

Abstract
WEE1 kinase regulates the G2-M cell-cycle checkpoint, a critical mechanism for DNA repair in cancer cells that can confer resistance to DNA-damaging agents. We previously reported a series of pyrazolopyrimidinones based on AZD1775, a known WEE1 inhibitor, as an initial investigation into the structural requirements for WEE1 inhibition. Our lead inhibitor demonstrated WEE1 inhibition in the same nanomolar range as AZD1775, and potentiated the effects of cisplatin in medulloblastoma cells, but had reduced single-agent cytotoxicity. These results prompted the development of a more comprehensive series of WEE1 inhibitors. Here, we report a series of pyrazolopyrimidinones and identify a more potent WEE1 inhibitor than AZD1775 and additional compounds that demonstrate that WEE1 inhibition can be achieved with reduced single-agent cytotoxicity. These studies support that WEE1 inhibition can be uncoupled from the potent cytotoxic effects observed with AZD1775, and this may have important ramifications in the clinical setting where WEE1 inhibitors are used as chemosensitizers for DNA-targeted chemotherapy.
Keywords: DNA damage, inhibitor, kinase, structure-activity relationship, WEE1
Graphical Abstract
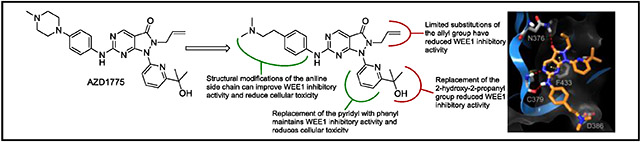
Wee changes impact activity profiles:
The activity of WEE1 kinase is critical for DNA repair in cancer cells and can confer resistance to DNA-targeted agents. Here, we report that structural modifications to the WEE1 inhibitor AZD1775 can result in improved inhibitory potency against WEE1 kinase activity with limited cytotoxicity. These studies may have important implications in the clinical setting where WEE1 inhibitors are used as chemosensitizers for DNA-targeted chemotherapy.
Introduction
WEE1 is a nuclear kinase that regulates the G2-M cell-cycle checkpoint by modulating the activity of cyclin-dependent kinase 1 (CDK1) and prevents entry to mitosis in response to DNA damage.[1] In normal cells, DNA damage can be repaired at either the G1 or the G2 checkpoint to preserve genomic integrity; however, cancer cells often have deficiencies in the G1 checkpoint as a means to acquire and propagate pro-oncogenic mutations, which results in an overreliance of DNA repair at the G2-M checkpoint.[2] The G2-M cell-cycle checkpoint is a critical mechanism for DNA repair in cancer cells and can confer resistance to DNA-damaging agents; therefore, the molecular components of the G2-M checkpoint, such as WEE1, are attractive targets for anticancer drug development.[3] Ataxia-telangiectasia-mutated (ATM) kinase and ataxia-telangiectasia-related (ATR) kinase mediate the repair of DNA double-strand and DNA single-strand breaks, respectively.[1, 4] ATR phosphorylates the checkpoint kinase CHK1 in response to DNA damage, which in-turn phosphorylates and activates WEE1 (Figure 1). The inhibitory phosphorylation of Tyr15 on CDK1 by WEE1 impedes CDK1/cyclin B function, resulting in cell-cycle arrest at G2-M and DNA-damage repair.[1] WEE1 is expressed at high levels in a number of cancers and is associated with poor clinical outcome,[5] suggesting that these cancers are heavily dependent on this mechanism for survival.
Figure 1. WEE1 and the G2-M cell-cycle checkpoint.
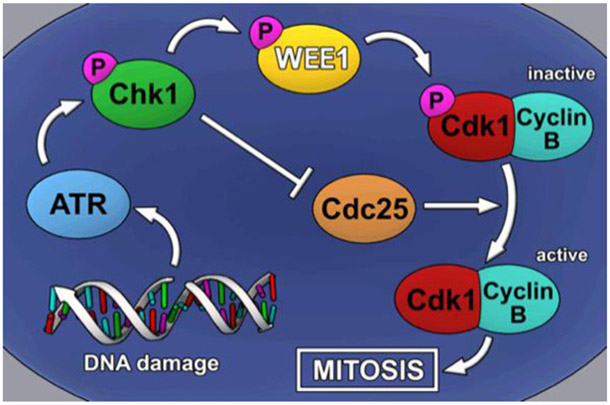
Schematic representation of the G2-M cell-cycle checkpoint, in which DNA damage detected by ATR triggers phosphorylation of CHK1, leading to the inhibition of the phosphatase Cdc25 and subsequent phosphorylation of WEE1 and the CDK1-CyclinB complex, resulting in mitotic arrest. Inhibition of WEE1 is sufficient to prevent cell cycle arrest, forcing the cell to undergo mitosis.
The pyrazolopyrimidinone AZD1775 (formerly MK1775) is a potent, ATP-competitive inhibitor of WEE1 kinase activity.[6] Inhibition of WEE1 promotes unscheduled mitotic entry through CDK1 activation, leading to loss of genome integrity, and ultimately mitotic catastrophe and apoptosis.[7] In addition, WEE1 inhibition has been shown to induce replication stress through CDK1/2-dependent aberrant firing of replication origins and subsequent nucleotide shortage.[8] Therefore, due to the critical role of WEE1 in regulating cell-cycle events, the incorporation of AZD1775 into radiation therapy and/or DNA-damaging agent regimens have emerged as an attractive strategy for combination therapy that is currently being evaluated in multiple clinical trials in a range of cancer types (www.ClinicalTrials.gov). Although AZD1775 has been reported to be well-tolerated in clinical trial, there have been few preclinical and clinical studies examining the effect of AZD1775 as a single-agent. A recent report from an ongoing Phase I study evaluating the safety and maximum tolerated dose (MTD) of AZD1775 in advanced refractory solid tumors (NCT01748825) disclosed supraventricular tachyarrhythmia and myelosuppression as dose-limiting toxicities.[9] However, many of the ongoing Phase II trials using various combination therapies are using dosing strategies below the established MTD for AZD1775. In preclinical studies, AZD1775 demonstrated potent cytotoxicity in a broad panel of cancer cell lines and led to xenograft tumor growth inhibition;[10] however, these anticancer activities cannot be attributed to WEE1 inhibition alone since AZD1775 is known to inhibit other kinases and may have off-target effects outside of the kinome.[6, 11] The combination of WEE1 inhibitors with DNA-damaging agents has been proposed to be more effective in p53-mutant cancer cells that rely on DNA repair at the G2-M checkpoint. However, several preclinical studies have demonstrated that AZD1775 has anticancer activity independent of p53 function, and this has also been attributed to the alternative functions of WEE1 and/or the off-target effects of AZD1775.[5d, 10]
In our previous study, we performed an initial examination of the structural components of AZD1775 that are required for WEE1 inhibition.[12] We used a computational-based model of AZD1775 docked into the WEE1 crystal structure to guide the design of a series of 14 pyrazolopyrimidinones as candidate WEE1 inhibitors. From this series, we identified a WEE1 inhibitor (CM-061, 1, Figure 2) that demonstrated inhibition of in vitro WEE1 kinase activity in the same nanomolar range as AZD1775, maintained comparable synergistic activity with cisplatin in medulloblastoma cells, but exhibited reduced single-agent cytotoxicity.[12] From our initial structure-activity relationship (SAR) study we concluded that AZD1775 has off-target effects that result in its potent cellular toxicity, side chain modifications on the pyrazolopyrimidinone core of AZD1775 can have marked and independent effects on WEE1 inhibition and cytotoxicity, and WEE1 inhibition can be achieved without potent cytotoxicity. This initial SAR study had limited scope, that focused on the sequential increase in complexity of the N-methylpiperazine group of the lead compound without systematically testing interactions within the WEE1 ATP-binding domain. In this manuscript, we perform a more extensive analysis of the predicted binding mode of AZD1775 to describe a more diverse, rationally designed series of pyrazolopyrimidinones as small molecule inhibitors of WEE1 kinase activity to allow for a more comprehensive understanding of the SAR for WEE1 inhibition with limited cytotoxicity.
Figure 2. Structure of WEE1 inhibitors AZD1775 and CM-061 (1) and predicted WEE1-AZD1775 interactions.

(A) Ribbon representation (blue ribbon, grey amino acid residues; with clipped pane molecular surface colored dark grey) of the ATP-binding site of WEE1 displaying the predicted binding mode of AZD1775 (cyan). Red dashed lines indicate H-bonds and yellow dashed lines indicate π-π interactions. (B) A stick display style representation of AZD1775 (cyan) and key interacting amino acid residues and target residues (grey) for inhibitor design. Red dashed lines indicate H-bonds and yellow dashed lines indicate π-π interactions. (C) Ligand interaction diagram of the predicted binding mode of AZD1775 in the ATP-binding site of WEE1. Red residues are charged negative, purple residues are charged positive, green residues are hydrophobic, and blue residues are polar, purple arrows indicate H-bonds, green lines indicate π-π interactions, and grey circles indicate areas of solvent exposure. (D) The chemical structure of the WEE1 inhibitors AZD1775 and CM-061 (1).
Results
Computational-based design of WEE1 inhibitors
To further develop our understanding of the SAR for WEE1 inhibition, and to improve the activity of this class of compounds, we re-examined our computational model of the binding conformation of AZD1775 in the ATP-binding site of WEE1. As expected from an ATP-competitive inhibitor, the core pyrazolopyrimidinone heterocycle forms H-bond interactions with the hinge residues C379 and N376 (Figure 2). Focusing on the aniline side chain, we observed that the aniline 3-position was oriented adjacent to the backbone nitrogen of G382 (Figure 2A), and the introduction of moieties capable of acting as H-bond acceptors in this region could improve binding to WEE1. Thus, we proposed to synthesize a series of compounds bearing dimethylamine and dimethylamide groups at the aniline 3-position (25-27), as well as a 3-cyanoaniline analog, to vary both the direction and distance of the H-bond acceptor in this region. Interestingly, the N-methylpiperazine moiety of AZD1775 was not predicted to make any substantive interactions (Figure 2A), due to the rigidity of the ring system; however, both E303 and D386 are in close proximity to the N-methylpiperazine moiety (~5-6Å) and have the potential for interaction. We hypothesized that substitution with a more flexible side chain containing a terminal basic group to interact with E303 and/or D386 could enhance binding and potency (28 and 29). Finally, as an extension to our previous studies into the structural complexity required to confer potency at this position,[12] the 6-amino (22) and unsubstituted aniline (24) analogs of AZD1775 were included in the proposed compound series.
From our previous study,[12] the hydroxyl of the pyridyl side chain was preferred for WEE1 binding, through interaction with D463 or the Mg2+ ion; however, the methyl groups may impede binding due to steric hinderance with the protein surface. The removal of the methyl groups may alleviate this potential clash and allow additional flexibility for the hydroxyl to form an H-bond interaction with D463. To evaluate this hypothesis, the pyridylmethanol analog (30) was proposed, and a phenyl analog of AZD1775 (31) as there were no apparent interactions involving the pyridine nitrogen. The final region of the molecule we investigated was the N2-allyl group, which does not appear to be critical for the binding of AZD1775 to WEE1 beyond hydrophobic interactions with V313, A326, and K328; however, the allyl group may be a metabolic liability via CYP450-mediated conversion to the diol or primary alcohol. Furthermore, we hypothesized that its removal may allow deeper access into the ATP-binding domain for tighter binding. To investigate the necessity for the allyl group, both the N2-methyl (32) and unsubstituted (35) analogs of AZD1775 were proposed. At the time of writing this manuscript, the 1.9Å co-crystal structure of the human WEE1 kinase domain in complex with AZD1775 was published (PDB: 5V5Y).[13] The proposed binding orientation of AZD1775 in the ATP-binding site of WEE1 in our computational model was in good agreement with the co-crystal structure (Supporting Information, Figure S4), with only minor differences observed in the orientation of the side chains. Importantly, both our computational model and the WEE1-AZD1775 co-crystal structure equally support the proposed structural modifications to AZD1775 described above.
Chemistry
AZD1775 analogs were prepared as outlined in Scheme 1 and described in the Experimental Section and Supporting Information. Briefly, tert-butylcarbazate was protected through reaction with phthalic anhydride (2), after which the carbamate nitrogen was functionalized with the necessary alkyl group through either nucleophilic substitution or Mitsunobu reaction. Removal of the phthalamide protecting group with methyl hydrazine gave the key tert-butyl alkylcarbazates (7-9), which were reacted with ethyl 4-chloro-2-methylthio-5-pyrimidinecarboxylate (10) in the presence of TFA to form the core pyrazolopyrimidin-3-one scaffolds (11-13). An Ullman-type aryl amination with the relevant functionalized pyridines (14-16) gave the penultimate pyrazole products (17-21), after which activation of the thioether with m-CPBA and reaction with relevant anilines or amines afforded the desired AZD1775 analogs (22-33). Surprisingly, a desired 3-cyanoaniline analog was inaccessible via this route, with no reaction occurring under ambient conditions, and degradation evident under increased temperature, and so this compound was omitted from the series.
Scheme 1. Synthesis of analogs.

Synthetic strategy for the preparation of proposed WEE1 inhibitor analogs of AZD1775 (22–35). Reagents and conditions: a) t-butyl carbazate, toluene, reflux, 18 h; b) alkyl halide, BnNEt3CI, K2CO3, MeCN, RT-50°C, 18–48 h or PMB-OH, DEAD, PPh3, THF, RT, 16 h; c) MeNHNH2, THF, RT, 18 h; d) i) DIPEA, THF, reflux, 72 h, ii) TFA, RT-70°C, 2 h or TFA, DCM, RT, 18 h; e) N,N’-diethylethylenediamine, Cul, K2CO3, 1,4-dioxane, 95°C, 18 h; f) i) mCPBA, toluene, RT, 1 h, ii) aniline /amine, DIPEA, toluene, RT, 18 h; g) TFA, reflux, 16 h; h) 4-ToISO2Na, Pd(PPh3)4, MeOH/THF, RT, 2 h.
The proposed synthesis of the N2-unsubstituted analog of AZD1775 (35) through TFA-mediated removal of the para-methoxybenzyl (PMB) protecting group did not afford the desired compound. In addition to removal of the PMB group, an acid-mediated elimination of the 2-propanol hydroxyl moiety adorning the pyridine ring resulted in formation of the isopropenyl analog (34) observed by NMR and MS. This compound was included in the kinase activity assay to further the scope of the SAR. As an alternative approach to the desired compound the allyl group of AZD1775 was directly removed through reaction with sodium para-toluenesulfinate tetrahydrate. As it was anticipated that the acid mediated elimination side reaction would occur during the TFA-mediated conversion of the PMB-protected compound (23) to the 6-aminopyrimidine analog (22), the desired unsubstituted amine was introduced through the reaction of pyrazolopyrimidinone (18) with ammonia. The PMB-protected compound was also included in the kinase activity assay to gain additional insight into the SAR for WEE1 inhibition.
In vitro WEE1 kinase activity assay
All of the synthesized compounds and relevant synthetic intermediates were tested for their inhibitory activity against recombinant WEE1 in the established TR-FRET activity assay,[12] and the resultant IC50 values are detailed in Table 1. For modification of the pyrazolopyrimidinone 6-position (Table 1; R3), the presence of an aniline did not appear to be critical for activity, with the aminopyrimidine 22 (IC50 = 17.7 ± 7.8 nm) exhibiting approximate equipotency when compared to the unsubstituted aniline 24 (IC50 = 9.6 ± 2.6 nm) and only a modest loss in activity with respect to AZD1775 (IC50 = 5.1 ± 0.9 nm). From our data, it appears that the steric constraints in this region of the ATP-binding domain limit the extent of substitution tolerated at the aniline 3-position. This is supported by the sequential decrease in activity from the dimethylaniline 25 (IC50 = 4.7 ± 1.7 nm) to the dimethylbenzylamine 26 (IC50 = 9.1 ± 1.2 nm) and the 3-dimethylamide analog 27 (IC50 = 27.3 ± 6.4 nm). The equipotency of 25 with AZD1775 in the absence of the N-methylpiperazine moiety, previously shown to be favorable for WEE1 binding,[12] would suggest that the 3-dimethylamino group improves binding with WEE1, likely through the proposed H-bond with G382. The inhibitory activity of the compounds designed to remove the conformational restraints imparted by the N-methylpiperazine ring of AZD1775 appear to support our hypothesis that this would improve interactions with either E303 or D386. However, the distance between the aniline ring and the basic dimethylamine plays a role, as evidenced by the reduction in inhibitory activity for compound 29 (IC50 = 7.8 ± 0.4 nm) as opposed to the improvement in activity for 28 (IC50 = 1.7 ± 0.9 nm) when compared to AZD1775.
Table 1.
Structures and inhibitory activities of pyrazolopyrimidinone WEE1 inhibitors.
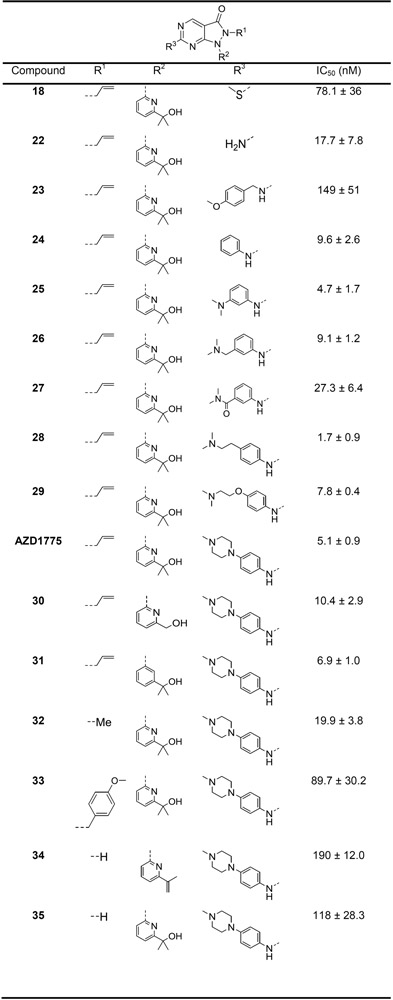 |
Inhibitory activity for the synthesized compounds from a recombinant WEE1 TR-FRET binding assay are given as the half maximal inhibitory concentration (IC50) in nm.
The removal of the methyl groups from the pyridyl side chain resulted in a small reduction in activity (30; IC50 = 10.4 ± 2.9 nm), with only negligible effects of lipophilic efficiency (LipE; AZD1775 = 6.11; 30 = 6.06). These methyl groups may act as a conformational lock that optimizes the position of the hydroxyl group for a distant H-bond interaction with D463 or the Mg2+ ion, mitigating an entropic penalty to binding that is incurred upon their removal. The pyridine nitrogen was not necessary for activity as equipotency with AZD1775 was obtained for compound 31 (IC50 = 6.9 ± 1.0 nm).
The substitution of the N2-allyl group with a methyl group resulted in a near 4-fold reduction in activity (32; IC50 = 19.9 ± 3.8 nm), while its removal in the desired compound 35 (IC50 = 118 ± 28 nm) and the synthetic side product 34 (IC50 = 190 ± 12 nm) reduced activity. We hypothesize that these substitutions may alter the electronics around the pyrazole carbonyl and thus the ability to act as an H-bond acceptor with N376. To test this hypothesis, we examined the electrostatic and hydrophobic field properties of the three compounds and found that, compared to AZD1775, both analogs exhibited an attenuated negative electrostatic field in this region, with the alterations in the unsubstituted species predicted to be particularly pronounced (Supporting Information, Figure S1).
WEE1 inhibitor effects on cellular viability and cytotoxicity
Previously, we utilized an MTS assay to determine the single-agent effects of WEE1 inhibitors on the cell viability of ONS-76 (p53 wild-type) and Daoy (p53 mutant) medulloblastoma cell lines.[12] All of the WEE1 inhibitors described in this manuscript with potent WEE1 kinase inhibitory activity in the in vitro TR-FRET assay (IC50 < 10 nm) were selected for evaluation in the MTS assay. In ONS-76 cells only three compounds exhibited an appreciable effect on cellular metabolic activity within the concentration range of the assay, to the degree that EC50 values could be determined (Figure 3A). These 3 compounds included AZD1775, that has been previously shown to be a potent inhibitor of cellular metabolic activity with an EC50 of 159 ± 31 nm in this assay,[12] 28 (EC50 = 252 ± 9 nm), and 29 (EC50 = 203 ± 40 nm). Interestingly, 28 demonstrated improved recombinant WEE1 kinase inhibitory activity over AZD1775, but had a decreased effect on cell viability. There was a significant lack of impact on cell viability for 31 (57% effect at 1.16 μm) although inhibition of WEE1 kinase activity was comparable to AZD1775, and the only structural modification was removal of the pyridine ring nitrogen. In Daoy cells, the disparity between WEE1 inhibtion and cell viability was further pronounced (Figure 3B). The p53 mutant status of Daoy cells would be anticipated to show greater correlation between WEE1 inhibition and single-agent WEE1 inhibitor effects on cell viability. However, only AZD1775 (EC50 = 179 ± 16 nm) was sufficiently active to allow an EC50 to be determined. The WEE1 inhibitors that demonstrated potent inhibition of cell viability in ONS-76 cells were also the most active inhibitors of Daoy cell viability (28 = 84% effect at 600 nm; 29 = 81% effect at 600 nm), but their effect was significantly diminished with respect to AZD1775. There was no effect on cellular viability with any of the additional WEE1 inhibitors within the concentration range of the assay.
Figure 3. Effect of single-agent WEE1 inhibitors on cell viability and cytotoxicity.
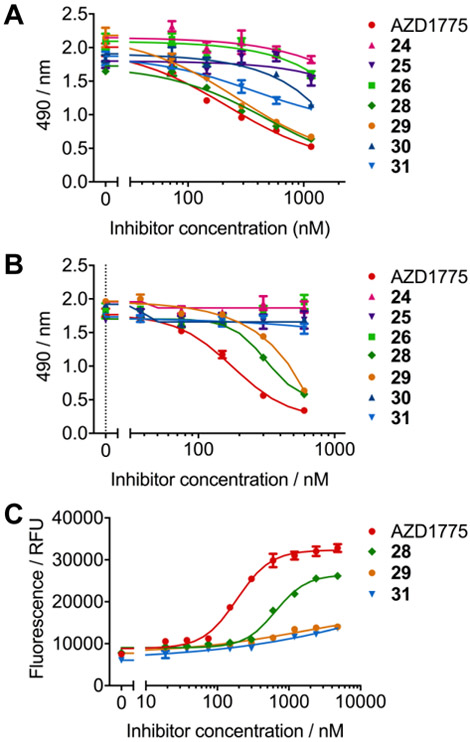
(A) Dose-response for ONS-76 cells treated with a concentration range of WEE1 inhibitors for 72 hours in an MTS assay. AZD1775 (red) inhibits cellular metabolic viability most potently (EC50 = 159 ± 31 nm), with 29 (orange, EC50 = 203 ± 40 nm) and 28 (green, EC50 = 252 ± 9 nm) displaying comparable results. Limited effects were observed for other inhibitors within the series (n=3, error bars/± = S.D.). (B) Dose-response for Daoy cells treated with a concentration range of WEE1 inhibitors for 72 hours in an MTS assay. AZD1775 (red, EC50 = 179 ± 16 nm) exhibits significantly more potent effects on cellular metabolic viability than 29 (orange, 81% activity at 600 nm) and 28 (green, 84% activity at 600 nm). No other analogs displayed any reduction in MTS signal up to 600 nm inhibitor dose (n=3, error bars/± = S.D.). (C) Dose-response for Daoy cells treated with a concentration range of WEE1 inhibitors for 72 hours in a CellTox green cytotoxicity assay. AZD1775 (red, EC50 = 188 ± 26 nm) exhibits significantly more potent cytotoxic effects than 28 (green, EC50 = 651 ± 16 nm). Sigmoidal dose-response curves were not obtained for compounds 29 and 31 up to 2400 nm inhibitor dose (n=3, error bars/± = S.D.).
The CellToxTM Green Cytotoxicity Assay was used to determine the cytotoxic effect of the WEE1 inhibitors. This assay uses an asymmetric cyanine dye that is non-toxic and excluded from viable cells; however, changes in membrane integrity that occur as a result of cell death allows the dye to enter the compromised cells and bind DNA, enhancing the dyes flurorescent properties. Using this assay we evaluated the cytotoxic effects of AZD1775 and compounds 28, 29, and 31 in Daoy cells over 72 hours (Figure 3C and Supporting Information, Figures S2 and S3). Overall, the cytotoxicity of the WEE1 inhibitors were concentration- and exposure time-dependent, but EC50 values could only be determined for AZD1775 (EC50 = 188 ± 26 nm) and 28 (EC50 = 651 ± 16 nm) after 72 hours. The ranking of the potency of cytotoxicity for these compounds correlated with the cell vaibility assay. AZD1775 had the most potent cytotoxic properties and demonstrated cytotoxic effects within 24 hours at concentrations >600 nm. After 72 hours AZD1775 demonstrated cytotoxic effects at concentrations >75 nm. Although 28 showed some cytotoxicty within 24 hours at 1.2 μm, it was significantly less cytotoxic than AZD1775 over the treatment period. Compounds 29 and 31 did not display sufficient cytotoxicity for EC50 determination and did not demonstrate any appreciable cytotoxicity until 48 hours and only at concentrations >300 nm.
Effect of WEE1 inhibitors on cellular pCDK1
All of the WEE1 inhibitors that demonstrated activity in the TR-FRET assay were advanced to determine their effect on the cellular concentration of phosphorylated CDK1 at Tyr15, an effective biomarker of cellular WEE1 activity.[14] Initially, Daoy cells were treated with all compounds at a single concentration of 220 nm, which we previously determined as the EC50 for AZD1775 in this assay (Figure 4A).[12] Of the compounds tested, only AZD1775 (p<0.001), 28 (p<0.001), and 29 (p<0.01) showed a statistically significant decrease in pCDK1 levels when compared to DMSO control, and were selected for further testing across a concentration range. Compound 31 did not cause a significant reduction in pCDK1 levels compared with DMSO, but the level of pCDK1 was observed to decrease to a level not statistically different from the AZD1775 treatment group. Interestingly, both 24 (p<0.05) and 25 (p<0.01) resulted in significant increases in pCDK1 levels with respect to DMSO, suggesting an off-target activation of the DNA damage response (DDR) pathway. The selected compounds were administered to cells at concentrations between 1 μm and 62 nm, and the resultant cell extracts were used to establish a dose-response in the pCDK1 ELISA (Figure 4B). Although the dose-response curves did not permit the determination of EC50 values, AZD1775, 28, and 29 demonstrated comparable effects on pCDK1 levels within the concentration range of the assay. This is unsurprising given the similar IC50 values for these compounds from the TR-FRET assay and that both 28 and 29 were the most potent of the novel compounds in the MTS assay in both ONS-76 and Daoy cells. However, in the MTS and CellTox assays using Daoy cells, the effect of 28 and 29 on cell viability and cytotoxicity were significantly reduced when compared to AZD1775, supporting that the inhibition of cellular WEE1 is not directly responsible for the potent cytotoxic effects of AZD1775. Although these compounds were not designed to address this observation, these findings for compounds 28, 29, and moreover 31 corroborate our previous research, where we first proposed that WEE1 inhibtion is uncoupled from the potent cytotoxic effects of AZD1775.[12]
Figure 4. WEE1 inhibitor effects on cellular pCDK1(Tyr15) concentrations.
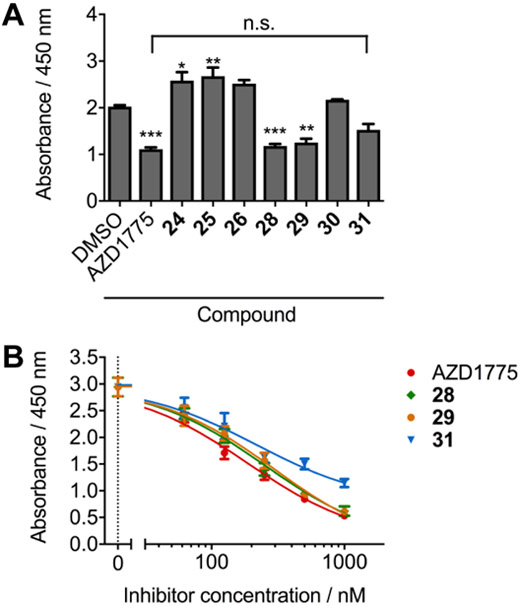
(A) Quantitative ELISA determination of pCDK1(Tyr15) levels in Daoy cell lysates (0.05 mg/mL total protein) after treatment for 24 hours with a single 220 nm dose of all active WEE1 inhibitors. A decrease in pCDK1 concentration when compared to DMSO control was observed for AZD1775, 28 and 29, whereas the decrease in pCDK1 upon treatment with 31 was statistically the same as that for AZD1775 (n=3, error bars = S.D., compared with DMSO; * = p<0.05, ** = p<0.01, *** = p<0.001; compared to AZD1775; n.s. = no significance). (B) pCDK1(Tyr15) ELISA dose response for Daoy cell lysates (0.05 mg/mL total protein) after treatment for 24 hours with AZD1775 (red), 28 (green), 29 (orange) and 31 (blue) (n=3, error bars/± = S.D.).
Discussion
In this manuscript, we have described a series of pyrazolopyrimidinones to further our understanding of the SAR for WEE1 inhibition. The evaluation of this series of pyrazolopyrimidinones against WEE1 kinase activity has allowed us to evaluate our computational model as a basis for WEE1 inhibitor design. We have identified several compounds bearing modification of the aniline substituent that resulted in equipotency with AZD1775 in terms of inhibition of WEE1 kinase activity. These data support our previous findings,[12] that the N-methylpiperazine of AZD1775 is amenable to modification. Furthermore, we have identified compound 28 that is a more potent inhibitor of WEE1 kinase activity compared to AZD1775 (IC50 = 1.7 ± 0.9 nm versus IC50 = 5.1 ± 0.9 nm). From the computational-based molecular docking of compound 28 into the ATP-binding site of WEE1 we found that the additional flexibility in the alkylamino side chain allowed improved interaction with D386, and this may be responsible for the observed improvement in inhibitory activity (Figure 5). We proposed replacement of the allyl group at the pyrazole N2 position in order to engineer out potential metabolic liabilities; however, substitution of this group may be limited. A significant loss of WEE1 inhibitory potency was observed with methyl N2 substitution (32; IC50 = 19.9 ± 3.8 nm) and complete loss of activity with removal of the N2 substitution altogether (35; IC50 = 118 ± 28 nm), likely due to the loss in hydrophobic interactions and electronic effects on the pyrazole ring (Supporting Information, Figure S1). The WEE1-AZD1775 co-crystal structure indicates that the allyl group may have a hydrophobic contact with Lys328.[11c] Further compounds may need to be developed to more thoroughly investigate the scope of replacing the allyl group to include substituents that maintain the electronic distribution of negative electrostatic and hydrophobic fields and that can be accommodated in the small hydrophobic pocket in the ATP-binding site of WEE1.
Figure 5. Predicted WEE1-compound 28 interactions.

(A) Ribbon representation (blue ribbon, grey amino acid residues; with clipped pane molecular surface colored dark grey) of the ATP-binding site of WEE1 displaying the predicted binding mode of 28 (orange). Red dashed lines indicate H-bonds and yellow dashed lines indicate π-π interactions. (B) Ligand interaction diagram of the predicted binding mode of 28 in the ATP-binding site of WEE1. Red residues are charged negative, purple residues are charged positive, green residues are hydrophobic, and blue residues are polar, purple arrows indicate H-bonds, green lines indicate π-π interactions, and grey circles indicate areas of solvent exposure.
Following the testing protocol that we have previously reported,[12] compounds that showed activity in the in vitro WEE1 kinase activity assay were tested as single agents in both ONS-76 (p53 wild-type) and Daoy (p53 mutant) cells to determine their effect on cell viability. Similarly, to what we have previously reported, there was a disparity between WEE1 inhibitory potency and the effect on cell viability in an MTS assay. Only the WEE1 inhibitors 28 and 29 possessed an effect on cellular viability over the concentration range examined, and even these effects were less potent than AZD1775, particularly in Daoy cells. This is interesting observation, as it would be assumed that the effect of WEE1 inhibitors would not only be more potent in Daoy cells due to the mutant status of p53, but that the cellular effects of WEE1 inhibitors as single agents would more closely correlate with inhibitory potency. We also examined the cytotoxic effects of the WEE1 inhibitors on Daoy cells using the CellTox assay. The cytotoxicity of the WEE1 inhibitors correlated with their effects on cell viability by MTS assay. AZD1775 demonstrated the most potent single-agent cytotoxic effects within 24 hours and over the 72 hour treatment period, supporting our previous data using xCELLigence Real-Time Cell Analysis.[12] The WEE1 inhibitor 28 displayed significantly reduced cytotoxicity compared with AZD1775, and 29 and 31 had negligible cytotoxic efffects over the 72 hour treatment period.
In order to determine the effect of compounds on cellular WEE1 activity, Daoy cell extracts were probed for pCDK1(Tyr15) concentration in an ELISA assay. Compounds 28, 29, and 31 reduced cellular pCDK1 levels, and hence inhibited cellular WEE1 activity, to statistically comparable levels observed with AZD1775 treatment. Interestingly, both 24 and 25 increased cellular pCDK1, suggesting that treatment with these compounds results in activation of the DDR, either through direct compound mediated DNA-damage or through effects on other members of the CHK1/WEE1 pathway. This observation may warrant further investigation to more thoroughly determine the significance of this effect and the mode of action, as any compounds within this class possessing such effects would be highly undesirable. The dose-response effects of AZD1775 and compounds 28, 29, and 31 on pCDK levels in Daoy cells were similar, despite less comparable effects on cell viabilityand cytotoxicity. We have previously shown that compounds, such as CM-061 (Figure 2, 1), have little effect on cell viability as single agents, but can inhibit cellular WEE1 activity and this accompanies its ability to synergize with DNA-damaging agents such as cisplatin.[12] Therefore, despite compounds 28, 29, and 31 exhibiting reduced inhibition of cell viability and cytotoxicity as single agents, these compounds are more suitable for use as potentiators of DNA-damaging agent chemotherapy. These findings may be of critical importance in evaluating WEE1 inhibition as a therapeutic strategy as the toxicity of AZD1775 may exacerbate therapy-related adverse effects in patients with medulloblastoma and other cancers.
Prior to our initial study,[12] there was limited SAR data for AZD1775 in the context of WEE1 inhibition. A screening program conducted by Bayu Pharmaceutical Co., that focused on the identification of WEE1 inhibitors from a small chemical compound library led to the discovery of AZD1775.[6] In a kinase profile of 223 kinases, AZD1775 demonstrated >80% inhibition of 8 other kinases at 1.0 μm in an in vitro radiolabeled ATP kinase assay.[6] The IC50 values determined for 7 of these kinases were 10-fold less potent compared with WEE1 (IC50 = 5 nm), and 2- to 3-fold less potent against the proto-oncogene Src family tyrosine kinase YES1 (IC50 = 14 nm). The identity of the other kinases from this profile screen have yet to be reported; however, it was stated in the same study that AZD1775 showed >100-fold selectivity for WEE1 over MYT1, but since WEE1 has only a ~30% sequence identity with MYT1 this difference in selectivity was not surprising. A kinome interaction network study identified ABL1, LCK, LRRK2, TNK2, and SYK as targets of AZD1775 with Ki values below 1 μM.[11a] Recently, it has been reported that AZD1775 has potent nanomolar activity against PLK1, and that AZD1775 should now be considered a dual WEE1 and PLK1 inhibitor.[11b, 11c] A full kinome profile for AZD1775 across a panel of 468 human kinases at a concentration of 500 nm identified PLK1, FLT3 (D835V), JAK3, GCN2 (S808G), ABL1 (H396P), and JAK2 as additional kinase targets.[11c] Furthermore, the authors suggested that WEE1 and PLK1 were the most relevant targets for mediating the anticancer effects of AZD1775; however, our studies support that WEE1 inhibition can be achieved with little impact on cytotoxicity. Therefore, we examined the effect of AZD1775 and the WEE1 inhibitors 28, 29, and 31 on PLK1 activity in a TR-FRET assay (Supporting Information, Table S1). AZD1775 was the most potent PLK1 inhibitor in this assay (IC50 77.6 ± 9.3 nm), followed by 29 (IC50 161 ± 10 nm), 28 (IC50 270 ± 29 nm), and then 31 (IC50 394 ± 37 nm). The ranking of PLK1 inhibitory potency by these compounds does not correlate with the rank order of cytotoxicity and it is unknown, but unlikely that a 3.5 to 5-fold difference in PLK1 inhibition would significantly impact cytotoxicity.
Overall, kinome profiling studies support that AZD1775 has narrow-spectrum activity and considering it interacts with an ATP-binding site its kinome selectivity profile is amirable; however, what is concerning is the potent single-agent cytotoxicity of AZD1775. A recent study presented data supporting our findings that AZD1775 induces apoptosis independent of WEE1 inhibition, and demonstrated potent single-agent effects of AZD1775 on cell viability in a panel of NSCLC cells.[11b] In order to identify the molecular targets of AZD1775 an immobilized analog, termed i-AZD1775, was developed to conduct a proteome-wide search of H322 cells and confirmed PLK1 as a AZD1775 target. [11b] A short-coming of this immobilized analog approach was that the effect of i-AZD1775 on cell viability was not determined. i-AZD1775 has an alkyl amino linker from the N-piperazine of AZD1775 and may not interact with the molecular target(s) that results in the potent cytotoxicity observed with AZD1775, as our previous work has shown that substitution of the N-piperazine reduces cytotoxicity.[12] Based on the results from the kinome profiling studies, we would propose that the potent cytotoxicity of AZD1775 is due to a combination of interactions within the kinome and molecular targets outside of the kinome and this is the focus of on-going investigations.
Conclusions
In summary, we have developed WEE1 inhibitors based on the pyrazolopyrimidinone core of AZD1775 and, combined with our previous results, we definitively show, principally with compound 31, that the potent cytotoxicity of AZD1775 can be uncoupled from WEE1 inhibition. Furthermore, we have identified compound 28 as a more potent inhibitor of WEE1 than AZD1775, that demonstrates WEE1 inhibition both in vitro and in cells, but has a reduced single-agent effect on cell viability and cytotoxicity compared to AZD1775. We present potent WEE1 inhibitors, compounds 28, 29, and 31, with limited cytotoxic activity that we are transitioning into advanced preclinical evaluation. The identification of potent WEE1 inhibitors with diminished single-agent cytotoxic effects is an important feature when developing chemosensitization agents, as this may allow for the safe administration of WEE1 inhibitor and dose reductions of DNA-damaging agents while maintaining clinical efficacy and reducing therapy-related adverse effects.
Experimental Section
Chemistry
General:
Where appropriate, reactions were carried out under an inert atmosphere of nitrogen with dry solvents, unless otherwise stated. Dry solvents were obtained directly from manufacturer and used without further purification. Yields refer to chromatographically and spectroscopically pure isolated yields. Reagents were purchased at the highest commercial-quality and used without further purification, unless otherwise stated. Reactions were monitored by thin-layer chromatography carried out on 0.25 mm E. Merck silica gel plates (60 F254) using ultraviolet light as visualizing agent. All melting points were determined using a Stuart Scientific SMP40 melting point apparatus and are uncorrected. 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained as solutions in deuterated solvents DMSO-d6 or CDCl3 using a Bruker Avance III 400 spectrometer recording at 400 MHz. Chemical shifts (δ) are reported in parts per million (ppm) and the spin-multiplicity abbreviated as: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sept (septet), m (multiplet), or br (broad), with coupling constants (J) given in Hertz (Hz). Mass Spectrometry (MS) was carried out on an API 4000. Fourier Transform Infrared (FTIR) spectra were obtained using a Bruker Alpha Platinum-ATR as a neat sample.
General procedure for the synthesis of substituted pyrazolopyrimidinones
Detailed synthetic methods and compound characterization for all compounds are provided in Supporting Information.
General Procedure for the alkylation of tert-butyl (1,3-dioxoisoindolin-2-yl)carbamate (4-6).
To a suspension of the carbamate (3) (1.0 equiv.) in MeCN (2 mL/mmol) was added benzyltriethylammonium chloride (0.1-0.2 equiv.), K2CO3 (4.0 equiv.) and the relevant alkylhalide (1.5-5.0 equiv.) sequentially. The reaction mixture was stirred at RT or 50°C for 18-48 h, before water (2 mL/mmol) was added and the organic phase was extracted with Et2O (2 × 5 mL/mmol). The combined organic extracts were dried (MgSO4) and evaporated to dryness and were purified by chromatography on silica.
General procedure for the removal of phthlalimide protecting groups (7-9).
Methylhydrazine (1.25 equiv.) was added to an ice cooled solution of phthalimide 4-6 (1.0 equiv.) in THF (2 mL/mmol). The reaction mixture was allowed to warm to RT and was stirred for 18 h. The resultant suspension was passed through a filter, and the filtrate was concentrated in vacuo. A mixture of Hexanes:EtOAc (3:1, 1 mL/mmol) was added, and the precipitate formed was removed via filtration. This process was repeated a further 2 times, and the final filtrate was concentrated to give the target compound.
General procedure for the synthesis of pyrazolopyrimidinones (11-13).
DIPEA (2.5 equiv.) and the relevant hydrazine 7-9 (1.05 equiv.) were added to a solution of ethyl 4-chloro-2-methylthio-5-pyrimidinecarboxylate (10; 1.0 equiv.) in THF (3 mL/mmol). The reaction mixture was heated at reflux for 72 h, before being concentrated in vacuo. Et2O (1 mL/mmol) was added to the residue, and the resultant precipitate was collected by filtration. The filtrate was evaporated to dryness, and the residue was cooled in an ice bath, after which TFA (1 mL/mmol) was added. The resultant solution was stirred at RT for 1 h, followed by 70°C for 1 h. The solvent was removed in vacuo and the residue was dissolved in EtOH (1 mL/mmol) and cooled in an ice bath, after which 6m NaOH (2 mL/mmol) was added. The resultant solution was stirred at RT for 15 min, before being acidified (pH 3) via the addition of conc. HCl. The solution was evaporated to dryness and the resultant residue was partitioned between chloroform (2 mL/mmol) and water (2 mL/mmol), and the organic phase was washed with brine (1 mL/mmol), dried (MgSO4), and concentrated in vacuo to afford the target compound.
General procedure for the preparation of pyridyl pyrazolopyrimidinones (17-21).
N,N’-Dimethylethylenediamine (2.0 equiv.) was added to a solution of the relevant pyrazolopyrimidinone 11-13 (1.0 equiv.), the relevant bromopyridine (14-16, 1.3 equiv.), copper iodide (1.0 equiv.) and K2CO3 (1.4 equiv.) in 1,4-dioxane (2 mL/mmol) at 80°C. The resultant suspension was heated at 95°C for 18 h, over which time a color change of orange to dark green occurred. The reaction mixture was cooled to RT and diluted with NH4OH (10 mL/mmol) before being extracted with EtOAc (2 × 10 mL/mmol). The combined organic extracts were washed with brine (10 mL/mmol), dried (MgSO4) and evaporated to dryness before the crude material was purified via chromatography on silica.
General procedure for the preparation of aniline pyridyl pyrazolopyrimidinones (22-33).
mCPBA (1.1 equiv.) was added to a solution of pyrazolopyrimidinones 17-21 (1.0 equiv.) in toluene (10 mL/mmol) and the resulting mixture was stirred at RT for 1 h. DIPEA (5.2 equiv.) and the relevant substituted aniline 39-43 or amine (1.3 equiv.) were added, and the reaction mixture was stirred at RT for 18 h. Saturated NaHCO3 solution (15 mL/mmol) was added, and the mixture was extracted with EtOAc (2 × 20 mL/mmol). The combined organic extracts were washed with brine (5 mL/mmol), dried (MgSO4) and concentrated in vacuo. The resultant residues were purified via chromatography on silica to give the target compounds (12-89%).
Compound characterization
Synthesis of tert-butyl (1,3-dioxoisoindolin-2-yl)carbamate (3).
The desired product was obtained as a white crystalline solid (16.1 g, 61.4 mmol, 91%). Rf 0.68 (1:1 Hexane:EtOAc); M.p. 191-194°C (Lit. = 186°C);[15] IR (cm−1) 3316, 2979, 1796, 1730, 1614, 1490; 1H NMR (400 MHz, DMSO-d6) 1.45 (9H, s, -OC(CH3)3), 7.87-8.04 (4H, m, H-4/5/6/7), 9.86 (1H, s, NH); 13C NMR (100 MHz, DMSO-d6) 28.3 (C(CH3)3), 81.6 (C(CH3)3), 124.2 (Ar-C), 129.8 (Ar-C), 135.8 (Ar-C), 154.4 (C=O), 165.9 (C=O).
Synthesis of tert-butyl (1,3-dioxoisoindolin-2-yl)(methyl)carbamate (4).
The desired product was obtained as a white crystalline solid (93 mg, 0.34 mmol, 89%). Rf 0.38 (1:1 Hexanes:EtOAc); M.p. 118-120°C (Lit. = 123°C);[15] IR (cm−1) 2972, 2934, 1791, 1723, 1609; 1H NMR (400 MHz, CDCl3) 1.34 (5.1H, s, C(CH3)3-major), 1.53 (3.9H, s, C(CH3)3-minor), 3.29 (1.7H, s, N-CH3-major), 3.32 (1.3H, s, N-CH3-minor), 7.74-7.93 (4H, m, H-4/5/6/7); 13C NMR (100 MHz, CDCl3) 27.9 (C(CH3)3-major), 28.1(C(CH3)3-minor), 36.5 (N-CH3-major), 38.1 (N-CH3-minor), 82.2 (C(CH3)3-major), 82.9 (C(CH3)3-minor), 123.8 (Ar-C), 129.9 (Ar-C), 130.1 (Ar-C), 134.6 (Ar-C), 134.7 (Ar-C), 153.6 (C=O-major), 153.8 (C=O-minor), 165.0 (C=O-major), 165.3 (C=O-minor); MS [M+H]+ m/z 276.8.
Synthesis of tert-butyl allyl(1,3-dioxoisoindolin-2-yl)carbamate (5).
The desired product was obtained as a white crystalline solid (15.7 g, 52.1 mmol, 85%) with no further purification needed. Rf 0.52 (4:1 Hexane:EtOAc); M.p. 72-75°C (Lit. = 76-78°C);[15] IR (cm−1) 2978, 2936, 1792, 1719, 1641; 1H NMR (400 MHz, DMSO-d6) 1.25 & 1.46 (9H, s, C(CH3)3), 4.19 (2H, dapp, J = 6.1 Hz, N-CH2), 5.10-5.17 (1H, m, allyl C-Htrans), 5.27 (1H, dd, J = 17.3, 1.3 Hz, allyl C-Hcis), 5.78-5.93 (1H, m, allyl C-H), 7.93-8.02 (4H, m, H-4/5/6/7); 13C NMR (100 MHz, DMSO-d6) 27.9 (C(CH3)3-major), 28.1 (C(CH3)3-minor), 51.7 (N-CH2-major), 53.7 (N-CH2-minor), 82.1 (C(CH3)3-major), 82.8 (C(CH3)3-minor), 119.1 (allyl-CH2-major), 119.7 (allyl-CH2-minor), 124.3, 124.4, 129.5, 129.6, 132.8 (Ar-C), 133.3 (Ar-C), 135.9 (Ar-C), 136.0 (Ar-C), 153.0 (C=O-major), 153.1 (C=O-minor), 165.3 (C=O-major), 165.5 (C=O-minor).
Synthesis of tert-butyl (1,3-dioxoisoindolin-2-yl)(4-methoxybenzyl)carbamate (6).
The desired product was obtained as a pale orange solid observed to be a pair of rotamers by NMR (65 mg, 0.17 mmol, 89%). Rf 0.42 (3:1 Hexanes:EtOAc); M.p. 106-108°C; IR (cm−1) 3003, 2979, 2962, 2934, 2836, 1793, 1737, 1715, 1610, 1511; 1H NMR (400 MHz, CDCl3) 1.37 (5.4H, s, C(CH3)3-major), 1.55 (3.6H, s, C(CH3)3-minor), 3.77 (1.8H, s, OCH3-major), 3.78 (1.2H, s, OCH3-minor), 4.80 (0.8H, s, benzyl CH2-minor), 4.83 (1.2H, s, benzyl CH2-major), 6.82 (2H, dd, J = 10.0, 8.5 Hz, H-4/7), 7.31 (2H, dd, J = 10.0, 8.5 Hz, H-5/6), 7.72-7.77 (2H, m, benzyl H-3/5), 7.80-7.86 (2H, m, benzyl H-2/6); 13C NMR (100 MHz, CDCl3) 27.9 (C(CH3)3-major), 28.2 (C(CH3)3-minor), 52.0 (benzyl-CH2-major), 53.9 (benzyl-CH2-minor), 55.2 (OCH3), 82.4 (C(CH3)3-major), 83.2 (C(CH3)3-minor), 113.7 (Ar-C), 123.7 (Ar-C), 127.1 (Ar-C), 129.8 (Ar-C), 130.0 (Ar-C), 130.5 (Ar-C), 134.5 (Ar-C), 153.5 (C=Ominor), 159.3 (C=Omajor), 165.0 (C=Omajor), 165.4 (C=Ominor); MS [M+NH4]+ m/z 400.2.
Synthesis of tert-butyl 1-methylhydrazine-1-carboxylate (7).
The desired product was obtained as a pale-yellow oil (0.338 g, 2.31 mmol, 77%). Rf 0.20 (1:1 Hexanes:EtOAc); IR (cm−1) 3247, 2924, 2854, 1697, 1640, 1568; 1H NMR (400 MHz, CDCl3) 1.47 (9H, s, C(CH3)3), 3.05 (3H, s, N-CH3), 4.10 (2H, br s, NH2).
Synthesis of tert-butyl 1-allylhydrazine-1-carboxylate (8).
The desired product was obtained as a pale-yellow oil (8.47 g, 49.2 mmol, 96%). Rf 0.22 (4:1 Hexane:EtOAc); IR (cm−1) 3336, 2977, 2932, 1690; 1H NMR (400 MHz, DMSO-d6) 1.40 (9H, s, -C(CH3)3), 3.85 (2H, ddd, J = 5.5, 1.4, 1.4 Hz, N-CH2), 4.46 (2H, s, NH2), 5.06-5.09 (1H, m, allyl C-Htrans), 5.11 (1H, br, allyl C-Hcis), 5.74-5.86 (1H, m, allyl C-H); 13C NMR (125 MHz, DMSO-d6) 28.5 (C(CH3)3), 53.6 (N-CH2), 79.4 (C(CH3)3), 116.2 (allyl-CH2), 134.6 (allyl-CH), 156.5 (C=O).
Synthesis of tert-butyl 1-(4-methoxybenzyl)hydrazine-1-carboxylate (9).
The desired product was obtained as a pale yellow oil (0.275 g, 1.09 mmol, 76%). Rf 0.24 (4:1 Hexane:EtOAc); IR (cm−1) 3336, 2975, 2933, 2836, 1688, 1612, 1511; 1H NMR (400 MHz, CDCl3) 1.51 (9H, s, C(CH3)3), 3.81 (3H, s, OCH3), 4.04 (2H, br s, NH2), 4.50 (2H, s, N-CH2), 6.88 (2H, d, J = 8.4 Hz, H-3/5), 7.24 (2H, d, J = 8.4 Hz, H-2/6); 13C NMR (100 MHz, CDCl3) 28.5 (C(CH3)3), 53.7 (OCH3), 55.3 (NCH2), 80.7 (C(CH3)3), 113.9 (Ar-C), 129.3 (Ar-C), 130.0 (Ar-C), 156.8 (Ar-C), 159.0 (C=O); MS [M+H]+ m/z 253.2.
Synthesis of 1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-2-methyl-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (11).
The desired product was obtained as a yellow solid (0.302 g, 1.54 mmol, 75%). Rf 0.23 (4:1 DCM:MeOH); M.p. 256-265°C (decomposed); IR (cm−1) 3336, 3024, 2940, 1683, 1638, 1587; 1H NMR (400 MHz, DMSO-d6) 2.53 (3H, s, SCH3), 3.36 (3H, s, N2-CH3), 8.68 (1H, s, H-4), 12.60 (1H, br s, N1-H); 13C NMR (100 MHz, DMSO-d6) 13.9 (SCH3), 31.0 (N2-CH3), 103.8, 158.1; MS [M+H]+ m/z 196.8.
Synthesis of 2-allyl-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (12).
The desired product was obtained as a yellow solid (5.44 g, 24.5 mmol, 51%). Rf 0.45 (9:1 DCM:MeOH); M.p. 125-128°C; IR (cm−1) 3032, 2979, 2926, 2659, 1656, 1615, 1566, 1514; 1H NMR (400 MHz, DMSO-d6) 2.53 (3H, s, SCH3), 4.38 (2H, dapp, J = 5.2 Hz, N2-CH2), 5.06-5.20 (2H, m, allyl C-Hcis/trans), 5.87 (1H, ddt, J = 17.2, 10.5, 5.3 Hz, alkene C-H), 8.67 (1H, s, H-4), 12.65 (1H, br, N1-H); MS [M+H]+ m/z 223.1.
Synthesis of 2-(4-methoxybenzyl)-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (13).
The desired product was obtained as a yellow solid (0.142 g, 0.47 mmol, 50%). Rf 0.41 (9:1 DCM:MeOH); M.p. 209-212°C; IR (cm−1) 3034, 2930, 1609, 1577, 1510; 1H NMR (400 MHz, CDCl3) 2.53 (3H, s, SCH3), 3.78 (3H, s, OCH3), 5.03 (2H, s, N2-CH2), 6.84 (2H, d, J = 8.5 Hz, benzyl H-3/5), 7.28 (2H, d, J = 8.5 Hz, benzyl H-2/6), 8.66 (1H, s, H-4); 13C NMR (100 MHz, DMSO-d6) 13.8 (SCH3), 46.8 (N2-CH2), 55.5 (OCH3), 103.7, 114.0, 114.1, 114.3, 129.5, 130.5, 158.3, 159.1; MS [M+H]+ m/z 303.2.
Synthesis of 2-(6-bromopyridin-2-yl)propan-2-ol (14).
The desired product was obtained as a yellow oil (0.365 g, 1.69 mmol, 85%). Rf 0.60 (1:1 Hexane:EtOAc); IR (cm−1) 3420, 2975, 2930, 1731, 1701, 1580, 1553; 1H NMR (400 MHz, DMSO-d6) 1.42 (6H, s, C(CH2)2), 5.33 (1H, s, OH), 7.47 (1H, dd, J = 7.7, 0.9 Hz, H-5), 7.67 (1H, dd, J = 7.7, 0.9 Hz, H-3), 7.73 (1H, dd, J = 7.7, 7.7 Hz, H-4); 13C NMR (100 MHz, DMSO-d6) 30.9 (C(CH2)2), 72.6 (C(CH2)2), 118.5 (Ar-C), 126.0 (Ar-C), 140.4 (Ar-C), 140.5 (Ar-C), 170.8 (Ar-C).
Synthesis of 1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-2-methyl-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (17).
The desired product was obtained as a white solid (0.215 g, 0.65 mmol, 72%). Rf 0.34 (19:1 DCM:MeOH); M.p. 155-158°C; IR (cm−1) 3432, 2973, 2928, 1683, 1604, 1562; 1H NMR (400 MHz, DMSO-d6) 1.46 (6H, s, C(CH3)2), 2.56 (3H, s, SCH3), 3.49 (3H, s, N2-CH3), 5.35 (1H, s, OH), 7.67 (1H, dapp, J = 7.7 Hz, H-5’), 7.79 (1H, dapp, J = 8.2 Hz, H-3’), 8.06 (1H, ddapp, J = 8.2, 7.7 Hz, H-4’), 9.00 (1H, s, H-4); 13C NMR (100 MHz, DMSO-d6) 14.4 (SCH3), 30.9 (C(CH3)2), 32.8 (N2-CH3), 72.8 (C(CH3)2), 104.8, 116.6, 117.5, 139.7, 146.8, 154.7, 158.3, 160.4, 168.4, 175.9; MS [M+H]+ m/z 332.6.
Synthesis of 2-allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (18).
The desired product was obtained as a white solid (0.653 g, 1.82 mmol, 81%). Rf 0.63 (9:1 DCM:MeOH); M.p. 108-111°C; IR (cm−1) 3337, 3081, 2966, 2924, 1663, 1601, 1559; 1H NMR (400 MHz, CDCl3) 1.61 (6H, s, C(CH3)2), 2.61 (3H, s, S-CH3), 3.77 (1H, s, OH), 4.82 (2H, dapp, J = 5.9 Hz, N2-CH2), 4.95 (1H, dapp, J = 16.9 Hz, alkene C-Htrans), 5.08 (1H, dapp, J = 10.3 Hz, alkene C-Hcis), 5.72 (1H, ddt, J = 16.9, 10.3, 5.9 Hz, alkene C-H), 7.42 (1H, dapp, J = 7.7 Hz, H-5’), 7.78 (1H, dapp, J = 8.0 Hz, H-3’), 7.93 (1H, dd, J = 8.0, 7.7 Hz, H-4’), 8.96 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 14.5 (SCH3), 30.5 (C(CH3)2), 47.5 (N2-CH2), 72.5 (C(CH3)2), 116.4 (Ar-C), 116.6 (Ar-C), 119.3 (allyl-CH2), 131.2, 139.2, 147.0 (Ar-C), 154.3 (Ar-C), 159.2 (C=O), 161.0 (Ar-C), 166.1 (Ar-C), 177.0 (Ar-C); MS [M+H]+ m/z 359.3.
Synthesis of 2-allyl-1-(3-(2-hydroxypropan-2-yl)phenyl)-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (19).
The desired product was obtained as a colorless oil (0.245 g, 0.68 mmol, 76%). Rf 0.26 (19:1 DCM:MeOH); IR (cm−1) 3400, 3077, 2973, 2928, 2871, 1676, 1594, 1561; 1H NMR (400 MHz, CDCl3) 1.64 (6H, s, C(CH3)2), 2.51 (3H, s, SCH3), 4.45 (2H, dapp, J = 6.0 Hz, N2-CH2), 4.99 (1H, dapp, J = 17.0, allyl C-Htrans), 5.14 (1H, dapp, J = 10.2 Hz, allyl C-Hcis), 5.71 (1H, ddt, J = 17.0, 10.2, 6.0 Hz, allyl C-H), 7.29 (1H, dapp, J = 7.2 Hz, H-6’), 7.47-7.56 (2H, m, H-4’/5’), 7.58 (1H, sapp, H-2’), 8.92 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 14.3 (SCH3), 31.9 (C(CH3)2), 46.2 (N2-CH2), 72.3 (C(CH3)2), 104.1, 119.4, 121.5, 123.2, 124.3, 129.3, 130.8, 135.4, 151.1, 154.3, 160.4, 161.6, 177.0; MS [M+H]+ m/z 357.2.
Synthesis of 2-allyl-1-(6-(hydroxymethyl)pyridin-2-yl)-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (20).
The desired product was obtained as a white solid (0.201 g, 0.61 mmol, 68%). Rf 0.23 (19:1 DCM:MeOH); M.p. 105-107°C; IR (cm−1) 3361, 3239, 2924, 2838, 1695, 1666, 1590, 1559; 1H NMR (400 MHz, CDCl3) 2.60 (3H, s, SCH3), 3.04 (1H, br, OH), 4.76-4.86 (4H, m, N2-CH2/CH2OH), 4.97 (1H, dapp, J = 17.1, allyl C-Htrans), 5.09 (1H, dapp, J = 10.3 Hz, allyl C-Hcis), 5.73 (1H, ddt, J = 17.1, 10.3, 6.2 Hz, allyl C-H), 7.30 (1H, dapp, J = 8.0 Hz, H-5’), 7.80 (1H, dapp, J = 8.1 Hz, H-3’), 7.92 (1H, ddapp, J = 8.1, 8.0 Hz, H-4’), 8.96 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 14.5 (SCH3), 47.5 (N2-CH2), 64.4 (CH2OH), 104.5, 117.0, 118.4, 119.3, 131.2, 138.9, 147.8, 154.3, 159.2, 161.0, 177.0; MS [M+H]+ m/z 330.0.
Synthesis of 1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-2-(4-methoxybenzyl)-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (21).
The desired product was obtained as an off-white solid (84 mg, 0.19 mmol, 74%). Rf 0.26 (1:1 Hexanes:EtOAc); M.p. 143-145°C; IR (cm−1) 3349, 2972, 2929, 2829, 1691, 1601, 1560; 1H NMR (400 MHz, CDCl3) 1.65 (6H, s, C(CH3)2), 2.55 (3H, s, SCH3), 3.73 (3H, s, OCH3), 5.34 (2H, s, N2-CH2), 6.68 (2H, d, J = 8.4 Hz, benzyl H-3/5), 6.83 (2H, d, J = 8.4 Hz, benzyl H2/6), 7.44 (1H, dapp, J = 7.5 Hz, H-5’), 7.56 (1H, dapp, J = 8.0 Hz, H-3’), 7.87 (1H, ddapp, J = 8.0, 7.5 Hz, H-4’), 8.95 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 14.4 (SCH3), 30.6 (C(CH3)2), 47.9 (N2-CH2), 55.2 (OCH3), 72.6 (C(CH3)2), 104.5, 114.0, 116.6, 127.3, 129.4, 139.2, 146.9, 154.3, 158.9, 159.4, 161.3, 166.1, 176.9; MS [M+H]+ m/z 438.2.
Synthesis of N,N-dimethyl-1-(3-nitrophenyl)methanamine (36).
The desired product was obtained as a yellow oil (0.601 g, 3.34 mmol, 72%). Rf 0.28 (1:1 Hexanes:EtOAc); IR (cm−1) 2976, 2944, 2859, 2820, 2774, 1523; 1H NMR (400 MHz, CDCl3) 2.28 (6H, s, N(CH3)2), 3.53 (2H, s, ArCH2), 7.51 (1H, dd, J = 8.0, 7.9 Hz, H-5), 7.68 (1H, dapp, J = 7.9 Hz, H-6), 8.13 (1H, dd, J = 8.0, 2.0 Hz, H-4), 8.21 (1H, sapp, H-2); 13C NMR (100 MHz, CDCl3) 45.4 (N(CH3)2), 63.4 (NCH2), 122.2 (Ar-C), 123.7 (Ar-C), 129.2 (Ar-C), 135.0 (Ar-C), 141.4 (Ar-C), 148.4 (Ar-C).
Synthesis of N,N-dimethyl-3-nitrobenzamide (37).
The desired product was obtained as a pale-yellow oil/low-melting solid (0.468 g, 2.41 mmol, 81%). Rf 0.23 (1:1 Hexanes:EtOAc); IR (cm−1) 3081, 3027, 2929, 2869, 1625, 1527; 1H NMR (400 MHz, CDCl3) 3.01 (3H, s, NCH3), 3.14 (3H, s, NCH3), 7.62 (1H, ddapp, J = 8.0, 7.8 Hz, H-5), 7.77 (1H, ddd, J = 7.8, 1.3, 1.2 Hz, H-6), 8.24-8.29 (2H, m, H-2/4); 13C NMR (100 MHz, CDCl3) 35.5 (NCH3), 39.5 (NCH3), 112.3 (Ar-C), 124.4 (Ar-C), 129.7 (Ar-C), 133.1 (Ar-C), 137.9 (Ar-C), 148.0 (Ar-C), 168.9 (C=O).
Synthesis of N,N-dimethyl-2-(4-nitrophenoxy)ethan-1-amine (38).
The desired product was obtained as a yellow oil (0.860 g, 4.09, 100%). Rf 0.27 (19:1 DCM:MeOH); IR (cm−1) 3114, 3084, 2945, 2824, 2774, 1737, 1591, 1508; 1H NMR (400 MHz, CDCl3) 2.36 (6H, s, N(CH3)2), 2.78 (2H, t, J = 5.6 Hz, OCH2CH2), 4.17 (2H, t, J = 5.6 Hz, OCH2CH2), 6.99 (2H, d, J = 9.2 Hz, H-2/6), 8.20 (2H, d, J = 9.2 Hz, H-3/5); 13C NMR (100 MHz, CDCl3) 45.9 (N(CH3)2), 57.9 (OCH2CH2), 66.8 (OCH2CH2), 114.5 (Ar-C), 125.9 (Ar-C), 141.6 (Ar-C), 163.8 (Ar-C).
Synthesis of N1,N1-dimethylbenzene-1,3-diamine (39).
The desired product was obtained as a red oil (0.328 g, 2.40 mmol, 80%). Rf 0.44 (1:1 Hexanes:EtOAc); IR (cm−1) 3343, 3220, 2878, 2800, 1606, 1579, 1501; 1H NMR (400 MHz, CDCl3) 2.94 (6H, s, N(CH3)2), 3.62 (2H, br s, NH2), 6.10-6.16 (2H, m, H-2/6), 6.24 (1H, dd, J = 8.1, 2.3 Hz, H-4), 7.07 (1H, dd, J = 8.1, 7.9 Hz, H-5); 13C NMR (100 MHz, CDCl3) 40.6 (N(CH3)2), 99.6 (Ar-C), 103.8 (Ar-C), 104.3 (Ar-C), 129.9 (Ar-C), 147.3 (Ar-C), 151.9 (Ar-C).
Synthesis of 3-((dimethylamino)methyl)aniline (40).
The desired product was obtained as a pale red oil (0.347 g, 2.31 mmol, 72%). Rf 0.16 (19:1 DCM:MeOH); IR (cm−1) 3270, 3147, 3079, 2974, 2942, 2858, 2816, 2774, 1666, 1610, 1552; 1H NMR (400 MHz, CDCl3) 2.26 (6H, s, N(CH3)2), 3.35 (2H, s, ArCH2), 3.65 (2H, br s, NH2), 6.59-6.62 (1H, m, H-4), 6.69-6.72 (2H, m, H-2/6), 7.12 (1H, dd, J = 8.0, 7.9 Hz, H-5); 13C NMR (100 MHz, CDCl3) 45.3 (N(CH3)2), 64.1 (ArCH2), 118.9 (Ar-C), 120.5 (Ar-C), 125.0 (Ar-C), 128.9 (Ar-C), 138.1 (Ar-C), 139.6 (Ar-C).
Synthesis of 3-amino-N,N-dimethylbenzamide (41).
The desired product was obtained as an off-white solid (0.327 g, 1.99 mmol, 85%). Rf 0.26 (19:1 DCM:MeOH); M.p. 87-89°C; IR (cm−1) 3419, 3345, 3240, 2928, 2850, 1649, 1579; 1H NMR (400 MHz, CDCl3) 2.99 (3H, s, NCH3), 3.11 (3H, s, NCH3), 3.76 (2H, br s, NH2), 6.68-6.80 (3H, m, H-2/4/6), 7.18 (1H, dd, J = 7.7, 7.6 Hz, H-5); 13C NMR (100 MHz, CDCl3) 35.2 (NCH3), 39.5 (NCH3), 113.5 (Ar-C), 116.0 (Ar-C), 116.9 (Ar-C), 129.2 (Ar-C), 137.5 (Ar-C), 146.6 (Ar-C), 171.8 (C=O).
Synthesis of 4-(2-(dimethylamino)ethyl)aniline (42).
The desired product was obtained as a yellow oil (0.422 g, 2.57 mmol, 59% - 2 steps). Rf 0.16 (9:1 DCM:MeOH); IR (cm−1) 3317, 3018, 2771, 2705, 2448, 1612, 1518; 1H NMR (400 MHz, CDCl3) 2.33 (6H, s, N(CH3)2), 2.50-2.55 (2H, m, ArCH2CH2), 2.68-2.73 (2H, m, ArCH2CH2), 3.55 (2H, br s, NH2), 6.65 (2H, d, J = 8.5 Hz, H-2/6), 7.01 (2H, d, J = 8.5 Hz, H-3/5); 13C NMR (100 MHz, CDCl3) 33.2 (ArCH2CH2), 45.3 (N(CH3)2), 61.8 (ArCH2CH2), 115.3 (Ar-C), 129.4 (Ar-C), 130.0 (Ar-C), 144.2 (Ar-C).
Synthesis of 4-(2-(dimethylamino)ethoxy)aniline (43).
The desired product was obtained as a brown oil (0.673 g, 3.74 mmol, 92%). Rf 0.38 (9:1 DCM:MeOH); IR (cm−1) 3335, 3216, 2943, 2867, 2822, 2774, 1627, 1508; 1H NMR (400 MHz, CDCl3) 2.35 (6H, s, N(CH3)2), 2.70 (2H, t, J = 5.8 Hz, OCH2CH2), 3.47 (2H, br s, NH2), 4.00 (2H, t, J = 5.8 Hz, OCH2CH2), 6.64 (2H, d, J = 8.8 Hz, H-3/5), 6.78 (2H, d, J = 8.8 Hz, H-2/6); 13C NMR (100 MHz, CDCl3) 45.8 (N(CH3)2), 58.4 (OCH2CH2), 66.6 (OCH2CH2), 115.8 (Ar-C), 116.4 (Ar-C), 140.1 (Ar-C), 152.0 (Ar-C).
Synthesis of 1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-2-methyl-6-((4-(4-methylpiperazin-1-yl)phenyl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (32).
The desired target compound was obtained as a yellow solid (0.16 mmol, 56%). Rf 0.39 (9:1 DCM:MeOH); M.p. 192-195°C; IR (cm−1) 3265, 3184, 3090, 2972, 2928, 2810, 1668, 1619, 1536, 1512; 1H NMR (400 MHz, DMSO-d6) 1.46 (6H, s, C(CH3)2), 2.23 (3H, s, N(CH2CH2)2NCH3), 2.44-2.49 (4H, m, N(CH2CH2)2NMe), 3.08-3.13 (4H, m, N(CH2CH2)2NMe), 3.42 (3H, s, N2-CH3), 5.32 (1H, s, OH), 6.93 (2H, d, J = 9.0 Hz, H-3’’/5’’), 7.59 (1H, dapp, J = 7.9 Hz, H-5’), 7.62 (2H, d, J = 9.0 Hz, H-2’’/6’’), 7.80 (1H, dapp, J = 7.5 Hz, H-3’), 8.08 (1H, dd, J = 7.9, 7.5 Hz, H-4’), 8.81 (1H, s, H-4). 10.11 (1H, br s, C6-NH); 13C NMR (100 MHz, DMSO-d6) 30.9 (C(CH3)2), 33.0 (N2-CH3), 46.3 (piperazine N-CH3), 49.0 (piperazine-CH2), 55.1 (piperazine-CH2), 72.8 (C(CH3)2), 116.0, 116.7, 121.5, 131.4, 139.3, 147.6, 156.2, 160.9, 161.8, 168.1; MS [M+H]+ m/z 475.2.
Synthesis of 2-allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((4-(4-methylpiperazin-1-yl)phenyl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (AZD-1775).
The desired target compound was obtained as a yellow solid (87 mg, 0.17 mmol, 55%).Rf 0.25 (9:1 DCM:MeOH); M.p. 170-174°C; IR (cm−1) 3420, 2969, 2810, 2364, 1639, 1602, 1541, 1512; 1H NMR (400 MHz, DMSO-d6) 1.47 (6H, s, C(CH3)2), 2.23 (3H, s, N-CH3), 2.42-2.50 (4H, m, N-(CH2CH2)2-NMe), 3.05-3.14 (4H, m, N-CH2CH2-NMe), 4.69 (2H, dapp, J = 5.9 Hz, N2-CH2), 4.83 (1H, dd, J = 17.1, 1.3 Hz, alkene C-Htrans), 5.00 (1H, dd, J = 10.3, 1.3 Hz, alkene C-Hcis), 5.32 (1H, s, OH), 5.67 (1H, ddt, J = 17.1, 10.3, 5.9 Hz, alkene C-H), 6.93 (2H, d, J = 9.1 Hz, H-3’’/5’’), 7.54-7.60 (1H, m, H-5’), 7.61 (2H, d, J = 9.1 Hz, H-2’’/6’’), 7.76 (1H, dapp, J = 8.1 Hz, H-3’), 8.06 (1H, dd, J = 8.1, 7.3 Hz, H-4’), 8.83 (1H, s, H-4), 10.1 (1H, br, C6-NH); 13C NMR (100 MHz, DMSO-d6) 30.9 (C(CH3)2), 46.2 (N-CH3), 47.1 (N2-CH2), 48.9 (piperazine-CH2), 55.1 (piperazine-CH2), 72.8 (C(CH3)2), 116.0, 116.7, 118.7, 121.6, 131.3, 132.7, 139.3, 147.6, 156.5, 161.0, 161.6, 168.0; MS [M+H]+ m/z 501.4.
Synthesis of 1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-2-(4-methoxybenzyl)-6-((4-(4-methylpiperazin-1-yl)phenyl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (33).
The desired target compound was obtained as a yellow solid (0.188 g, 0.32 mmol, 89%). Rf 0.46 (9:1 DCM:MeOH); M.p. 196-199°C; IR (cm−1) 3275, 3186, 2963, 2936, 2838, 1684, 1603, 1536, 1512; 1H NMR (400 MHz, CDCl3) 1.64 (6H, s, C(CH3)2), 2.40 (3H, s, N-CH3), 2.62-2.67 (4H, m, N-(CH2CH2)2-NMe), 3.20-3.25 (4H, m, N-CH2CH2-NMe), 3.72 (3H, s, OCH3), 5.29 (2H, s, N2-CH2), 6.67 (2H, d, J = 8.6 Hz, benzyl H-2/6), 6.85 (2H, d, J = 8.6 Hz, benzyl H-3/5), 6.91 (2H, d, J = 8.7 Hz, H-3’’/5’’), 7.37 (1H, dapp, J = 7.8 Hz, H-5’), 7.43 (2H, d, J = 8.7 Hz, H-2’’/6’’), 7.57 (1H, dapp, J = 8.1 Hz, H-3’), 7.82 (1H, dd, J = 8.1, 7.8 Hz, H-4’), 8.83 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 30.6 (C(CH3)2), 46.0 (N-CH3), 48.1 (N2-CH2), 49.4 (piperazine-CH2), 55.0 (OCH3), 55.2 (piperazine-CH2), 72.5 (C(CH3)2), 113.9, 116.0, 116.3, 116.5, 122.1, 127.7, 129.5, 130.4, 138.8, 147.4, 148.1, 156.3, 159.2, 161.0, 162.5, 165.8; MS [M+H]+ m/z 581.4.
Synthesis of 2-allyl-1-(3-(2-hydroxypropan-2-yl)phenyl)-6-((4-(4-methylpiperazin-1-yl)phenyl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (31).
The desired target compound was obtained as a pale yellow solid (53 mg, 0.11 mmol, 64%). Rf 0.39 (9:1 DCM:MeOH); M.p. 164-167°C; IR (cm−1) 3287, 2972, 2935, 2838, 2798, 1701, 1668, 1606, 1542, 1512; 1H NMR (400 MHz, CDCl3) 1.64 (6H, s, C(CH3)2), 2.39 (3H, s, N-CH3), 2.60-2.64 (4H, m, N-(CH2CH2)2-NMe), 3.18-3.23 (4H, m, N-CH2CH2-NMe), 4.40 (2H, dapp, J = 6.1 Hz, N2-CH2), 5.00 (1H, dapp, J = 17.2, alkene C-Htrans), 5.12 (1H, dapp, J = 10.1 Hz, alkene C-Hcis), 5.73 (1H, ddt, J = 17.2, 10.1, 6.1 Hz, alkene C-H), 6.90 (2H, d, J = 8.8 Hz, H-3’’/5’’), 7.29-7.34 (1H, m, H-5’), 7.45 (2H, d, J = 8.8 Hz, H-2’’/6’’), 7.49 (2H, dapp, J = 4.8 Hz, H-4’/6’), 7.60 (1H, sapp, H-2’), 8.83 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 31.9 (C(CH3)2), 46.1 (N-CH3), 46.5 (N2-CH2), 49.4 (piperazine-CH2), 55.0 (piperazine-CH2), 72.4 (C(CH3)2), 116.6, 119.2, 123.7, 129.1, 130.7, 131.1, 136.2, 150.9, 156.3, 162.7; MS [M+H]+ m/z 500.2.
Synthesis of 2-allyl-1-(6-(hydroxymethyl)pyridin-2-yl)-6-((4-(4-methylpiperazin-1-yl)phenyl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (30).
The desired target compound was obtained as a yellow solid (0.134 g, 0.29 mmol, 61%). Rf 0.34 (9:1 DCM:MeOH); M.p. 197-200°C; IR (cm−1) 3253, 3176, 3065, 2939, 2818, 1687, 1674, 1610; 1H NMR (400 MHz, CDCl3) 2.39 (3H, s, N-CH3), 2.59-2.64 (4H, m, N-(CH2CH2)2-NMe), 3.19-3.25 (4H, m, N-CH2CH2-NMe), 4.73 (2H, dapp, J = 6.0 Hz, N2-CH2), 4.82 (2H, s, CH2OH), 4.98 (1H, dapp, J = 17.2, alkene C-Htrans), 5.07 (1H, dapp, J = 10.2 Hz, alkene C-Hcis), 5.73 (1H, ddt, J = 17.2, 10.2, 6.0 Hz, alkene C-H), 6.94 (2H, d, J = 8.6 Hz, H-3’’/5’’), 7.24 (1H, dapp, J = 7.5 Hz, H-5’), 7.47 (2H, d, J = 8.6 Hz, H-2’’/6’’), 7.77 (1H, dapp, J = 8.1 Hz, H-3’), 7.87 (1H, dd, J = 8.1, 7.5 Hz, H-4’), 8.84 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 46.1 (N-CH3), 47.8 (N2-CH2), 49.5 (piperazine-CH2), 55.1 (piperazine-CH2), 64.3 (CH2OH), 116.4, 116.7, 117.8, 119.1, 122.0, 130.4, 131.6, 138.6, 148.2, 148.4, 156.3, 158.9, 161.4, 162.4; MS [M+H]+ m/z 473.2.
Synthesis of 2-allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((4-methoxybenzyl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (23).
The desired target compound was obtained as an off-white solid (55 mg, 0.12 mmol, 22%). Rf 0.38 (19:1 DCM:MeOH); M.p. 136-138°C; IR (cm−1) 3442, 3219, 2972, 2922, 1667, 1614, 1593, 1546; 1H NMR (400 MHz, CDCl3) 1.60 (6H, s, C(CH3)2), 3.82 (3H, s, OCH3), 3.98 (1H, s, OH), 4.55-4.61 (2H, m, NHCH2), 4.71-4.78 (2H, m, N2-CH2), 4.96 (1H, dapp, J = 16.4 Hz, alkene C-Htrans), 5.06 (1H, dapp, J = 9.9 Hz, alkene C-Hcis), 5.72 (1H, ddt, J = 16.4, 9.9, 6.2 Hz, alkene C-H), 6.89 (2H, d, J = 8.5 Hz, benzyl H-3/5), 7.26 (2H, d, J = 8.5 Hz, benzyl H-2/6), 7.33 (1H, dapp, J = 7.7 Hz, H-5’), 7.73 (1H, dapp, J = 8.1 Hz, H-3’), 7.85 (1H, ddapp, J = 8.1, 7.7 Hz, H-4’), 8.73 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 30.5 (C(CH3)2), 42.3 (benzyl CH2), 47.8 (N2-CH2), 55.3 (OCH3), 72.4 (C(CH3)2), 114.1, 115.7, 115.9, 119.0, 129.0, 130.0, 131.7, 138.8, 147.6, 156.3, 159.2, 161.6, 162.7, 163.4, 166.6; MS [M+H]+ m/z 447.4.
Synthesis of 2-allyl-6-amino-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (22).
The desired target compound was obtained as a white solid (13 mg, 0.04 mmol, 29%). Rf 0.53 (KP-NH - 19:1 DCM:MeOH); M.p. 195-197°C; IR (cm−1) 3325, 3187, 2979, 2924, 2856, 1667, 1649, 1616, 1563; 1H NMR (400 MHz, DMSO-d6) 1.46 (6H, s, C(CH3)2), 4.62 (2H, dapp, J = 5.6 Hz, N2-CH2), 4.81 (1H, dapp, J = 17.1 Hz, alkene C-Htrans), 4.98 (1H, dapp, J = 10.0 Hz, alkene C-Hcis), 5.32 (1H, s, OH), 5.64 (1H, ddt, J = 17.1, 10.0, 5.6 Hz, alkene C-H), 7.53 (2H, br s, NH2), 7.59 (1H, dapp, J = 7.7 Hz, H-5’), 7.71 (1H, dapp, J = 8.1 Hz, H-3’), 7.95 (1H, dapp, J = 8.1, 7.7 Hz, H-4’), 8.70 (1H, s, H-4); 13C NMR (100 MHz, DMSO-d6) 30.9 (C(CH3)2), 47.1 (N2-CH2), 72.8 (C(CH3)2), 98.9, 116.4, 116.6, 118.6, 132.7, 139.2, 147.8, 156.8, 161.9, 162.0, 165.4, 168.0; MS [M+H]+ m/z 327.2.
Synthesis of 2-allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-(phenylamino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (24).
The desired target compound was obtained as a white solid (35 mg, 0.09 mmol, 31%). Rf 0.50 (19:1 DCM:MeOH); M.p. 153-155°C; IR (cm−1) 3245, 3191, 3080, 3056, 2975, 2929, 1671, 1615, 1540; 1H NMR (400 MHz, CDCl3) 1.61 (6H, s, C(CH3)2), 3.97 (1H, s, OH), 4.78 (2H, dapp, J = 6.2 Hz, N2-CH2), 4.96 (1H, dd, J = 17.0, 1.1 Hz, alkene C-Htrans), 5.07 (1H, dd, J = 10.2, 1.1 Hz, alkene C-Hcis), 5.73 (1H, ddt, J = 17.0, 10.2, 6.2 Hz, alkene C-H), 7.15 (1H, dd, J = 7.4, 7.3 Hz, H-4’’), 7.36-7.41 (3H, m, H-5’/3’’/5’’), 7.63 (2H, dapp, J = 7.8 Hz, H-2’’/6’’), 7.79 (1H, dapp, J = 7.9 Hz, H-3’), 7.91 (1H, ddapp, J = 7.9, 7.8 Hz, H-4’), 8.90 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 30.6 (C(CH3)2), 47.6 (N2-CH2), 72.5 (C(CH3)2), 101.1, 116.2, 116.3, 119.1, 120.6, 124.0, 128.9, 131.5, 138.2, 138.9, 147.4, 156.3, 161.0, 161.3, 162.0, 165.9; MS [M+H]+ m/z 403.4.
Synthesis of 2-allyl-6-((3-(dimethylamino)phenyl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (25).
The desired target compound was obtained as a pale yellow/green solid (29 mg, 0.06 mmol, 23%). Rf 0.30 (19:1 DCM:MeOH); M.p. 81-84°C; IR (cm−1) 3407, 3219, 3080, 2963, 2926, 1694, 1605, 1572, 1548; 1H NMR (400 MHz, CDCl3) 1.61 (6H, s, C(CH3)2), 2.95 (6H, s, N(CH3)2, 3.92 (1H, s, OH), 4.76 (2H, dapp, J = 6.0 Hz, N2-CH2), 4.95 (1H, dapp, J = 17.2 Hz, alkene C-Htrans), 5.06 (1H, dapp, J = 10.1 Hz, alkene C-Hcis), 5.73 (1H, ddt, J = 17.2, 10.1, 6.0 Hz, alkene C-H), 6.54 (1H, dd, J = 8.4, 2.0 Hz, H-4’’), 6.87 (1H, br s, H-2’’), 7.05 (1H, dapp, J = 7.9 Hz, H-6’’), 7.23 (1H, dd, J = 8.4, 7.9 Hz, H-5’’), 7.37 (1H, dapp, J = 7.6 Hz, H-5’), 7.50 (1H, br s, N-H), 7.81 (1H, dapp, J = 7.9 Hz, H-3’), 7.87 (1H, ddapp, J = 7.9, 7.6 Hz, H-4’), 8.88 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 30.5 (C(CH3)2), 40.6 (N(CH3)2), 47.6 (N2-CH2), 72.5 (C(CH3)2), 101.0, 104.8, 108.7, 109.2, 116.1, 116.5, 119.0, 129.4, 131.6, 138.9, 139.0, 147.5, 151.3, 156.3, 161.2, 161.4, 162.1, 165.9; MS [M+H]+ m/z 446.2.
Synthesis of 2-allyl-6-((3-((dimethylamino)methyl)phenyl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (26).
The desired target compound was obtained as an off-white solid (35 mg, 0.08 mmol, 27%). Rf 0.18 (EtOAc); M.p. 121-123°C; IR (cm−1) 3407, 3230, 2975, 2927, 2772, 1665, 1610, 1542; 1H NMR (400 MHz, CDCl3) 1.61 (6H, s, C(CH3)2), 2.29 (6H, s, N(CH3)2), 3.47 (2H, s, ArCH2), 4.78 (2H, dapp, J = 6.1 Hz, N2-CH2), 4.96 (1H, dapp, J = 17.1 Hz, alkene C-Htrans), 5.07 (1H, dapp, J = 10.1 Hz, alkene C-Hcis), 5.73 (1H, ddt, J = 17.1, 10.1, 6.1 Hz, alkene C-H), 7.10 (1H, dapp, J = 7.5 Hz, H-4’’), 7.33 (1H, ddapp, J = 7.9, 7.5 Hz, H-5’’), 7.39 (1H, dapp, J = 7.6 Hz, H-5’), 7.54-7.62 (2H, m, H-2’’/6’’), 7.82 (1H, dapp, J = 7.9 Hz, H-3’), 7.92 (1H, ddapp, J = 7.9, 7.6 Hz, H-4’), 8.89 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 30.5 (C(CH3)2), 45.4 (N(CH3)2), 47.6 (N2-CH2), 64.3 (Ar-CH2), 72.5 (C(CH3)2), 101.2, 116.1, 116.4, 119.1, 119.2, 120.8, 124.6, 128.8, 131.6, 138.3, 138.9, 139.7, 147.4, 156.3, 161.1, 161.2, 162.0, 165.9; MS [M+H]+ m/z 460.0.
Synthesis of 3-((2-allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-3-oxo-2,3-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N,N-dimethylbenzamide (27).
The desired target compound was obtained as a white solid (16 mg, 0.03 mmol, 12%). Rf 0.34 (19:1 DCM:MeOH); M.p. 93-96°C; IR (cm−1) 3405, 3270, 2972, 2929, 1673, 1615, 1541; 1H NMR (400 MHz, CDCl3) 1.60 (6H, s, C(CH3)2), 2.97 (3H, s, NCH3), 3.16 (3H, s, NCH3), 3.97 (1H, s, OH), 4.79 (2H, dapp, J = 6.0 Hz, N2-CH2), 4.95 (1H, dapp, J = 17.0 Hz, alkene C-Htrans), 5.06 (1H, dapp, J = 10.3 Hz, alkene C-Hcis), 5.72 (1H, ddt, J = 17.0, 10.3, 6.0 Hz, alkene C-H), 7.14 (1H, dapp, J = 7.5 Hz, H-4’’), 7.35-7.41 (2H, m, H-5’/5’’), 7.48 (1H, dapp, J = 8.1 Hz, H-6’’), 7.82 (1H, dapp, J = 8.1 Hz, H-3’), 7.94 (1H, br s, N-H), 7.98-8.05 (2H, m, H-4’/2’’), 8.89 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 30.5 ((C(CH3)2), 35.4 (NCH3), 39.6 (NCH3), 47.6 N2-CH2), 72.5 (C(CH3)2), 101.5, 116.3, 116.6, 118.8, 119.1, 120.9, 121.9, 128.9, 131.5, 137.2, 138.6, 139.6, 147.2, 156.4, 160.8, 161.0, 161.8, 165.8, 171.2; MS [M+H]+ m/z 474.2.
Synthesis of 2-allyl-6-((4-(2-(dimethylamino)ethyl)phenyl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (28).
The desired target compound was obtained as an off-white solid (36 mg, 0.08 mmol, 18%). Rf 0.14 (9:1 DCM:MeOH); M.p. 116-119°C; IR (cm−1) 3252, 3191, 3099, 2968, 2929, 2855, 2827, 2782, 1745, 1672, 1603, 1568; 1H NMR (400 MHz, CDCl3) 1.61 (6H, s, C(CH3)2), 2.33 (6H, s, N(CH3)2), 2.53-2.59 (2H, m, ArCH2CH2), 2.76-2.82 (2H, m, ArCH2CH2), 4.77 (2H, dapp, J = 5.8 Hz, N2-CH2), 4.96 (1H, dapp, J = 17.2 Hz, alkene C-Htrans), 5.06 (1H, dapp, J = 10.3 Hz, alkene C-Hcis), 5.73 (1H, ddt, J = 17.2, 10.3, 5.8 Hz, alkene C-H), 7.21 (2H, d, J = 8.2 Hz, H-3’’/5’’), 7.39 (1H, dapp, J = 7.7 Hz, H-5’), 7.53 (2H, d, J = 8.2 Hz, H-2’’/6’’), 7.78 (1H, dapp, J = 7.9 Hz, H-3’), 7.90 (1H, dd, J = 7.9, 7.7 Hz, H-4’), 8.87 (1H, s, H-4); 13C NMR (100 MHz, CDCl3) 30.6 (C(CH3)2), 33.8 (ArCH2CH2), 45.5 (N(CH3)2), 47.6 (N2-CH2), 61.5 (ArCH2CH2), 72.5 (C(CH3)2), 101.1, 116.2, 116.3, 119.1, 120.6, 129.0, 131.6, 136.1, 136.2, 138.9, 147.5, 156.4, 161.1, 161.3, 162.1, 165.9; MS [M+H]+ m/z 474.4.
Synthesis of 2-allyl-6-((4-(2-(dimethylamino)ethoxy)phenyl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (29).
The desired target compound was obtained as an off-white solid (47 mg, 0.11 mmol, 38%). Rf 0.28 (9:1 DCM:MeOH); M.p. 123-126°C; IR (cm−1) 3248, 3081, 2976, 2937, 2870, 2821, 2773, 1680, 1614, 1512; 1H NMR (400 MHz, DMSO-d6) 1.47 (6H, s, C(CH3)2), 2.28 (6H, s, N(CH3)2), 2.69 (2H, t, J = 5.2 Hz, OCH2CH2), 4.06 (2H, t, J = 5.2 Hz, OCH2CH2), 4.69 (2H, dapp, J = 5.4 Hz, N2-CH2), 4.83 (1H, dapp, J = 17.1 Hz, alkene C-Htrans), 5.00 (1H, dapp, J = 10.3 Hz, alkene C-Hcis), 5.33 (1H, s, OH), 5.67 (1H, ddt, J = 17.1, 10.3, 5.4 Hz, alkene C-H), 6.94 (2H, d, J = 8.4 Hz, H-3’’/5’’), 7.59-7.66 (3H, m, H-5’/2’’/6’’), 7.75 (1H, dapp, J = 7.4 Hz, H-3’), 8.05 (1H, dd, J = 7.8, 7.4 Hz, H-4’), 8.85 (1H, s, H-4), 10.19 (1H, br s, C6-NH); 13C NMR (100 MHz, DMSO-d6) 30.9 (C(CH3)2, 45.8 (N(CH3)2), 47.0 (N2-CH2), 58.1 (OCH2CH2), 66.2 (OCH2CH2), 72.8 (C(CH3)2), 114.8, 116.8, 118.7, 132.7, 139.3, 147.5, 154.9, 156.6, 161.6, 168.1; MS [M+H]+ m/z 490.4.
Synthesis of 6-((4-(4-methylpiperazin-1-yl)phenyl)amino)-1-(6-(prop-1-en-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (34).
The desired target compound was obtained as a pale yellow solid (39 mg, 0.09 mmol, 91%). Rf 0.36 (9:1 DCM:MeOH); M.p. 260-270°C (decomposed); IR (cm−1) 3245, 3175, 2933, 2836, 2791, 1691, 1611; 1H NMR (400 MHz, DMSO-d6) 2.20 (3H, s, CCH3), 2.25 (3H, s, N-CH3), 2.47-2.51 (4H, m, N-(CH2CH2)2-NMe), 3.09-3.13 (4H, m, N-CH2CH2-NMe), 5.37 (1H, s, alkene C-H), 6.12 (1H, s, alkene C-H), 6.93 (2H, d, J = 8.9 Hz, H-3’’/5’’), 7.45 (1H, dapp, J = 7.5 Hz, H-3’), 7.67 (2H, d, J = 8.9 Hz, H-2’’/6’’), 7.98 (1H, dd, J = 7.8, 7.5 Hz, H-4’), 8.09-8.16 (1H, m, H-5’), 8.83 (1H, s, H-4), 9.88 (1H, s, C6-NH), 11.95 (1H, br s, N2-H); 13C NMR (100 MHz, DMSO-d6) 20.7 (C(CH2)CH3), 46.1 (N-CH3), 49.1 (piperazine-CH2), 55.1 (piperazine-CH2), 113.4, 116.2, 116.6, 117.3, 121.5, 132.1, 139.4, 142.5, 147.2, 154.7, 156.7, 157.4, 160.3; MS [M+H]+ m/z 443.4.
Synthesis of 1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((4-(4-methylpiperazin-1-yl)phenyl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one (35).
The desired target compound was obtained as an off-white solid (36 mg, 0.08 mmol, 80%). Rf 0.21 (1:1 DCM:MeOH); M.p. >350°C; IR (cm−1) 3248, 3168, 2974, 2936, 2791, 1612, 1535; 1H NMR (400 MHz, DMSO-d6) 1.52 (6H, s, C(CH3)2), 2.23 (3H, s, N-CH3), 2.45-2.49 (4H, m, N-(CH2CH2)2-NMe), 3.05-3.09 (4H, m, N-CH2CH2-NMe), 5.83 (1H, s, OH), 6.91 (2H, d, J = 9.1 Hz, H-3’’/5’’), 7.19 (1H, dapp, J = 7.5 Hz, H-5’), 7.69 (2H, d, J = 9.1 Hz, H-2’’/6’’), 7.80 (1H, dd, J = 7.9, 7.5 Hz, H-4’), 8.13 (1H, dapp, J = 7.9 Hz, H-3’), 8.45 (1H, s, H-4), 9.23 (1H, s, C6-NH); 13C NMR (100 MHz, DMSO-d6) 31.2 (C(CH3)2), 46.3 (N-CH3), 49.5 (piperazine-CH2), 55.2 (piperazine-CH2), 72.3 (C(CH3)2), 116.4, 120.5, 133.5, 138.6, 146.4, 150.5, 152.5, 159.1, 166.4; MS [M+H]+ m/z 461.2.
Biological Assays
Cells and Reagents:
Daoy and ONS-76 cells (Medulloblastoma) were obtained from ATCC and were passaged for <6 months following resuscitation. Cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Sigma Aldrich), 1 mm sodium pyruvate (Gibco), 1X penicillin/streptomycin solution (Cellgro) and 1X non-essential amino acids (Sigma Aldrich) at 37°C in an incubator humidifier with 95% air and 5% CO2. Seeded cells were allowed to adhere for 24 h prior to use in all assays. In all treatment conditions, the final DMSO concentration did not exceed 0.5%. All chemical reagents were purchased from the Aldrich Chemical Company, Fisher Chemicals or Alfa Aesar Chemicals and were of the highest available purity.
WEE1 Recombinant Kinase Inhibition Assay:
LanthaScreen™ Eu time-resolved fluorescence resonance energy transfer (TR-FRET) kinase binding assays (Invitrogen) were performed in 384-well, low-volume plates (Corning) using recombinant WEE1 kinase, Kinase Tracer 178 and LanthaScreen™ Eu-anti-GST antibody (Invitrogen). Assays were performed at 25°C in a reaction mixture consisting of 5 μL serially diluted inhibitor solution, 5 μL Kinase Tracer 178 solution, and 5 μL kinase/antibody solution. All reagents were prepared as solutions in 1X kinase buffer A (Invitrogen) at 3X final desired concentration. Inhibitor solutions were prepared such that final DMSO concentrations did not exceed 0.5%, which was shown to have no effect on kinase activity. Inhibitors were assayed in the final concentration range of 0.04 nm to 10 μm. Kinase Tracer 178 was used at a final concentration of 150 nm and the antibody and kinase were used at final concentrations of 3 nm and 5 nm, respectively. All reagents were incubated together for 1 h at room temperature and read using a PerkinElmer Envision 2104 Multilabel Reader enabled for TR-FRET (Excitation = 340 nm; Tracer emission = 665 nm; Antibody emission = 615 nm; Delay = 100 μs; Integration = 200 μs). Emission ratios (665 nm/615 nm) were determined for each inhibitor concentration and the data analyzed using a non-linear regression analysis of the log dose-response curve to determine IC50 values.
PLK1 Recombinant Kinase Inhibition Assay.
LANCE® Eu time-resolved fluorescence resonance energy transfer (TR-FRET) kinase assay (PerkinElmer) was performed in 384-well OptiPlates (Corning) using recombinant PLK1 kinase (Carna), ULight™-p70 S6K (Thr389) substrate (PerkinElmer) and ATP (Sigma) according to supplier protocols. All reagents were prepared in kinase buffer (2 mm DTT, 50 mm HEPES, 1 mm EGTA, 10 mm MgCl2, 0.01% Tween20, pH 7.5) and inhibitor solutions were prepared such that the final DMSO concentration did not exceed 0.5%, which was shown to have no effect on kinase activity. PLK1 was used at a final concentration of 50 nm, ULight™-CREBtide substrate was used at a final concentration of 50 nm and ATP was used at a final concentration of 5 μm. Assays were performed at 25°C in a reaction mixture consisting of 2.5 μL serially diluted inhibitor solution, 2.5 μL kinase, 2.5 μL ATP and 2.5 μL substrate. Reagents were incubated for 1 h before the reaction was halted through the addition of EDTA (10 mm) after which Eu anti-phospho-p70 S6K (Thr389) antibody (PerkinElmer) was added at a final concentration of 2 nm for 1 h. The plate was read using a Biotek Synergy H1 Hybrid plate reader enabled for TR-FRET (Excitation = 340 nm; Substrate emission = 665 nm; Antibody emission = 615 nm; Delay = 100 μs; Integration = 200 μs). Emission ratios (665 nm/615 nm) were calculated for each well and half-maximal inhibitory concentration (IC50) values were determined for each inhibitor through non-linear regression analysis of the log dose-response curve.
Cellular Metabolic Viability Assay:
Daoy and ONS-76 cells were seeded into sterile 96-well plates (Corning Inc.) at 2000 cells/well in 100 μL media. Inhibitors were administered at the MTS EC50 of AZD1775 (Daoy; EC50 = 150 nm, ONS-76; EC50 = 290 nm) and 2-fold concentrations above and below the EC50. Cells were incubated for 72 h with 50 μL of each diluted inhibitor solution. Cell viability was measured by 2 h incubation with 30 μL CellTiter 96® AQueous One Cell Proliferation reagent (Promega) and formazan concentration assessed through colorimetric analysis using a BioTek Synergy H1 plate reader (Absorption = 490 nm).
CellTox Green Cytotoxicity Assay:
Daoy cells were seeded into sterile 96-well plates (Corning Inc.) at 5000 cells/well in 100 μL media containing 1:500 dilution of CellTox™ Green dye (Promega). Inhibitor solutions (50 μL) were administered over a concentration range from 4800 nm to 18.75 nm. Cells were incubated for a total of 72 h and fluorescence was measured at 1, 24, 48, and 72 h using a BioTek Synergy H1 plate reader (Excitation = 485 nm; Emission = 520 nm).
Quantification of p-CDK1 (Tyr15) Levels:
Daoy cells were plated in sterile 6-well plates at 200,000 cells/well and treated with active inhibitors at a dose of 220 nm and incubated for 24 h prior to preparing cell lysates. Any compounds that were found to inhibit cellular p-CDK1 levels at this concentration when compared to DMSO control were tested across a concentration range from 1000 nm to 62.5 nm. Following drug incubation, the media was aspirated from cells before cells were trypsinized and resuspended in TES/SB buffer containing protease inhibitors. Cells were lysed on ice through sonication, and cell lysates were diluted with ELISA Pathscan® sample diluent to a final volume of 100 μL and protein concentration of 0.05 mg/ml prior to use. The relative concentration of p-CDK1 Tyr15 was determined using an enzyme-linked immunosorbent assay according to recommended protocol (Cell Signaling, ELISA Pathscan® phosphor-Cdc2 (Tyr15)).
Statistical Analysis:
All experiments were repeated in triplicate. Statistical analyses were conducted using GraphPad Prism 5.0 and the error bars in each figure represent the mean and standard deviation (S.D.). Results were considered statistically significant if p < 0.05 by one- or two-way ANOVA as appropriate with Tukey’s multiple comparison test.
Molecular Modeling
The predicted binding orientation of AZD1775 within the kinase domain of WEE1 (PDB ID: 1X8B) [16] was reported previously.[12] AZD1775 and 28 were docked into the ATP-binding site of the WEE1 crystal structure using the GLIDE module within Schrödinger (Release 2017-4, Schrödinger, LLC, NY).[17] Molecular comparisons of electrostatic and hydrophobic field properties between AZD1775 and selected analogs were conducted using Forge™ 10.4.2 (Cresset, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/forge/) [18].
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH) under Award Number R21NS084084. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Molecular modeling studies were conducted at the University of Colorado Computational Chemistry and Biology Core Facility, which is funded in part by NIH/NCATS Colorado CTSA Grant Number UL1 TR001082.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References:
- [1].Do K, Doroshow JH, Kummar S, Cell Cycle 2013, 12(19), 3159–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a Chen T, Stephens PA, Middleton FK, Curtin NJ, Drug Discovery Today 2012, 17(5–6), 194–202; [DOI] [PubMed] [Google Scholar]; b Bucher N, Britten CD, British Journal of Cancer 2008, 98(3), 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Matheson CJ, Backos DS, Reigan P, Trends Pharmacol Sci 2016, 37(10), 872–881. [DOI] [PubMed] [Google Scholar]
- [4].Puigvert JC, Sanjiv K, Helleday T, FEBS J 2016, 283(2), 232–245. [DOI] [PubMed] [Google Scholar]
- [5].a Masaki T, Shiratori Y, Rengifo W, Igarashi K, Yamagata M, Kurokohchi K, Uchida N, Miyauchi Y, Yoshiji H, Watanabe S, Omata M, Kuriyama S, Hepatology 2003, 37(3), 534–543; [DOI] [PubMed] [Google Scholar]; b Iorns E, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, Mackay A, Mein CA, Natrajan R, Savage K, Tamber N, Reis-Filho JS, Turner NC, Ashworth A, PLoS One 2009, 4(4), e5120; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Magnussen GI, Hellesylt E, Nesland JM, Trope CG, Florenes VA, Holm R, BMC Cancer 2013, 13, 288; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mueller S, Hashizume R, Yang X, Kolkowitz I, Olow AK, Phillips J, Smirnov I, Tom MW, Prados MD, James CD, Berger MS, Gupta N, Haas-Kogan DA, Neuro Oncol 2014, 16(3), 352–360; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, Van Tilborg AA, Zwinderman AH, Geerts D, Kaspers GJ, Peter Vandertop W, Cloos J, Tannous BA, Wesseling P, Aten JA, Noske DP, Van Noorden CJ, Wurdinger T, Cancer Cell 2010, 18(3), 244–257; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Music D, Dahlrot RH, Hermansen SK, Hjelmborg J, de Stricker K, Hansen S, Kristensen BW, J Neurooncol 2016, 127(2), 381–389; [DOI] [PubMed] [Google Scholar]; g Harris PS, Venkataraman S, Alimova I, Birks DK, Balakrishnan I, Cristiano B, Donson AM, Dubuc AM, Taylor MD, Foreman NK, Reigan P, Vibhakar R, Mol Cancer 2014, 13, 72; [DOI] [PMC free article] [PubMed] [Google Scholar]; h Tibes R, Bogenberger JM, Chaudhuri L, Hagelstrom RT, Chow D, Buechel ME, Gonzales IM, Demuth T, Slack J, Mesa RA, Braggio E, Yin HH, Arora S, Azorsa DO, Blood 2012, 119(12), 2863–2872; [DOI] [PMC free article] [PubMed] [Google Scholar]; i Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, Zaberezhnyy V, Patel PR, Gao D, Tan AC, DeGregori J, Leukemia 2012, 26(6), 1266–1276; [DOI] [PMC free article] [PubMed] [Google Scholar]; j Magnussen GI, Holm R, Emilsen E, Rosnes AK, Slipicevic A, Florenes VA, PLoS One 2012, 7(6), e38254; [DOI] [PMC free article] [PubMed] [Google Scholar]; k Slipicevic A, Holth A, Hellesylt E, Trope CG, Davidson B, Florenes VA, Gynecol Oncol 2014, 135(1), 118–124. [DOI] [PubMed] [Google Scholar]
- [6].Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, Itadani H, Takahashi-Suzuki I, Fukasawa K, Oki H, Nambu T, Jiang J, Sakai T, Arakawa H, Sakamoto T, Sagara T, Yoshizumi T, Mizuarai S, Kotani H, Mol Cancer Ther 2009, 8(11), 2992–3000. [DOI] [PubMed] [Google Scholar]
- [7].De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T, Clin Cancer Res 2011, 17(13), 4200–4207. [DOI] [PubMed] [Google Scholar]
- [8].a Beck H, Nahse V, Larsen MS, Groth P, Clancy T, Lees M, Jorgensen M, Helleday T, Syljuasen RG, Sorensen CS, J Cell Biol 2010, 188(5), 629–638; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuasen RG, Sorensen CS, Mol Cell Biol 2012, 32(20), 4226–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S, J Clin Oncol 2015, 33(30), 3409–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, Zawel L, Fawell SE, Gilliland DG, Shumway SD, Mol Cancer Ther 2013, 12(8), 1442–1452. [DOI] [PubMed] [Google Scholar]
- [11].a Metz JT, Johnson EF, Soni NB, Merta PJ, Kifle L, Hajduk PJ, Nat Chem Biol 2011, 7(4), 200–202; [DOI] [PubMed] [Google Scholar]; b Wright G, Golubeva V, Remsing Rix LL, Berndt N, Luo Y, Ward GA, Gray JE, Schonbrunn E, Lawrence HR, Monteiro ANA, Rix U, ACS Chem Biol 2017, 12(7), 1883–1892; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhu JY, Cuellar RA, Berndt N, Lee HE, Olesen SH, Martin MP, Jensen JT, Georg GI, Schonbrunn E, J Med Chem 2017, 60(18), 7863–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Matheson CJ, Venkataraman S, Amani V, Harris PS, Backos DS, Donson AM, Wempe MF, Foreman NK, Vibhakar R, Reigan P, ACS Chem Biol 2016, 11(4), 921–930. [DOI] [PubMed] [Google Scholar]
- [13].Zhu J-Y, Cuellar RAD, Berndt N, Lee HE, Olesen SH, Martin MP, Jensen JT, Georg GI, Schönbrunn E, Journal of Medicinal Chemistry 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].O’Farrell PH, Trends in Cell Biology 2001, 11(12), 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brosse N, Pinto M-F, Jamart-Grégoire B, The Journal of Organic Chemistry 2000, 65(14), 4370–4374. [DOI] [PubMed] [Google Scholar]
- [16].Squire CJ, Dickson JM, Ivanovic I, Baker EN, Structure 2005, 13(4), 541–550. [DOI] [PubMed] [Google Scholar]
- [17].a Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS, J Med Chem 2004, 47(7), 1739–1749; [DOI] [PubMed] [Google Scholar]; b Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, J Med Chem 2006, 49(21), 6177–6196; [DOI] [PubMed] [Google Scholar]; c Halgren TA, J Chem Inf Model 2009, 49(2), 377–389. [DOI] [PubMed] [Google Scholar]
- [18].Cheeseright T, Mackey M, Rose S, Vinter A, Journal of Chemical Information and Modeling 2006, 46(2), 665–676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.