

Abstract
Hepatocellular carcinoma (HCC) remains a leading cause of cancer-related deaths, warranting better therapies. Restoration of tumor-suppressive microRNAs depleted in hepatocellular carcinoma represents a new therapeutic strategy. Herein, we sought to identify a potent microRNA (miRNA) agent that could alleviate HCC tumor burden and improve survival. Among a collection of bioengineered noncoding RNA molecules produced through bacterial fermentation, we identified let-7c agent as the most potent inhibitor of HCC cell viability. Bioengineered let-7c selectively modulated target gene expression (Lin-28 homolog B [LIN28B], AT-rich interactive domain-containing protein 3B [ARID3B], B cell lymphoma-extra large [Bcl-xl], and c-Myc) in HCC cells, and consequently induced apoptosis and inhibited tumorsphere growth. When formulated with liposomal-branched polyethylenimine polyplex, bioengineered let-7c exhibited serum stability up to 24 h. Furthermore, liposomal polyplex-formulated let-7c could effectively reduce tumor burden and progression in orthotopic HCC mouse models, while linear polyethyleneimine-formulated let-7c to a lower degree, as revealed by live animal and ex vivo tissue imaging studies. This was also supported by reduced serum α-fetoprotein and bilirubin levels in let-7c-treated mice. In addition, lipopolyplex-formulated let-7c extended overall survival of HCC tumor-bearing mice and elicited no or minimal immune responses in healthy immunocompetent mice and human peripheral blood mononuclear cells. These results demonstrate that bioengineered let-7c is a promising molecule for advanced HCC therapy, and liposomal polyplex is a superior modality for in vivo RNA delivery.
Keywords: microRNA, bioengineering, hepatocellular carcinoma, lipopolyplex, xenograft
Graphical Abstract
Introduction
Hepatocellular carcinoma (HCC) is the most common malignancy of liver with an increasing incidence, a 5-year survival rate of 18%,1 and an overall median survival of 29.8 months without improvement over time.2 Given the molecular heterogeneity and complex etiology of HCC, development of pharmacological interventions has been slow.
Although nivolumab, and anti-PD1 monoclonal antibody, has recently been suggested as a promising therapy for HCC,3 the only molecularly targeted drugs indicated for HCC are the multi-kinase inhibitors, sorafenib and regorafenib, and these show only modest (approximately 3 months) improvements in overall survival.4, 5 Over the past two decades, the discovery of aberrant microRNA (miRNA or miR) expression in cancer cells has evoked new targeted strategies for HCC therapy.6, 7, 8, 9, 10, 11, 12 For example, tumor-suppressive miRNAs (e.g., miR-124, -199a, and -206) depleted in HCC tissues may be reintroduced into cancer cells to manage HCC progression.13, 14, 15, 16, 17, 18 Because miRNA replacement presents a promising approach for cancer therapy,7 efforts to translate this strategy into clinical pharmacological interventions is warranted to improve upon current standards of care.
To investigate miRNA-based cancer therapies, we have developed a novel noncoding RNA (ncRNA) bioengineering platform to produce recombinant miRNA agents via bacterial fermentation,19 based upon a stable hybrid tRNA/pre-miRNA molecule (e.g., tRNAMet/pre-miR-34a) identified in our lab.20, 21 By replacing the miRNA (e.g., miR-34a) duplexes with target miRNA (e.g., let-7c) or small interfering RNA (siRNA; e.g., Nrf2-siRNA), we have produced a set of biologic miRNA or siRNA agents containing a common tRNA/pre-miR-34a molecular scaffold, which were all expressed at high levels in a common strain of Escherichia coli HST08 and purified to a high degree of homogeneity by anion exchange fast protein liquid chromatography (FPLC).21 These bioengineered miRNA agents are produced and folded in living cells, without or with minimal posttranscriptional modifications, and thus distinguished from conventional miRNA mimics or molecules that are produced by chemical synthesis and typically consisted of extensive degrees and various types of ribose and phosphate backbone modifications.19, 22 Further studies have demonstrated that bioengineered noncoding RNA (ncRNA) agents are selectively processed to target miRNA or siRNA molecules in human cells, modulate target gene expression, and control cellular processes.20, 21, 23, 24, 25, 26
Systemic RNA therapy is hampered by the susceptibility of RNA molecules to serum RNases and the ability to cross the membrane barrier, warranting proper delivery systems. Polyethylenimine (PEI)-RNA complexes offer high delivery efficiency; however, PEI is cytotoxic with the increase of doses.27 Lipidation of PEI-RNA polyplexes can reduce the toxicity of polyplexes28 as the resultant lipopolyplexes (LPPs) exhibit more favorable biocompatibilities.29, 30, 31 Using a bioengineered GFP siRNA agent as model molecule, we have found that PEI-based cationic LPP nanocomplex offers efficient delivery of bioengineered RNA molecules in orthotopic HCC xenograft mouse models, leading to more consistent knockdown of target gene expression than polyplex in tumor tissue.32
Herein, we present our findings on the identification of bioengineered let-7c as the most potent inhibitor against HCC cell viability among a small collection of recombinant miRNA or siRNA agents that are known to exhibit anti-proliferative activities. Following the validation and delineation of mechanistic actions of let-7c on target gene expression, as well as HCC cell stemness and apoptosis, our results demonstrate the utility of intravenously (i.v.) administered LPP/let-7c nanotherapeutics to reduce tumor progression and improve overall survival in orthotopic HCC xenograft mouse models. In addition, LPP-formulated let-7c treatment is well tolerated in mice, showing no or minimal immunogenicity in human peripheral blood mononuclear cells (PBMCs) and immunocompetent mice.
Results
Bioengineered let-7c Is the Most Potent Inhibitor against HCC Cell Proliferation among a Collection of ncRNA Agents
Screening of a small collection of bioengineered miRNA agents was predictive for their anti-proliferative activities (Figure 1A), in which the truncated RNA, namely MSA (methionine tRNA with Sephadex aptamer), just containing the tRNA portion that was proven as a good control to define the actions of tRNA-carried miRNAs20, 21 consistently yielded the least inhibition of cell viability. Several miRNA agents, including miR-298, miR-124, let-7c, miR-328, miR-144, and miR-126, showed greater antiproliferative activities in Huh7 cells, and thus were pursued for dose-response studies (Figure 1B). Let-7c was revealed as the most potent ncRNA, with the lowest EC50 value (0.51 nM) in the inhibition of Huh7 cell proliferation (Figure 1C). Furthermore, let-7c was as pure (>97%, by high-performance liquid chromatography [HPLC]) as other tested ncRNAs purified by the same anion exchange FPLC method21 and had a low endotoxin level (Figure S1), suggesting minimal interference by impurities.
Figure 1.

Bioengineered let-7c Molecule Is Identified as the Most Potent Inhibitor against HCC Cell Proliferation among a Small Collection of ncRNAs
(A) Antiproliferative activities of a collection of bioengineered ncRNA agents (5 nM) against luciferase/GFP-expressing Huh7 cells were examined by luminometric ATP assay. Values were normalized to Lipofectamine 3000 transfection reagent/vehicle control (0% inhibition). (B) Dose-response curves of the top ranked ncRNA agents were further determined, and (C) their pharmacodynamic parameters were estimated, which indicate that let-7c is the most potent inhibitor of HCC cell viability in this collection of ncRNA agents. Values are mean ± SD (n = 3 per group). *p < 0.05, compared with MSA control.
Bioengineered let-7c Reduces Protein Levels of Target Genes
To verify the actions of recombinant let-7c prodrug, we first examined protein levels of several known let-7c targets important in cancer. Lin-28 homolog B (LIN28B), a canonical target of let-7 family miRNAs,33 was reduced by biologic let-7c over 60% and 85% in Huh7 and Hep3B cells, respectively (Figure 2A). This was associated with much higher levels of let-7c in cells treated with biologic let-7c- than control MSA (data not shown). Furthermore, AT-rich interactive domain-containing protein 3B (ARID3B) protein level34 was suppressed consistently by bioengineered let-7c prodrug in both Huh7 and Hep3B cells, whereas downregulation of B cell lymphoma-extra large (Bcl-xl)35 and c-Myc36 was obvious only in Huh7 cells at 72 h post-transfection (Figure 2A). Interestingly, significant downregulation of c-Myc in let-7c-transfected Hep3B cells was observed in Hep3B cells at 48 h after transfection (data not shown). In addition, the expression of HMGA2,37, 38 as detected by immunofluorescence, was reduced significantly by biologic let-7c in both Huh7 and Hep3B cells (Figure 2B).
Figure 2.

Suppression of Protein Levels of Target Genes by Bioengineered let-7c Agent in HCC Cell Lines
(A) Immunoblot analyses of let-7c targeted ARID3B, c-MYC, LIN28B, and Bcl-xl protein levels and (B) immunofluorescent analysis of HMGA2 in Huh7 and Hep3B cells treated with 15 nM let-7c or control MSA. Immunoblot intensity was normalized to total protein and Lipofectamine 3000/vehicle control for comparison between groups. ap < 0.05 compared with vehicle; bp < 0.05 compared with MSA (one-way ANOVA with Bonferroni’s post hoc test). Immunofluorescent intensity of HMGA2 staining was normalized to total DAPI-positive cells. *p < 0.05 compared with MSA (Student’s t test).
Induction of Apoptosis of HCC Cells by Bioengineered let-7c
Let-7 family miRNAs have been shown to induce apoptosis via targeting of Bcl-xl, among other mechanisms.35 Likewise, our data showed that a low dose (5 nM) of biologic let-7c prodrug induced a modest yet robust increase in early and late apoptotic cell populations in Huh7 cells (Figure 3), compared with either vehicle or MSA control treatments. Additionally, necrotic cell populations were not altered following the transfection of let-7c and MSA.
Figure 3.

Apoptotic Cell Death Is Significantly Induced by Bioengineered let-7c Agent
Huh7 cells were transfected with 5 nM Lipofectamine 3000-carried MSA or let-7c for 48 h, stained with propidium iodide and Annexin V-FITC, and counted by a flow cytometer with a total cell gate of 10,000 events. A significant shift of the total population toward early and late apoptotic cells was observed, whereas total necrotic population showed no difference. Values are mean ± SD (n = 3 per group). *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA with Bonferroni’s post hoc test).
Biologic let-7c Suppresses HCC Cell Stemness
Negative feedback between let-7 and LIN28 influences the stemness of cancer cells,33 which can have important impact on therapeutic efficacy. As such, we evaluated cancer stem cell (CSC) growth using a tumorsphere assay in Huh7 cells. Following transfection in adherent conditions and subsequent growth in ultra-low-attachment, serum-free conditions, we observed a significant half-diameter reduction in primary tumorsphere size, but not tumorsphere count, in let-7c-treated cells (Figure 4). Upon subsequent dissociation, transfection, and growth in ultra-low-attachment, serum-free conditions to form secondary tumorspheres, a similar 50% reduction in diameter was observed in biologic let-7c-treated cells. Although sphere formation efficiency from primary tumorspheres was not significantly reduced, sphere count and individual cell number were significantly reduced in secondary tumorspheres by let-7c treatment (Figure 4).
Figure 4.

Bioengineered let-7c Prodrug Sharply Reduces Tumorsphere Growth
Following transfection with Lipofectamine 3000-formulated MSA or let-7c in adherent conditions, an equal number of Huh7 cells were grown in serum-free, ultra-low-attachment conditions for 7 days to yield primary tumorspheres. Primary tumorspheres were then digested to single cell, transfected again, and grown for another 7 days in serum-free, ultra-low attachment conditions to yield secondary tumorspheres. let-7c treatment resulted in smaller primary and secondary tumorspheres. Values are mean ± SD (n = 3 per group). *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA with Bonferroni’s post hoc test).
Preparation and Characterization of Biologic let-7c-Loaded LPP Nanocomplex
We thus employed LPP to load bioengineered let-7c molecule (Figure 5A) toward therapy study in animal models because our recent study demonstrated the effectiveness of LPP to deliver biologic RNA molecules to liver tissues.32 The size of biologic let-7c-loaded LPP was 98.35 ± 5.11 nm with a zeta potential value of 43.9 ± 2.2 mV, which was complemented by transmission electron microscopy (TEM) examination (Figure 5B). Control MSA was formulated in the same manner, and LPP/MSA nanocomplex showed similar size (102.4 ± 5.9 nm) and zeta potential (45.1 ± 1.2 mV) (Figure S2). In addition, LPP could effectively protect bioengineered let-7c agent from degradation in both FBS and human serum up to 6 h, whereas modest degradation had occurred by 24 h (Figures S3A and S3B).
Figure 5.

Biologic let-7c Is Efficiently Delivered into HCC Cells by LPP Nanocomplex to Control Cell Proliferation
(A) Schematic illustration of let-7c-loaded lipopolyplex (LPP). (B) TEM image of let-7c-loaded LPP (indicated by arrows) nanocomplex, as well as the size and zeta potential measured by dynamic light scattering. Scale bar indicates 500 nm. (C) Efficient delivery of let-7c (15 nM) into Huh7 cells led to (D) sharp suppression of cell growth. Lipofectamine 3000 (LF3000) treatments were used for comparison. Values are mean ± SD of triplicate treatments (n = 3 per group). **p < 0.01; ***p < 0.001 (one- or two-way ANOVA with Bonferroni’s post hoc test).
LPP Efficiently Delivers Biologic let-7c Prodrug into HCC Cells to Elicit Inhibition of Cell Growth
We further assessed in vitro delivery efficiency by LPP in Huh7 cells, in parallel to Lipofectamine 3000 (LF3000). Our data demonstrated that biologic let-7c prodrug was efficiently delivered into Huh7 cells by LPP nanocomplex, as manifested by the increase in comparable levels of let-7c as those delivered by LF3000 (Figure 5C), which led to a sharp suppression of cell proliferation (Figure 5D). These data were also complemented by efficient delivery of another bioengineered ncRNA molecule, GFP-siRNA,20, 21 by LPP nanoparticles, as indicated by the knockdown of target GFP levels in GFP/luciferase-expressing Huh7 cells (Figure S4).
Bioengineered let-7c Prodrug Significantly Reduces HCC Tumor Progression in Orthotopic Xenograft Mouse Models and Is Well Tolerated
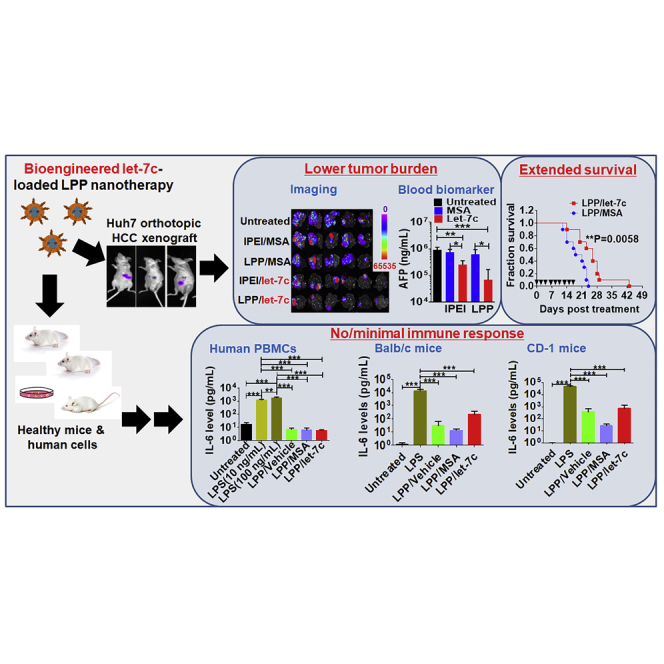
We thus established orthotopic HCC xenograft mouse models with luciferase/GFP-expressing Huh7 cells to investigate the efficacy of let-7c therapy (Figure 6A). As revealed by bioluminescent imaging in live animals (Figure 6B), HCC tumor burden was inhibited by approximately 50% by both i.v. administered LPP- and in vivo-jetPEI (IPEI)-formulated let-7c prodrug, compared with untreated mice, whereas control MSA had no impact. Ex vivo imaging of liver tumoral GFP signals (Figure 6C) further demonstrated the effectiveness of let-7c for the control of HCC, which was reduced over 70% by LPP/let-7c and around 33% by IPEI/let-7c as compared with untreated mice. Suppression of HCC was associated with higher levels of let-7c in both healthy livers and tumors isolated from let-7c-treated mice (Figure 6D). Moreover, the efficacy of biologic let-7c therapy in the inhibition of orthotopic HCC was supported by significantly lower serum α-fetoprotein (AFP) levels in let-7c-treated mice (Figure 6E), as well as less HCC tumors in let-7c-treated mouse liver slices with variable degrees of necrosis determined by histopathological analyses (Figure 6F). In addition, immunohistochemistry (IHC) studies on HCC xenograft tissues demonstrated that, although there is no significant difference in the level of cell proliferation (i.e., Ki-67) among various treatments, biologic let-7c prodrug therapy, especially the LPP formulation, led to an obvious higher degree of apoptosis (i.e., active-caspase-3), as compared with MSA control (Figure S5). It is also noteworthy that LPP/let-7c nanotherapeutics was efficient in the control of HCC, as indicated by a more ubiquitous and significantly greater degree of reduction of tumor burden determined by multiple measurements (Figure 6).
Figure 6.

Systemic Administration of LPP/let-7c Nanotherapeutics Largely Reduces Tumor Growth in Orthotopic HCC Huh7 Xenograft Mouse Models
(A) Timeline of establishment of HCC xenograft mouse models and drug treatment. (B) Suppression of orthotopic HCC progression by LPP/let-7c was demonstrated by live animal imaging of luciferase bioluminescent signals. In vivo-jetPEI (IPEI)-formulated biologic let-7c and MSA were used for comparison. (C) Ex vivo GFP fluorescence images of HCC-bearing livers further demonstrated the effectiveness of bioengineered let-7c prodrug, which was associated with high levels of tumoral let-7c (D). (E) Serum AFP levels were significantly reduced in let-7c-treated mice. (F) Representative H&E staining images (×40) of tumor-bearing liver tissues and quantitative measurement of the percentage of tumors in corresponding liver slices. Areas circled in blue lines are necrotic areas, and the red lines are applied to distinguish tumors from healthy liver (L) tissues. Values are mean ± SD (n = 4–6 in each group). *p < 0.05; ***p < 0.001 (one- or two-way ANOVA with Bonferroni’s post hoc test).
All treatments were well tolerated as body weights of all mice showed steady increases over time (Figure S6A). To further investigate the safety of bioengineered let-7c prodrug, blood biochemistry profiles were determined (Figures S6B–S6F). Biomarkers of hepatic and renal functions including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine were all within the normal ranges. To our surprise, blood total bilirubin levels in untreated and MSA-treated mice were highly variable and inclined to be above normal range, whereas they retained within normal range in biologic let-7c-treated mice, which may be another indication of the effectiveness of let-7c prodrug therapy in the control of HCC.
LPP/let-7c Nanotherapeutics Significantly Improves the Overall Survival of Orthotopic HCC Tumor-Bearing Mice
A separate cohort of orthotopic HCC Huh7 xenograft mice was established and employed to define the magnitude of benefit of i.v. administered LPP/let-7c nanotherapeutics on overall survival. After the development of HCC was confirmed by quantitative bioluminescence imaging of live mice, subjects showing the same degrees of tumor burden were randomized for LPP/let-7c and control LPP/MSA treatments (Figure 7A). Survival analysis showed that, compared with LPP/MSA, LPP/let-7c therapy significantly improved overall survival of HCC tumor-bearing mice (Figure 7B). This was also indicated by a longer median survival of LPP/let-7c-treated mice (26.0 days) than LPP/MSA controls (19.5 days). In agreement with the safety profiles of let-7c treatment in the other therapy study (Figure S6), LPP/let-7c treatment did not alter mouse body weights compared with LPP/MSA (Figure 7C).
Figure 7.

LPP/let-7c Nanotherapeutics Significantly Improves Overall Survival of Orthotopic HCC Xenograft Tumor-Bearing Mice
(A) Bioluminescence images of HCC tumor-bearing animals before the i.v. treatment with LPP/let-7c and control LPP/MSA, and quantitative measurement of bioluminescent intensities. (B) Survival analysis showed that LPP/let-7c-treated mice lived much longer than the control (**p < 0.01; n = 10 per group; log rank [Mantel-Cox] test). The median survival was 26.0 days for LPP/let-7c-treated mice and 19.5 days for LPP/MSA-treated animals. (C) Mouse body weights during the treatment. ▼ indicates days on which mice received treatments.
LPP/let-7c Produces No or Minimal Immunogenicity in Human PBMCs and Immunocompetent Mice
Lastly, we assessed whether LPP/let-7c nanotherapeutics induces any immune response in human PBMCs and two different strains of healthy immunocompetent mice (BALB/c and CD-1). As expected, LPS treatment provoked a robust cytokine release in both human PBMCs (Figure 8A) and BALB/c (Figure 8B) and CD-1 (Figure 8C) mice, as indicated by a significantly sharp 1,000- to 30,000-fold elevation of interleukin (IL)-6 levels, as well as increase in tumor necrosis factor alpha (TNF-α) and IL-10 levels. By contrast, LPP/let-7c treatment did not alter the levels of IL-6, IL-4, or IL-10 in human PBMCs, whereas it slightly increased TNF-α levels, which is not statistically significantly different from untreated cells. Although LPP/vehicle, LPP/MSA, and LPP/let-7c all caused a mild increase in serum IL-6 levels in BALB/c and CD-1 mice, as compared with untreated mice, the elevated IL-6 levels were two to three orders of magnitude lower than those induced by LPS, indicating that LPP/let-7c shows minimal immunogenicity compared with LPS.
Figure 8.

Safety of LPP/let-7c Nanotherapeutics
LPP/let-7c has no or limited impact on cytokine release in human PBMCs (A) and two stains of immunocompetent mice (B and C). LPS was used as positive control to induce cytokine release storm, whereas untreated and LPP vehicle-treated mice or cells were considered as negative controls. Values are mean ± SD. For BALB/c and CD-1 mice, three females and three males were included in each group (n = 6). Blood was harvested 1 h after i.v. administration of formulated RNA molecules, and serum samples were prepared for cytokine measurement. For human PBMCs, each treatment was conducted in triplicate (n = 3), and cell culture medium was collected 24 h post-treatment. **p < 0.01; ***p < 0.001 (two-way ANOVA with Bonferroni’s post hoc test). n.d., non-detectable.
Discussion
miRNA replacement therapy represents a promising strategy for the control of tumor progression given the loss of expression of tumor-suppressive miRNAs in cancerous cells, including miR-122 and let-7 family miRNAs (e.g., let-7c and let-7a) in human HCC samples.35, 39, 40 However, due to the complexity in dysregulation of miRNAs, as well as other regulatory factors and pathways, reintroduction of functional miRNAs may not necessarily coincide with optimal efficacy. As such, although miR-122 is the most abundant hepatic miRNA and is frequently attenuated in HCC,41 we found that bioengineered let-7c showed the highest antiproliferative activity against HCC cells among a small collection of ncRNA agents including miR-122. Although the in vivo efficacy of other miRNAs is not compared with let-7c herein, this screening method is predictive of potential benefits of let-7c in relieving in vivo HCC tumor burden and improving overall survival revealed in this study.
Current miRNA research and drug development primarily uses synthetic miRNA mimics, which carry extensive and variable chemical modifications expected to improve systemic stability.19 Although these miRNA agents are assumed to retain the same “sequences” because of the presence of identical nucleobases, they are chemically different molecules and inevitably have distinct higher-order structures, as well as physiochemical and biological activities. Moreover, synthetic RNA agents pose risk for the induction of cytokine release syndrome.42, 43 This is also in contrast to protein research and therapy, wherein recombinant or bioengineered proteins expressed in living cells have been preferably utilized instead of synthetic polypeptides and proteins. Bioengineered miRNA molecules presented herein represent a novel class of biologic miRNA agents, which are folded and tolerated in living cells, and thus may better capture the properties of cellular RNA macromolecules.19, 22 With minimal natural modifications and exhibiting favorable stability in human cells,25, 44 recombinant miRNA agents are selectively processed to target mature miRNAs that rewrite cellular miRNome profile and execute regulatory functions.21
The pleiotropic nature of miRNA-controlled gene regulation behind cancer cellular processes warrants extensive validation. The interplay between LIN28 and let-7 family miRNAs33, 45 is a critical component in the regulation of pluripotency as well as HCC and other liver diseases.46 LIN28, shown to be upregulated in stem-like cells, can reprogram cells into an undifferentiated state,47 and thus LIN28 may be a druggable target for the suppression of CSCs and tumor initiation. By contrast, LIN28-regulatory let-7 family miRNAs shown to inhibit pluripotency and favor differentiation may be employed to manage CSC maintenance and replication.48, 49 This study demonstrated a consistent action of bioengineered let-7c prodrug in the inhibition of tumorsphere growth, which is likely attributable to the strong suppression of LIN28B expression in Huh7 cells that is consistently shown in Hep3B cells as well. Moreover, induction of apoptosis is a common mechanism of antineoplastic agents, and resistance to apoptosis is a common feature of CSCs. let-7 family miRNAs have also been shown to either induce or sensitize cells to apoptosis via attenuation of anti-apoptotic proteins, including Bcl-xl.35 In this study, we found the suppression of Bcl-xl expression by biologic let-7c in Huh7 cells, which is consistent with the induction of apoptotic, but not necrotic, cell populations by a low dose of let-7c prodrug. It is also interesting to observe variable effects of let-7c on different targets (e.g., c-MYC versus LIN28B) in the same cell line and/or the same target (e.g., Bcl-xl) in different cell lines (e.g., Huh7 versus Hep3B). This is understandable because a specific gene is often regulated by multiple regulatory mechanisms; among them, miRNA-controlled posttranscriptional regulation may have certain degree of contribution to that gene. In addition, genotypes and regulatory signaling pathways, as well as miRNA machinery, may be variable in different HCC cell lines, highlighting the challenges in molecular therapy and demands for precision medication.
RNA drugs for systemic administration currently under clinical investigation are mainly delivered by lipid-based systems (e.g., liposomes), given their excellent biocompatibility and favorable lipid composition.50, 51, 52 For example, a phase I trial is under way to evaluate a small activating, double-stranded RNA targeting the transcription factor C/EBP-α formulated in SMARTICLES liposomal nanoparticle for advanced HCC (https://clinicaltrials.gov/ct2/show/NCT02716012). Among lipid-based delivery systems, LPPs convey the favorable properties of both liposomes and polyplexes.28, 29, 53 Our recent studies have demonstrated that IPEI is able to deliver biologic RNAs to livers to achieve target gene knockdown,20 as well as tumor tissues to control disease.21, 23, 26, 54 In the present study, we identified a more favorable serum stability of let-7c formulated in LPP nanocomplex, which was improved to even greater degrees as compared with IPEI,32 owing to the outer PEGylated lipid coating of polyplex. As a result, LPP showed high in vitro delivery efficiency in HCC cell lines. Most notably, LPP/let-7c provided a significantly greater extent of suppression of orthotopic HCC tumor burden in vivo, consistently supported by multiple independent endpoints including ex vivo GFP reporter intensity, serum AFP level, and histological tumor area. In addition, we revealed that LPP/let-7c nanotherapeutics significantly improved the median survival of orthotopic HCC mice by 6.5 days. Although modest, the HCC xenograft used in this study is incredibly aggressive, occupying more than 50% of total liver tissue after 2 weeks.
Consistent with previous findings,25, 26 this study demonstrated that bioengineered RNAs were well tolerated in HCC tumor-bearing immunodeficient mice and caused no or minimal degree of cytokine release in immunocompetent mice. Interestingly, serum bilirubin level, an indicator of liver damage, fell within normal range in let-7c-treated mice only. This is likely attributable to the effectiveness of let-7c therapy in the control of HCC tumor growth, suppressing further liver damage, and highlighting the aggressive nature of this HCC model.55 Moreover, the present study showed for the first time that bioengineered RNAs show minimal immunogenicity in human PBMCs, an addition to the safety profile of recombinant miRNA molecules that are produced and folded in living cells.
In conclusion, we have demonstrated the efficacy of LPP/let-7c nanotherapeutics in an aggressive orthotopic HCC tumor mouse model, showing marginal immunogenicity in mice and human PBMCs. The first-in-class biologic let-7c prodrug was identified as the most potent among a small set of miRNAs in inhibiting HCC cell viability via interference of specific targets and critical cellular functions. Our findings suggest that LPP-formulated biologic let-7c may serve as an effective and safe treatment for HCC, warranting clinical verification.
Materials and Methods
Materials
In vivo-jetPEI (linear 22-kDa PEI; in vivo-jetPEI polyethylenimine [lPEI]) was purchased from Polyplus Transfection (Illkirch, France). Branched PEI with molecular weight 10,000 Da (bPEI10k) was bought from Alfa Aesar (Wardhill, MA, USA). 1,2-Di-O-octadecenyl-3-trimethylammonium propane (DOTMA) and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL, USA). 1,2-Dimyristoyl-sn-glycerol, methoxypolyethylene glycol (DMG-PEG2000) was purchased from NOF America Corporation (White Plains, NY, USA). LF3000, TRIzol RNA isolation reagent, and bicinchoninic acid assay (BCA) protein assay kit were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Direct-zol RNA MiniPrep Kit was from Zymo Research (Irvine, CA, USA). Celltiter-Glo assay was purchased from Promega (Madison, WI, USA). Matrigel was purchased from Corning (Corning, NY, USA). Human AFP ELISA kit was purchased from R&D Systems (San Diego, CA, USA). All other chemicals and organic solvents of analytical grade were purchased from Sigma-Aldrich or Thermo Fisher Scientific.
Cell Culture
Huh7 cells were purchased from the Japanese Collection of Research Bioresources and grown in DMEM (GIBCO, Grand Island, NY, USA), and Hep3B and Sk-Hep-1 cells were obtained from American Type Culture Collection and grown in Eagle’s minimal essential medium (Cellgro, Manassas, VA, USA). All of the cell lines were supplemented with 10% fetal bovine serum (GIBCO, Grand Island, NY, USA) and 1% antibiotic/antimycotic (Cellgro, Manassas, VA, USA). GFP/luciferase-expressing cell lines were established after transduction of parental cells with pCCLc-Luc-EGFP lentiviral constructs (Vector Core; UC Davis Medical Center, Sacramento, CA, USA). All cells were transfected by using the LF3000 (Invitrogen, Carlsbad, CA, USA) per the manufacturer’s instructions for in vitro RNA delivery, unless otherwise indicated.
Production of Recombinant miRNA Agents
Bioengineering of miRNA molecules was conducted as described recently.21 In brief, inserts encoding target miRNA- or siRNA-containing ncRNAs were cloned into a pBSMrna vector using In-Fusion cloning technology and transformed into HST08 Escherichia coli. Recombinant ncRNAs were purified by anion exchange FPLC21, 24 to >96% purity as determined by a HPLC assay.25 Bioengineered let-7c less than 95% pure was re-purified by the same FPLC method to reach >96% homogeneity (Figure S1). Endotoxin activity was examined with Pyrogent-5000 kinetic LAL assay (Lonza, Walkersville, MD, USA).
Cell Viability Assay
GFP/luciferase-expressing Huh7 cells (5,000 cells/well) were seeded in 96-well plates and grown overnight. Biologic let-7c or MSA was administered in triplicate with either LF3000 or LPP. Cell viability was measured using Cell Titer-Glo kit. Inhibition of cell viability was determined as relative to vehicle control (0% inhibition), and pharmacodynamics parameters were estimated by fitting the data to a normalized dose-response equation with variable slope:
Given low efficacy, MSA (Emax, Emin = 100, 20.64%) and miR-144 (Emax, Emin = 44.42, 17.27%) in Huh7 cells were best fit to the full dose-response equation:
Immunoblot and Immunofluorescence Analyses
Huh7 and Hep3B cells were seeded in a six-well plate at 300,000 cells/well and transfected with 15 nM RNA. After 72 h, cells were harvested and lysed in radio immunoprecipitation assay (RIPA) buffer with protease inhibitor (Pierce, Rockford, IL, USA). Protein levels were determined by BCA assay (Pierce, Rockford, IL, USA). After separation on a 12% SDS-PAGE gel (Bio-Rad, Hercules, CA, USA), proteins were transferred onto a polyvinylidene difluoride membrane and blocked in 5% milk/0.1% Tween 20 in Tris-buffered saline. Total immobilized protein was imaged per the manufacturer’s instructions. Membranes were incubated with primary antibodies (Bcl-xl rabbit monoclonal antibody [mAb] [CST 2764], c-Myc rabbit mAb [CST 13987], and LIN28B rabbit mAb [CST 11965] from Cell Signaling; ARID3B rabbit polyclonal antibody [pAb] [AB 92328] from Abcam) overnight at 4°C at 1:1,000 dilution in 5% BSA in TBS-T, followed by horseradish peroxidase-conjugated goat-anti-rabbit secondary antibody (1:10,000 dilution [111-035-003] from Jackson Immunoresearch) for 2 h at room temperature prior to chemiluminescent imaging with Clarity ECL (Bio-Rad, Hercules, CA, USA). Relative band intensity was normalized to total immobilized protein.
To assay HMGA2 expression, Huh7 and Hep3B cells were grown on glass chamber slides and transfected with MSA or let-7c. After 72 h, cells were fixed with 10% formalin, permeabilized with 0.3% Triton X-100, and incubated with HMGA2 rabbit mAb (CST 8179) (1:400 dilution in 5% BSA) overnight at 4°C. Antigen was detected with Alexa 488-conjugated anti-rabbit IgG Fab fragment (CST 4412), and nuclei were counterstained with DAPI (CST 4803).
Flow Cytometry
Huh7 cells were plated in six-well plates at a density of 150,000 cells/well and transfected with 5 nM RNA. After 48 h, cells were stained with propidium iodide (PI) and Annexin V-FITC (fluorescein isothiocyanate) per manufacturer’s instructions (Trevigen, Gaithersburg, MD, USA). Cell count and fluorophore intensity were measured using a BD Biosciences Fortessa 20 color cytometer. Total event count was gated at 10,000 events, and quadrant gating was set relative to vehicle control. Healthy cells were defined by low PI/low Annexin V staining, early apoptotic cells were defined by low PI/high Annexin V, late apoptotic cells were defined by high PI/high Annexin V, and necrotic cells were defined by high PI/low Annexin V intensity.
Tumorsphere Assay
Huh7 cells were seeded under adherent conditions at a density of 300,000 cells/well in six-well plates and transfected with 15 nM RNA. After 48 h, live cells were transferred to 24-well ultra-low-attachment plates (Corning, Kennebunk, ME, USA) at a density of 2,500 cells/well and grown in DMEM/F12+B27 with penicillin/streptomycin, GlutaMAX (GIBCO, Grand Island, NY, USA), 20 ng/mL human epidermal growth factor, and 10 ng/mL human basic fibroblast growth factor (PeproTech, Rocky Hill, NJ, USA). After 7 days, primary tumorspheres (>10 μm diameter) were counted, and sphere diameter was measured in ImageJ and dissociated with trypsin to a single cell. After all cells were transferred to new wells in ultra-low-attachment, serum-free conditions, cells were transfected again with 15 nM RNA. After 7 days, secondary tumorspheres were again counted, and diameter was measured and dissociated to count individual cells. Sphere formation efficiency (%) was calculated relative to total single cells seeded from the previous generation.
Formulation and Characterization of LPP Nanocomplex
Details of formulation and characterization of LPP nanocomplex, as well as serum stability and in vitro delivery efficiency, are provided in the Supplemental Methods and as previously described.29
Therapy Studies in Orthotopic HCC Xenograft Mouse Models
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of California, Davis.
Establishment of Orthotopic HCC Xenograft Mouse Model
Luciferase/GFP-expressing Huh7 cells were mixed with Matrigel to a final concentration of 1 × 108 cells/mL. Four-week-old male athymic nude mice (Jackson Laboratory, Bar Harbor, ME, USA) were anesthetized, and an incision (∼1 cm) along the linea alba in the midline of the abdominal muscle layer was made. 20 μL of Huh7 cells in Matrigel suspension (2 × 106 cells) was injected into the left lobe of liver. Successful engraftment of Huh7 cells was confirmed by bioluminescent imaging using ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA), following the intraperitoneal injection of D-luciferin (150 mg/kg) (BioVision, Milpitas, CA, USA).
Tumor Progression Study
One week post-inoculation, mice were assigned into five groups (untreated, lPEI/MSA, LPP/MSA, lPEI/let-7c, and LPP/let-7c) according to tumor sizes determined by in vivo bioluminescence imaging, and treated i.v. (40 μg RNA) three times per week through tail vein injection. Mice were imaged once per week to monitor tumor growth. Mice were sacrificed 2 h after the last dose on day 15. Livers with engrafted tumors were harvested and imaged for GFP fluorescence using ChemiDoc MP Imaging System. RNA was extracted from healthy livers and HCC tumors, and the levels of let-7c were quantified using stem loop qRT-PCR using gene-selective primers (Table S1). Blood was collected for blood chemistry profiling (Comparative Pathology Laboratory, UC Davis), and serum AFP was examined by ELISA (R&D Systems, Minneapolis, MN, USA). H&E histopathological study was performed by the Clinical Immunohistochemistry Laboratory at Roswell Park Cancer Institute (Buffalo, NY, USA).
Survival Study
One week post-inoculation, a separate cohort of tumor-bearing mice was assigned into two groups (LPP/MSA and LPP/let-7c) according to tumor sizes determined by bioluminescence imaging, and treated i.v. with 40 μg RNA three times per week for 3 weeks. Body weights were recorded twice a week to assess animal health. Survival was analyzed by Kaplan-Meier method and compared by log rank (Mantel-Cox) test.
Induction of Cytokine Release
Human PBMCs were purchased from Lonza (Walkerville, MD, USA) and maintained in RPMI 1640 supplemented with 10% human AB serum (Sigma, St. Louis, MO, USA). PBMCs were seeded onto a 96-well plate at a density of 2 × 105 cells/well and allowed to grow overnight. Cells were treated with 10 ng/mL or 100 ng/mL LPS (positive control), LPP/MSA (5 nM), LPP/let-7c (5 nM), or LPP vehicle. Twenty-four hours post-treatment, medium was harvested and cell debris was removed by centrifugation. IL-6, TNF-α, IL-4, and IL-10 levels were quantified using corresponding human cytokine ELISA assay kit (Invitrogen, Carlsbad, CA, USA).
Healthy BALB/c and CD-1 mice (5–6 weeks old) were randomly assigned into different groups (three females and three males per group) and injected i.v. with 40 μg of either MSA or let-7c-loaded LPP nanocomplex, 20 μg of LPS (positive control), or LPP vehicle (negative control). Blood was collected 1 h post-treatment, and serum IL-6 and IL-4 levels were quantified using a mouse IL-6 and IL-4 ELISA assay kit (Invitrogen, Carlsbad, CA, USA).
Statistical Analysis
All values are mean ± SD. Statistical analysis was performed using one- or two-way ANOVA with Bonferroni’s post hoc test or Student’s t test where appropriate (GraphPad Prism, San Diego, CA, USA).
Author Contributions
Conceptualization, A.-M.Y., J.L.J., Q.-Y.Z., and M.-J.T.; Methodology, J.L.J., Q.-Y.Z., M.-J.T., P.Y.H., Z.D., and A.-M.Y.; Resources, A.-M.Y., J.L.J., Q.-Y.Z., P.Y.H., and M.-J.T.; Investigation, J.L.J., Q.-Y.Z., P.Y.H., M.-J.T., and J.-X.Q.; Writing, J.L.J., Q.-Y.Z., M.-J.T., A.-M.Y., and all other authors; Visualization, J.L.J., Q.-Y.Z., J.-X.Q., and A.-M.Y.; Funding Acquisition, A.-M.Y.; Supervision, A.-M.Y.
Acknowledgments
This study was supported by the National Institute of General Medical Sciences (grant R01GM113888 to A.-M.Y.) and in part by the National Cancer Institute (grants U01CA175315 and R01CA225958 to A.-M.Y.) of the NIH. J.L.J. was supported by an NIGMS-funded Pharmacology Training Program grant (T32GM099608). The authors appreciate the access to the Flow Cytometry and Molecular Pharmacology Shared Resources funded by the UC Davis Comprehensive Cancer Center Support Grant (CCSG) awarded by the National Cancer Institute (P30CA093373).
Footnotes
Supplemental Information includes Supplemental Methods, one table, and six figures and can be found with this article online at https://doi.org/10.1016/j.omtn.2019.01.007.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Kim N.G., Nguyen P.P., Dang H., Kumari R., Garcia G., Esquivel C.O., Nguyen M.H. Temporal trends in disease presentation and survival of patients with hepatocellular carcinoma: a real-world experience from 1998 to 2015. Cancer. 2018;124:2588–2598. doi: 10.1002/cncr.31373. [DOI] [PubMed] [Google Scholar]
- 3.Iñarrairaegui M., Melero I., Sangro B. Immunotherapy of hepatocellular carcinoma: facts and hopes. Clin. Cancer Res. 2018;24:1518–1524. doi: 10.1158/1078-0432.CCR-17-0289. [DOI] [PubMed] [Google Scholar]
- 4.Bruix J., Qin S., Merle P., Granito A., Huang Y.H., Bodoky G., Pracht M., Yokosuka O., Rosmorduc O., Breder V., RESORCE Investigators Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. doi: 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- 5.Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F., de Oliveira A.C., Santoro A., Raoul J.L., Forner A., SHARP Investigators Study Group Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 6.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 7.Bader A.G., Brown D., Winkler M. The promise of microRNA replacement therapy. Cancer Res. 2010;70:7027–7030. doi: 10.1158/0008-5472.CAN-10-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klingenberg M., Matsuda A., Diederichs S., Patel T. Non-coding RNA in hepatocellular carcinoma: mechanisms, biomarkers and therapeutic targets. J. Hepatol. 2017;67:603–618. doi: 10.1016/j.jhep.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Kota J., Chivukula R.R., O’Donnell K.A., Wentzel E.A., Montgomery C.L., Hwang H.W., Chang T.C., Vivekanandan P., Torbenson M., Clark K.R. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 11.Yu A.M., Tian Y., Tu M.J., Ho P.Y., Jilek J.L. MicroRNA pharmacoepigenetics: posttranscriptional regulation mechanisms behind variable drug disposition and strategy to develop more effective therapy. Drug Metab. Dispos. 2016;44:308–319. doi: 10.1124/dmd.115.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie M., Ma L., Xu T., Pan Y., Wang Q., Wei Y., Shu Y. Potential regulatory roles of microRNAs and long noncoding RNAs in anticancer therapies. Mol. Ther. Nucleic Acids. 2018;13:233–243. doi: 10.1016/j.omtn.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callegari E., D’Abundo L., Guerriero P., Simioni C., Elamin B.K., Russo M., Cani A., Bassi C., Zagatti B., Giacomelli L. miR-199a-3p modulates MTOR and PAK4 pathways and inhibits tumor growth in a hepatocellular carcinoma transgenic mouse model. Mol. Ther. Nucleic Acids. 2018;11:485–493. doi: 10.1016/j.omtn.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu X., Rivera A., Tao L., De Geest B., Zhang X. Construction of an oncolytic herpes simplex virus that precisely targets hepatocellular carcinoma cells. Mol. Ther. 2012;20:339–346. doi: 10.1038/mt.2011.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandbothe M., Buurman R., Reich N., Greiwe L., Vajen B., Gürlevik E., Schäffer V., Eilers M., Kühnel F., Vaquero A. The microRNA-449 family inhibits TGF-β-mediated liver cancer cell migration by targeting SOX4. J. Hepatol. 2017;66:1012–1021. doi: 10.1016/j.jhep.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Wu H., Tao J., Li X., Zhang T., Zhao L., Wang Y., Zhang L., Xiong J., Zeng Z., Zhan N. MicroRNA-206 prevents the pathogenesis of hepatocellular carcinoma by modulating expression of met proto-oncogene and cyclin-dependent kinase 6 in mice. Hepatology. 2017;66:1952–1967. doi: 10.1002/hep.29374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J., Yang Y., Yang T., Yuan S., Wang R., Pan Z., Yang Y., Huang G., Gu F., Jiang B. Double-negative feedback loop between microRNA-422a and forkhead box (FOX)G1/Q1/E1 regulates hepatocellular carcinoma tumor growth and metastasis. Hepatology. 2015;61:561–573. doi: 10.1002/hep.27491. [DOI] [PubMed] [Google Scholar]
- 18.Lu Y., Yue X., Cui Y., Zhang J., Wang K. MicroRNA-124 suppresses growth of human hepatocellular carcinoma by targeting STAT3. Biochem. Biophys. Res. Commun. 2013;441:873–879. doi: 10.1016/j.bbrc.2013.10.157. [DOI] [PubMed] [Google Scholar]
- 19.Yu, A.M., Jian, C., Yu, A.H., and Tu, M.J. RNA therapy: are we using the right molecules? Pharmacol. Ther. Published online December 4, 2018. 10.1016/j.pharmthera.2018.11.011. [DOI] [PMC free article] [PubMed]
- 20.Chen Q.X., Wang W.P., Zeng S., Urayama S., Yu A.M. A general approach to high-yield biosynthesis of chimeric RNAs bearing various types of functional small RNAs for broad applications. Nucleic Acids Res. 2015;43:3857–3869. doi: 10.1093/nar/gkv228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho P.Y., Duan Z., Batra N., Jilek J.L., Tu M.J., Qiu J.X., Hu Z., Wun T., Lara P.N., DeVere White R.W. Bioengineered noncoding RNAs selectively change cellular miRNome profiles for cancer therapy. J. Pharmacol. Exp. Ther. 2018;365:494–506. doi: 10.1124/jpet.118.247775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho P.Y., Yu A.M. Bioengineering of noncoding RNAs for research agents and therapeutics. Wiley Interdiscip. Rev. RNA. 2016;7:186–197. doi: 10.1002/wrna.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jian C., Tu M.J., Ho P.Y., Duan Z., Zhang Q., Qiu J.X., DeVere White R.W., Wun T., Lara P.N., Lam K.S. Co-targeting of DNA, RNA, and protein molecules provides optimal outcomes for treating osteosarcoma and pulmonary metastasis in spontaneous and experimental metastasis mouse models. Oncotarget. 2017;8:30742–30755. doi: 10.18632/oncotarget.16372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li P.C., Tu M.J., Ho P.Y., Jilek J.L., Duan Z., Zhang Q.Y., Yu A.X., Yu A.M. Bioengineered NRF2-siRNA is effective to interfere with NRF2 pathways and improve chemosensitivity of human cancer cells. Drug Metab. Dispos. 2018;46:2–10. doi: 10.1124/dmd.117.078741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W.P., Ho P.Y., Chen Q.X., Addepalli B., Limbach P.A., Li M.M., Wu W.J., Jilek J.L., Qiu J.X., Zhang H.J. Bioengineering novel chimeric microRNA-34a for prodrug cancer therapy: high-yield expression and purification, and structural and functional characterization. J. Pharmacol. Exp. Ther. 2015;354:131–141. doi: 10.1124/jpet.115.225631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tu M.J., Ho P.Y., Zhang Q.Y., Jian C., Qiu J.X., Kim E.J., Bold R.J., Gonzalez F.J., Bi H., Yu A.M. Bioengineered miRNA-1291 prodrug therapy in pancreatic cancer cells and patient-derived xenograft mouse models. Cancer Lett. 2019;442:82–90. doi: 10.1016/j.canlet.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv H., Zhang S., Wang B., Cui S., Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 28.Rezaee M., Oskuee R.K., Nassirli H., Malaekeh-Nikouei B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J. Control. Release. 2016;236:1–14. doi: 10.1016/j.jconrel.2016.06.023. [DOI] [PubMed] [Google Scholar]
- 29.Ewe A., Panchal O., Pinnapireddy S.R., Bakowsky U., Przybylski S., Temme A., Aigner A. Liposome-polyethylenimine complexes (DPPC-PEI lipopolyplexes) for therapeutic siRNA delivery in vivo. Nanomedicine (Lond.) 2017;13:209–218. doi: 10.1016/j.nano.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Ewe A., Schaper A., Barnert S., Schubert R., Temme A., Bakowsky U., Aigner A. Storage stability of optimal liposome-polyethylenimine complexes (lipopolyplexes) for DNA or siRNA delivery. Acta Biomater. 2014;10:2663–2673. doi: 10.1016/j.actbio.2014.02.037. [DOI] [PubMed] [Google Scholar]
- 31.Schäfer J., Höbel S., Bakowsky U., Aigner A. Liposome-polyethylenimine complexes for enhanced DNA and siRNA delivery. Biomaterials. 2010;31:6892–6900. doi: 10.1016/j.biomaterials.2010.05.043. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Q.Y., Ho P.Y., Tu M.J., Jilek J.L., Chen Q.X., Zeng S., Yu A.M. Lipidation of polyethylenimine-based polyplex increases serum stability of bioengineered RNAi agents and offers more consistent tumoral gene knockdown in vivo. Int. J. Pharm. 2018;547:537–544. doi: 10.1016/j.ijpharm.2018.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nam Y., Chen C., Gregory R.I., Chou J.J., Sliz P. Molecular basis for interaction of let-7 microRNAs with Lin28. Cell. 2011;147:1080–1091. doi: 10.1016/j.cell.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao T.T., Hsu W.H., Ho C.H., Hwang W.L., Lan H.Y., Lo T., Chang C.C., Tai S.K., Yang M.H. let-7 modulates chromatin configuration and target gene repression through regulation of the ARID3B complex. Cell Rep. 2016;14:520–533. doi: 10.1016/j.celrep.2015.12.064. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu S., Takehara T., Hikita H., Kodama T., Miyagi T., Hosui A., Tatsumi T., Ishida H., Noda T., Nagano H. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J. Hepatol. 2010;52:698–704. doi: 10.1016/j.jhep.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 36.Sampson V.B., Rong N.H., Han J., Yang Q., Aris V., Soteropoulos P., Petrelli N.J., Dunn S.P., Krueger L.J. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–9770. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 37.Lee Y.S., Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayr C., Hemann M.T., Bartel D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gramantieri L., Ferracin M., Fornari F., Veronese A., Sabbioni S., Liu C.G., Calin G.A., Giovannini C., Ferrazzi E., Grazi G.L. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 40.Hou J., Lin L., Zhou W., Wang Z., Ding G., Dong Q., Qin L., Wu X., Zheng Y., Yang Y. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19:232–243. doi: 10.1016/j.ccr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Nakao K., Miyaaki H., Ichikawa T. Antitumor function of microRNA-122 against hepatocellular carcinoma. J. Gastroenterol. 2014;49:589–593. doi: 10.1007/s00535-014-0932-4. [DOI] [PubMed] [Google Scholar]
- 42.Robbins M., Judge A., MacLachlan I. siRNA and innate immunity. Oligonucleotides. 2009;19:89–102. doi: 10.1089/oli.2009.0180. [DOI] [PubMed] [Google Scholar]
- 43.Yu H., Wang Z., Sun G., Yu Y. Recognition of nucleic acid ligands by toll-like receptors 7/8: importance of chemical modification. Curr. Med. Chem. 2012;19:1365–1377. doi: 10.2174/092986712799462603. [DOI] [PubMed] [Google Scholar]
- 44.Li M.-M., Addepalli B., Tu M.-J., Chen Q.-X., Wang W.-P., Limbach P.A., LaSalle J.M., Zeng S., Huang M., Yu A.M. Chimeric microRNA-1291 biosynthesized efficiently in Escherichia coli is effective to reduce target gene expression in human carcinoma cells and improve chemosensitivity. Drug Metab. Dispos. 2015;43:1129–1136. doi: 10.1124/dmd.115.064493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo Y., Chen Y., Ito H., Watanabe A., Ge X., Kodama T., Aburatani H. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene. 2006;384:51–61. doi: 10.1016/j.gene.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 46.McDaniel K., Hall C., Sato K., Lairmore T., Marzioni M., Glaser S., Meng F., Alpini G. Lin28 and let-7: roles and regulation in liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2016;310:G757–G765. doi: 10.1152/ajpgi.00080.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 48.Reinhart B.J., Slack F.J., Basson M., Pasquinelli A.E., Bettinger J.C., Rougvie A.E., Horvitz H.R., Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 49.Viswanathan S.R., Daley G.Q. Lin28: a microRNA regulator with a macro role. Cell. 2010;140:445–449. doi: 10.1016/j.cell.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 50.Kim H.J., Kim A., Miyata K., Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 2016;104:61–77. doi: 10.1016/j.addr.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 51.Sullenger B.A., Nair S. From the RNA world to the clinic. Science. 2016;352:1417–1420. doi: 10.1126/science.aad8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xue H.Y., Guo P., Wen W.C., Wong H.L. Lipid-based nanocarriers for RNA delivery. Curr. Pharm. Des. 2015;21:3140–3147. doi: 10.2174/1381612821666150531164540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia Y., Tian J., Chen X. Effect of surface properties on liposomal siRNA delivery. Biomaterials. 2016;79:56–68. doi: 10.1016/j.biomaterials.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Y., Tu M.J., Yu Y.F., Wang W.P., Chen Q.X., Qiu J.X., Yu A.X., Yu A.M. Combination therapy with bioengineered miR-34a prodrug and doxorubicin synergistically suppresses osteosarcoma growth. Biochem. Pharmacol. 2015;98:602–613. doi: 10.1016/j.bcp.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carr B.I., Guerra V., Giannini E.G., Farinati F., Ciccarese F., Ludovico Rapaccini G., Di Marco M., Benvegnù L., Zoli M., Borzio F., Italian Liver Cancer (ITA.LI.CA) Group Association of abnormal plasma bilirubin with aggressive hepatocellular carcinoma phenotype. Semin. Oncol. 2014;41:252–258. doi: 10.1053/j.seminoncol.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.