
Bhan et al (Bhan et al. 2014) have shown that mammary gland EZH2 is regulated transcriptionally, and the EZH2 promoter has several functional EREs. Recent work from one of our laboratories (Nanjappa et al. Submitted) indicates that estrogen may also regulate EZH2 expression transcriptionally in mouse uteri. In addition to xenoestrogen effects on EZH2, xenoestrogen-induced increases in phosphorylated and active AK are capable of phosphorylating and changing the activity of EZH2 and thus altering histone methylation and ultimately gene activity (Fig. 4) (Bredfeldt et al. 2010; Greathouse et al. 2012). DNA methylation profiles can also be modified by estrogens and xenoestrogens.
Fig. 4. Membrane estrogen receptor activation can have effects on enhancer of zeste homolog 2 (EZH2) and epigenetic imprinting.
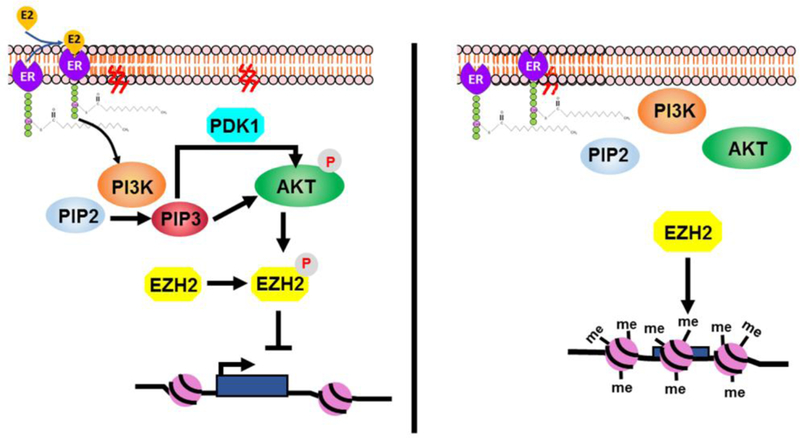
Left panel: Binding of E2, other estrogens or EDC can activate the phosphoinositol 3-kinase/ protein kinase B (PI3K/AKT) pathway, resulting in phosphorylation EZH2, which inactivates it and blocks it from methylating histone 3 (H3) proteins at certain sites (e.g., H3K27me3), resulting in a more open chromatin (euchromatin) arrangement that favors gene transcription. Right panel: In contrast, lack of mESR1 activation results in decreased phosphorylation of EZH2, which causes more extensive methylation of histones by EZH2 and a more compacted chromatin (heterochromatin) that is less transcriptionally active.
Extensive work has shown that maternal BPA exposure during gestation can modify DNA methylation in target genes in the offspring (Dolinoy et al. 2007). Maternal or perinatal exposure to BPA (Bromer et al. 2010; Cheong et al. 2018) or other EDC such as diethylhexyl phthalate (DEHP) (Manikkam et al. 2013) can also decrease estrogen target gene methylation in uterus, leading to increased gene expression in response to estrogen. In addition to epigenetic effects which alter methylation and activity of estrogen target genes, EDC such as BPA can also have epigenetic effects on ESR1 and ESR2 expression (Doshi et al. 2011), and thus alter estrogen responsiveness. Transgenerational effects described for various endocrine disruptors (Hiyama et al. 2011) likely result from these types of effects on DNA methylation, histone protein modifications, or alterations in heritable microRNAs (Brieno-Enriquez et al. 2015; Singh and Li 2012), although it is not clear how enzymes catalyzing DNA methylation and histone protein modification confer memory-specificity across generations (Ptashne 2013). While work on epigenetic BPA effects typically focus on females, changes in DNA and histone methylation signatures following developmental BPA exposure could also alter prostate cancer susceptibility in males (Prins et al. 2017; Wang et al. 2016).
13. Conclusions
It is increasingly apparent that a wide variety of EDC, including non-classic xenoestrogens, can signal through mESR. The fact that EDC, at low concentrations (nM and pM range) that are environmentally relevant, can stimulate rapid effects through mESR suggests that the non-genomic actions of these xenoestrogens are important and thus merit further study. In addition to short-term effects mediated through mESR, signaling through mESR also has the potential to induce epigenetic marks that can be permanent and transgenerationally propagated.These results collectively indicate that EDC acting through mESR may induce both rapid physiological changes and longstanding epigenetic modifications. Clearly, a full assessment of the effects of EDC must take the totality of their actions mediated through both ESR signaling pathways into account.
Select data indicate that EDC may have more potent effects relative to E2 through mESR than through nESR. These provocative findings indicate that assessments of the relative potency of various EDC based solely on binding to nESR or in stimulating genomic effects may underestimate their overall potential to disrupt endocrine signaling. Further work is needed in this area to not only confirm these initial reports but also to develop a plausible mechanistic explanation for such potential differences in signaling potency through ESR that should be structurally identical. In addition to direct effects through mESR, EDC can alter palmitoylation and the amount of mESR through effects on both palmitoylation and CAV, and these effects could have significant impacts on the ultimate response of a cell or tissue to a particular EDC
The development of various transgenic mice with selective ablation of mESR or nESR provide useful tools to understand the comprehensive non-genomic and genomic effects of both E2 and EDC. Work is currently underway in our laboratories to examine effects of DES in these mouse models. Other investigators are encouraged to consider testing the effects of other EDC in these mouse models. Additional work is also needed in these mouse models to determine how removal of one or both ESR forms affects associated regulatory pathways, including enzymes regulating palmitoylation, DHHC-7 and -21, and depalmitoylation, APT-1 and -2. Future results in this rapidly developing area should provide a more complete picture of short- and long-term (epigenetic) effects of both endogenous and exogenous estrogens occurring through mESR signaling and how such changes underpin clinical disorders.
Highlights:
Estrogens can signal through membrane (m) and nuclear (n) estrogen receptor 1 (ESR1).
mESR1 is from same transcript as nESR1 but directed to membrane via palmitoylation.
E2 and xenoestrogen signaling through mESR1 induces rapid cellular changes.
Endocrine disrupting chemicals (EDC) might affect shuttling of ESR to membrane.
Mice lacking m SR1 or nESR1 can be used to test EDC-effects via mESR1 +/− nESR1.
Acknowledgments
Funding: This work was supported in part by NIH grants R03 HD087528, R21 HD088006, and R01 PR015540 (to . S. Cooke). C.S. Rosenfeld was supported by NIH grant R01 ES025547.
Abbreviations
- 2-Br
2-bromohexadecanoic acid
- AKT
protein kinase B
- APT
acyl protein thioesterase(s), e.g. APT-1 and -2
- BPA
bisphenol A
- BPS
bisphenol S
- BPA
bovine serum albumin
- CAV1/Cav1
caveolin-1
- Cav3
caveolin-3
- CREB
cAMP response element-binding protein
- DDT
1,1-trichloro-2,2-bis[4-chlorophenyl]ethane
- DE-71
penta-bromodiphenyl
- DE-79
octa-bromodiphenyl
- DEHP
diethylhexyl phthalate
- DES
diethylstilbestrol
- E2
17β-estradiol
- E2BP
estradiol-binding protein
- EDC
endocrine disrupting chemical(s)
- EE2
17α-ethinyl estradiol
- EGF
epidermal growth factor
- Emem
transgene human ESR1 E-domain, a N-terminal fragment of the CNS protein neuromodulin
- ERE
estrogen response element
- ERK
extracellular signal-regulated kinase
- ESR
estrogen receptor(s)
- Esr1KO
estrogen receptor 1 knockout
- EZH2
enhancer of Zeste homolog 2
- GPCR
G protein-coupled receptors
- GPER
G protein-coupled estrogen receptor
- H2NES
transgenic mice that retain non-genomic ESR1 signaling but lack genomic ESR1 signaling due to mutation in the hinge region of ESR1
- H3
histone H3
- JNK
c-jun-N-terminal kinase
- MAPK
mitogen-activated protein kinase
- mESR
membrane estrogen receptor(s)
- MOER
membrane-only ESR1
- nESR
nuclear estrogen receptor(s)
- NOER
nuclear-only ESR1
- OCP
organochlorine pesticides
- PAT
palmitoyl-acyltransferase(s)
- e.g.
DHHC-7 and -21
- PBDE
polybrominated diphenyl ether
- PI3K
phosphoinositol 3-kinase
- PK
protein kinase(s)
- PRC2
polycomb repressive complex 2
- PRR5L/Prr5l
proline rich 5 like
- SHBG
sex hormone binding globulins
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- Acconcia F, Bocedi A, Ascenzi P, Marino M. 2003. Does palmitoylation target estrogen receptors to plasma membrane caveolae? IUBMB Life 55:33–35. [DOI] [PubMed] [Google Scholar]
- Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. 2004. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun 316:878–883. [DOI] [PubMed] [Google Scholar]
- Adlanmerini M, Solinhac R, Abot A, Fabre A,Raymond-LetronI Guihot AL,et al. 2014. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A 111:E283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Laribi O, Ropero AB, Fuentes E, Ripoll C, Soria B, et al. 2005. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ Health Perspect 113:969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck R, Bertolino S, Abbot SE, Aaronson PI, Smirnov SV. 1998. Modulation of arachidonic acid release and membrane fluidity by albumin in vascular smooth muscle and endothelial cells. Circ Res 83:923–931. [DOI] [PubMed] [Google Scholar]
- Belcher SM, Chen Y, Yan S, Wang HS. 2012. Rapid estrogen receptor-mediated mechanisms determine the sexually dimorphic sensitivity of ventricular myocytes to 17β-estradiol and the environmental endocrine disruptor bisphenol . Endocrinology 153:712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhan A, Hussain I,Ansari KI, Bobzean SA, Perrotti LI, Mandal SS. 2014. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-A and diethylstilbestrol. J Mol Biol 426:3426–3441. [DOI] [PubMed] [Google Scholar]
- Bhandari RK, Deem SL, Holliday DK, Jandegian CM, Kassotis CD, Nagel SC, et al. 2015. Effects of the environmental estrogenic contaminants bisphenol A and 17α-ethinyl estradiol on sexual development and adult behaviors in aquatic wildlife species. Gen Comp Endocrinol 214:195–219. [DOI] [PubMed] [Google Scholar]
- Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. 2009. Low doses of bisphenol promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect 117:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramwell L, Glinianaia SV, Rankin J, Rose M, Fernandes A, Harrad S, et al. 2016. Associations between human exposure to polybrominated diphenyl ether flame retardants via diet and indoor dust, and internal dose: A systematic review. Environ Int 92-93:680–694. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. 2010. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol 24:993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieno-Enriquez MA, Garcia-Lopez J, Cardenas DB, Guibert S, Cleroux E, Ded L, et al. 2015. Exposure to endocrine disruptor induces transgenerational epigenetic deregulation of microRNAs in primordial germ cells. PLOS One 10:e0124296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromer JG, Zhou Y, Taylor MB, Doherty L, Taylor HS.2010Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J 24:2273–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulayeva NN, Watson CS. 2004. Xenoestrogen-induced ERK-1 and ERK-2 activation via multiple membrane-initiated signaling pathways. Environ Health Perspect 112:1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KA, Li Y, Arao Y, Petrovich RM, Korach KS. 2011. Selective mutations in estrogen receptor alpha D-domain alters nuclear translocation and non-estrogen response element gene regulatory mechanisms. J Biol Chem 286:12640–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KA, Li Y, Liu L, Korach KS. 2014. Research resource: comparison of gene profiles from wild-type ERα and ERα hinge region mutants. Mol Cell Endocrinol 28:1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. 2008. Exposure of the .U.S. Population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect 116:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell DJ, Mastrocco F, Nowak E, Johnston J, Yekel H, Pfeiffer D, et al. 2010. An assessment of potential exposure and risk from estrogens in drinking water. Environ Health Perspect 118:338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Calafat AM, Doerge DR, Umbach DM, Bernbaum JC, Twaddle NC, et al. 2009. Isoflavones in urine, saliva, and blood of infants: Data from a pilot study on the estrogenic activity of soy formula. J Expo Sci Environ Epidemiol 19:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, et al. 2010. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest 120:2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss KL, Barrera J, Umetani M, Umetani J, Kim SH, Madak-Erdogan Z, et al. 2016Nonnuclear estrogen receptor activation improves hepatic steatosis in female mice. Endocrinology 157:3731–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang Y, Xu F, Wei X, Zhang J, Wang C, et al. 2017. The rapid effect of bisphenol-A on long-term potentiation in hippocampus involves estrogen receptors and ERK activation. Neural Plast 2017:5196958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong A, Johnson SA, Howald EC, Ellersieck MR, Camacho L, Lewis SM, et al. 2018. Gene expression and DNA methylation changes in the hypothalamus and hippocampus of adult rats developmentally exposed to bisphenol A or ethinyl estradiol: A CLARITY-BPA Consortium study. Epigenetics:1–17. [DOI] [PMC free article] [PubMed]
- Chopra AK, Sharma MK, Chamoli S. 2011. Bioaccumulation of organochlorine pesticides in aquatic system--an overview. Environ Monit Assess 173:905–916. [DOI] [PubMed] [Google Scholar]
- Cosentino C, Di Domenico M, Porcellini A, Cuozzo C, De Gregorio G, Santillo MR, et al. 2007P85 regulatory subunit of PI3K mediates cAMP-PKA and estrogens biological effects on growth and survival. Oncogene 26:2095–2103. [DOI] [PubMed] [Google Scholar]
- De Coster S, van Larebeke N. 2012. Endocrine-disrupting chemicals: Associated disorders and mechanisms of action. J Environ Public Health 2012:713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty LF, Bromer JG, Zhou Y, Aldad TS, Taylor HS. 2010. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: An epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer 1:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. 2007. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 104:13056– 13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi T, Mehta SS, Dighe V, Balasinor N, Vanage G. 2011. Hypermethylation of estrogen receptor promoter region in adult testis of rats exposed neonatally to bisphenol A. Toxicology 289:74–82. [DOI] [PubMed] [Google Scholar]
- Dufy B, Vincent JD, Fleury H, Du Pasquier P, Gourdji D, Tixier-Vidal A. 1979. Membrane effects of thyrotropin-releasing hormone and estrogen shown by intracellular recording from pituitary cells. Science 204:509–511. [DOI] [PubMed] [Google Scholar]
- Environment Canada. 2008. Screening assessment for the challenge phenol, 4,4’ -(1-methylethylidene)bis-(bisphenol A) chemical abstracts service registry number 80-05–7. [Google Scholar]
- Galloway T, Cipelli R, Guralnick J, Ferrucci L, Bandinelli S, Corsi AM, et al. 2010. Daily bisphenol A excretion and associations with sex hormone concentrations: Results from the InCHIANTI adult population study. Environ Health Perspect 118:1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzo P, Ascenzi P, Bulzomi P, Marino M. 2008. The nutritional flavanone naringenin triggers antiestrogenic effects by regulating estrogen receptor alpha-palmitoylation. Endocrinology 149:2567–2575. [DOI] [PubMed] [Google Scholar]
- Garcia-Morales P, Saceda M, Kenney N, Kim N, Salomon DS, Gottardis MM, et al. 1994. Effect of cadmium on estrogen receptor levels and estrogen-induced responses in human breast cancer cells. J Biol Chem 269:16896–16901. [PubMed] [Google Scholar]
- GrandViewResearch. 2014. Global bisphenol A (BPA)market by appliation (appliances, automotive, consumer, construction, electrical & electronics) expected to reach usd 20.03 billion by 2020 Http://www.Digitaljournal.Com/pr/2009287.
- Greathouse KL, Cook JD, Lin K, Davis BJ, Berry TD, Bredfeldt TG, et al. 2008. Identification of uterine leiomyoma genes developmentally reprogrammed by neonatal exposure to diethylstilbestrol. Reprod Sci 15:765–778. [DOI] [PubMed] [Google Scholar]
- Greathouse KL, Bredfeldt T, Everitt JI, Lin K, Berry T, Kannan K, et al. 2012. Environmental estrogens differentially engage the histone methyltransferase EZH2 to increase risk of uterine tumorigenesis. Mol Cancer Res 10:546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignard E, Lapenna S, Bremer S. 2012. Weak estrogenic transcriptional activities of bisphenol A and bisphenol S. Toxicol In Vitro 26:727–731. [DOI] [PubMed] [Google Scholar]
- Harlid S, Adgent M, Jefferson WN, Panduri V, Umbach DM, Xu Z, et al. 2017. Soy formula and epigenetic modifications: Analysis of vaginal epithelial cells from infant girls in the IFED study. Environ Health Perspect 125:447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, et al. 2006. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol 20:491–502. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Ulfelder H, Poskanzer DC. 1971. Adenocarcinoma of the vagina: Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med 284:878–881. [DOI] [PubMed] [Google Scholar]
- Hill LW, Stuart HC. 1929. soy bean food preparation for feeding infants with milk idosyncrasy. J Am Med Assoc 93:985–987. [Google Scholar]
- Hines CJ, Christianson AL, Jackson MV, Ye X, Pretty JR, Arnold JE, et al. 2018. An evaluation of the relationship among urine, air, and hand measures of exposure to bisphenol A (BPA) in US manufacturing workers. Ann Work Exp Health 62:840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiyama M, Choi EK, Wakitani S, Tachibana T, Khan H, Kusakabe KT, et al. 2011. Bisphenol-A (BPA) affects reproductive formation across generations in mice. J Vet Med Sci 73:1211–1215. [DOI] [PubMed] [Google Scholar]
- Huang CY, Liang CM, Chu CL, Liang SM. 2009. Albumin fibrillization induces apoptosis via integrin/FAK/Akt pathway. BMC Biotechnol 9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff MO, Todd SL, Smith AL, Elpers JT, Smith AP, Murphy RD, et al. 2016. Arsenite and cadmium activate MAPK/ERK via membrane estrogen receptors and G-protein coupled estrogen receptor signaling in human lung adenocarcinoma cells. Toxicol Sci 152:62–71. [DOI] [PubMed] [Google Scholar]
- Iqbal MJ, Dalton M, Sawers RS. 1983. Binding of testosterone and oestradiol to sex hormone binding globulin, human serum albumin and other plasma proteins: Evidence for non-specific binding of oestradiol to sex hormone binding globulin. Clinical Sci 64:307–314. [DOI] [PubMed] [Google Scholar]
- Jensen EV, Desombre ER, Kawashima T, Suzuki T, Kyser K, Jungblut PW. 1967. Estrogen-binding substances of target tissues. Science 158:529–530. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, Chepko G, et al. 2003. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med 9:1081–1084. [DOI] [PubMed] [Google Scholar]
- Kallen B, Mastroiacovo P, Lancaster PA, Mutchinick O, Kringelbach M, Martinez-Frias ML, et al. 1991. Oral contraceptives in the etiology of isolated hypospadias. Contraception 44:173–182. [DOI] [PubMed] [Google Scholar]
- Ke FC, Ramirez VD. 1987. Membrane mechanism mediates progesterone stimulatory effect on LHRH release from superfused rat hypothalami in vitro. Neuroendocrinology 45:514–517. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Moss RL, Dudley CA. 1977. The effects of microelectrophoretically applied estrogen, cortisol and acetylcholine on medial preoptic-septal unit activity throughout the estrous cycle of the female rat. Exp Brain Res 30:53–64. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Loose MD, Ronnekleiv OK. 1992. Estrogen suppresses mu-opioid- and GABAB-mediated hyperpolarization of hypothalamic arcuate neurons. J Neurosci 12:2745–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike E, Yanagisawa R, Takigami H, Takano H. 2014. Penta- and octa-bromodiphenyl ethers promote proinflammatory protein expression in human bronchial epithelial cells in vitro. Toxicol In Vitro 28:327–333. [DOI] [PubMed] [Google Scholar]
- Kojima H, Takeuchi S, Uramaru N, Sugihara K, Yoshida T, Kitamura S. 2009. Nuclear hormone receptor activity of polybrominated diphenyl ethers and their hydroxylated and methoxylated metabolites in transactivation assays using chinese hamster ovary cells. Environ Health Perspect 117:1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GGJM, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, et al. 1998. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 139:4252–4263. [DOI] [PubMed] [Google Scholar]
- Kwekel JC, Burgoon LD, Burt JW, Harkema JR, Zacharewski TR. 2005. A cross-species analysis of the rodent uterotrophic program: Elucidation of conserved responses and targets of estrogen signaling. Physiol Genomics 23:327–342. [DOI] [PubMed] [Google Scholar]
- Kwekel JC, Forgacs AL, Williams KJ, Zacharewski TR .2013o-p′-DDT-mediated uterotrophy and gene expression in immature C57BL/6 mice and Sprague-Dawley rats. Toxicol Appl Pharmacol 273:532–541. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski CF, Bolden AL, Liroff RA, Rochester JR, Vandenbergh JG. 2016. Twenty-five years of endocrine disruption science: Remembering Theo olborn. Environ Health Perspect 124:A151–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa P, Pesiri V, Leclercq G, Marino M, Acconcia F. 2012. Palmitoylation regulates 17β-estradiol-induced estrogen receptor-alpha degradation and transcriptional activity. Mol Endocrinol 26:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathe R, Houston DR. 2018. Fatty-acylation target sequence in the ligand-binding domain of vertebrate steroid receptors demarcates evolution from estrogen-related receptors. J Steroid Biochem Mol Biol pii: S0960-0760(18)30383–2. [DOI] [PubMed] [Google Scholar]
- Laws SC, Carey SA, Ferrell JM, Bodman GJ, Cooper RL. 2000. Estrogenic activity of octylphenol, nonylphenol, bisphenol A and methoxychlor in rats. Tox Sci 54:154–167. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Suh HN, Han HJ. 2009. ffect of BSA-induced ER stress on SGLT protein expression levels and alpha-MG uptake in renal proximal tubule cells. Am J Physiol Renal Physiol 296:F1405–1416. [DOI] [PubMed] [Google Scholar]
- Lehmler HJ, Liu B, Gadogbe M, Bao W. 2018. Exposure to bisphenol A, bisphenol F, and bisphenol S in U.S. Adults and children: The national health and nutrition examination survey 2013–2014. ACS Omega 3:6523–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon-Olea M, Martyniuk CJ, Orlando EF, Ottinger MA, Rosenfeld CS, Wolstenholme J, et al. 2014. Current concepts in neuroendocrine disruption. Gen Comp Endocrinol 203:158–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Washburn KA, Moore R, Uno T, Teng C, Newbold RR, et al. 1997. Developmental exposure to diethylstilbestrol elicits demethylation of estrogen-responsive lactoferrin gene in mouse uterus. Cancer Res 57:4356–4359. [PubMed] [Google Scholar]
- Li X, Gao Y, Guo LH, Jiang G. 2013. Structure-dependent activities of hydroxylated polybrominated diphenyl ethers on human estrogen receptor. Toxicology 309:15–22. [DOI] [PubMed] [Google Scholar]
- Li Y, Hamilton KJ, Lai AY, Burns KA, Li L, Wade PA, et al. 2014. Diethylstilbestrol (DES)-stimulated hormonal toxicity is mediated by ERα alteration of target gene methylation patterns and epigenetic modifiers (DNMT3A, MBD2, AND HDAC2) in the mouse seminal vesicle. Environ Health Perspect 122:262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hamilton KJ, Wang T, Coons LA, Jefferson WN, Li R, et al. 2018a. DNA methylation and transcriptome aberrations mediated by ERα in mouse seminal vesicles following developmental DES exposure. Proc Natl Acad Sci U S A 115:E4189–e4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Perera L, Coons LA, Burns KA, Tyler Ramsey J, Pelch KE, et al. 2018b. Differential in vitro biological action, coregulator interactions, and molecular dynamic analysis of bisphenol A (BPA), BPAF, and BPS ligand-ERα complexes. Environ Health Perspect 126:017012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AH, Li RW, Ho EY, Leung GP, Leung SW, Vanhoutte PM, et al. 2013. Differential ligand binding affinities of human estrogen receptor-alpha isoforms. PLoS One 8:e63199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Conibear E. 2015. Enzymatic protein depalmitoylation by acyl protein thioesterases. Biochem Soc Trans 43:193–198. [DOI] [PubMed] [Google Scholar]
- Liu Z, Yu X, Shaikh ZA. 2008. Rapid activation of ERK1/2 and AKT in human breast cancer cells by cadmium. Toxicol Appl Pharmacol 228:286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud AM, Al-Alem U, Ali MM, Bosland MC. 2015. Genistein increases estrogen receptor beta expression in prostate cancer via reducing its promoter methylation. Steroid Biochem Mol Biol 152:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malliari E, Kalantzi OI. 2017. Children’s exposure to brominated flame retardants in indoor environments - a review. Environ Int 108:146–169. [DOI] [PubMed] [Google Scholar]
- Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. 2013. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLOS One 8:e55387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M, Ascenzi P 2006. Steroid hormone rapid signaling: The pivotal role of S-palmitoylation. IUBMB life 58:716–719. [DOI] [PubMed] [Google Scholar]
- Martin MB, Reiter R, Pham T, Avellanet YR, Camara J, Lahm M, et al. 2003. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology 144:2425–2436. [DOI] [PubMed] [Google Scholar]
- McLachlan JA. 2016. Environmental signaling: From environmental estrogens to endocrine‐ disrupting chemicals and beyond. Andrology 4:684–694. [DOI] [PubMed] [Google Scholar]
- Mean F, Pellaton M, Magrini G. 1977. Study on the binding of dihydrotestosterone, testosterone and oestradiol with sex hormone binding globulin. Clinica Chimica Acta 80:171–180. [DOI] [PubMed] [Google Scholar]
- Meerts IA, Letcher RJ, Hoving S, Marsh G, Bergman A, Lemmen JG, et al. 2001. In vitro estrogenicity of polybrominated diphenyl ethers, hydroxylated PDBEs, and polybrominated bisphenol A compounds. Environ Health Perspect 109:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitzen J, Luoma JI, Boulware MI, Hedges VL, Peterson BM, Tuomela K, et al. 2013Palmitoylation of estrogen receptors is essential for neuronal membrane signaling. Endocrinology 154:4293–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitzen J, Britson KA, Tuomela K, Mermelstein PG. 2017. The expression of select genes necessary for membrane-associated estrogen receptor signaling differ by sex in adult rat hippocampus. Steroids pii: S0039–128X(17)30179–4. doi: 10.1016/j.steroids.2017.09.012:epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menazza S, Sun J, Appachi S, Chambliss KL, Kim SH, Aponte A, et al. 2017. Non-nuclear estrogen receptor alpha activation in endothelium reduces cardiac ischemia-reperfusion injury in mice. J Mol Cell Cardiol 107:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado-Feliciano M, Bigsby RM. 2008. The polybrominated diphenyl ether mixture DE-71 is mildly estrogenic. Environ Health Perspect 116:605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Becker JB, Surmeier J. 1996. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci 16:595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Molina JM, Amaya E, Grimaldi M, Saenz JM, Real M, Fernandez MF, et al. 2013. In vitro study on the agonistic and antagonistic activities of bisphenol-S and other bisphenol-A congeners and derivatives via nuclear receptors. Toxicol Appl Pharmacol 272:127–136. [DOI] [PubMed] [Google Scholar]
- Nanjappa MK, Hess RA, Medrano TI, Locker SH, Levin ER, Cooke PS. 2016. Membrane-localized estrogen receptor 1 is required for normal male reproductive development and function in mice. Biol Reprod 157:2909–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanjappa MK, Mesa AM, Medrano TI, Jefferson WN, DeMayo FJ, Williams CJ, et al. Submitted. The histone methyltransferase EZH2 is required for normal uterine development and function in mice. Biol Reprod [DOI] [PMC free article] [PubMed]
- Newbold RR, Padilla-Banks E, Jefferson WN. 2006. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology 147:S11–17. [DOI] [PubMed] [Google Scholar]
- O’Malley BW, McGuire WL. 1968. Studies on the mechanism of estrogen-mediated tissue differentiation: Regulation of nuclear transcription and induction of new RNA species. Proc Natl Acad Sci U S A 60:1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JR, Herbst AL, Noller KL, Boggs DA, Troisi R, Titus-Ernstoff L, et al. 2009. Urogenital abnormalities in men exposed to diethylstilbestrol in utero: A cohort study. Environ Health Perspect 8:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore MB, Landeros RV, Ramadoss J, Magness.2018.Structuralanalysisofestrogen receptors: Interaction between estrogen receptors and Cav-1 within the caveolae. Biol Reprod doi: 10.1093/biolre/ioy188.epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Levin ER. 2006. Nature of functional estrogen receptors at the plasma membrane. Mol Cell Endocrinol 20:1996–2009. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. 2007. conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem 282:22278–22288. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Kim JK, O’Mahony F, Lee EYHP, Luderer U, et al. 2009. Developmental phenotype of a membrane only estrogen receptor α (MOER) mouse. J Biol Chem 284:3488–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Deschenes RJ, Levin ER. 2012. DHHC-7 and -21 are palmitoylacyltransferases for sex steroid receptors. Mol Biol Cell 23:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M, O’Mahony F, Harvey H, Harvey BJ, Levin ER. 2013. Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Science Signaling 6:ra36. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Lewis M, Hammes S, Levin ER. 2014. Membrane-localized estrogen receptor alpha is required for normal organ development and function. Dev Cell 29:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Blumberg B, Levin ER. 2016. Membrane and nuclear estrogen receptor alpha collaborate to suppress adipogenesis but not triglyceride content. FASEB J 30:230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras RJ, Szego CM. 1975. Endometrial cell calcium and oestrogen action. Nature 253:357–359. [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Szego CM. 1977. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature 265:69–72. [DOI] [PubMed] [Google Scholar]
- Pietron WJ, Malagocki P. 2017. Quantification of polybrominated diphenyl ethers (PBDES) in food. A review. Talanta 167:411–427. [DOI] [PubMed] [Google Scholar]
- Prifti S, Mall P, Rabe T. 2003. Synthetic estrogen-mediated activation of ERK 2 intracellular signaling molecule. Gynecol Endocrinol 17:423–428. [DOI] [PubMed] [Google Scholar]
- Prins GS, Ye SH, Birch L, Zhang X, Cheong A, Lin H, et al. 2017. Prostate cancer risk and DNA methylation signatures in aging rats following developmental BPA exposure:A dose-response analysis. Environ Health Perspect 125:077007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M 2013. Epigenetics: Core misconcept. Proc Natl Acad Sci U S A 110:7101–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada I, Fuentes E, Viso-Leon MC, Soria B, Ripoll C, Nadal A. 2002. Low doses of the endocrine disruptor bisphenol-A and the native hormone 17beta-estradiol rapidly activate transcription factor CREB. FASEB J 16:1671–1673. [DOI] [PubMed] [Google Scholar]
- Rambo CO, Szego CM. 1983. Estrogen action at endometrial membranes: Alterations in luminal surface detectable within seconds. J Cell Biol 97:679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER. 2003. Identification of a structural determinant necessary for the localization and function of estrogen receptor α at the plasma membrane. Mol Cell Biol 23:1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CS. 2017. Neuroendocrine disruption in animal models due to exposure to bisphenol analogues . Front Neuroendocrinol 47:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa SI, Pereira PA, Malikov V, Ferreira IM, Madeira MD. 2014. Role of plasma membrane estrogen receptors in mediating the estrogen induction of progesterone receptors in hypothalamic ventromedial neurons. J Comp Neurol 522:298–307. [DOI] [PubMed] [Google Scholar]
- Saito N, Kanasugi H, Kimura K, Suzuki T, Komiya Y, Ogawa H, et al. 1989. Estrogen-binding protein in blood and follicular fluid, and its biochemical properties in human females. Gynecol Obstetric Invest 28:87–93. [DOI] [PubMed] [Google Scholar]
- Schug TT, Janesick A, Blumberg B, Heindel JJ. 2011. Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol 127:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar TV, Foygel K, Massoud TF, Gambhir SS, Paulmurugan R. 2016. A transgenic mouse model expressing an ERα folding biosensor reveals the effects of bisphenol A on estrogen receptor signaling. Sci Rep 6:34788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setchell KDR, Zimmer-Nechemias L, Cai J, Heubi JE. 1997. Exposure of infants to phyto-oestrogens from soy-based infant formula. Lancet 350:23–27. [DOI] [PubMed] [Google Scholar]
- Sieli PT, Jasarevic E, Warzak DA, Mao J, Ellersieck MR, Liao C, et al. 2011. Comparison of serum bisphenol A concentrations in mice exposed to bisphenol A through the diet versus oral bolus exposure. Environ Health Perspect 119:1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Li SS. 2012. Epigenetic effects of environmental chemicals bisphenol A and phthalates. Int J Mol Sci 13:10143–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithells RW. 1981. Oral contraceptives and birth defects. Dev Med Child Neurol 23:369–372. [PubMed] [Google Scholar]
- Soltysik K, Czekaj P. 2013. Membrane estrogen receptors - is it an alternative way of estrogen action? J Physiol Pharmacol 64:129–142. [PubMed] [Google Scholar]
- Stefkovich ML, Arao Y, Hamilton KJ, Korach KS. 2018. Experimental models for evaluating non-genomic estrogen signaling. Steroids 133:34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. 1999. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology 140:5455–5458. [DOI] [PubMed] [Google Scholar]
- Stoica A, Katzenellenbogen BS, Martin MB. 2000. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol 14:545–553. [DOI] [PubMed] [Google Scholar]
- Subedi L, Ji E, Shin D, Jin J, Yeo JH, Kim SY. 2017. Equol, a dietary daidzein gut metabolite attenuates microglial activation and potentiates neuroprotection in vitro. Nutrients 9:pii:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szego CM, Davis JS.1967denosine3’,5’-monophosphate in rat uterus: Acute elevation by estrogen. Proc Natl Acad Sci U S A 58:1711–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabatadze N, Smejkalova Woolley CS. 2013. Distribution and posttranslational modification of synaptic ERα in the adult female rat hippocampus. Endocrinology 154:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi Y, Koslowski M, Bodenner DL. 2004. Binding of estrogen receptor with estrogen conjugated to bovine serum albumin (BSA). Nuclear Receptor 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple JL, Wray S. 2005. Bovine serum albumin-estrogen compounds differentially alter gonadotropin-releasing hormone-1 neuronal activity. Endocrinology 146:558–563. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J. 2005. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146:624–632. [DOI] [PubMed] [Google Scholar]
- Thompson LA, Darwish WS, Ikenaka Y, Nakayama SM, Mizukawa H, Ishizuka M. 2017. Organochlorine pesticide contamination of foods in Africa: Incidence and public health significance. J Vet Med Sci 79:751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson TL, Moss RL. 1994. Estrogen regulation of dopamine release in the nucleus accumbens: Genomic- and nongenomic-mediated effects. J Neurochem 62:1750–1756. [DOI] [PubMed] [Google Scholar]
- Timms BG, Howdeshell KL, Barton L, Bradley S, Richter CA, vom Saal FS. 2005. Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc Natl Acad Sci U S A 102:7014–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournaire M, Devouche E, Epelboin S, Cabau A,DunbavandA LevadouA. 2018Birth defects in children of men exposed in utero to diethylstilbestrol (DES). Therapie pii: S0040–5957(18) 30040–4. doi: 10.1016/j.therap.2018.02.007.epub. [DOI] [PubMed] [Google Scholar]
- Turi A, Kiss AL, Mullner N. 2001. Estrogen downregulates the number of caveolae and the level of caveolin in uterine smooth muscle. Cell Biol Int 25:785–794. [DOI] [PubMed] [Google Scholar]
- Valera MC, Fontaine C, Lenfant F, Cabou C, Guillaume M, Smirnova N, et al. 2015. Protective hematopoietic effect of estrogens in a mouse model of thrombosis: Respective roles of nuclear versus membrane estrogen receptor alpha. Endocrinology 156:4293–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. 2007. Human exposure to bisphenol A (BPA). Reprod Toxicol 24:139–177. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. 2009. Bisphenol- and the great divide: A review of controversies in the field of endocrine disruption. Endocr Rev 30:75–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg LN, Chahoud I, Padmanabhan V, Paumgartten FJ, Schoenfelder G. 2010. Biomonitoring studies should be used by regulatory agencies to assess human exposure levels and safety of bisphenol A. Environ Health Perspect 118:1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigezzi L, Ramos JG, Kass L, Tschopp MV, Munoz-de-Toro M, Luque EH, et al. 2016. A deregulated expression of estrogen-target genes is associated with an altered response to estradiol in aged rats perinatally exposed to bisphenol A. Mol Cell Endocrinol 426:33–42. [DOI] [PubMed] [Google Scholar]
- Viñas R, Watson S. 2013a. Mixtures of xenoestrogens disrupt estradiol-induced non-genomic signaling and downstream functions in pituitary cells. Environ Health 12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viñas R, Watson CS. 2013b. Bisphenol S disrupts estradiol-induced nongenomic signaling in a rat pituitary cell line: Effects on cell functions. Environ Health Perspect 121:352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vom Saal FS, Akingbemi BT, Belcher SM, Birnbaum LS, Crain DA, Eriksen M, et al. 2007. Chapel Hill bisphenol A expert panel consensus statement: Integration of mechanisms, effects in animals and potential to impact human health at current levels of exposure. Reprod Toxicol 24:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong AM, Yolton K, Dietrich KN, Braun JM, Lanphear BP, Chen A. 2018. Exposure to polybrominated diphenyl ethers (PBDES) and child behavior: Current findings and future directions. Horm Behav 101:94–104. [DOI] [PubMed] [Google Scholar]
- Wang Q, Trevino LS, Wong RL, Medvedovic M, Chen J, Ho SM, et al. 2016. Reprogramming of the epigenome by MLL1 links early-life environmental exposures to prostate cancer risk. Mol Endocrinol 30:856–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CS, Gametchu B. 2003. Proteins of multiple classes may participate in nongenomic steroid actions. Exp Biol Med (Maywood) 228:1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CS, Jeng YJ, Kochukov MY. 2010. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci 115:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CS, Jeng YJ, Guptarak J. 2011. Endocrine disruption via estrogen receptors that participate in nongenomic signaling pathways. J Steroid Biochem Mol Biol 127:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Song X, Shaikh ZA. 2015. Cadmium promotes the proliferation of triple-negative breast cancer cells through EGFR-mediated cell cycle regulation. Toxicol Appl Pharmacol 289:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ. 2017. Soy-based therapeutic baby formulas: Testable hypotheses regarding the pros and cons. Front Nutr 3:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SJ, Cheung See Kit M, Martin BR. 2018. Protein depalmitoylases. Crit Rev Biochem Mol Biol 53:83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RL, Walker CL. 2013. Molecular pathways: Environmental estrogens activate nongenomic signaling to developmentally reprogram the epigenome. Clin Cancer Res 19:3732–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak AL, Bulayeva NN, Watson CS. 2005. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-alpha-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ Health Perspect 113:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LH, Zhang XM, Wang F, Gao CJ, Chen D, Palumbo JR, et al. 2018. Occurrence of bisphenol S in the environment and implications for human exposure: A short review. Sci Total Environ 615:87–98. [DOI] [PubMed] [Google Scholar]
- Xu F, Wang X, Wu N, He S, Yi W, Xiang S, et al. 2017. Bisphenol A induces proliferative effects on both breast cancer cells and vascular endothelial cells through a shared GPER-dependent pathway in hypoxia. Environ Pollut 231:1609–1620. [DOI] [PubMed] [Google Scholar]
- Xue J, Wan Y, Kannan K. 2016. Occurrence of bisphenols, bisphenol A diglycidyl ethers (BADGES), and novolac glycidyl ethers (NOGES) in indoor air from Albany, New York, USA, and its implications for inhalation exposure. Chemosphere 151:1–8. [DOI] [PubMed] [Google Scholar]
- Yanagihara N, Toyohira Y, Shinohara Y. 2008. Insights into the pharmacological potential of estrogens and phytoestrogens on catecholamine signaling. Ann N Y Acad Sci 1129:96–104. [DOI] [PubMed] [Google Scholar]
- Yu L, Ham K, Gao X, Castro L, Yan Y, Kissling GE,et al. 2016Epigenetic regulation of transcription factor promoter regions by low-dose genistein through mitogen-activated protein kinase and mitogen-and-stress activated kinase 1 nongenomic signaling. J Cell Commun Signal 14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Filardo EJ, Shaikh ZA. 2010. The membrane estrogen receptor GPR30 mediates cadmium-induced proliferation of breast cancer cells. Toxicol Appl Pharmacol 245:83–90. [DOI] [PubMed] [Google Scholar]
- Zang Y, Odwin-Dacosta S, Yager JD. 2009. Effects of cadmium on estrogen receptor mediated signaling and estrogen induced DNA synthesis in T47D human breast cancer cells. Toxicol Lett 184:134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J, Manthey D, Bayatti N, van der Burg B, Goodenough S, Behl. 2002. Estrogen receptor alpha-mediated silencing of caveolin gene expression in neuronal cells. J Biol Chem 277:38772–38780. [DOI] [PubMed] [Google Scholar]