

Abstract
Direct sampling mass spectrometry (MS) has been advancing aggressively, showing immense potential in translating MS into the clinical field. Unlike traditional MS analysis involving extensive sample preparation and chromatographic separation, quick and simple procedures with minimal sample pretreatment or purification became available with direct sampling. An overview of the development in this field is provided, including some representative ambient ionization and fast extraction methods. Quantitative applications of these methods are emphasized and their efficacy are highlighted from a clinical aspect; non-quantitative applications in clinical analysis are also discussed. This review also discusses the integration of direct sampling MS with miniature mass spectrometers and its future outlook as an emerging clinical tool for point-of-care analysis.
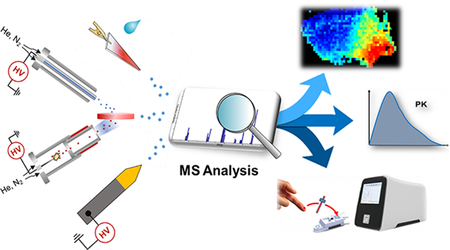
Graphical Abstract
Direct sampling mass spectrometry enables high-performance clinical analysis, such as imaging, drug monitoring and point-of-care testing.
1. Introduction
Mass spectrometry (MS) is an indispensable analytical technology for the modern healthcare industry. Due to its high sensitivity and selectivity, MS is exceptionally suitable for quantitative evaluations. Utilization of MS coupled with chromatography, especially liquid chromatography (LC), has been the golden standard for quantitative analysis of various compounds. The use of chromatography for analysis of complex clinical samples could provide desirable analytical performances but requires sample preparation that is typically labor intense and time consuming. Therefore, chromatography-MS methods are limited to use in clinical laboratories. For application in resource-limited or regulatorily restricting settings, the overall lab procedures for MS analysis pose a huge obstacle that cannot be easily overcome. The development of direct sampling methods has drastically changed this situation, enabling a wide adoption of MS for clinical research and diagnostics.
Direct sampling MS encompasses a wide range of MS methods that aim for quick and simple analysis requiring minimal pre-treatment of biological samples. The most important feature is that samples are directly introduced into mass spectrometers without chromatographic separation. Ambient ionization represents a major direction in research and technical development for direct sampling MS, originating with the development of desorption electrospray ionization (DESI)1 and direct analysis in real time (DART).2 In the past decade, a large number of ambient ionization methods have been developed, such as atmospheric pressure solid analysis probe3, laser ablation electrospray ionization (LAESI),4 low temperature plasma (LTP)5, dielectric barrier discharge ionization6, extractive electrospray ionization,7 and paper spray ionization (PSI).8
With the advancement of direct sampling techniques, quantitative analysis plays a significant role in clinical applications such as disease diagnostics and routine screening which have been discussed in several reviews.9–11 Quantitative analysis using direct sampling MS remains challenging due to the complexity of the matrices and lack of appropriate methods to incorporate internal standards. For assessing the quantitative performance of direct sampling MS methods, criteria generally used for bioanalytical method validation can be used, such as the linearity of matrix-matched calibration curve, lower limit of quantitation (LOQ), and relative standard deviation (RSD). Selectivity, reproducibility and stability of the developed methods should also be characterized. For non-quantitative applications such as imaging and chemical profiling, utilizing a relative signal intensity or analyte ratios paired with statistical tests enable efficient clinical analysis.
Another important factor for future clinical MS analysis is the availability of suitable mass spectrometry systems. Samples can be collected and shipped to clinical laboratories for analysis; however, to fully take advantage of direct sampling analysis, point-of-care (POC) systems represent more sensible solutions. Samples can be analyzed at the point of collection, providing immediate feedback to assist decision making. Issues with method compatibility and automation are significant hurdles that must be overcome to provide unique information that can only be obtained at the POC. The common denominator in all clinical aspects is a mass spectrometer that fulfills several POC criteria. A POC MS system should consist of a robust, easy-to-use mass spectrometer and use sampling cartridges with sampling procedures familiar to clinical practitioners, thus can be operated by healthcare personnel to obtain reliable results.12
In this review, we will discuss the applications of a variety of direct sampling methods for MS analysis related to clinical applications, both quantitative and qualitative aspects are presented. For quantitative methods, we discuss several ambient ionization techniques as well as methods to improve overall quantitative performance; for non-quantitative methods, we discuss mass spectrometry imaging (MSI), chemical profiling methods and emerging intra-surgical tools for in vivo MS analysis. We also highlight the development of cartridge technologies and miniature mass spectrometers for future POC MS systems.
2. Direct Sampling MS for Quantitative Analysis
Quantitative applications for direct sampling methods seek to target diagnostic screening or routine sampling of drugs of abuse or therapeutic drug monitoring. Furthermore, the quantitation of metabolites can also provide clinically relevant diagnostic information which has been reported in several methods. This section introduces three significant ambient ionization methods for quantitation: paper spray ionization, desorption electrospray ionization, and direct analysis in real time.
2.1. Paper Spray Ionization (PSI)
Since its introduction in 20108, PSI has been increasingly attracting interest in the field of ambient ionization MS. This method presents the ultimate simplicity for directly analyzing samples as complex as blood or tissues. In a typical experiment, chromatography paper is cut into a triangle and a few microliters of liquid sample, such as blood, is dropped onto the paper to form a dried sample spot (Fig. 1A-C). The paper triangle is then held in front of the MS inlet and a small amount (5–30 μL) of solvent is applied directly onto the triangle (Fig. 1D). The solvent elutes the analytes from the sample spot; when a high voltage (about 3 kV) is applied on the wetted paper, an electrospray is generated to produce ions to be analyzed by MS. For analyzing complex biological samples, the cellulose paper serves as a substrate retaining interfering species including salts, cell debris and proteins. Due to this clean-up effect, PSI is particularly suitable for quantitation of dried sample spots, especially from biofluid samples such as blood and urine.13 PSI is also applicable to analysis of solid samples, such as tissues.14 The simplicity of the PSI allows for experiments to be easily conducted in various settings, and has inspired novel methodologies such as leaf spray15.
Fig. 1.

(A) Schematic of paper spray. (B) Chemical structure and mass spectrum of imatinib in whole blood by paper spray-MS/MS. (C) Quantitative analysis of imatinib in whole blood samples by using imatinib-D8 as the internal standard (Reprinted with permission8. Copyright 2010 Wiley-VCH). (D) Spatial characterization of the paper spray intensity in relation to mass spectrometry inlet (Reprinted with permission13. Copyright 2010 American Chemical Society).
A significant amount of effort has been made on the improvement of reproducibility, ionization efficiency, as well as the reduction of the background noise to enable quantitative analysis by PSI-MS methods. It is well known that incorporation of the IS is critical for MS quantitation and even more so for direct MS analysis methods. For PSI-MS, the IS can be spiked into the sample or preprinted onto the paper substrate.16, 17 The porous nature of the cellulose paper makes it possible to load certain amounts of compounds that are to be co-eluted later by solvents during paper spray. The incorporation of the IS can also be combined with sample collection processes. For example, in one study the IS was pre-coated onto the inner wall of a sampling capillary, which was used to collect and transfer blood sample onto the paper substrate for PSI-MS.17 Improved reproducibility (RSDs less than 8%) was obtained for therapeutic drug analysis.
The application of PSI for drugs of abuse analysis has attracted a considerable amount of interest9 due to its simplicity and broad applicability. The quantitation of drugs of abuse and their metabolites in dried blood spots (DBS) was demonstrated by PSI-MS/MS.18 Wide linear dynamic ranges were obtained using IS premixed with the samples, showing LOQs below screening cut-off levels.19 Espy et al. compared the quantitation of eight drugs of abuse in whole blood using prototype PSI cartridges and extraction spray ionization.20 Similar results satisfactory for drugs of abuse analysis were obtained using these two methods. Besides DBS, PSI can also be applied for analysis of samples collected in other forms, for example, fingerprints. Costa et al. reported the analysis of drugs of abuse in fingerprints collected on paper by quantitative PSI-MS. The results by analyzing 239 fingerprint samples from drugs of abuse user and control groups showed a 99% true-positive rate and a 2.5% false-positive rate for detection of cocaine.21 The advantages of PSI, including extremely low cost, short analysis time and simple procedures, highlight the potential of implementing PSI-MS screening of drugs of abuse for law enforcement.
Another important clinical application demonstrated with PSI is the quantitation of therapeutic drugs in blood. A wide variety of therapeutic drugs have been monitored through PSI-MS, such as anticancer drugs or immunosuppressants.9 A comprehensive characterization was done with imatinib, an anticancer drug. Whole blood samples were prepared by spiking imatinib at a series of concentrations and imatinib-d8 at 1 μg/mL as the IS. For analysis, only 0.4 μL blood sample was spotted on the paper substrate and let dry.8 A linear dynamic range of 62.5 ng/mL to 4 μg/mL was obtained, covering the entire therapeutic range. Manicke et al. investigated the quantitative performance of PSI for therapeutic drug monitoring (TDM), for 15 hydrophobic and weakly basic drugs.22 Calibration standards and quality control (QC) samples were prepared for sitamaquine and amitriptyline. For sitamaquine, isotope-labeled IS was spiked in wet blood, allowing a linear range of 5–1000 ng/mL; the QC results showed an RSD less than 5 % and an imprecision within 2%. In the case of amitriptyline, isotope-labeled IS was deposited onto the paper prior to loading blood sample. The imprecision was about 10% for all concentrations tested except for 0.9 ng/mL (imprecision 22%). By comparing quantitation results for blood samples from different donors, it was concluded that this method was not affected by relative matrix effects.
Although the quantitative performance of PSI has been extensively explored for different kinds of therapeutic drugs, it remains to be further validated with real clinical samples. Shi et al. evaluated tacrolimus, an immunosuppressive drug that has a high clinical demand for TDM, utilizing an automated PSI source and cartridge designed at Purdue University12, 23 and later commercialized by Prosolia24, 25. To establish clinical relevance, PSI was also cross-validated with FDA-approved immunoassays and LC-MS/MS methods. Significant correlations between PSI-MS and other methods, including two immunoassays and two LC-MS/MS assays were established (Fig. 2). Using a similar approach, they also extended the method by simultaneously quantifying cyclosporine and sirolimus, showing that PSI was a novel and effective means for TDM of immunosuppressive drugs.25 In another study, quantification of imatinib in plasma was performed by using field-assisted PS, where a stainless steel needle was mounted axially to the paper triangle to increase the field strength.26 Nineteen gastrointestinal stromal tumors patients treated with 400 mg/day of imatinib were enrolled in a study to evaluate the quantitation capability of PSI-MS/MS, in comparison with traditional LC-MS/MS. The results showed a correlation of r = 0.783 in terms of drug concentrations in blood samples. In order to further evaluate the robustness of PSI, Yannell et al. compared PSI-MS analysis of DBS using different devices to collect blood samples in-field.27 Blood samples were collected in a remote area in Vietnam using disposable pipettes with precut Whatman 31ET paper substrates for PSI, DBS collection cards, and PSI cartridges. Imatinib and its major metabolite N-desmethyl-imatinib were measured from blood samples collected in-field from 4 patients for 5 consecutive days. Good linearities in concentration were identified for all devices. The RSDs for the analysis were about 31% at average, which can be attributed to the difficulty in control of the small volumes for blood samples collected and dispensed on paper.
Fig. 2.

Method comparisons of PSI to two immunoassays (A) and (B), and two LC-MSMS assays (C) and (D) (Reprinted with permission24. Copyright 2010 Elsevier).
2.2. Desorption Electrospray Ionization (DESI)
As one of the pioneering methods for ambient ionization, DESI has been widely applied in biomedical, pharmaceutical and forensics fields.28 The methodology behind DESI utilizes an charged solvent stream angled towards the sample surface to extract analytes and propel the resulting ions towards the MS inlet. Unlike conventional methods like punch out or cutting (PSI), DESI has the advantage of instantly analyzing DBS without additional steps and enabling point-by-point ionization for imaging, covered later in the review. Siebenhaar et al. used DESI to quantify aspirin in DBS by measuring its concentration in whole blood at multiple time points (to 300 min) after dosing a healthy volunteer with aspirin.29 The reported limit of detection (LOD) for aspirin was 8 μg/mL and LOQ was 10 μg/mL, with a linear range from 10 to 2000 μg/mL with an RSD less than 14%. Wiseman et al. reported direct analysis of DBS with DESI, quantifying sitamaquine, terfenadine, and prazosin using verapamil as internal standard.30 LODs were found to be 10 ng/mL and linear ranges from 10–10000 ng/mL were achieved. Quantitative analysis of drugs in urine was performed by coupling solid-phase microextraction (SPME) with DESI, LODs below 50 ng/mL in urine was achieved for all tested drugs except for meprobamate, whose LOD was found to be 160 ng/mL.31 The nature of DESI makes it very suitable for high speed screening. Such methodology could potentially be applied to high-throughput impurity profiling and process control in the pharmaceutical industry.32 For example, Chen et al. reported the semi-quantitative measurement of a simulated process failure in pharmaceutical production line, at an analysis speed of 2–3 samples/s, where thermal degradation of loratadine was clearly detected.
2.3. Direct Analysis in Real Time
Unlike PSI or DESI where ions are generated by a solvent-based electrospray, direct analysis in real time (DART) utilizes a corona discharge that results in the generation ions which are then exposed to a sample. Initial applications of DART have shown amenability to various polar and nonpolar analytes in raw samples.2 Although most quantitative application of DART were about food safety,33–35 the forensics and security applications of DART were also demonstrated for the analysis of drugs of abuse and explosives.36 An early application of DART-MS/MS for quantitative small molecule analysis was reported by Zhao et al.37, about 80% of the tested commercial compounds were found to have LODs below 5 ng/mL in plasma, linear ranges were 0.5–2000 ng/mL for verapamil and loperamide, and 1–2000 ng/mL for bufuralol with acceptable precision and accuracy. Yu et al.38 reported the implementation of DART for quantitation of drugs in biological matrices. LOD of verapamil in rat plasma was found to be 0.1 ng/mL, a linear range of 2–20000 ng/mL was achieved although IS was not incorporated during the analysis. DART can be coupled with sample preparation steps such as SPME for quantitation of drugs in blood samples.39 LOD of 0.3 μg/mL was obtained for analyzing diazepam in blood samples of 5 μL. Hsieh et al.40 reported a workflow for quantitation of endogenous cholesterol in human serum with DART where serum were loaded onto chromatography paper. Quantitation was performed by spotting cholesterol-d6 onto the serum sample spot on paper. It has been validated in 63 serum samples from 21 ultramarathon runners at three different time points, the results were compared to that obtained from fluorometric-enzymatic assay tests.
2.4. Coated Blade Spray
Coated blade spray (CBS) was developed as a method that integrates SPME and substrate-based spray ionization (Fig. 3).41, 42 Similar to the modifications of paper spray, stainless steel sheets were coated with materials such as C18-polyacrylonitrile. CBS retains the simplicity of PSI while avoiding the binding of matrix to a porous substrate. The coated blades had a larger surface area than SPME resulting in a larger loading capacity, while a washing step removes loosely-bound substances to minimize the matrix effects. A typical procedure includes preconditioning of the device, sample extraction, washing and MS analysis. Although several extra steps were added, the whole process could be finished within 3 min while achieving a LOQ at the parts-per-trillion level for cocaine spiked in phosphate buffered saline solutions.
Fig. 3.

(A) Experimental setup for blade-spray extraction and desorption/ionization. Quantitative analysis by CBS-MS, B) plasma spiked with cocaine and its isotopologue [D3] cocaine, C) urine spiked with diazepam and its isotopologue [D5] diazepam (Reprinted with permission41. Copyright 2014 Wiley-VCH).
CBS was applied for high-throughput screening and quantitation of both controlled substances and therapeutic drugs in biofluids.43 A holder for 96 CBS device was constructed and used in conjunction with a robotic sample preparation system for preconditioning, extraction and washing. LOQs of 0.1–10 ng/mL for plasma and 0.25–10 ng/mL for urine were achieved for 18 compounds. Although a longer extraction time was required compared to manual mode, automation of CBS enabled the analysis time per sample to be reduced to less than a minute. Using the same automated sample preparation system, quantitation of immunosuppressive drugs was demonstrated in whole blood samples.44 It should be noted that relatively large amounts of samples (> 100 μL) were required for CBS. In order to minimize the sample amount, biofluid samples can also spotted directly on blades coated with hydrophobic-lipophilic balance particles,45 which only requires 10 μL of biofluid sample. LOQs of 1–5 ng/mL in plasma and 1–10 ng/mL in blood were achieved for 17 drugs.
2.5. Improvement of Quantitative Performances
Clinical applications such as diagnostic screening or routine sampling heavily emphasize the improvement of quantitative performances of sensitivity and reproducibility. With the current frontrunning direct sampling methods mentioned above, several constraints stemming from complex matrices or poor desorption/ionization lead to suppressed sensitivity that can affect quantitative analysis. To overcome this hurdle, fast extraction methods and modifications of traditional ambient ionization methods are presented as solutions to improve quantitative analysis.
2.5.1. Slug-flow Microextraction (SFME)
Liquid-liquid extraction is a common sample extraction method used in analytical procedures. Because conventional liquid-liquid extraction demands large amounts of solvent, efforts have been devoted to lowering the sampling volume. Some examples include dispersive liquid-liquid microextraction46, hollow-fiber liquid-phase microextraction47 and electromembrane extraction48. In order to couple fast liquid microextraction with direct MS analysis, a highly efficient slug-flow microextraction (SFME) method was developed (Fig. 4A).49 The movement of the immiscible solvent and biofluid plugs within the glass capillary introduced a turbulence that facilitates the transfer of analytes to the extraction solvent, where equilibrium could be achieved after 5 cycles of slug-flow movements. SFME could be operated in either on-line or off-line mode; for on-line mode, the extraction was carried out within the nanotip that will be used for nanoESI; for off-line mode, the extraction was done using a glass capillary and then the extraction solvent is transferred to a nanotip for direct MS analysis. Both methods required a short amount of time. Considering that biofluid samples could be analyzed in its collected form, this method was especially suitable for point-of-care applications. By using just 5 μL of biofluid samples (whole blood or urine), LODs better than 1 ng/mL were achieved for most analytes. On-line derivatization was also demonstrated in this study, which improved the detection of analytes normally challenging for direct analysis, such as steroids in urine. Quantitative performance of SFME was demonstrated in the initial investigation: spiked methamphetamine and IS in blood showed linear response in the examined range of 1 ng/mL to 100 ng/mL (Fig. 4B). RSD within 10% was obtained for concentrations above 10 ng/mL.
Fig. 4.

(A) Schematic of SFME, and (B) calibration curve of obtained methamphetamine in bovine blood by SFME-MS through adding internal standard (IS) into the extraction solvent (Reprinted with permission49. Copyright 2014 Wiley-VCH). (C) Schematic of three-phase SFME, D) MS/MS spectra of glutamine (2 ng/mL) in the synthetic urine sample by three-phase SFME and direct MS analysis (Reprinted with permission50. Copyright 2018 Elsevier).
Since water-immiscible solvents were required as extraction solvents, SFME usually might not be efficient to polar compounds. Recently, a three-phase SFME method was also introduced as a suitable approach for analyzing polar compounds in biofluids (Fig. 4C), where a water-immiscible bridge solvent was placed between the biofluid and polar extraction solvent plugs.50 In a typical three-phase SFME system of biofluid-hexane-methanol/water, salts, cell debris and proteins in biofluid were blocked by the hexane, while polar analytes could be transferred into the extraction solvents cycle-by-cycle. By coupling with direct MS analysis, LODs of 2 ng/mL were obtained for amino acids in urine samples (Fig. 4D). The three-phase SFME method was also applied to hydrophilic drugs such as tenofovir-diphosphate (logP −4.6). Normal SFME was also inefficient in analyzing compounds that are suspended in low polarity matrices, such as oil samples. A reverse-phase SFME method was developed, where polar solvents such as methanol/water were used as the extraction solvents. This method was applied to analyze pesticide residues in vegetable oils, enabling highly sensitive and quantitative analysis.
2.5.2. Solid-phase Microextraction (SPME)
SPME is a sample preparation technology that combines sample preconcentration and sample cleanup. Commonly, SPME is coupled with chromatography methods for quantitative analysis of compounds in different kinds of matrices.51–54 The recent coupling of SPME with ambient MS has opened new opportunities for the field, as significant improvement in quantitative performance were achieved through this development.55
Theoretically, the fibers and other devices used in SPME-GC or -LC could be used directly for MS analysis. However, some aspects of the conventional SPME needs to be improved, especially the adsorption and desorption times under mild conditions. For fast sampling of analytes from blood samples, high-speed agitation with vortex was applied on commercially available SPME fibers. The adsorption process completed in 2 min, following with a quick rinse step. Finally, the fiber was inserted into a sharp tip filled with solvents for analyte desorption, nanoESI and MS analysis (Fig. 5A).56 The biocompatible coating on the SPME fiber did not cause fouling or absorption of proteins, minimizing clogging issues for the nanoESI emitter. Due to the preconcentration effect of SPME, analysis of relatively large sample volumes yielded LOQs as low as 100 pg/mL for methadone from 700 μL urine, and 100 pg/mL for amitriptyline from 300 μL whole blood. For small volume samples, a miniaturized SPME fiber coated with polypyrrole was used. Different from the SPME-nanoESI procedures discussed before, this device was much smaller and allowed static extractions from biofluid samples of 1–10 μL.57 Quantitative analysis of spiked biofluid samples (urine, plasma and whole blood) yielded LOQs at the low ng/mL levels with dynamic ranges encompassing two orders of magnitude, while keeping the total analysis time to less than 5 min.
Fig. 5.

(A) SPME-nanoESI (Reprinted with permission56. Copyright 2016 American Chemical Society); (B) SPME-OPP (Reprinted with permission58. Copyright 2017 American Chemical Society); (C) SPME-MOI (Reprinted with permission59. Copyright 2018 Future Science).
Another strategy of coupling SPME with MS was through open port probes (OPP), as reported by Gómez-Ríos et al.58 (Fig. 5B) After fast adsorption and rinsing steps, the SPME fiber was placed into the sample dome of the OPP where it was in touch with a continuous flow, desorbing and transferring the extracted analytes to an ionization source by aspiration. Quantitative performance of this method was characterized by analyzing fentanyl, buprenorphine and clenbuterol in urine samples, identifying accuracy greater than 90%. Tascon et al. adopted a microfluidic open interface (MOI) to couple SPME to MS, that featured a more contained desorption chamber than OPP (Fig. 5C).59 This method was applied to quantify several immunosuppressive drugs from 100 μL whole blood, reporting LOQs lower than 1 ng/mL.
2.5.3. Modified Paper Spray Ionization
Although various papers have concluded the applicability of PSI for clinical quantitative analysis, PSI’s sensitivity can be limited by several constraints such as matrix effects when analyzing complex biological samples; therefore, certain analyte sensitivities may be suppressed. To broaden the applicability of PSI and improve its sensitivity, many studies have sought to modify the paper substrates. Coating materials onto the paper substrates was shown to have significant improvements, including the use of graphene,60 zirconia,61 metal-organic framework62 as the coating materials. Paper substrates (Whatman grade SG81) coated with commercially available silica could be used directly for PSI. For analysis of DBS, 5 to 50-fold increases in LOQs were observed for lidocaine, amitriptyline, sunitinib, verapamil and citalopram in comparison to 31ET chromatography paper.63 Another study reported very low LOQs of 0.004–0.084 ng/mL in biofluids by coating with polystyrene microspheres.64 RSDs less than 5% were obtained for different analytes in DBS at concentrations of 10 ng/mL. By modifying the paper properties, novel features such as online extraction through silane treated hydrophobic paper were enabled (Fig. 6A-B).65 The treatment of the paper prevented aqueous-based biological samples from wetting the substrates while an organic solvent with low surface tension could. Thus, online liquid-liquid extraction could occur and allow for biofluids to be analyzed directly without interfering matrix effects. Based-on this hydrophobic paper design, the same group developed a new blood collection strategy where dried blood spheroid instead of traditional DBS were collected. The benefits include stabilization of analytes, easier sample extraction and minimization of negative effects that are common for DBS (e.g. hematocrit effects). Quantitation of drugs in dried blood spheroids was carried out using PSI.66 In a very recent work, polystyrene-impregnated paper substrates were fabricated and applied in PSI-MS of proteins and peptides (Fig. 6C).67 With significant improvement of extraction/ionization efficiency, some proteins and peptides (e.g angiotensin II and myoglobin) could be well detected even at very low concentration with LOQs in the range of 1–5 ng/mL.
Fig. 6.

(A) PSI-MS by the hydrophobic paper substrate, and (B) tandem MS analysis of drugs extracted from urine and blood. 3.9 ng/mL methamphetamine in blood and 3.9 ng/mL benzoylecgonine transitions in urine (Reprinted with permission65. Copyright 2016 American Chemical Society). (C) Comparison of mass spectra of several proteins by filter paper and PS-impregnated paper substrates-based PSI-MS (Reprinted with permission67. Copyright 2018 Royal Society of Chemistry).
Besides comprehensive modifications on the paper substrate, paper-based microfluidics is another rapidly developing field with smarter devices opening new opportunities for low-cost diagnostics.68, 69 Wax printing is a very simple method to imprint microfluidics structures on paper.70 Integration of microfluidic channels on paper substrate by wax printing for PSI, as demonstrated by Damon et al, could provide enhanced signal due to more stable electrospray and slower solvent evaporation.71 A paper-based MS immunoassay platform was reported by the same group, where a cleavable mass reporter was conjugated with antibody and ionized on a wax-printed paper substrate for MS analysis, quantitation of protein was achieved.72 By carefully design the microfluidic channels on paper substrate, new applications of PSI are yet to be explored.
3. Non-Quantitative Direct Sampling MS Methods for Clinical Analysis
Outside of quantitative analysis, direct sampling MS methods have also showed unique capabilities in other clinical applications such as imaging, direct clinical profiling and real-time analyte monitoring of tissue samples. These methods utilize semi to non-quantitative methods such as relative analyte ratios or correlations between analyte concentration and signal intensity to verify its diagnostic value, eliminating IS use and variability. These methods facilitate the detection of valuable in vivo chemical information that can only be obtained after extensive pretreatment or in its native state.
3.1. Ambient Ionization MS Imaging
MSI has been well established as a clinically significant tool to generate molecular-specific images from tissue samples. Ambient ionization-based MSI enables imaging of chemical information on untreated samples under atmospheric pressure outside the mass spectrometer.73, 74 Here we highlighted some ambient ionization which has been well applied for MSI of biomedical samples, including DESI, nanospray desorption electrospray ionization (nano-DESI), and LAESI.
DESI holds a significant advantage in providing spatial information from tissue samples. DESI MSI has been implemented regularly for clinical analysis. Some recent applications include lipid and metabolite profiling for discrimination of gray matter, white matter and different types of tumor tissues (Fig. 7A),75 intraoperative mapping of oncometabolite 2-hydroxyglutarate (2-HG, a biomarker for isocitrate dehydrogenase I and II mutations) to guide brain tumor surgery76, and discrimination of malignant and benign microscopic skin tumors77. Pirro et al. reported assessment of tumor margin during tumor resection using DESI, by normalizing signal intensities of 2-HG to total ion counts, the authors were able to correlate the normalized intensity to 2-HG concentration.78 In another study, Banerjee et al. reported using glucose/citrate ion ratios to predict cancer in prostate. This information can be collected in less than a minute after quick sample preparation and predict cancerous state.79 By using statistical analysis to identify significant metabolites in an independent set of samples, the generated model differentiated healthy and cancerous prostate tissue sections with nearly 90% accuracy.
Fig. 7.

(A) DESI-MS PCA projection and H&E stain of human brain tissue. A clear distinction can be made between glioma and white matter tissue with calculated relative tumor cell concentration. (Reprinted with permission75. Copyright 2015 National Academy of Sciences). (B) Nano-DESI quantitative images of endogenous PC and their absolute abundance in various sections of brain tissue. (Reprinted with permission83. Copyright 2014 American Chemical Society). (C) LAESI imaging paired with high resolution MS for clear differentiation of species with identical nominal mass. (Reprinted with permission86. Copyright 2010 American Chemical Society).
Nano-DESI is different from DESI in that it separates desorption and ionization where analytes were extracted in a liquid bridge formed between primary and secondary capillaries, and transferred to a secondary capillary for nanospray.80 Nano-DESI was applied to tissue imaging and resulted a resolution of better than 12 μm.81 A significant advantage for nano-DESI is the simultaneous mapping, identification and quantitation of analytes. By incorporation of IS into nano-DESI solvent, absolute quantitation can be achieved. This was realized in several reports; one of which added deuterated nicotine to nano-DESI solvent to quantify nicotine in rat brain tissue.82 The same strategy was expanded and applied to quantitation of endogenous phosphatidylcholines in brain tissue sections (Fig. 7B),83 quantitation of small molecule neurotransmitters in rat brain tissue sections84, and more recently, quantitation of prostaglandins in mouse uterus tissue section85.
LAESI is a combinatory method, where a laser is used to generate neutral species and ionized by charged droplets. Combination of laser desorption and ESI was first reported in 2005 (i.e. electrospray-assisted laser desorption ionization, ELDI), where post-ionization of laser-desorbed neutral species generates the ions.87 As an extension of ELDI, LAESI utilizes mid-infrared (mid-IR) laser to desorb neutral species from sample which then post-ionized by ESI, such method is applicable to water-rich samples due to the strong absorption of mid-IR by water molecules. In the initial publication, LAESI was applied for quantitation of verapamil and reserpine in 50% methanol solutions acidified with 0.1% acetic acid, linear dynamic range of four orders of magnitude was achieved without addition of internal standard. LODs were found to be 8 and 25 fmol for verapamil and reserpine, respectively. Although quantitation in complex matrix was not reported, LAESI was applied to direct analysis of urine, whole blood and serum samples, as well as in vivo profiling of metabolites in a French marigod seedling.4 The same group later reported depth profiling of leaf tissue and in vivo molecular imaging on a leaf, the spatial resolution of LAESI in molecular imaging was estimated to be 200–300 μm.88 Other imaging applications of LAESI were reported for tissue analysis, such as imaging of small metabolites and lipids in rat brain tissue (Fig. 7C)86 and top-down MSI of intact proteins in a mouse lung tissue section using commercialized LAESI source89.
3.2. Direct Profiling of Clinical Samples
In addition to imaging, there are various MS-based methods that can be applied for clinical applications such as biomarker or disease analysis. High-throughput direct sampling methods could facilitate biomarker discovery and easily transfer into routine disease screening methods. The presences of target molecules can provide general guidelines for further analysis and clinical verification, eliminating the dependence on an IS. Thus, relative quantitation based upon ion intensity or profiling (e.g. lipid/metabolite profiling) provides sufficient evidence for diagnostics. In this section, we discussed several methods that have been used for profiling of chemical information of clinical samples, including liquid extraction surface analysis (LESA), liquid microjunction surface sampling probe (LMJ-SSP), and touch spray. It should be noted that LESA90 and LMJ-SSP91 have also shown potential for MSI.
LESA is a direct sampling method developed in 2010,94 where a droplet of solvent is placed onto a sample surface for a fast liquid-surface extraction and ionized by nanoESI. Kertesz and Van Berkel described the adaption of a commercially available Advion NanoMate chip-based infusion nanoESI system to perform fully-automated LESA. The system was demonstrated for MALDI spot, DBS and tissue analysis. Quantitation of verapamil was realized in MALDI spots with a linear range of 1–200 ng/mL. Furthermore, LESA was also utilized in a limited imaging capacity, mapping drug distribution in rat brain tissue. By monitoring several analyte compounds across tissue sections, images were assessing the penetration of xenobiotics across the blood-brain barrier at a 1 mm spatial resolution.95 Edwards et al. reported LESA of DBS for analysis of hemoglobin (Hb) variants.96 Griffiths and Cooper later demonstrated the analysis of intact hemoglobin complex sampled from mouse liver tissue sections, which provide a tool for native MS imaging.97 The same group also reported combination of field asymmetric waveform ion mobility (FAIMS) with LESA of DBS, FAIMS enabled the separation of hemoglobin α- and β- units from lipid species, hence the detection of lipids were possible.98 By combining analyte or biomolecule profiling overlaid on histological stains, LESA has shown to be a significant profiling tool to examine xenobiotics in tissue sections.
LMJ-SSP utilizes the liquid microjunction formed between the sample surface and the probe to extract analytes, and then transfer the liquid for ionization under atmospheric conditions.99 Although similar to nano-DESI, the difference is that LMJ-SSP utilizes a dual coaxial tube. Gaissmaier et al. reported using a commercialized version of LMJ-SSP, Flowprobe (Prosolia), for TDM with DBS samples.100 The LOQ for acetaminophen was determined to be 5 μg/mL, although the sensitivity of method was limited, it was still appropriate for acetaminophen (10–20 μg/mL), covering the entire therapeutic range. LMJ-SSP was also coupled with online photochemical Paternò-Büchi reaction to perform rapid in situ determination of lipid C=C location isomers in tissue samples (Fig. 8A).92, 101 LMJ-SSP was used to extract unsaturated lipids and then react with acetone under UV irradiation; the reaction products were ionized and analyzed in MS and MS/MS mode. Relative quantitation was carried out based on diagnostic ion intensities of two isomers.
Fig. 8.

(A) Photochemical reactions paired with LMJ-SSP for the profiling of lipid isomers in tissue. (Reprinted with permission92. Copyright 2018 American Chemical Society). (B) Touch Spray MS for intraoperative assessment of human glioma. (Reprinted with permission93. Copyright 2017 Royal Society of Chemistry).
Touch spray MS is closely related to probe electrospray ionization in that it utilizes a metal probe and similar to PSI for it is also substrate-based spray ionization.102 For touch spray, a teasing needle is used to “touch” solid sample, then solvent and high voltage are applied to the needle to ionize the analytes in front of MS inlet. In the initial investigation, touch spray was demonstrated for a variety of applications: detection of lipids in mouse brain tissue, ex vivo detection of prostate cancer, in vitro detection of bacteria, detection of illicit drugs in DBS, quantitation of therapeutic drugs in whole blood and detection of fungicides in oranges.103 Touch spray was then utilized to differentiate human prostate cancer from normal tissue based-on their lipids profiles.104 Similar methodology was also applied to analysis of human kidney tissues to distinguish renal cell carcinomas from healthy tissue.105 Swab touch spray was reported for direct detection of strep throat causing bacterium106 and drug analysis from oral fluid107. Pirro et al. also reported assessment of surgical margins and oncometabolites using human glioma tissue sections, which demonstrates the potential of swab touch spray for intraoperative MS analysis (Fig. 8B).93
3.3. In vivo MS Analysis
Stemming from the success of MS-based direct sampling biomolecule analysis, in vivo diagnostics translates these methods into a comprehensive system that can instantly show positive/negative results, a critical component in time-sensitive environments like surgery. Direct sampling permits these methods to be used outside of analytical laboratories, while relative quantitative techniques eliminate IS complications. Paired with statistical analyses, real-time diagnostic information can be obtained with consideration to biocompatibility for in vivo diagnostics. This section reviews two novel real-time analyte monitoring methods, Rapid evaporative ionization MS (REIMS) and MasSpec Pen, that have reported the differentiation of diseased and healthy tissue.
REIMS utilizes rapid thermal evaporation to generate ions, therefore can be applied directly to living tissues. By using principle-component analysis, REIMS was able to differentiate healthy and cancerous breast tissue (Fig. 9A).108 Intraoperative analysis of tumor in vivo was realized with REIMS109 and REIMS-based endoscopic method was also developed for tissue identification110. The utilization of REMIS to collect MS profile and data analysis to obtain diagnostic information was coined “iKnife”, which is now commercialized by Waters Corporation. Identification of clinically relevant microorganism was realized by placing microbial biomass directly on the bipolar forceps.111 The methodology has been adapted and used for automatic high-throughput identification of microorganism. Similar results were obtained for hand-held bipolar probe REIMS and high-throughput REIMS, with speciation accuracies of 96.3% and 93.9%, respectively.112
Fig. 9.

(A) REIMS for intraoperative tissue identification. (Reprinted with permission109. Copyright 2017 American Association for the Advancement of Sciences). (B) MasSpec Pen for in vivo cancer diagnostics. (Reprinted with permission113. Copyright 2017 American Association for the Advancement of Sciences).
Alongside REIMS, a significant emerging tool for in vivo MS-based diagnostics is the MasSpec Pen (Fig. 9B).113 Similar to LESA, DESI and nano-DESI, the MasSpec pen is an automated sampling probe that utilizes liquid-solid interaction for in vivo and ex vivo tissue analysis. The probe generates a water droplet to contact the tissue for several seconds and transports it to a high-resolution MS for analysis. Unlike other direct sampling techniques, the MasSpec Pen method targets in vivo applications and takes biocompatibility into consideration. Ex vivo analysis of human tissue samples and in vivo cancer diagnosis during surgery of a mouse model was demonstrated. Afterwards, statistical analysis was applied to build models and compare to various other MS-based techniques, identifying 87.5% sensitivity and 100% specificity for cancer as well as 95.6% accuracy for breast cancer. Further statistical algorithms utilizing machine learning and validated molecular information shows an overall sensitivity, specificity and accuracy at 96.4%, 96.2% and 96.3% for cancer, respectively.
4. Translation of Direct Sampling MS to POC Applications
The benefits of direct sampling MS can only be fully appreciated when applied in a POC settings, where quick feedback of testing result is crucial for decision making in disease diagnosis or treatment. Although direct sampling ionization has largely simplified the operation of MS analysis, the over procedures can still be too complicated for users without professional training as analytical chemists. Therefore, it is critical to develop easy-to-use and cost-effective technologies that are amenable to the current healthcare system.
4.1. Development of Cartridges
Disposable sample cartridges are ideal for POC applications because they are easy to use, inexpensive to produce, and preventing cross-contamination. Due to its simplicity, PSI has great potential for cartridge development. A preliminary PS cartridge was fabricated using SLA (stereolithography apparatus) process; the cartridge was made of SLA resin Nanoform 15120 and consisted of holder, lid and electrode.23 With short analysis time, cartridge-based PSI would also be amenable for high throughput analysis, as previously reported.114 A platform was made for holding and moving 14 PS tips (Fig. 10A) for analysis. An automated source was designed at Purdue University and later commercialized by Prosolia Inc. (Indianapolis, IN, USA).24, 25, 27
Fig. 10.

(A) High through-put PSI (Reprinted with permission114. Copyright 2013 Elsevier); (B) PSI cartridge with integrated SPE (Reprinted with permission115. Copyright 2015 American Chemical Society); (C) 3D printed cartridge for targeted protein detection (Reprinted with permission116. Copyright 2017 American Chemical Society).
Various designs of cartridges also enabled integration of other functions with direct sampling methods. Zhang and Manicke fabricated a PSI cartridge with integrated solid phase extraction (SPE) (Fig. 10B).115 The SPE column significantly improved the signal by selectively enriching target compounds and removing the sample matrices. The sensitivity toward five drugs was improved by 14 to 70 times in comparison with direct PSI. Bills and Manicke developed a PSI cartridge with blood fractionation capabilities using different membranes integrated into the cartridge, so plasma samples instead of whole blood samples could be analyzed with PSI-MS when desired.117 Quantitative analysis of atenolol and carbamazepine by this novel PSI cartridge was compared with PSI analysis of plasma samples obtained by centrifugation, which showed similar results. Salentijn et al. reported a 3D printed PSI cartridge with fast wetting and continuous solvent supply features, allowing for a continuous spray of over ten minutes compared to the one minute spray time using the original PSI.118 They also reported another two PSI cartridge designs fabricated via 3D printing, integrating desolvation and ion optics.119 In both designs, sheath gas was used to improve the signals, while the second design included an electrostatic lens to focus the spray. Another cartridge designed by Zhang et al. utilized antibody columns to selectively enrich proteins from biofluids, and used a carbon nanotube coated porous polyethylene tip to generate electrospray (Fig. 10C).116 The device could be used for detection of apolipoprotein C1 T45S variant, hemoglobin species, wild-type transthyretin and transthyretin mutants in human plasma samples.
4.2. Miniature MS Systems
The miniaturization of MS systems is another important aspect for transferring MS methods into clinical and POC applications. The POC MS systems must meet a set of new criteria that are different from those used for evaluating traditional, in-lab mass spectrometers. Generally, operations of low cost, low power consumption, high portability, robustness and sufficient analytical performance are expected for POC applications.10, 11, 120, 121
One of the most significant barriers to miniaturize mass spectrometers is the limited pumping capacity due to size reduction, which could have a significant impact on the intake of analyte ions and thereby the analysis sensitivity. One solution to this limitation was to modify the continuous interface system using differential pumping as introduced in 2015 by Zhai et al122. Several modifications were also made to the system, including in-vacuum plasma ionization123, integration of an ion funnel124, and laserspray ionization for biological samples.125
Another solution to this barrier is the discontinuous atmospheric pressure interface (DAPI) that allowed a most significant reduction of pumping system while maintaining good sensitivity for the analysis.126 A pinch valve was used to keep the vacuum system closed and only opens briefly for sample introduction. This allowed the vacuum chamber to maintain a 10−5 Torr pressure only using small pumps and a characteristic pressure curve during operation. The interesting pressure behavior introduced by DAPI led to a new ionization method named synchronized discharged ionization, which could potentially be used for solid biological samples such as tissue sections or biopsies.127, 128 A series of miniature mass spectrometers using ion trap were developed with DAPI, including Mini 11129, Mini 12 (Fig. 11A-B)12, a backpack Mini130 and Mini β.131 LTP analysis of melamine in complex matrices such as milk and synthetic urine was reported using Mini 10.5.132 Depending on different matrices, the LODs were ranged from 0.03 μg/mL to 0.25 μg/mL.
Fig. 11.

(A) A simplified operation protocol of Mini 12 MS system. (B) Configuration of Mini 12 system. (C) MS5 mass spectrum for 20 ppm clenbuterol in 50/50 MeOH/H2O, by using nanoESI-Mini 12 (inset shows the isolated peak of ions with m/z 168). (D) Calibration curve showing ratio of amitriptyline/amitriptyline-d6 in blood, by extraction spray ionization and Mini 12 (product ion m/z of 233 was monitored) (Reprinted with permission12. Copyright 2014 American Chemical Society). (E) Schematic of paper capillary spray, (F) calibration curve of sitagliptin by paper capillary spray MS analysis (Reprinted with permission128. Copyright 2016 Springer). (G) Schematic of fast blood drug analysis by Culex autosampler, SFME and Mini 12, (H) shows the whole blood concentration profile of sitagliptin by the system (Reprinted with permission133. Copyright 2017 Future Science).
Some direct sampling methods can be coupled directly to the Mini 12.12 The system consisted of a rectilinear ion trap, it was demonstrated for tandem MS capabilities up to MS5 (Fig. 11C). Direct quantitative analysis was performed with amitriptyline spiked in whole blood using paper spray as the ionization source, reporting LODs of at least 7.5 ng/mL (Fig. 11D). Ma et al. reported the use of PSI coupling to Mini 12 for analysis of synthetic cannabinoids in biofluids134. The LOQs for the tested synthetic cannabinoids in blood or urine were between 10 and 20 ng/mL. Kirby et al. also demonstrated the analysis of drug of abuse in urine in conjunction with digital microfluidics135, where extraction of dried urine spot was performed on a home-built digital microfluidics system and analyzed using Mini 12. The LOQ was 40 ng/mL for cocaine.
To better couple the PSI with miniature MS system, an alternative design was proposed as the paper capillary spray. A fused silica capillary was inserted into the cut filter paper to act as the sprayer (Fig. 11E-F).128 The paper capillary spray combines PSI’s simple sampling methodology with the stable electrospray of nanoESI. As a result, paper capillary spray generated a single, stable Taylor cone and has a more uniform droplet formation in comparison to PSI. Quantitation of sitagliptin in bovine whole blood was demonstrated, showing a wide linear range and strong reproducibility.
As an exploration of miniature MS system for drug discovery and clinical applications, a pharmacokinetics study was performed using SFME sampling and mini-MS quantitation of therapeutic drugs in whole blood samples (Fig. 11G-H).133 In this study, an automated blood sampler was used to draw whole blood samples from freely moving rats that were dosed with therapeutic drugs. SFME was used to extract the drug compounds from the whole blood samples that were then analyzed by Mini 12 using nanoESI. Analysis of spiked samples using SFME and Mini 12 yielded high levels of accuracy and precision that met the recommended criteria while plotting TDM curves.
5. Conclusion and Future Outlook
The development of ambient ionization has promoted the application of mass spectrometry in clinical analysis. Besides direct profiling of chemical information from tissue samples and imaging of disease state of organs, quantitative analysis by direct sampling-based MS also plays an important role in disease diagnostics and drug analysis. Although many direct sampling methods have been developed and some of have been demonstrated in clinical samples, the translation of direct sampling MS into clinical practices is still challenging and needs further improvement.
Since ionization is often performed in the presence of matrices, the ionization efficiency and reproducibility can be easily affected. Modifications of the paper substrate with materials such as polystyrene spheres or carbon nanotubes have showed improved sensitivity to a number of compounds. Sample extraction with SFME has shown to further improve quantitative performance for biofluids analysis. Some extraction methods were also developed to further Other methods such as SPME and CBS can achieve superior sensitivity and reproducibility for biofluid analysis, but are limited by the required preconditioning, sampling and rinsing steps. Due to varying ionization efficiencies across samples and methods, the incorporation of IS is still necessary for calibration or application of other relative quantitative methods. The IS can be spiked into the sample directly, but this may pose a problem when only a small volume of clinical samples is available. Quantitation based on metabolite or lipid isomer ratios can also be employed in direct sampling MS methods. Numerous studies have shown that ratios of some metabolites can be used as biomarkers for some diseases, such as the use of glucose/citrate ion signal ratio for diagnosis of prostate cancer79, and ratios of lipid double bond isomers136 or sn- isomers137.
Direct sampling methods have also showed good performance in MS based imaging and profiling of chemical information in cinical analysis. DESI is most suitable for clinical imaging and high-throughput screening, where quantitation is also possible but does not show very high sensitivity. As an improvement to DESI, nano-DESI has shown potential for quantitative imaging, a novel aspect that can enable more specific biomolecule or xenobiotic characterization that could be useful in triaging or imaging drug distribution throughout an organ. Similarly, the rapid extraction nature of liquid extraction sampling methods (LMJ-SSP, LESA, nano-DESI) could accelerate the analysis, but inevitably sacrifices extraction efficiency. LAESI MSI imaging shows a wider compatibility to analytes and is suitable for water-rich samples. However, these methods may not be applicable to low-content analytes in matrices or analytes that are incompatible with certain solvents. And further improvements in automation and commercialization of these sources could help lower this barrier and allow more clinical research to be done. With demonstrated compatibility for ex and in vivo cancer diagnostics, technologies such as REIMS and MasSpec pen have taken the first step in translating MS for intra-surgical applications. Although numerous direct sampling MS methods have been developed and evaluated for the analysis of different kinds of compounds such as drugs, metabolites and lipids, only a few researches explored the analysis of other important classes of biological molecules such as proteins, which are usually important biomarkers in clinical analysis. Another limitation is the lack of standardization throughout each direct sampling method; various papers on the same method have reported differing experimental conditions such as IS introduction or biofluid samples, resulting differing ionization efficiencies and varying sensitivities. Many studies show novel proof-of-concept or improvements to existing methods, but implementation rather than innovation is key in the near-saturated field of direct sampling MS. A solution to this is the development of cartridges. In order to apply direct sampling methods into clinical analysis, cartridges should be developed according to clinical practices. For example, sample collecting, storage and bio-safety are criteria that should be considered along with costs and ease-of-use. Currently, cartridges of paper spray and coated blade spray are commercially available.
The other aspect of developing a comprehensive POC MS-system for clinical and in-field applications is the miniaturization of MS. It is worth noting that miniature MS-based POC or in-field quantitative analysis are mainly for targeted applications, which are very different with normal MS methods seeking best performances and wide applications. We have seen several miniature MS systems that are capable of analyzing biofluid and tissue samples by coupling with direct sampling methods, such as PSI coupled miniature MS system, that provides users with significant levels of sensitivity and specificity. In the next few years, we hope we can see more integrated miniature MS systems with smaller sizes and improved performances.
Acknowledgements
The authors would like to thank financial supports from the National Natural Science Foundation of China (Project 21627807) and the National Institutes of Health (Project 1R01AI122298 and R01AI122932).
Footnotes
Conflicts of interest
Z. O. is the founder of PURSPEC Technologies Inc.
References
- 1.Takats Z, Wiseman JM, Gologan B and Cooks RG, Science, 2004, 306, 471–473. [DOI] [PubMed] [Google Scholar]
- 2.Cody RB, Laramee JA and Durst HD, Anal. Chem, 2005, 77, 2297–2302. [DOI] [PubMed] [Google Scholar]
- 3.McEwen CN, McKay RG and Larsen BS, Anal. Chem, 2005, 77, 7826–7831. [DOI] [PubMed] [Google Scholar]
- 4.Nemes P and Vertes A, Anal. Chem, 2007, 79, 8098–8106. [DOI] [PubMed] [Google Scholar]
- 5.Harper JD, Charipar NA, Mulligan CC, Zhang X, Cooks RG and Ouyang Z, Anal. Chem, 2008, 80, 9097–9104. [DOI] [PubMed] [Google Scholar]
- 6.Na N, Zhao M, Zhang S, Yang C and Zhang X, J. Am. Soc. Mass Spectrom, 2007, 18, 1859–1862. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Venter A and Cooks RG, Chem. Commun, 2006, 2042–2044. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Liu J, Cooks RG and Ouyang Z, Angew. Chem. Int. Ed, 2010, 49, 877–880. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira CR, Yannell KE, Jarmusch AK, Pirro V, Ouyang Z and Cooks RG, Clin. Chem, 2016, 62, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma X and Ouyang Z, TrAC, Trends Anal. Chem, 2016, 85, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Wang X, Xia Y and Ouyang Z, Theranostics, 2017, 7, 2968–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Chen TC, Ren Y, Hendricks PI, Cooks RG and Ouyang Z, Anal. Chem, 2014, 86, 2909–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Wang H, Manicke NE, Lin JM, Cooks RG and Ouyang Z, Anal. Chem, 2010, 82, 2463–2471. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Manicke NE, Yang Q, Zheng L, Shi R, Cooks RG and Ouyang Z, Anal. Chem, 2011, 83, 1197–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Wang H, Cooks RG and Ouyang Z, Anal. Chem, 2011, 83, 7608–7613. [DOI] [PubMed] [Google Scholar]
- 16.Manicke NE, Yang Q, Wang H, Oradu S, Ouyang Z and Cooks RG, Int. J. Mass Spectrom, 2011, 300, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J, Cooks RG and Ouyang Z, Anal. Chem, 2013, 85, 5632–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manicke NE, Bills BJ and Zhang C, Bioanalysis, 2016, 8, 589–606. [DOI] [PubMed] [Google Scholar]
- 19.Su Y, Wang H, Liu J, Wei P, Cooks RG and Ouyang Z, Analyst 2013, 138, 4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Espy RD, Teunissen SF, Manicke NE, Ren Y, Ouyang Z, van Asten A and Cooks RG, Anal. Chem, 2014, 86, 7712–7718. [DOI] [PubMed] [Google Scholar]
- 21.Costa C, Webb R, Palitsin V, Ismail M, de Puit M, Atkinson S and Bailey MJ, Clin. Chem, 2017, 63, 1745–1752. [DOI] [PubMed] [Google Scholar]
- 22.Manicke NE, Abu-Rabie P, Spooner N, Ouyang Z and Cooks RG, J. Am. Soc. Mass Spectrom, 2011, 22, 1501–1507. [DOI] [PubMed] [Google Scholar]
- 23.Yang Q, Wang H, Maas JD, Chappell WJ, Manicke NE, Cooks RG and Ouyang Z, Int. J. Mass Spectrom, 2012, 312, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi RZ, El Gierari el TM, Manicke NE and Faix JD, Clin. Chim. Acta, 2015, 441, 99–104. [DOI] [PubMed] [Google Scholar]
- 25.Shi RZ, El Gierari el TM, Faix JD and Manicke NE, Clin. Chem, 2016, 62, 295–297. [DOI] [PubMed] [Google Scholar]
- 26.D’Aronco S, Dall’Armi M, Crotti S, Calandra E, Traldi P, Di Marco V, Buonadonna A, Corona G, Giodini L, Marangon E, Posocco B, Toffoli G and Agostini M, J. Mass Spectrom, 2017, 52, 283–289. [DOI] [PubMed] [Google Scholar]
- 27.Yannell KE, Kesely KR, Chien HD, Kissinger CB and Cooks RG, Anal. Bioanal. Chem, 2017, 409, 121–131. [DOI] [PubMed] [Google Scholar]
- 28.Cooks RG, Ouyang Z, Takats Z and Wiseman JM, Science, 2006, 311, 1566–1570. [DOI] [PubMed] [Google Scholar]
- 29.Siebenhaar M, Kullmer K, Fernandes NM, Hullen V and Hopf C, Anal. Bioanal. Chem, 2015, 407, 7229–7238. [DOI] [PubMed] [Google Scholar]
- 30.Wiseman JM, Evans CA, Bowen CL and Kennedy JH, Analyst 2010, 135, 720. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy JH, Aurand C, Shirey R, Laughlin BC and Wiseman JM, Anal. Chem, 2010, 82, 7502–7508. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, Talaty NN, Takats Z and Cooks RG, Anal. Chem, 2005, 77, 6915–6927. [DOI] [PubMed] [Google Scholar]
- 33.Guo T, Yong W, Jin Y, Zhang L, Liu J, Wang S, Chen Q, Dong Y, Su H and Tan T, Mass Spectrom. Rev, 2017, 36, 161–187. [DOI] [PubMed] [Google Scholar]
- 34.Gross JH, Anal. Bioanal. Chem, 2014, 406, 63–80. [DOI] [PubMed] [Google Scholar]
- 35.Albert A, Shelley JT and Engelhard C, Anal. Bioanal. Chem, 2014, 406, 6111–6127. [DOI] [PubMed] [Google Scholar]
- 36.Pavlovich MJ, Musselman B and Hall AB, Mass Spectrom. Rev, 2018, 37, 171–187. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y, Lam M, Wu D and Mak R, Rapid Commun. Mass Spectrom, 2008, 22, 3217–3224. [DOI] [PubMed] [Google Scholar]
- 38.Yu S, Crawford E, Tice J, Musselman B and Wu JT, Anal. Chem, 2009, 81, 193–202. [DOI] [PubMed] [Google Scholar]
- 39.Mirnaghi FS and Pawliszyn J, Anal. Chem, 2012, 84, 8301–8309. [DOI] [PubMed] [Google Scholar]
- 40.Hsieh HY, Li LH, Hsu RY, Kao WF, Huang YC and Hsu CC, Anal. Chem, 2017, 89, 6146–6152. [DOI] [PubMed] [Google Scholar]
- 41.Gomez-Rios GA and Pawliszyn J, Angew. Chem. Int. Ed, 2014, 53, 14503–14507. [DOI] [PubMed] [Google Scholar]
- 42.Gomez-Rios GA, Tascon M and Pawliszyn J, Bioanalysis, 2018, 10, 257–271. [DOI] [PubMed] [Google Scholar]
- 43.Tascon M, Gomez-Rios GA, Reyes-Garces N, Poole J, Boyaci E and Pawliszyn J, Anal. Chem, 2017, 89, 8421–8428. [DOI] [PubMed] [Google Scholar]
- 44.Gomez-Rios GA, Tascon M, Reyes-Garces N, Boyaci E, Poole JJ and Pawliszyn J, Anal. Chim. Acta, 2018, 999, 69–75. [DOI] [PubMed] [Google Scholar]
- 45.Gomez-Rios GA, Tascon M, Reyes-Garces N, Boyaci E, Poole J and Pawliszyn J, Sci. Rep, 2017, 7, 16104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F and Berijani S, J. Chromatogr. A, 2006, 1116, 1–9. [DOI] [PubMed] [Google Scholar]
- 47.Pedersen-Bjergaard S and Rasmussen KE, Anal. Chem, 1999, 71, 2650–2656. [DOI] [PubMed] [Google Scholar]
- 48.Pedersen-Bjergaard S and Rasmussen KE, J. Chromatogr. A, 2006, 1109, 183–190. [DOI] [PubMed] [Google Scholar]
- 49.Ren Y, McLuckey MN, Liu J and Ouyang Z, Angew. Chem. Int. Ed, 2014, 53, 14124–14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren Y, Zhang W, Lin Z, Bushman LR, Anderson PL and Ouyang Z, Talanta, 2018, 189, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bojko B, Cudjoe E, Gomez-Rios GA, Gorynski K, Jiang R, Reyes-Garces N, Risticevic S, Silva EA, Togunde O, Vuckovic D and Pawliszyn J, Anal. Chim. Acta, 2012, 750, 132–151. [DOI] [PubMed] [Google Scholar]
- 52.Souza-Silva ÉA, Reyes-Garcés N, Gómez-Ríos GA, BoyacƖ E, Bojko B and Pawliszyn J, TrAC, Trends Anal. Chem, 2015, 71, 249–264. [Google Scholar]
- 53.Gorynski K, Gorynska P, Gorska A, Harezlak T, Jaroch A, Jaroch K, Lendor S, Skobowiat C and Bojko B, J. Pharm. Biomed. Anal, 2016, 130, 55–67. [DOI] [PubMed] [Google Scholar]
- 54.Reyes-Garces N, Gionfriddo E, Gomez-Rios GA, Alam MN, Boyaci E, Bojko B, Singh V, Grandy J and Pawliszyn J, Anal. Chem, 2018, 90, 302–360. [DOI] [PubMed] [Google Scholar]
- 55.Fang L, Deng J, Yang Y, Wang X, Chen B, Liu H, Zhou H, Ouyang G and Luan T, TrAC, Trends Anal. Chem, 2016, 85, 61–72. [Google Scholar]
- 56.Gomez-Rios GA, Reyes-Garces N, Bojko B and Pawliszyn J, Anal. Chem, 2016, 88, 1259–1265. [DOI] [PubMed] [Google Scholar]
- 57.Piri-Moghadam H, Ahmadi F, Gomez-Rios GA, Boyaci E, Reyes-Garces N, Aghakhani A, Bojko B and Pawliszyn J, Angew. Chem. Int. Ed, 2016, 55, 7510–7514. [DOI] [PubMed] [Google Scholar]
- 58.Gomez-Rios GA, Liu C, Tascon M, Reyes-Garces N, Arnold DW, Covey TR and Pawliszyn J, Anal. Chem, 2017, 89, 3805–3809. [DOI] [PubMed] [Google Scholar]
- 59.Tascon M, Alam MN, Gomez-Rios GA and Pawliszyn J, Anal. Chem, 2018, 90, 2631–2638. [DOI] [PubMed] [Google Scholar]
- 60.Ji J, Nie L, Liao L, Du R, Liu B and Yang P, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci, 2016, 1015–1016, 142–149. [DOI] [PubMed] [Google Scholar]
- 61.Zheng Y, Wang Q, Wang X, Chen Y, Wang X, Zhang X, Bai Z, Han X and Zhang Z, Anal. Chem, 2016, 88, 7005–7013. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Zheng Y, Wang T, Xiong X, Fang X and Zhang Z, Anal. Methods, 2016, 8, 8004–8014. [Google Scholar]
- 63.Zhang Z, Xu W, Manicke NE, Cooks RG and Ouyang Z, Anal. Chem, 2012, 84, 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang T, Zheng Y, Wang X, Austin DE and Zhang Z, Anal. Chem, 2017, 89, 7988–7995. [DOI] [PubMed] [Google Scholar]
- 65.Damon DE, Davis KM, Moreira CR, Capone P, Cruttenden R and Badu-Tawiah AK, Anal. Chem, 2016, 88, 1878–1884. [DOI] [PubMed] [Google Scholar]
- 66.Damon DE, Yin M, Allen DM, Maher YS, Tanny CJ, Oyola-Reynoso S, Smith BL, Maher S, Thuo MM and Badu-Tawiah AK, Anal. Chem, 2018, 90, 9353–9358. [DOI] [PubMed] [Google Scholar]
- 67.Li J, Zheng Y, Mi W, Muyizere T and Zhang Z, Anal. Methods, 2018, 10, 2803–2811. [Google Scholar]
- 68.Martinez AW, Phillips ST, Butte MJ and Whitesides GM, Angew. Chem. Int. Ed, 2007, 46, 1318–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yetisen AK, Akram MS and Lowe CR, Lab. Chip, 2013, 13, 2210–2251. [DOI] [PubMed] [Google Scholar]
- 70.Carrilho E, Martinez AW and Whitesides GM, Anal. Chem, 2009, 81, 7091–7095. [DOI] [PubMed] [Google Scholar]
- 71.Damon DE, Maher YS, Yin M, Jjunju FP, Young IS, Taylor S, Maher S and Badu-Tawiah AK, Analyst, 2016, 141, 3866–3873. [DOI] [PubMed] [Google Scholar]
- 72.Chen S, Wan Q and Badu-Tawiah AK, J. Am. Chem. Soc, 2016, 138, 6356–6359. [DOI] [PubMed] [Google Scholar]
- 73.Wu C, Dill AL, Eberlin LS, Cooks RG and Ifa DR, Mass Spectrom. Rev, 2013, 32, 218–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vaysse PM, Heeren RMA, Porta T and Balluff B, Analyst, 2017, 142, 2690–2712. [DOI] [PubMed] [Google Scholar]
- 75.Jarmusch AK, Pirro V, Baird Z, Hattab EM, Cohen-Gadol AA and Cooks RG, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Santagata S, Eberlin LS, Norton I, Calligaris D, Feldman DR, Ide JL, Liu X, Wiley JS, Vestal ML, Ramkissoon SH, Orringer DA, Gill KK, Dunn IF, Dias-Santagata D, Ligon KL, Jolesz FA, Golby AJ, Cooks RG and Agar NY, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, 11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Margulis K, Chiou AS, Aasi SZ, Tibshirani RJ, Tang JY and Zare RN, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, 6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pirro V, Alfaro CM, Jarmusch AK, Hattab EM, Cohen-Gadol AA and Cooks RG, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 6700–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Banerjee S, Zare RN, Tibshirani RJ, Kunder CA, Nolley R, Fan R, Brooks JD and Sonn GA, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 3334–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roach PJ, Laskin J and Laskin A, Analyst, 2010, 135, 2233–2236. [DOI] [PubMed] [Google Scholar]
- 81.Laskin J, Heath BS, Roach PJ, Cazares L and Semmes OJ, Anal. Chem, 2012, 84, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lanekoff I, Thomas M, Carson JP, Smith JN, Timchalk C and Laskin J, Anal. Chem, 2013, 85, 882–889. [DOI] [PubMed] [Google Scholar]
- 83.Lanekoff I, Thomas M and Laskin J, Anal. Chem, 2014, 86, 1872–1880. [DOI] [PubMed] [Google Scholar]
- 84.Bergman HM, Lundin E, Andersson M and Lanekoff I, Analyst, 2016, 141, 3686–3695. [DOI] [PubMed] [Google Scholar]
- 85.Duncan KD, Fang R, Yuan J, Chu RK, Dey SK, Burnum-Johnson KE and Lanekoff I, Anal. Chem, 2018, 90, 7246–7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nemes P, Woods AS and Vertes A, Anal. Chem, 2010, 82, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shiea J, Huang MZ, Hsu HJ, Lee CY, Yuan CH, Beech I and Sunner J, Rapid Commun. Mass Spectrom, 2005, 19, 3701–3704. [DOI] [PubMed] [Google Scholar]
- 88.Nemes P, Barton AA, Li Y and Vertes A, Anal. Chem, 2008, 80, 4575–4582. [DOI] [PubMed] [Google Scholar]
- 89.Kiss A, Smith DF, Reschke BR, Powell MJ and Heeren RM, Proteomics, 2014, 14, 1283–1289. [DOI] [PubMed] [Google Scholar]
- 90.Eikel D, Vavrek M, Smith S, Bason C, Yeh S, Korfmacher WA and Henion JD, Rapid Commun. Mass Spectrom, 2011, 25, 3587–3596. [DOI] [PubMed] [Google Scholar]
- 91.Kertesz V, Ford MJ and Van Berkel GJ, Anal. Chem, 2005, 77, 7183–7189. [DOI] [PubMed] [Google Scholar]
- 92.Tang F, Guo C, Ma X, Zhang J, Su Y, Tian R, Shi R, Xia Y, Wang X and Ouyang Z, Anal. Chem, 2018, 90, 5612–5619. [DOI] [PubMed] [Google Scholar]
- 93.Pirro V, Llor RS, Jarmusch AK, Alfaro CM, Cohen-Gadol AA, Hattab EM and Cooks RG, Analyst, 2017, 142, 4058–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kertesz V and Van Berkel GJ, J. Mass Spectrom, 2010, 45, 252–260. [DOI] [PubMed] [Google Scholar]
- 95.Swales JG, Tucker JW, Spreadborough MJ, Iverson SL, Clench MR, Webborn PJ and Goodwin RJ, Anal. Chem, 2015, 87, 10146–10152. [DOI] [PubMed] [Google Scholar]
- 96.Edwards RL, Creese AJ, Baumert M, Griffiths P, Bunch J and Cooper HJ, Anal. Chem, 2011, 83, 2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Griffiths RL and Cooper HJ, Anal. Chem, 2016, 88, 606–609. [DOI] [PubMed] [Google Scholar]
- 98.Griffiths RL, Dexter A, Creese AJ and Cooper HJ, Analyst, 2015, 140, 6879–6885. [DOI] [PubMed] [Google Scholar]
- 99.Van Berkel GJ, Kertesz V, Koeplinger KA, Vavrek M and Kong AN, J. Mass Spectrom, 2008, 43, 500–508. [DOI] [PubMed] [Google Scholar]
- 100.Gaissmaier T, Siebenhaar M, Todorova V, Hullen V and Hopf C, Analyst, 2016, 141, 892–901. [DOI] [PubMed] [Google Scholar]
- 101.Ma X and Xia Y, Angew. Chem. Int. Ed, 2014, 53, 2592–2596. [DOI] [PubMed] [Google Scholar]
- 102.Hiraoka K, Nishidate K, Mori K, Asakawa D and Suzuki S, Rapid Commun. Mass Spectrom, 2007, 21, 3139–3144. [DOI] [PubMed] [Google Scholar]
- 103.Kerian KS, Jarmusch AK and Cooks RG, Analyst, 2014, 139, 2714–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kerian KS, Jarmusch AK, Pirro V, Koch MO, Masterson TA, Cheng L and Cooks RG, Analyst, 2015, 140, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alfaro CM, Jarmusch AK, Pirro V, Kerian KS, Masterson TA, Cheng L and Cooks RG, Anal. Bioanal. Chem, 2016, 408, 5407–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jarmusch AK, Pirro V, Kerian KS and Cooks RG, Analyst, 2014, 139, 4785–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pirro V, Jarmusch AK, Vincenti M and Cooks RG, Anal. Chim. Acta, 2015, 861, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schafer KC, Denes J, Albrecht K, Szaniszlo T, Balog J, Skoumal R, Katona M, Toth M, Balogh L and Takats Z, Angew. Chem. Int. Ed, 2009, 48, 8240–8242. [DOI] [PubMed] [Google Scholar]
- 109.Balog J, Sasi-Szabo L, Kinross J, Lewis MR, Muirhead LJ, Veselkov K, Mirnezami R, Dezso B, Damjanovich L, Darzi A, Nicholson JK and Takats Z, Sci. Transl. Med, 2013, 5, 194ra193. [DOI] [PubMed] [Google Scholar]
- 110.Balog J, Kumar S, Alexander J, Golf O, Huang J, Wiggins T, Abbassi-Ghadi N, Enyedi A, Kacska S, Kinross J, Hanna GB, Nicholson JK and Takats Z, Angew. Chem. Int. Ed, 2015, 54, 11059–11062. [DOI] [PubMed] [Google Scholar]
- 111.Strittmatter N, Rebec M, Jones EA, Golf O, Abdolrasouli A, Balog J, Behrends V, Veselkov KA and Takats Z, Anal. Chem, 2014, 86, 6555–6562. [DOI] [PubMed] [Google Scholar]
- 112.Bolt F, Cameron SJ, Karancsi T, Simon D, Schaffer R, Rickards T, Hardiman K, Burke A, Bodai Z, Perdones-Montero A, Rebec M, Balog J and Takats Z, Anal. Chem, 2016, 88, 9419–9426. [DOI] [PubMed] [Google Scholar]
- 113.Zhang J, Rector J, Lin JQ, Young JH, Sans M, Katta N, Giese N, Yu W, Nagi C, Suliburk J, Liu J, Bensussan A, DeHoog RJ, Garza KY, Ludolph B, Sorace AG, Syed A, Zahedivash A, Milner TE and Eberlin LS, Sci. Transl. Med, 2017, 9, eaan3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shen L, Zhang J, Yang Q, Manicke NE and Ouyang Z, Clin. Chim. Acta, 2013, 420, 28–33. [DOI] [PubMed] [Google Scholar]
- 115.Zhang C and Manicke NE, Anal. Chem, 2015, 87, 6212–6219. [DOI] [PubMed] [Google Scholar]
- 116.Zhang C, Glaros T and Manicke NE, J. Am. Chem. Soc, 2017, 139, 10996–10999. [DOI] [PubMed] [Google Scholar]
- 117.Bills BJ and Manicke NE, Clin. Mass Spectrom, 2016, 2, 18–24. [Google Scholar]
- 118.Salentijn GI, Permentier HP and Verpoorte E, Anal. Chem, 2014, 86, 11657–11665. [DOI] [PubMed] [Google Scholar]
- 119.Salentijn GI, Oleschuk RD and Verpoorte E, Anal. Chem, 2017, 89, 11419–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou X, Liu J, Cooks RG and Ouyang Z, Bioanalysis, 2014, 6, 1497–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Snyder DT, Pulliam CJ, Ouyang Z and Cooks RG, Anal. Chem, 2016, 88, 2–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhai Y, Feng Y, Wei Y, Wang Y and Xu W, Analyst, 2015, 140, 3406–3414. [DOI] [PubMed] [Google Scholar]
- 123.Zhai Y, Jiang T, Huang G, Wei Y and Xu W, Analyst, 2016, 141, 5404–5411. [DOI] [PubMed] [Google Scholar]
- 124.Zhai Y, Zhang X, Xu H, Zheng Y, Yuan T and Xu W, Anal. Chem, 2017, 89, 4177–4183. [DOI] [PubMed] [Google Scholar]
- 125.Zhai Y, Liu S, Gao L, Hu L and Xu W, Anal. Chem, 2018, 90, 5696–5702. [DOI] [PubMed] [Google Scholar]
- 126.Gao L, Cooks RG and Ouyang Z, Anal. Chem, 2008, 80, 4026–4032. [DOI] [PubMed] [Google Scholar]
- 127.Chen TC and Ouyang Z, Anal. Chem, 2013, 85, 1767–1772. [DOI] [PubMed] [Google Scholar]
- 128.Ren Y, Chiang S, Zhang W, Wang X, Lin Z and Ouyang Z, Anal. Bioanal. Chem, 2016, 408, 1385–1390. [DOI] [PubMed] [Google Scholar]
- 129.Gao L, Sugiarto A, Harper JD, Cooks RG and Ouyang Z, Anal. Chem, 2008, 80, 7198–7205. [DOI] [PubMed] [Google Scholar]
- 130.Hendricks PI, Dalgleish JK, Shelley JT, Kirleis MA, McNicholas MT, Li L, Chen TC, Chen CH, Duncan JS, Boudreau F, Noll RJ, Denton JP, Roach TA, Ouyang Z and Cooks RG, Anal. Chem, 2014, 86, 2900–2908. [DOI] [PubMed] [Google Scholar]
- 131.Guo X, Bai H, Lv Y, Xi G, Li J, Ma X, Ren Y, Ouyang Z and Ma Q, Talanta, 2018, 180, 182–192. [DOI] [PubMed] [Google Scholar]
- 132.Huang G, Xu W, Visbal-Onufrak MA, Ouyang Z and Cooks RG, Analyst, 2010, 135, 705–711. [DOI] [PubMed] [Google Scholar]
- 133.Pu F, Zhang W, Bateman KP, Liu Y, Helmy R and Ouyang Z, Bioanalysis, 2017, 9, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ma Q, Bai H, Li W, Wang C, Cooks RG and Ouyang Z, Talanta, 2015, 142, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kirby AE, Lafreniere NM, Seale B, Hendricks PI, Cooks RG and Wheeler AR, Anal. Chem, 2014, 86, 6121–6129. [DOI] [PubMed] [Google Scholar]
- 136.Ma X, Chong L, Tian R, Shi R, Hu TY, Ouyang Z and Xia Y, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Paine MRL, Poad BLJ, Eijkel GB, Marshall DL, Blanksby SJ, Heeren RMA and Ellis SR, Angew. Chem. Int. Ed, 2018, 57, 10530–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]