

Abstract
Pd(0)-catalyzed Mizoroki-Heck reactions traditionally exhibit poor reactivity with polysubstituted, unbiased alkenes. Intermolecular reactions with simple, all-carbon tetrasubstituted alkenes are unprecedented. Here we report that pendant carboxylic acids, combined with bulky monophospine ligands on palladium, can direct the arylation of tri- and tetrasubstituted olefins. Quaternary carbons are established at high Fsp3 attached-ring junctures and the carboxylate directing group can be removed after coupling. Carboxylate directivity prevents over-arylation of the new, less substituted alkene, which can be diversified in subsequent reactions.
Keywords: Mizoroki-Heck, cross-coupling, carboxylic acid, directing group effects
Graphical Abstract
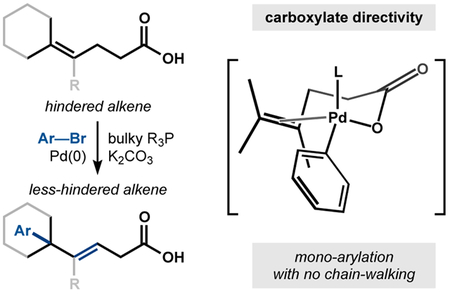
The Mizoroki-Heck reaction1 has become a staple of cross coupling and has transformed how molecules are synthesized. A large volume of research in the decades since its discovery has identified some limitations in its scope.2 For example, electronically unbiased, cyclic and sterically hindered olefins remain challenging substrates with low inherent reactivity and selectivity3 in traditional Heck arylations.4 Intermolecular Heck reactions are unprecedented with all-carbon tetrasubstituted olefins, with the exception of strained hydrocarbons5 like bicyclopropylidene.6 Here we show that potassium carboxylates serve as directing groups to enable single Heck reactions of triand tetrasubstituted alkenes, including hindered cyclic motifs. Products avoid polyarylation and include sp2-sp3 attached-ring motifs and diversifiable unsaturated building blocks, otherwise inaccessible by cross-coupling methods.
Many different approaches have been applied to extend the classical Heck reaction to more traditionally challenging substrates (Figure 1a). Early work by Grigg7 and Overman8 demonstrated the utility of intramolecular Heck reactions to overcome the reactivity barrier for highly substituted substrates, with their groups and others forging various spiro- and polycyclic systems from tri- and some tetrasubstiuted olefins. The reactivity enhancement from intramolecular delivery of the PdII-Ar species was leveraged in several directed Heck reactions.9 Notably, the groundbreaking work of Hallberg10 and Carretero11 used tethered functional groups bearing basic nitrogens to alter arylation regiochemistry and relay stereochemical information, with the former able to form quaternary centers from a fully-substituted, albeit polarized substrate. A seminal report from White and coworkers in 2008 established that oxidative Heck conditions could engage unbiased terminal olefins using weakly directing functional groups to yield terminally arylated products,12 and Sigman in 2014 extended the asymmetric redox-relay Heck reaction to trisubstituted olefins with oxidative conditions.13,14 With the exception of enelactams, redox-relay Heck reactions do not tolerate cyclic substrates, which tend to yield mixtures of regioisomers.15
Figure 1:

Approaches to the Heck reaction on challenging substrates.
Heck reactions with sterically hindered and unbiased olefins remain non-trivial in many cases, evidenced by recent syntheses of κ-opioid receptor agonists 20-nor-SalA16 and O6C-20-nor-SalA.17 A late-stage Heck arylation on a hindered, unbiased olefin could not be achieved using traditional or oxidative Heck conditions. However, incorporation of a carboxylic acid close to the alkene significantly accelerated arylation relative to deactivation of the palladium catalyst or decomposition of the aryl halide, 3-bromofuran (Figure 1c).16 Reactivity enhancement from the direction of carboxylic acids had been observed in the C-H activation literature, but until now has not been established in Heck arylation.18 We wondered if carboxylate-directivity could be applied to traditionally unreactive Heck substrates.19 Here we report that carboxylic acids accelerate and direct the intermolecular Heck reaction of tri- and tetrasubstituted olefins (Figure 1d) in substrate types (unbiased, cyclic, hindered) that are unrepresented in the Heck literature, directed or undirected. The reactivity enhancement of the potassium carboxylate allows the formation of quaternary centers from linear, cyclic, bicyclic, aliphatic, and hindered styrenyl substrates. Carboxylate-directivity seems to inhabit a ‘goldilocks region’ among Mizoroki-Heck regimes: cationic-Pd conditions favor chain-walking into a terminating group13,14 and chelating directing groups prevent β-hydride elimination altogether.19 In addition, previously-established directing groups like amines and esters are ineffective in promoting the coupling. Carboxylate-directivity promotes regioselective engagement of hindered alkenes yet allows regio- and stereoselective β-hydride elimination without iterative arylation of the new, less hindered alkene.
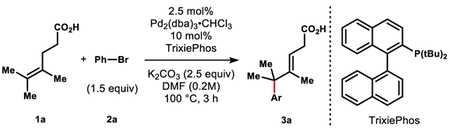
We identified tetrasubstituted alkene 1a as a model substrate to explore and optimize carboxylate directivity (for a full table of optimization, see the Supporting Information). Type of base significantly influenced yield: lithium, sodium and cesium cations were inferior to potassium, and ammonium cation was ineffective. A strong influence of cation has previously been observed in carboxylate-directed C-H insertion chemistry, where a proposed κ2 binding of alkali cation is suggested to induce a κ1 Pd-carboxylate binding mode.18 The effect here can alternatively be explained by altered aggregation states of different alkali cations,20 or perturbed Lewis basicity of the alkali carboxylates. Choice of phosphine proved crucial for efficiency and breadth of scope. Bisphosphines were ineffective, likely due to occupancy of the Pd valence necessary for carboxylate coordination (entries 4–5). Rapidly dissociating, larger bite-angle bisphosphines delivered small amounts of product (entry 6). A bulky mono-phosphine proved optimal for the reaction, either favoring an L1Pd-Ar species with open binding sites for both carboxylate and alkene, or dissociating from Pd to promote a cationic pathway.21 Interestingly, XPhos performed poorly, although it offered the highest yields in 20-nor-SalA.16 While simple triphenylphosphine was capable of promoting the reaction, it was outcompeted by the bulkier TrixiePhos, especially with electron rich arenes (entry 9 vs. 10). Interestingly, iodoarenes reacted to low conversion despite their greater propensity towards oxidative addition. No reaction was observed in the absence of palladium.
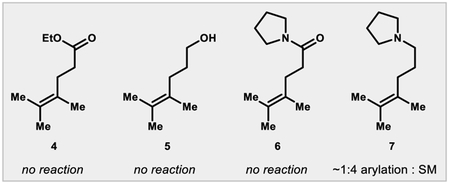
The arylation exhibits high regio- and stereoselectivity, delivering the E-alkene and the arene distal to the directing group, which is consistent with a 6-membered chelate of the carboxylate to the Pd||-alkyl intermediate (see Scheme 2, below). In contrast to redox-relay Heck reactions of trisubstituted olefins, the unsaturation does not migrate into conjugation22 with the carboxylic acid but remains adjacent to the newly formed quaternary center. Additionally, the alkene isomer preferentially formed is internal and trisubstituted rather than terminal and disubstituted.23 The carboxylic acid was essential for reaction: substrates containing weakly Lewis basic esters, alcohols, and amides were completely unreacted under the optimized conditions after 16 hours. Tertiary amine 7 reacted to low conversion and gave a complex mixture of arylated materials.
Scheme 2.

Mechanistic model for regio- and stereochemistry.
The carboxylate-directed Heck was successfully applied to a wide range of tri- and tetrasubstituted olefins in modest to good yields after a second esterification step (Table 2). Unless otherwise noted, stereoselectivity was excellent (>20:1 E/Z by 1H NMR) and regiocontrol was excellent in all cases except a cyclopentane substrate (3t-w). Only olefins proximal to the carboxylic acid underwent reaction; the distal olefin of 3d was unreactive. In addition to simple linear aliphatic substrates, cyclic and styrenyl tri- and tetrasubstituted olefins were competent Heck partners. The intrinsic electronic bias of the styrene was completely overridden in the cases of 3e, 3f, and 3m, giving valuable diarylated quaternary centers. Rearrangement of the styrene substitution did not change delivery of the arene electrophile (see 3s). The use of carbo- and heterocyclic olefins produced challenging all-carbon quaternary centers at Csp3-Csp2 attached-ring motifs, including the bicycle 3n and cyclododecane 3o. These motifs represent high fraction-sp3 (Fsp3) equivalents of biaryls and valuable scaffolds for medicinal chemistry.24 For example, phenyl substituted cyclohexane 8, an intermediate in the synthesis of S1P1 agonists, could be prepared in a concise and higher-yielding 3 step route as compared to the reported 6 step sequence.25 Electron-rich bromoarenes performed best, in contrast to nickel-catalyzed hydroarylations,26 which also form quaternary carbons at attached-ring bridgeheads, but usually prefer electron-deficient haloarenes.27 Diastereoselectivity in the Heck reaction could be effected by: substitution α to the directing carboxylic acid, (3w), existing stereocenters on cyclic substrates (3k), or bridging-ring topology (3n). While most reactions in Table 2 were performed on small scale (0.1 mmol), scale-up proved uneventful and allowed over 500 mg of 3u to be prepared in one batch from the tetrasubstituted cyclopentene.28
Table 2.
Olefin Scope


|
0.1 mmol scale, isolated yield over 2 steps, reaction time 16 h. Esterification conditions unless otherwise noted: (COCl)2 (2 eq), DCM (0.1M); MeOH (excess).
1H NMR Yield of crude material.
1 eqivalent of ArBr used.
Esterification: DIC, DMAP, MeOH, DCM.
Esterification: N,N’-diiPr-O-tBu isourea, DCM. [f] Ratio of endocyclic to exocyclic olefin products.
Aryl scope is explored in Table 3 using tetrasubstituted substrate 1a. Para- and meta- substitution is well tolerated, and mono-, di-, and trisubstituted arenes could be installed with similar ease. Electron rich and electron neutral arenes outperform electron poor, with electron rich aryl bromides noted to complete fastest. Even highly electron-rich trimethoxyphenyl 3ag was delivered in good yield. Aryl bromides with strongly electron-withdrawing groups in the para position were found to stall at low conversion from apparent catalyst deactivation (see Scheme 2 and Supporting Information). Heterocycles were acceptable coupling partners in this chemistry, exemplified by benzodioxole 3ah and protected indole 3aj.
Table 3.
Aryl halide scope with a tetrasubstituted alkene.

|
0.1 mmol scale, isolated yield over 2 steps, reaction time 16 h. Esterification conditions unless otherwise noted: (COCl)2 (2 eq), DCM (0.1M); MeOH (excess)
Esterification conditions: DIC, DMAP, MeOH, DCM
12:1 ratio of isomers favoring the internal [d] Combined yield including deprotected product.
The products of carboxylate-directed Heck reaction exhibit high synthetic utility by virtue of the olefin and carboxylic acid functional groups, both of which are versatile synthetic handles. To demonstrate this versatility, we diversified 3u to form the densely functionalized products of Scheme 1. The olefin was capable of functionalization by Drago-Mukaiyama hydration,29 epoxidation, and dihydroxylation, and treatment of the dihydroxylation product with oxalyl chloride in methanol furnished 5-membered lactone 12. Formation of redox active ester in place of the methyl ester allowed straightforward decarboxylation or decarboxylative arylation.30 As a result, motifs that were previously inaccessible by Mizoroki-Heck chemistry can now be unmasked and retrosynthetically transformed to unsaturated carboxylates.
Scheme 1.

Product Diversification[a]
[a] Conditions: a) R = NHPI, Ph2Zn, Fe(acac)2, dppBz (34%); b) R = NHPI, Zn0, PhSiH3, NiCl2·6H2O (39%; c) R = Me, Mn(acac)2, PhSiH3, PPh3, O2 (64%, 2:1 dr); d) R = Me, i. NMO, OsO4; ii. (COCl)2, MeOH (65%, 6:1 dr); e) R = Me, m-CPBA (100%, 2:1 dr)
Several interesting mechanistic features are also worth noting. Although less-substituted olefins were generated, over-arylation of products in Table 2 only occurred rarely and only in small quantities. The adjacent quaternary carbons were not solely responsible for enforcing single arylation: a homologated analog of the products in Tables 2 and 3 underwent facile carboxylate-directed Heck reaction (14→15, Scheme 2a) but the less hindered β,γ-unsaturated product (15) did not undergo arylation. Therefore, transition state geometry may play a role in effecting monoarylation. A low-energy, pseudo-chair conformation might be involved, which fits the relative regiochemistry of arylation and stereochemistry generated in 3w (Scheme 2b). This assembly would be geometrically unfavorable for β,γ-unsaturated products (15 and Tables 2 and 3) and distant alkenes (3d). Additionally, β-hydride elimination generates an olefin out of conjugation with the carboxylic acid, which suggests that chain-walking processes cannot occur. The preference for tetrasubstituted substrates to form the internal, trisubstituted olefin over the terminal 1,1-disubstituted olefin (see Table 3, ca. 10:1 on average)23,31 is striking given that statistics should favor β-hydride elimination to the terminal position.32,33 That the observed alkene isomer avoids the thermodynamic sink of conjugation and the statistically favored product suggests that the carboxylate directing group has a role enforcing β-hydride elimination, kinetically or thermodynamically, in addition to directing the regiochemistry of arene insertion. The superiority of TrixiePhos to engage hindered olefins was unexpected given its large size among monodentate phosphine ligands and may suggest that dissociation occurs prior to alkene coordination. Alternatively, the monoligated palladium L1Pd-Ar favored by bulky phosphines34 may be necessary to allow coordination of both the carboxylate and the alkene. Another surprising feature was the poor performance of both aryl iodides and electron deficient aryl bromides, which typically couple well in Heck reactions but in our system led to catalyst deactivation. Analysis of the crude reaction mixture in reactions of electron poor arenes revealed formation of biaryls and LCMS peaks corresponding to substrate dehydrogenation. Catalyst deactivation possibly occurs downstream of reductive homocoupling to generate biaryls and inactive Pd|| salts,35 a process that is bimolecular in XPd||-Ar and would be disproportionality favored by rapid oxidative addition compared to the productive reaction pathway (Scheme 2c). This hypothesis also accounts for the superiority of sterically bulky ligands, which may prevent Pd||-Ar association leading to deactivation — an observation made previously in styrenyl Heck reactions.36
In conclusion, carboxylate-directivity advances the Heck reaction to a new milestone: the intermolecular coupling of tetrasubstituted alkenes. In addition to providing products of high synthetic value, the success of this strategy underscores the profound rate enhancement imparted by carboxylate directing groups.18 The reactivity trend in counter-cation is noteworthy and distinguishes the carboxylate from neutral protecting groups like amines or esters, which are ineffective to promote the reaction. The absence of chain walking and the regioselectivity of β-hydride elimination may also point to a role of the carboxylate downstream of arene insertion.37,38 The observations here invite further mechanistic inquiry and the insights gained from this work may be translatable to other modes of olefin cross-coupling.
Supplementary Material
Table 1.
Reaction Optimization
| Entry | Variations from above | % yield[a] |
|---|---|---|
| none | ||
| Li2CO3 / Na2CO3 / Cs2CO3 vs K2CO3 | ||
| Et3N vs K2CO3 | ||
| BINAP vs TrixiePhos | ||
| dppe vs TrixiePhos | ||
| dppf vs TrixiePhos | ||
| XPhos vs TrixiePhos | ||
| PPh3 vs TrixiePhos | ||
| 4-OMe-PhBr vs PhBr | ||
| 4-OMe-PhBr vs PhBr, PPh3 vs TrixiePhos | ||
| PhI vs PhBr |
0.1 mmol scale, yield determined by quantitative LCMS.
Ar = 4-OMe-Ph,1H NMR yield after 1 h.
Acknowledgements
Generous support was provided by the National Institutes of Health (R35 GM122606), Nanjing University of Science & Technology (scholarship to Y. W.) and Westfälische Wilhelms Universität Münster (scholarship to A. E.). We thank Zefeng Chen for early contributions to substrate scope and Jeremy J. Roach to early discovery efforts. Professors Keary Engle and Jin-Quan Yu are acknowledged for helpful input.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Mizoroki T, Mori K, Ozaki A, Bull. Chem. Soc. Jpn 1971, 44, 581; [Google Scholar]; b) Heck RF; Nolley JP, J. Org. Chem 1972, 37, 2320. [Google Scholar]
- [2].For selected reviews and books, see:Heck RF, Acc. Chem. Res 1979, 12, 146;Cabri W, Candiani I, Acc. Chem. Res 1995, 28, 2;Beletskaya IP, Cheprakov AV, Chem. Rev 2000, 100, 3009;Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem. Int. Ed 2005, 44, 4442;Karimi B, Behzadnia H, Elhamifar D, Akhavan PF, Esfahani FK, Zamani A, Synthesis, 2010, 1399.
- [3].Dieck HA, Heck RF, Am Chem J. Soc. 1974, 96, 1133. [Google Scholar]
- [4].a) Ref. 2a;Bates R, Organic synthesis using transition metals. John Wiley & Sons, 2012, pp. 153–190;Zheng C, Stahl SS, Chem. Commun 2015, 51, 12771.
- [5].Dyker G, Körning J, Jones PG, Bubenitschek P, Angew. Chem. Int. Ed. Engl 1993, 32, 1733. [Google Scholar]
- [6].Bräse S, de Meijere A, Angew. Chem. Int. Ed 1995, 34, 2545. [Google Scholar]
- [7].Grigg R, Sridharan V, Stevenson P, Worakun T, J. Chem. Soc., Chem. Commun 1986, 1697. [Google Scholar]
- [8].Abelman MM, Oh T, Overman LE, J. Org. Chem, 1987, 52, 4130. [Google Scholar]
- [9].Oestreich M, Eur. J. Org. Chem 2005, 783. [Google Scholar]
- [10].a) Andersson C-M, Larsson J, Hallberg A, J. Org. Chem 1990, 55 5757; [Google Scholar]; b) Larhed M, Andersson C-M, Hallberg A, Acta Chem. Scand 1993, 47, 212; [Google Scholar]; c) Larhed M, Andersson C-M, Hallberg A, Tetrahedron 1994, 50, 285. [Google Scholar]; d) Nilsson P, Larhed M, Hallberg A, J. Am. Chem. Soc 2003, 125, 3430. [DOI] [PubMed] [Google Scholar]
- [11].Buezo ND, Alonso I, Carretero JC, J. Am. Chem. Soc 1998, 120, 7129. [Google Scholar]
- [12].a) Delcamp JH, Brucks AP, White MC, J. Am. Chem. Soc 2008, 130, 11270; [DOI] [PubMed] [Google Scholar]; b) Delcamp JH, Gormisky PE, White MC, J. Am. Chem. Soc 2013, 135, 8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mei T-S, Patel HH, Sigman MS, Nature 2014, 508, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].For other work on migratory Heck reactions, see:Tamaru Y, Yamada Y, Yoshida Z-I, Tetrahedron, 1979, 35, 329.Larock RC, Leung W-Y, Stolz-Dunn S, Tetrahedron Lett, 1989, 30, 6629.Larock RC, Y.-d. Lu, A. C. Bain, J. Org. Chem 1991, 56, 4589.Oliveira CC, Pfaltz A, Correia CRD, Angew. Chem. Int. Ed 2015, 54, 14036.
- [15].Yuan Q, Sigman MS, J. Am. Chem. Soc 2018, 140, 6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Roach JJ, Sasano Y, Schmidt CL, Zaidi S, Katritch V, Stevens RC, Bohn LM, Shenvi RA, ACS Cent. Sci, 2017, 3, 1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hirasawa S, Cho M, Brust TF, Roach JJ, Bohn LM, Shenvi RA, Bioorganic Med. Chem. Lett 2018, 28, 2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Giri R, Yu J-Q, J. Am. Chem. Soc 2008, 130, 14082. [DOI] [PubMed] [Google Scholar]; b) Engle KM, Mei T-S, Wasa M, Yu J-Q, Acc. Chem. Res 2012, 45, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].For use of 8-aminoquinoline directing groups for palladium catalyzed aryl-olefin cross-coupling, see:Yang K, Gurak JA Jr., Liu Z, Engle KM, J. Am. Chem. Soc 2016, 138, 14705;Liu Z, Zeng T, Yang KS, Engle KM, J. Am. Chem. Soc 2016, 138, 15122.
- [20].Algera RF, Ma Y, Collum DB, J. Am. Chem. Soc 2017, 139, 7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cabri W, Candiani I, Bedeschi A, J. Org. Chem 1993, 58, 7421. [Google Scholar]
- [22].Zhang C, Santiago CB, Kou L, Sigman MS, J. Am. Chem. Soc 2015, 137, 7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].The terminal, γ,δ-unsaturated olefin is not observed by 1H NMR in the crude reaction mixtures. However, a diarylated compound arising from its intermediacy is occasionally present as a ~10% impurity (see Supporting Information).
- [24].a) Lovering F, Bikker J, Humbolt C, J. Med. Chem 2009, 52, 6752; [DOI] [PubMed] [Google Scholar]; b) Brown DG, Boström J, J. Med. Chem 2016, 59, 4443. [DOI] [PubMed] [Google Scholar]
- [25].Das J, Soo Sung K, International Patent WO2012040532. 2012.
- [26].Green SA, Vásquez-Céspedes S, Shenvi RA, J. Am. Chem. Soc 2018, 140, 11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].For a recent exception using electron-rich iodoarenes, see:Wang X, Ma G, Peng Y, Pitsch CE, Moll BJ, Ly TD, Wang X, and Gong H, J. Am. Chem. Soc 2018, 140, 14490.
- [28].In some cases, excess aryl halide led to diarylation of γ,δ-unsaturated products, which could be lessened through fewer equivalents of arene.
- [29].Crossley SWM, Obradors C, Martinez RM, Shenvi RA, Chem. Rev 2016, 116, 8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Toriyama F, Cornella J, Wimmer L, Chen T-G, Dixon DD, Creech G, Baran PS, J. Am. Chem. Soc 2016, 138, 11132. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qin T, Malins LR, Edwards JT, Merchant RR, Novak AJE, Zhong JZ, Mills RB, Yan M, Yuan C, Eastgate MD, Baran PS, Angew. Chem. Int. Ed 2017, 56, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Small amounts of the alternative alkene regioisomer (1,1-disubstition) could be observed when electron-deficient arenes were used (3ac and 3ae), but the trisubstituted alkene still predominated 12:1.
- [32].Cf. Ref. 19. Professor Engle first noted the ‘goldilocks’ effect of carboxylate-directivity on β-hydride elimination: the alkyl-palladium is not too stable and not too labile.
- [33].Trost BM, Lautens M, Chan C, Jebaratnam DJ, Mueller T, J. Am. Chem. Soc 1991, 113, 636. [Google Scholar]
- [34].a) Hartwig JF, Angew. Chem., Int. Ed 1998, 37, 2046; [DOI] [PubMed] [Google Scholar]; b) Martin R, Buchwald SL, Acc. Chem. Res 2008, 41, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Attempts to rescue inactive Pd(II) using stoichiometric reductants proved unsuccessful.
- [36].van Strijdonck GPF, Boele MDK, Kamer PCJ, de Vries JG, van Leewen PWNM, Eur. J. Inorg. Chem 1999, 1073. [Google Scholar]
- [37].For carboxylate assistance in haloarene oxidative addition, see:Houpis IN, Van Hoeck J-P, Tilstam U, Synlett 2007, 14, 2179;Houpis IN, Huang C, Nettekoven U, Chen JG, Liu R, Canters M, Org. Lett 2008, 10, 5601.
- [38].For carboxylate assistance in iridium-catalyzed asymmetric alkene hydrogenation, see:Zhu S-F, Zhou Q-L, Acc. Chem. Res 2017, 50,988.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.