

Abstract
RfaH, member of the NusG/Spt5 family, activates virulence genes in Gram-negative pathogens. RfaH exists in two states, with its C-terminal domain (CTD) folded either as α-helical hairpin or β-barrel. In free RfaH, the α-helical CTD interacts with, and masks the RNA polymerase binding site on, the N-terminal domain, autoinhibiting RfaH and restricting its recruitment to opsDNA sequences. Upon activation, the domains separate and the CTD refolds into the β-barrel, which recruits a ribosome, activating translation. Using NMR spectroscopy, we show that only a complete ops-paused transcription elongation complex activates RfaH, probably via a transient encounter complex, allowing the refolded CTD to bind ribosomal protein S10. We also demonstrate that upon release from the elongation complex, the CTD transforms back into the autoinhibitory α-state, resetting the cycle. Transformation-coupled autoinhibition allows RfaH to achieve high specificity and potent activation of gene expression.
The antitermination factor RfaH adopts two functional states where its C-terminal domain is folded either as an α-helical hairpin or β-barrel. Here the authors employ solution state NMR measurements to show that the C-terminal domain transforms into the β-barrel only upon binding to the elongation complex and refolds back after dissociation.
Introduction
Multi-subunit RNA polymerases (RNAP) transcribe all cellular genomes and interact with a plethora of accessory proteins that modulate every step of RNA synthesis. Among them, NusG/Spt5 is the only regulator that is conserved across all domains of life1. NusG homologs control gene expression by reducing RNAP pausing and arrest to enhance its processivity2–4 and by enabling crosstalk between transcription and concomitant cellular processes. These proteins physically link elongating RNAP to a ribosome5 or transcription termination factor Rho6,7 in bacteria and to factors mediating mRNA capping8, histone modification9, and somatic hypermutation10 in eukaryotes.
The modular structure of NusG proteins underpins this bridging activity (Fig. 1a). The N-terminal domains (NTDs) exhibit mixed α/β topology and establish similar contacts to the two largest subunits of bacterial, archaeal, and eukaryotic RNAPs11–15. In bacteria, these contacts are mediated by the β′ clamp and the β lobe and protrusion domains13. The C-terminal domains (CTDs; one in bacteria and archaea, multiple in eukaryotes) contain a Kyrpides, Ouzounis, Woese motif16, fold into a five-stranded β-barrel that is flexibly connected to the NTD, and serve as interaction platform for various binding partners, making co-transcriptional contacts to cellular proteins that ultimately determine their effects on gene expression. In Escherichia coli, NusG-CTD interacts with Rho to inhibit synthesis of foreign and aberrant RNAs6,17 or with ribosome to couple transcription to translation5,18.
Fig. 1.
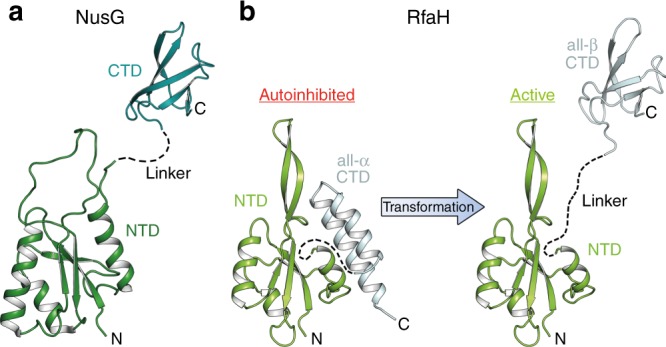
Structures of NusG and RfaH from E. coli. Structures of a NusG and b RfaH are in ribbon representation, the linker connecting the domains is indicated by a dashed line. PDB IDs: NusG-NTD, ‘2K06’; NusG-CTD, ‘2JVV’; RfaH, ‘5OND’; RfaH-CTD, ‘2LCL’
In addition to housekeeping factors that co-localize with elongating RNAP across most genes19–21, highly specialized NusG paralogs are present in ciliates22, plants23, and bacteria24. In order to avoid off-target recruitment, these paralogs must be specifically recruited to their target genes. This is particularly critical when their function is opposite to that of housekeeping NusG, as is the case of bacterial paralogs which silence Rho-dependent termination25.
RfaH, the best studied NusG paralog, activates expression of cell wall biosynthesis, conjugation, and virulence genes by inhibiting Rho26; mutations in rho and nusG suppress the loss of E. coli rfaH27. RfaH activates fewer than ten operons in E. coli, each containing an operon polarity suppressor (ops) element in their 5′ UTRs28. When RNAP pauses at the ops site, the non-template (NT) DNA strand in the transcription bubble forms a hairpin structure13,29. During recruitment, RfaH makes base-specific contacts with two flipped-out ops bases via its NTD. While these contacts explain sequence specificity of RfaH, off-target recruitment and competition with housekeeing NusG is additionally controlled by autoinhibition.
RfaH-NTD exhibits the mixed α/β topology typical for NusG proteins but, in contrast to all other known NusGs, the RfaH-CTD folds as an α-helical hairpin in free RfaH (all-α state; Fig. 1b). The CTD hairpin tightly interacts with the NTD, masking the RNAP-binding site and autoinhibiting RfaH30. The relief of autoinhibition requires domain dissociation, thought to be triggered by transient contacts to ops. Thereafter, the released NTD binds to the clamp helices of the β′ subunit (β′CH) and the gate loop of the β subunit (βGL) of the RNAP26,28, while the CTD spontaneously and completely refolds into a NusG-like β-barrel (all-β state; Fig. 1b) and recruits the ribosome via interactions with ribosomal protein S10, substituting for a missing Shine-Dalgarno sequence31. As striking as this transformation is, the lack of spurious RfaH recruitment at non-ops sites28 suggests that refolding may be reversible: following dissociation from RNAP at a terminator, RfaH must either perish or transform back into the autoinhibited state32 because activated RfaH does not require ops for recruitment30,33.
Here, we used NMR spectroscopy adapted to supramolecular, multicomponent systems in combination with functional studies to explore the conformational transitions that accompany RfaH binding to and dissociation from RNAP. Our results indicate that RfaH functions in a true cycle. We identify the ops-paused transcription elongation complex (EC) as a minimal activation signal for RfaH and demonstrate that, upon recruitment, RfaH-CTD refolds into the β-barrel that subsequently interacts with ribosomal protein S10. We further show that RfaH-CTD refolds into the α-helical state after RfaH release from the EC, thereby re-establishing the autoinhibited state. Our results demonstrate unmatched conformational and functional plasticity of RfaH, which refolds not once but twice during its functional cycle, as befits a transformer protein34.
Results
The ops-paused EC is necessary for RfaH activation
To elucidate the mechanism of RfaH recruitment to the EC and identify a signal that induces domain opening, we used a combination of solution-state NMR spectroscopy approaches which allow the characterization of protein:ligand interactions and structural transitions as well as the analysis of excited, low-populated states. In addition to uniformly 15N-labeled proteins, samples were employed where [1H,13C]-labeled methyl groups of Ile, Leu, and Val residues in perdeuterated proteins served as NMR-active probes ([I,L,V]-labeling); the latter method increases the sensitivity to enable studies of large complexes35.
In the methyl-transverse relaxation optimized spectroscopy (methyl-TROSY) spectrum of free [I,L,V]-RfaH we observed only signals of the autoinhibited form (Fig. 2a). To test if, nevertheless, free RfaH exists in equilibrium of the closed and open conformations, with the open state being only low-populated, we first carried out 15N-based chemical exchange saturation transfer (CEST) experiments (Supplementary Figure 1a-d; ref. 36). This method allows for the detection and characterization of ‘invisible’, i.e., sparsely populated, excited conformational states that are in slow chemical exchange with a visible ground-state conformation. In CEST experiments the saturation of 15N spins by a weak radio frequency field can be transferred between different conformational states if these states exchange on a timescale of 5–50 ms. If the transmitter frequency, which is stepped through the spectral region of 15N spins, coincides with the resonance frequency of a spin, the signal intensity is significantly decreased, causing a dip in the CEST profile (normalized intensity (I/I0) of a signal as function of the transmitter frequency of the saturation field). If the major state is in equilibrium with another (minor) state, the exchange between these states will be evidenced by a second dip in the CEST profile occurring at the resonance frequency of the minor state. In the CEST profiles of the RfaH-CTD residues that have well-separated signals in the all-α and the all-β state, no second dip at the expected chemical-shift position corresponding to the all-β state could be observed (Supplementary Figure 1a-d). Thus, within the detection limits of the CEST experiment (population > 0.2%, exchange rate 20–200 s−1), all free RfaH occupies the closed, autoinhibited state.
Fig. 2.

RfaH:ops interaction. a 2D [1H, 13C] methyl-TROSY spectra of 45 µM [I,L,V]-RfaH titrated with ops (concentration of stock solution: 1.3 mM). Inset: enlargement of boxed region. b Interaction of [I,L,V]-RfaH with ops. Methyl-TROSY-derived normalized chemical shift changes vs. sequence position of RfaH (corresponding spectra are depicted in a). Horizontal lines: significance levels of Δδnorm = 0.04 ppm, black; = 0.09 ppm, orange, = 0.14 ppm, red. Source data are provided as a Source Data file. c ops binding surface of RfaH as derived from the titration of [I,L,V]-RfaH with ops. Affected methyl groups are mapped onto the RfaH:ops9 structure (PDB ID: ’5OND’). RfaH is shown in ribbon (left) and surface (right) representation (RfaH-NTD, light gray; RfaH-CTD, dark gray), ops9 in ribbon representation (black) with nucleosides as sticks. The arrow indicates how the structures are rotated with respect to each other. Termini are labeled. For graphical representation of the interaction site the whole amino acid is colored. Ile, Leu, and Val residues are shown as sticks with the carbon atoms of the methyl groups as spheres. Slightly affected (0.04 ≤ Δδnorm < 0.09 ppm), yellow; moderately affected (0.09 ≤ Δδnorm < 0.14 ppm), orange; strongly affected (Δδnorm ≥ 0.14 ppm), red; unaffected, colored according to their domain; not assigned methyl groups, black. Two amino acids on either side of an affected Ile/Leu/Val residue are highlighted in beige unless they were unaffected Ile/Leu/Val residues
To test whether RfaH opens and closes on a timescale faster than accessible by CEST experiments, we performed Carr-Purcell-Meiboom-Gill sequence (CPMG) experiments. This analysis enables the measurement of the contribution of chemical exchange to the transverse relaxation rate (R2) of any nucleus for exchange processes in the range of ~200–2000 s−1 37. In brief, a series of refocusing 180° pulses is applied with different time intervals (τ180) between the pulses. During long time intervals, i.e., at low CPMG frequencies (1/2τ180), the chemical exchange can contribute to R2, resulting in an increase of R2. In the CMPG experiments of 15N-RfaH, we observed slightly enhanced R2 rates at lower CPMG frequencies for residues located in a loop in the DNA-binding region29 (T72, V75) as well as in the β-hairpin (S47), suggesting that these RfaH-NTD regions exhibit flexibility (Supplementary Figure 1e). In contrast, R2,eff did not change for RfaH-NTD residues in the domain interface (F51) or RfaH-CTD residues (F123, G135, M140, L145; Supplementary Figure 1e), indicating that the autoinhibited state is stable. Together, these findings argue against an equilibrium of the closed and open conformations of RfaH on a timescale faster than 0.5 ms–50 ms.
The DNA-binding site of RfaH is located on the RfaH-NTD, opposite the RfaH-CTD interaction surface13,29. A [1H,15N]-heteronuclear single quantum coherence (HSQC)-based NMR titration of 15N-RfaH with ops indicated that binding of RfaH to opsDNA does not induce domain separation29. Exploiting the high sensitivity of methyl groups, we next wanted to corroborate this result. Chemical-shift changes upon titration of [I,L,V]-RfaH with opsDNA were consistent with the DNA-binding site determined via the 15N-based titration and observed in the RfaH:ops9 crystal structure and in the cryo electron microscopy (EM) RfaH:ops-paused EC structure (Fig. 2; refs. 13,29). Signals corresponding to the all-β RfaH-CTD could not be observed during the titration, suggesting that binding to DNA alone cannot be a signal for domain opening.
RfaH weakly binds to free RNAP38. To test whether these contacts could activate RfaH, we measured one- and two-dimensional (1D, 2D) methyl-TROSY spectra of [I,L,V]-RfaH titrated with RNAP (Fig. 3a). The overall intensity of signals corresponding to autoinhibited RfaH decreased uniformly, but no changes in chemical shifts were observed and β-barrel CTD signals did not appear (Fig. 3a and Supplementary Figure 2), indicating that while RfaH can interact with RNAP, this binding does not induce domain dissociation/transformation. Adding an excess of NusG-NTD to the [I,L,V]-RfaH:RNAP complex recovered some of the intensity of [I,L,V]-RfaH signals (Fig. 3b), implying RfaH displacement by NusG-NTD. Since RfaH and NusG share binding sites13,28, this finding suggests that the closed RfaH binds near the final RfaH-NTD binding site on the EC.
Fig. 3.
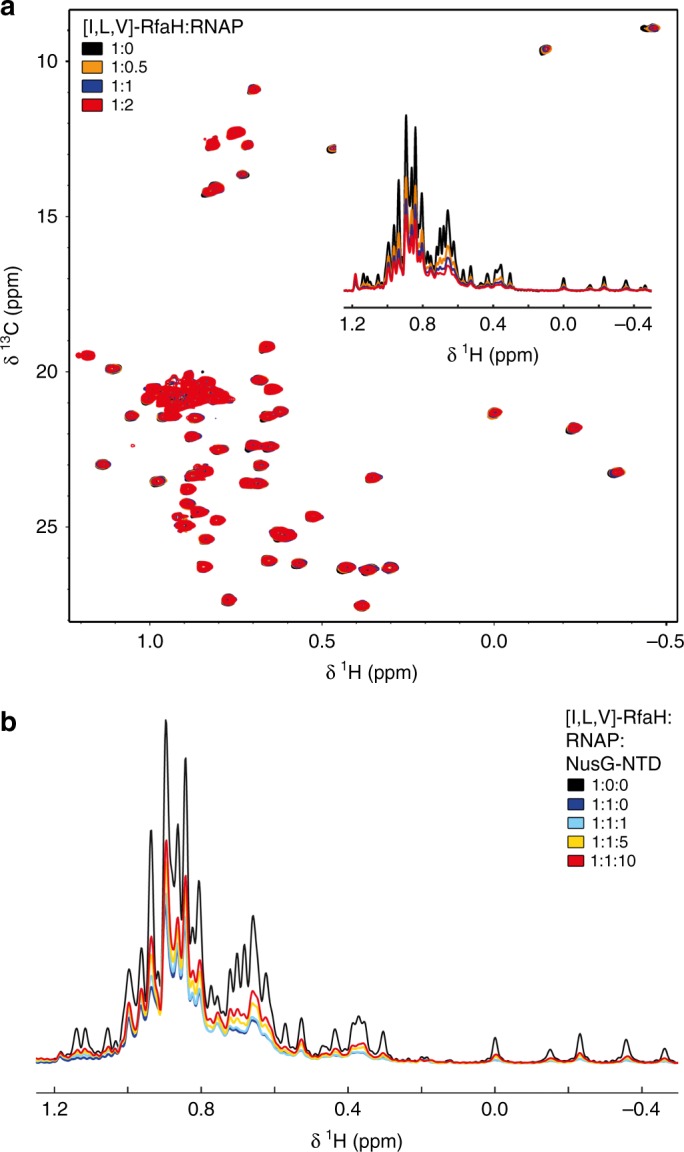
Binding of RfaH to RNAP. a 2D [1H, 13C] methyl-TROSY spectra of 15 µM [I,L,V]-RfaH titrated with RNAP (concentration of stock solution: 78 µM). Inset: normalized 1D [1H, 13C]-methyl-TROSY spectra, colored as 2D spectra. See also Supplementary Figure 2. b Displacement of RfaH from RNAP by NusG-NTD. Normalized 1D methyl-TROSY spectra of [I,L,V]-RfaH (30 μM, black), [I,L,V]-RfaH in the presence of equimolar RNAP (30 μM each, red), and [I,L,V]-RfaH upon titration of the [I,L,V]-RfaH:RNAP complex (30 μM each) with 712 µM NusG-NTD; molar ratio of [I,L,V]-RfaH:RNAP:NusG-NTD is indicated in color
These results show that neither DNA nor RNAP alone can relieve RfaH autoinhibition. To test if EC paused at the ops site (opsEC) is suffient to induce domain separation, we assembled opsEC with a nucleic-acid scaffold (Supplementary Figure 3) and perdeuterated RNAP. A methyl-TROSY-based titration of [I,L,V]-RfaH with the opsEC showed that signal intensity of [I,L,V]-RfaH methyl groups decreased non-uniformly, with only slight chemical shift changes (Fig. 4a). Signals of the α-helical RfaH-CTD disappeared while, concurrently, resonances corresponding to the β-barrel RfaH-CTD appeared and gradually intensified, indicating refolding of the RfaH-CTD (Fig. 4a and Supplementary Figure 4). Next we wanted to exclude the possibility that the RfaH-CTD refolding is due to cleavage of the RfaH linker caused by protease impurities or sample degradation during long-lasting NMR experiments. Both scenarios would lead to the release of the RfaH-CTD and its subsequent spontaneous transformation, as shown for an RfaH variant where a TEV protease cleavage site was introduced into the linker31. Thus, we performed translational diffusion experiments of [I,L,V]-RfaH and [I,L,V]-RfaH-CTD in the absence and presence of opsEC (Supplementary Figure 5a-e). The diffusion coefficient of [I,L,V]-RfaH:opsEC is significantly smaller than that of free [I,L,V]-RfaH or [I,L,V]-RfaH-CTD (Supplementary Table 1), confirming that the all-β signals in the [I,L,V]-RfaH:opsEC sample arise from RfaH bound to opsEC and that signals of the freed RfaH-CTD are visible even when RfaH is bound to the opsEC.
Fig. 4.

RfaH recruitment to the opsEC. a 2D [1H, 13C] methyl-TROSY spectra of [I,L,V]-RfaH in the absence or presence of opsEC assembled with 2H-RNAP (concentration of [I,L,V]-RfaH: 233 µM (1:0), 54 µM (1:0.5), 27 µM (1:1.25), 15 µM (1:2.4), 8 µM (1:5)). α and β indicate the all-α or all-β state of the RfaH-CTD. b Relative signal intensity of [I,L,V]-RfaH-NTD methyl groups with 0.5 equivalents of opsEC. Orange and red lines indicate thresholds for moderately (60% of average relative intensity) and strongly (30% of average relative intensity) affected methyl groups, respectively. Source data are provided as a Source Data file. c Mapping of affected methyl groups onto RfaH-NTD structure (ribbon representation; light gray; PDB ID: ‘5OND’). Ile, Leu, and Val residues are in stick representation with the carbon atom of the methyl groups as sphere. Termini and secondary structure elements are labeled. The representation was graphically extended by including the two flanking residues on each side of an affected residue (beige) as established39. d RfaH-NTD bound to the opsEC (PDB ID: ‘6C6S’). RfaH-NTD is in surface representation, color code as in c, DNA and selected elements of the RNAP are in ribbon representation and labeled. The arrow indicates how the structures are rotated with respect to each other
The decrease of RfaH-NTD methyl group signal intensity likely is a combination of two effects: a general decrease resulting from the increased molecular mass of [I,L,V]-RfaH upon complex formation and a non-uniform decrease due to slow or intermediate exchange on the chemical shift timescale. Thus we analyzed the signal intensity in certain titration steps quantitatively to identify affected residues as established39 (Fig. 4b). In brief, in each titration step, relative intensity, i.e., the ratio of remaining signal intensity to that in the spectrum of free [I,L,V]-RfaH, was determined. Residues with relative signal intensities below certain thresholds were classified as either strongly or moderately affected (for details see Methods section). Mapping of the relative signal intensity of the 1:0.5 complex on the three-dimensional structure of RfaH-NTD revealed a patch where signal intensity changed significantly (Fig. 4c). To aid visualization limited by a small number of NMR-active probes, we graphically extended the representation of affected residues by including the two flanking residues on each side, unless they were an unaffected Ile, Leu, or Val residue (beige in Fig. 4c). Comparing the affected regions with the cryo EM structure of the RfaH:opsEC13 shows that the main, high-affinity contacts with the β′CH are in good agreement (Fig. 4d). The HTTT motif in helix α2 of RfaH interacts with the βGL13,26, but since this motif lacks NMR-active probes and the closest labeled residues point to the interior of RfaH, no information on these contacts could be obtained (Fig. 4c). These results demonstrate that in the presence of opsEC RfaH domains dissociate, RfaH-NTD binds to the EC, and RfaH-CTD refolds into the β-state, confirming that the ops-paused EC is the relevant signal for RfaH recruitment.
EC-bound RfaH interacts with S10
In a subpopulation of the RfaH:opsEC complexes observed by cryo EM, the RfaH-CTD binds to the β-flap tip helix at the RNA exit channel13. To test if this interaction also occurs in solution, we performed a titration of [I,L,V]-RfaH-CTD with opsEC (Supplementary Figure 5f). In the 1D methyl-TROSY spectra signal intensity of [I,L,V]-RfaH-CTD decreases by ~25% upon addition of opsEC. This loss of intensity indicates complex formation as the molecular mass of [I,L,V]-RfaH-CTD increases upon opsEC binding, although this interaction might be weak. These observations are in agreement with the finding that the diffusion coefficient of [I,L,V]-RfaH-CTD is slightly decreased in the presence of opsEC (Supplementary Table 1). The finding that the signals of the all-β RfaH-CTD, i.e., the freed CTD, are visible when RfaH is bound to the opsEC strengthens the hypothesis that RfaH-CTD is only transiently bound to RNAP in the EC.
We argued that RfaH recruits a ribosome via interactions observed in a binary complex of isolated RfaH-CTD and S1031. To test if this contact is preserved when RfaH is bound to the opsEC, we performed an NMR-based titration of [I,L,V]-RfaH with S10 in the presence of the opsEC using S10 lacking the ribosome-binding loop (S10∆) in complex with NusB to increase stability40. Upon addition of protonated opsEC to [I,L,V]-RfaH in equimolar concentration, mainly signals of the β-barrel CTD were observable, showing that RfaH is bound to the opsEC and that the CTD is in the all-β state (Fig. 5a). Subsequent titration with S10∆:NusB decreased intensity of some of these signals significantly (Fig. 5a, b). Affected residues are located in β-strands 3 and 4 as well as in the connecting loop (Fig. 5c), in agreement with the binding site observed in the binary RfaH-CTD:S10∆ complex (Fig. 5d and Supplementary Figure 6). Thus, the S10 interaction site of RfaH-CTD is accessible in the opsEC:RfaH complex, consistent with the cryo EM structure of the RfaH:opsEC complex13 and our data that RfaH-CTD:S10 interaction is required for translation activation31.
Fig. 5.

Structural basis of translation activation by RfaH. a 2D [1H, 13C] methyl-TROSY spectra of [I,L,V]-RfaH alone (200 µM), in the presence of equimolar concentration of opsEC (23 µM), and upon titration of RfaH:opsEC with 234 µM S10Δ:NusB; molar ratio [I,L,V]-RfaH:opsEC:S10Δ:NusB is indicated in color. Resonances with significant intensity changes are labeled. b Methyl-TROSY-derived relative intensity of [I,L,V]-RfaH methyl groups after addition of one equivalent of opsEC and two equivalents of S10Δ:NusB vs. sequence position in RfaH. Orange and red lines indicate thresholds for moderately affected (80% of the average relative intensity) and strongly affected (65% of the average relative intensity) methyl groups, respectively. Source data are provided as a Source Data file. c Mapping of affected methyl groups onto RfaH-CTD structure (PDB ID: ‘2LCL’). RfaH (gray) is shown in ribbon (left) and surface (right) representation, methyl groups are shown as spheres and are color-coded. d Model of the RfaH-CTD:S10Δ complex based on the NusG-CTD:S10Δ complex (PDB ID: ‘3D3B’). S10Δ in ribbon and surface representation (blue), representation of RfaH-CTD as in c. The orientation of RfaH-CTD relative to c is indicated
RfaH is recycled upon release from the EC
The presence of RfaH-NTD is sufficient to induce the RfaH-CTD folding into an α-state that is energetically unfavorable in the isolated domain31,32,41, leading us to propose that RfaH transforms back into the autoinhibited state after the EC dissociates at a terminator32. Testing this hypothesis at a canonical terminator by NMR spectroscopy would be challenging because such a complex is unstable. Instead, we induced [I,L,V]-RfaH release from the opsEC by addition of a 10-fold molar excess of NusG-NTD and monitored RfaH displacement by recording methyl-TROSY spectra (Fig. 6a). The addition of protonated opsEC to [I,L,V]-RfaH in a 1:1 molar ratio led to the disappearance of signals corresponding to autoinhibited RfaH and mainly β-barrel CTD signals were observable, confirming RfaH recruitment and transformation. Upon titration of [I,L,V]-RfaH:opsEC with protonated NusG-NTD, all-β CTD signals were partially replaced by signals of autoinhibited RfaH (Fig. 6a), consistent with RfaH displacement from the opsEC followed by recycling into its autoinhibited state.
Fig. 6.

Recycling of RfaH. a 2D [1H, 13C] methyl-TROSY spectra of [I,L,V]-RfaH alone (200 µM), in the presence of equimolar concentration of opsEC (23 µM), and upon titration of RfaH:opsEC with NusG-NTD (concentration of stock solutions 240 µM and 486 µM); molar ratio [I,L,V]-RfaH:opsEC:NusG-NTD is indicated in color. α and β indicate the all-α or all-β state of the RfaH-CTD. b Experimental set-up to follow RfaH state using in vitro transcription assay. c Determination of RfaH effect on single-round RNA synthesis. The relevant RNA region is shown on the left, with the ops element highlighted in green. Prominent pause sites (U38, G39, and C40) are indicated. Halted α32P-labeled A24 ECs were chased in the presence of RfaH-NTD, RfaHFL, or supernatants from roadblocked (RB) or free (SN) first-round reactions on the WT or G35C (corresponds to G8C in the ops element) template. Reactions were quenched at the indicated times (in seconds) and analyzed on 10% denaturing acrylamide gels; a representative gel is shown. d The fractions of RNA species indicated were determined from 360-s time points. The ratios of RNA in the presence and in the absence of the RfaH variant indicated were determined from three independent biological replicates and are shown as mean ± standard deviation. Source data are provided as a Source Data file
We next wanted to probe the fate of RfaH released from RNAP in a more natural pathway, upon completion of RNA synthesis. The autoinhibited RfaH depends on wild-type (WT) ops site for recruitment and cannot act on a G8C ops template where the NT-DNA hairpin is disrupted29. By contrast, the isolated RfaH-NTD can bind to the EC at any site30 and we showed that the RfaH-NTD as well as RfaH variants locked in the open state due to substitutions at the NTD-CTD interface are recruited to RNAP transcribing the G8C template33. Here we used a two-step in vitro assay (Fig. 6b) to test if released RfaH regains its autoinhibited state, and thus dependence on ops for recruitment. In the first step, a linear DNA template containing T7A1 promoter and the ops element was immobilized on streptavidin beads via a biotin moiety. Transcription was carried out by E. coli RNAP in the presence of full-length RfaH (RfaHFL) and the supernatant containing released RfaH (RfaHSN) was collected. In the second step, RfaHSN was added to halted radiolabeled ECs formed on templates with either WT or G8C ops. Following the addition of NTP substrates, the RNA products collected at different times were analyzed by gel electrophoresis (Fig. 6c) and quantified.
On the WT ops template, RfaHFL reduced RNAP pausing at U38 ~4-fold and delayed RNAP escape from the ops site (G39 + C40 positions) ~4-fold (Fig. 6d). RfaH-NTD and RfaHSN had very similar effects. A control in which RNAP release was prevented by a protein roadblock (RB; see Methods section) demonstrated that under these conditions all RfaH was bound to RNAP, as no activity was present in the supernatant. Notably, at low GTP (5 µM) used in these experiments to enable manual sampling, RfaH-induced pause at G39 + C40 masks its antipausing effects downstream, and the run-off transcript yields do not increase in the presence of RfaH. As expected, RfaH-NTD stimulated productive RNA synthesis on the G8C ops template ~2.5-fold, whereas neither RfaHFL nor RfaHSN had any effect. These results suggest that RfaH regains the autoinhibited, ops-dependent state after the EC dissociates at the end of the linear DNA template, in support of the direct observation of reverse transformation by NMR spectroscopy (Fig. 6a).
Discussion
The results presented here support our earlier hypothesis that RfaH operates in a true cycle, which begins and ends with the inactive, autoinhibited state (Fig. 7). RfaH recruitment is unusually complex, with the opsEC serving as a minimal signal (Fig. 4); while RfaH can weakly interact with the opsDNA29 or core RNAP (Fig. 2), it binds to the opsEC with ~1000-fold higher affinity that matches its cellular concentrations38. When RNAP pauses at the ops site, the NT strand forms a hairpin, exposing the nucleotides in the loop region at the enzyme’s surface, allowing sequence-specific recognition by RfaH13,29. The delay of RNAP at the ops site is thought to provide a crucial time window during which autoinhibited RfaH locates its few genomic targets and establishes interactions with certain RNAP elements (likely the βGL) and the accessible ops nucleotides, stabilizing the NT-DNA hairpin and forming a transient encounter complex. In this complex, RfaH is positioned near its final binding site on the β′CH, but the high-affinity NTD:β′CH contacts are precluded by the α-helical CTD. As the autoinhibited state of RfaH does not exchange with an open conformation on the NMR timescale, the functional role of the encounter complex remains to be determined, although several possibilities are conceivable. (i) Contacts in the encounter complex could pre-orient RfaH and increase its local concentration near the β′CH, facilitating RfaH-NTD binding to the tip of the β′CH. (ii) The encounter complex could induce conformational changes that destabilize the interdomain interface and ultimately lead to transient domain opening. Although not being observable in our experiments, we cannot rule out that the binding of RfaH to opsDNA or RNAP alone may cause such changes in the RfaH-NTD:RfaH-CTD interface. However, as only the central ops bases are exposed on the surface of the RNAP13, RfaH will inevitably interact with certain RNAP elements as soon as it establishes contacts with the ops hairpin. Thus, we hypothesize that a combination of interactions with opsDNA and RNAP provokes the weakening of the RfaH domain interface. Although we do not observe either scenario, we favor a model where elements of both mechanisms underlie RfaH activation. Upon dissociation of the RfaH domains the encounter complex is converted into a stable RfaH:EC complex. The RfaH-NTD maintains interactions with the EC throughout elongation, increasing transcription processivity through stabilizing contacts with the NT-DNA and the upstream duplex DNA, as well as by blocking RNAP swiveling that occurs during pausing13,42. The released RfaH-CTD transforms and binds S10 (Fig. 5), converting RfaH into a potent activator of translation initiation31 and possibly coupling transcription to translation elongation, as proposed for NusG18. Finally, RfaH completes the cycle by transforming back into the autoinhibited state upon release from RNAP (Fig. 6).
Fig. 7.

Functional cycle of RfaH. Structural transformations of the interdomain interface and the RfaH-CTD underlie reversible switching between the autoinhibited and active states of RfaH
Observations that all NusG homologs promote productive transcription, with the NTD being sufficient for this activity30,43, led us and others to focus on the NTD-dependent modification of RNAP. Yet the regulatory diversity of NusG homologs is conferred by their CTDs, which interact with different partners to ensure coordination between RNA synthesis and posttranscriptional events. Comparison of RfaH- and NusG-CTDs reveals a combination of similar and distinct activities. Both CTDs interact similarly with S105,31, suggesting that they may bridge RNAP and the lead ribosome; the available evidence is consistent with RfaH recruitment of ribosome31 and with coupling by NusG18 but a systematic analysis remains to be done. In contrast, other interactions/roles are different. First, NusG-CTD binds to Rho, promoting termination at suboptimal sequences through favoring the closed, translocation-competent ring state6,7, whereas RfaH does not bind to Rho and inhibits Rho-dependent termination26. Second, RfaH-CTD could affect folding of the nascent RNA through transient contacts to the RNAP exit channel13, although the significance of this interaction remains to be determined. Finally, the RfaH-CTD prevents off-target recruitment and thus competition with NusG through autoinhibition mediated by transformation, maintaining the separation of the RfaH and NusG regulons.
Even though RfaH and NusG primary sequences are quite divergent33, only a few residues determine their key regulatory differences. Activation of Rho-dependent termination is determined by a 5-residue surface loop of NusG that, when grafted onto the RfaH-CTD, is sufficient for stimulation of Rho transition into the active conformation6. Similarly, the existence of the autoinhibited state is controlled by a few residues at the interface between the RfaH-NTD and RfaH-CTD; a single substitution of each of two key RfaH residues for their NusG counterparts disrupts the interface and alleviates the requirement for ops during recruitment33. In contrast, RfaH binding to the NT-DNA relies on readout of the primary sequence and the secondary structure of the ops hairpin by many RfaH residues, which are not conserved in NusG13,29.
Autoinhibition is a widespread regulatory mechanism in which intramolecular interactions between separate regions of a polypeptide negatively regulate its function, allowing temporal and spatial regulation of cellular processes by limiting activation to certain physiological conditions44. Autoinhibition modulates function of diverse proteins, from transcription factors45 to protein kinases46, and is implicated in virulence47 and disease48. Autoinhibition enables tight regulation, particularly when thousands of potential targets need to be distinguished, as is the case with E3 ubiquitin ligases49.
NusG cooperates with Rho to promote termination at suboptimal sites17, an essential function of NusG to silence foreign DNA50. Although being outnumbered by NusG 100:151, RfaH efficiently outcompetes NusG for binding to the EC32 and abolishes Rho-mediated termination26. To prevent interference with essential NusG, RfaH recruitment must be strictly limited to ops operons, making attainment of autoinhibition a key step in the specialization of RfaH. Contrasting other cases of autoinhibition, autoinhibition in RfaH is achieved not only by the interaction of two domains, but is coupled to the transformation of a whole domain into a conformation, the all-α form, that does not correspond to the structure of the isolated domain31,32.
It is not known whether other specialized NusG paralogs are autoinhibited and, if so, how they are activated and whether the CTD refolds similarly. In RfaH, transient contacts to the opsDNA hairpin and flanking RNAP regions are required to trigger domain dissociation, maybe via an encounter complex. While autoinhibited RfaH-like proteins could use analogous contacts to the NT-DNA strand and the paused EC for activation, other mechanisms could be envisioned, such as allosteric activation; e.g., small ligands could bind to either domain to weaken their interactions.
While in E. coli NusG, the domains move independently, and no intramolecular domain interactions can be detected52, autoinhibition has been observed for Thermotoga maritima NusG53. Here, in contrast to RfaH, autoinhibition is accomplished by the β-barrel CTD, which shields the RNAP-binding site on the NTD and most probably provides thermal stabilization54. Why does RfaH use an α-helical hairpin?
To answer this question, we generated a model of RfaH where the all-β CTD interacts with the NTD as in TmNusG (RfaHβCTD; Supplementary Figure 7). The linker is sufficiently long and RfaHβCTD can be easily integrated into a closed conformation without steric problems. Analysis of the domain interactions reveals that in RfaH the binding surface is larger than in RfaHβCTD (~900 Å2 vs. ~700 Å2), resulting in a significantly more negative solvation energy (~−14 kcal/mol vs. −8 kcal/mol). The stronger domain interaction in RfaH may be required to prevent an equilibrium between open and closed state, consistent with our present results, and thus uncontrolled activation.
We speculate that a need to tightly control the off-target recruitment necessitates the transformation-coupled autoinhibition in RfaH, especially as the use of the all-α CTD state imparts dual autoinhibition—the closed state lacks the binding sites for both RNAP and the ribosome, potentially further minimizing deleterious effects of spurious RfaH activation. Studies of structures and recruitment of NusG paralogs from other species will reveal their underlying specificity mechanisms.
The thermodynamic hypothesis states that, under physiological conditions, a protein adopts the three-dimensional structure which corresponds to the state of the lowest Gibbs free energy for the whole system. This so-called physiological state is solely determined by the totality of interactions and thus the amino acid sequence55. Most proteins exist as an ensemble of closely related equilibrium structures in their energetically stable state and thus follow this one-sequence, one-fold paradigm. However, more and more chameleonic/metamorphic proteins56 that defy this paradigm are found [see ref. 57 for a recent review]. In these metamorphic proteins, more than one distinct structural form is energetically favored.
In most cases, the metamorphic regions are small (5–14 residues) and metamorphosis involves either transitions between unstructured to structured states or conformational switching (α ↔ β). Transitions can be triggered by simple external cues (e.g., pH, temperature, salt concentration; refs. 58,59) may be driven by evolutionary pressure as observed in the Cro family of bacteriophage transcription factors60, or may underlie biological activity, such as regulation of chemotaxis by lymphotactin61, pore formation by lytic toxins62, regulation of the circadian clock by KaiB63, or photoreactivation in cytochromes64. As in classical metamorphoses, the observed changes are usually unidirectional, although reversible refolding has been reported61,63,65.
What sets RfaH apart from other metamorphic proteins are (i) the scale of the reversible transformation, in which the entire 50-residue domain refolds, (ii) distinct and essential biological functions of both alternative folds, and (iii) the fact that this dramatic behavior occurs in a member of the only universally conserved family of transcription factors. The fold of the CTD is solely determined by the presence or absence of the NTD32, i.e., the information which fold to adopt is encoded in RfaH itself. A major determinant of the NTD:CTD interaction is the salt bridge E48:R138 as its elimination leads to a coexistence of the autoinhibited state and the open form with the CTD in the β-barrel conformation31, turning RfaH into a NusG-like general transcription factor as the dependence on ops is abolished31. Arguments (i) and (ii) prompted us to name RfaH a transformer protein34. An α→β switch of a whole protein/domain is only known for amyloidogenic proteins, such as prions66, but the two states cannot coexist, the transition is irreversible, and the resulting β-aggregates are pathogenic.
In conclusion, metamorphosis is an increasingly recognized regulatory tool in nature, but the functional and conformational plasticity coupled with autoinhibition of RfaH sets new standards for regulation and suggests that similar principles are exploited by many transformer proteins awaiting discovery.
Methods
Cloning
The gene encoding RfaH was amplified from pIA238 using primers Fw_rfaH_pET19bmod and Rv_rfaH_pET19bmod (Supplementary Table 2) and cloned into pET19bmod, a variant of pET19b, via NdeI and BamHI restriction sites. The recombinant target protein carries a hexahistidine (His6) tag followed by a tobacco etch virus (TEV) protease cleavage site at its N-terminus.
Gene expression and protein purification
All E. coli strains used in gene expression were derivatives of E. coli B and grown at temperatures between 16 and 37 °C. Antibiotics were added to the medium as follows: ampicillin 100 µg/ml, carbenicillin 100 µg/ml, kanamycin 50 µg/ml, and chloramphenicol 34 µg/ml. The source organisms for all proteins used in this work are derivatives of E. coli K. All expression plasmids are listed in Supplementary Table 3.
RfaH for NMR studies was produced as described in ref. 31. In brief, E. coli BL21 (λ DE3) cells (Novagen, Madison, WI, USA) harboring plasmid pET19bmod_RfaH were grown in lysogeny broth (LB) medium supplemented with kanamycin to an optical density at 600 nm (OD600) of 0.6 at 37 °C. The temperature was lowered to 20 °C and gene expression was induced after 30 min by addition of 0.2 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG). Cells were harvested after overnight incubation by centrifugation (6000 × g), resuspended in buffer ARfaH (50 mM tris(hydroxymethyl)aminomethane (Tris)/HCl (pH 7.5), 300 mM NaCl, 5% (v/v) glycerol, 1 mM dithiothreitol (DTT)) supplemented with 10 mM imidazole, DNase I (AppliChem GmbH, Darmstadt, Germany), and 1/2 protease inhibitor tablet (cOmplete, EDTA-free, Roche Diagnostics, Mannheim, Germany) and lysed using a microfluidizer. The lysate was cleared by centrifugation and the soluble fraction was then applied to a HisTrap column (column volume 1–5 ml, GE Healthcare, Munich, Germany) that was subsequently washed with buffer ARfaH supplemented with 10 mM imidazole. A step gradient from 100 mM to 1 M imidazole in buffer ARfaH was used for elution. RfaH-containing fractions were combined and dialyzed against buffer BRfaH (50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 5% (v/v) glycerol, 1 mM DTT). After 2 h TEV protease was added and cleavage was carried out overnight at 4 °C. The solution was then again applied on a 5 ml HisTrap column (GE Healthcare, Munich, Germany). The target protein was collected in the flow-through, concentrated by ultrafiltration, flash-frozen in liquid nitrogen, and stored at −80 °C.
RfaH for transcription assays was produced similarly, except that plasmid pIA238 was used for the expression, resulting in E. coli RfaH with N-terminal His6-tag followed by a thrombin cleavage site. Thus, cleavage was carried out during overnight dialysis at room temperature in the presence of thrombin instead of TEV protease.
The production of RfaH-CTD was according to ref. 31 and the conditions were similar to the ones used for full-length RfaH. For expression E. coli BL21 (DE3) cells containing pETGB1a_EcrfaH-CTD(101-162) were used. The plasmid codes for E. coli RfaH-CTD with N-terminal His6-Gb1 tag followed by a TEV protease cleavage site. For purification a 5 ml Ni2+-HiTrap column (GE Healthcare, Munich, Germany) was used and buffer ARfaH-CTD consisted of 50 mM Tris/HCl (pH 7.5), 150 mM NaCl. The pure target protein was finally dialyzed against 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.5), 100 mM NaCl, concentrated by ultrafiltration, flash-frozen in liquid nitrogen, and stored at −80 °C.
For the production of NusG-NTD52 E. coli BL21 (λ DE3) cells harboring plasmid pET11a_EcNusG-NTD(1-124), which encodes E. coli NusG 1-124, were grown in ampicillin-containing LB medium to an OD600 of 0.8 at 37 °C. Overexpression was induced by addition of 1 mM IPTG. After 4 h cells were harvested by centrifugation (6000 × g), resuspended in buffer ANusG-NTD (50 mM Tris/HCl (pH 7.5), 150 mM NaCl) supplemented with DNase I (AppliChem GmbH, Darmstadt, Germany) and 1/4 protease inhibitor tablet (cOmplete, EDTA-free, Roche Diagnostics, Mannheim, Germany), and lysed with a microfluidizer. After centrifugation, nucleic acids were precipitated by addition of streptomycine sulfate (1% (w/v)). Upon centrifugation (NH4)2SO4 was added to the supernatant to a concentration of 50% (w/v), precipitating NusG-NTD. The precipitate was pelleted by centrifugation and dissolved in buffer BNusG-NTD (10 mM Tris/HCl (pH 7.5)). The solution was dialyzed against buffer BNusG-NTD before being applied to a 5 ml HeparinFF column (GE Healthcare, Munich, Germany). The column was washed with buffer BNusG-NTD and the target protein was eluted by a NaCl step gradient from 50 mM to 1 M in buffer BNusG-NTD. NusG-NTD containing fractions were combined, concentrated by ultrafiltration, and applied to a HiLoad S75 size exclusion column (GE Healthcare, Munich, Germany) equilibrated with buffer CNusG-NTD (25 mM HEPES (pH 7.5), 100 mM NaCl). NusG-NTD containing fractions were combined and the solution was concentrated by ultrafiltration and flash-frozen in liquid nitrogen before being stored at −80 °C.
The production of S10Δ:NusB was based on ref. 67. Briefly, E. coli BL21 (λ DE3) cells harboring the plasmids for either S10Δ (pGEX-6P_ecoNusEΔ; encodes E. coli S10Δ with N-terminal glutathione S-transferase (GST)-tag followed by PreScission protease cleavage site) or NusB (pET29b_ecoNusB; encodes E. coli NusB), were grown in LB medium containing ampicillin or kanamycin, respectively, at 37 °C to an OD600 of 0.5. The temperature was lowered to 20 °C and gene expression was induced after 30 min by addition of 0.5 mM IPTG. After overnight incubation cells were harvested by centrifugation (6000 × g). Cell pellets of S10Δ and NusB-containing cells, obtained from the same culture volume, were resuspended in buffer AS10Δ:NusB (50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1 mM DTT) and combined. Cells were subsequently lysed using a microfluidizer and the lysate was stirred for 30 min at 4 °C to ensure formation of the S10Δ:NusE complex. The extract was then cleared by centrifugation and applied to four coupled 5 ml GSTrap FF columns (GE Healthcare, Munich, Germany) equilibrated with buffer AS10Δ:NusB. After washing with buffer AS10Δ:NusB the complex was eluted with buffer AS10Δ:NusB containing 15 mM reduced glutathione. The S10Δ:NusB solution was supplemented with PreScission protease and dialyzed against buffer BS10Δ:NusB (50 mM Tris/HCl (pH 7.5), 1 mM DTT) overnight. The protein solution was applied to two 5 ml HiTrap Q XL columns (GE Healthcare, Munich, Germany) coupled to two HiTrap SP XL columns (GE Healthcare, Munich, Germany). Upon washing with buffer BS10Δ:NusB the HiTrap SP XL columns were disconnected and S10Δ:NusB were eluted with buffer BS10Δ:NusB containing 1 M NaCl. The solution was dialyzed against 25 mM HEPES (pH 7.5), 100 mM NaCl, concentrated by ultrafiltration, before being flash-frozen in liquid nitrogen, and stored at −80 °C.
RNAP for in vitro transcription assays was produced according to ref. 68. E. coli BL21 (λ DE3) cells harboring pVS10 (encoding E. coli RNAP subunits α, β, β′ with C-terminal His6 tag, and ω) were grown at 37 °C in carbenicillin-containing LB medium to an OD600 of 0.75 before overexpression was induced by 1 mM IPTG for 3 h. Cells were harvested by centrifugation (6000 × g) and resuspended in buffer ARNAP1 (50 mM Tris/HCl (pH 6.9), 500 mM NaCl, 5% (v/v) glycerol) supplemented with one protease inhibitors cocktail (Roche Applied Science) and 1 mg/ml lysozyme. Cell lysis was carried out by sonication and the cleared extract was supplemented with 20 mM imidazole before being loaded onto a His GraviTrap column (GE Healthcare Life Science). The column was washed with buffer ARNAP1 containing 20 mM imidazole and RNAP was eluted with buffer ARNAP1 containing 250 mM imidazole. The protein solution was dialyzed against buffer BRNAP1 (50 mM Tris/HCl (pH 6.9), 5% (v/v) glycerol, 0.5 mM EDTA, 1 mM DTT) supplemented with 75 mM NaCl and was then applied to a HiPrep Heparin FF column (GE Healthcare Life Science) to remove nucleic acids. The column was washed with buffer BRNAP1 containing 75 mM NaCl and RNAP was eluted with a constant NaCl gradient from 75 mM to 1.5 M in buffer BRNAP1. Target protein containing fractions were dialyzed against buffer BRNAP1 containing 75 mM NaCl and applied on a MonoQ column (GE Healthcare Life Science). Washing and elution were analogous to the Heparin affinity chromatography step. RNAP-containing fractions were combined, dialyzed against 10 mM Tris/HCl (pH 7.5), 100 mM NaCl, 50% (v/v) glycerol, 0.1 mM EDTA, 0.1 mM DTT, and stored at −20 °C.
The production of RNAP for NMR studies was based on ref. 69. Expression was carried out in E. coli BL21 (λ DE3) containing plasmid pVS10. Cells were grown in LB medium supplemented with ampicillin to an OD600 of 0.7. The temperature was lowered to 16 °C and overexpression was induced at OD600 = 0.8 with 0.5 mM IPTG. After overnight growth cells were harvested by centrifugation. The pellet was resuspended in buffer ARNAP2 (50 mM Tris/HCl (pH 6.9), 500 mM NaCl, 5% (v/v) glycerol, 1 mM β-mercaptoethanol (β-ME)) containing 10 mM imidazole, DNase I (AppliChem GmbH, Darmstadt, Germany), and 1/2 protease inhibitor tablet (cOmplete, EDTA-free, Roche Diagnostics, Mannheim, Germany). Cells were lysed using a microfluidizer and the lysate was cleared by centrifugation. The supernatant was applied to a 40 ml Ni2+-Chelating Sepharose column (GE Healthcare, Munich, Germany). After washing with buffer ARNAP2 containing 10 mM imidazole RNAP was eluted using an imidazole gradient from 90 mM to 1 M imidazole in buffer ARNAP2. RNAP-containing fractions were dialyzed against buffer BRNAP2 (50 mM Tris/HCl (pH 6.9), 5% (v/v) glycerol, 0.5 mM EDTA, 1 mM β-ME) containing 100 mM NaCl and then applied to two coupled 5 ml Heparin FF columns (GE Healthcare, Munich, Germany). The columns were washed with buffer BRNAP2 (containing 100 mM NaCl) and RNAP was eluted with a constant gradient from 100 mM to 1 M NaCl in buffer BRNAP2. The fractions containing core RNAP were dialyzed against buffer CRNAP2 (50 mM Tris/HCl (pH 6.9), 150 mM NaCl, 5% (v/v) glycerol, 0.5 mM EDTA, 1 mM β-ME) and subsequently concentrated by ultrafiltration. The concentrate was applied to a HiLoad S200 size exclusion column (GE Healthare, Munich, Germany) equilibrated with buffer CRNAP2 to remove inactive RNAP aggregates. Fractions containing pure, active enzyme were concentrated by ultrafiltration, glycerol was added to a final concentration of 50% (v/v) and the protein solution was stored at −20 °C.
Protein purity was checked by SDS-PAGE, the absence of nucleic acids was checked by recording UV/Vis spectra on a Nanodrop ND-1000 spectrometer (PEQLAB, Erlangen, Germany). Concentrations were determined by measuring the absorbance at 280 nm (A280) in a 10 mm quartz cuvette (Hellma, Müllheim, Germany) on a Biospectrometer basic (Eppendorf, Hamburg, Germany).
Isotopic labeling
15N- and 15N/13C-labeled proteins were produced by growing E. coli cells in M9 medium70,71 containing (15NH4)2SO4 and 13C-D-glucose. For the production of perdeuterated proteins, cells were grown in M9 medium70,71 prepared with increasing amounts of D2O (25% (v/v), 50% (v/v), 99.9% (v/v) D2O; Eurisotop, Saint-Aubin, France) with d7-glucose as carbon source. The site-specific [1H,13C]-labeling of Ile, Leu, and Val methyl groups in perdeuterated proteins was performed according to published protocols72, i.e., expression was carried out as described for the production of perdeuterated proteins, but the medium contained d7-glucose as carbon source and 60 mg/l 2-keto-3-d3-4-13C-butyrate and 100 mg/l 2-keto-3-methyl-d3-3-d1-4-13C-butyrate (both from Eurisotop, St. Aubin Cedex, France) were added 1 h prior to induction. Expression and purification were as described for the production of unlabeled proteins.
NMR spectroscopy
NMR experiments were performed on Bruker Avance 700 MHz, Bruker Ascend Aeon 900 MHz, and Bruker Ascend Aeon 1000 MHz spectrometers. All spectrometers were equipped with cryogenically cooled, inverse triple resonance probes. Processing of NMR data was carried out using in-house routines. 2D/3D spectra were visualized and analyzed by NMRViewJ (One Moon Scientific, Inc., Westfield, NJ, USA), 1D spectra by MatLab (The MathWorks, Inc., Version 7.1.0.183). Measurements were conducted at 15 °C. The initial sample volume was 500 µl, if not stated otherwise.
The resonance assignments for the backbone amide groups of RfaH and for the methyl groups of RfaH-CTD were taken from a previous study31. For resonance assignment of the RfaH methyl groups [13C, 15N]-RfaH in 25 mM HEPES (pH 7.5), 50 mM NaCl, 5% (v/v) glycerol, 1 mM DTT, 10% D2O and [I,L,V]-RfaH in 50 mM Na2HPO4/NaH2PO4 (pH 7.5), 50 mM KCl, 0.3 mM EDTA, 99.9% D2O were used. The assignment was based on standard double and triple resonance experiments on [13C,15N]-RfaH with (H)CCH-total correlation spectroscopy (TOCSY) and H(C)CH-TOCSY spectra allowing the non-sequence-specific identification of peaks belonging to the two methyl groups within individual Leu or Val side chains. Additionally, 3D CCH- and HCH-nuclear Overhauser effect spectroscopy (NOESY) spectra (mixing times: 250 and 200 ms, respectively) were obtained from [I,L,V]-RfaH. Combining the NOESY patterns with structural information from the crystal structure of the RfaH:ops9 complex (protein data bank (PDB) ID: ‘5OND’), and the identification of associated methyl groups finally allowed for the assignment of most non-overlapping resonances.
For interaction studies involving RNAP all components were in 50 mM Na2HPO4/NaH2PO4 (pH 7.5), 50 mM KCl, 0.3 mM EDTA, 99.9% (v/v) D2O.
Interaction studies with chemical shifts changes in the fast regime on the chemical-shift timescale were analyzed by calculating the normalized chemical-shift perturbation (Δδnorm) according to Eq. (1) for [1H,13C] correlation spectra.
| 1 |
where Δδ is the resonance frequency difference in ppm.
To analyze the signal intensity quantitatively in both 1D and 2D experiments, the intensity was normalized by the concentration of the labeled protein, the number of scans, the receiver gain, and the length of the 90° proton pulse. The ratio of remaining signal intensities and signal intensities in the spectrum of the free, labeled protein was calculated for each titration step, resulting in relative signal intensities. The determination of the interaction surface of opsEC and S10 on RfaH was carried out as described in ref. 39. In brief, the mean value of all relative signal intensities in each titration step was determined and experiment-specific thresholds of the mean value were defined. Residues with relative signal intensities below these thresholds were classified as either strongly or moderately affected and Leu and Val residues were considered as affected if at least one of the two signals showed a significant decrease in intensity. Only unambiguously assigned signals were used in the analysis. Affected residues were mapped on the three-dimensional structure of RfaH/RfaH-CTD and binding surfaces were graphically extended by (i) highlighting the complete amino acids instead of only the methyl group and (ii) highlighting the two amino acids on either side of an affected Ile, Leu, or Val residue unless they were unaffected/unassigned Ile, Leu, Val residues.
Translational diffusion coefficients (D) were determined using a stimulated echo (STE) experiment combined with a 1D [1H, 13C]-HMQC for selecting 13C-bound protons using an [I,L,V]-RfaH sample in D2O buffer73. Gradient pulses (δgrd/2) for de- and rephasing were 2.5 ms and the diffusion time (Δdiff) was set to 80 ms. Gradient strengths (g) were varied between 1 and 47 G cm−1. The decay of signal intensity (I) was fitted to Equation (2) using GraFit (Erithacus Software Ltd., Horley, UK, Version 6.0.12).
| 2 |
with I0 being the initial signal intensity, γH the gyromagnetic ratio of protons, and τ the recovery delay after the gradient pulses (200 μs).
CEST experiments were carried out at 298 K and 900 MHz 1H-frequency according to ref. 36, using a [2H, 13C, 15N]-RfaH sample in 25 mM HEPES (pH 7.5), 50 mM NaCl, 5% (v/v) glycerol, 1 mM DTT, 10% D2O. Saturation was achieved by a 35 Hz B1-field applied during an exchange period of 500 ms.
CPMG relaxation dispersion experiments were conducted at 288 K and a 700 MHz 1H-frequency using a [2H, 15N]-RfaH sample in 10 mM K2HPO4/KH2PO4 (pH 7.5), 50 mM KCl, 10% D2O. The constant time approach74 was applied with a total constant time period of 36 ms and νCPMG ranging from 30 to 2000 Hz.
Assembly of opsEC
Assembly of the ops-paused EC and design of the nucleic acids were based on published methods38. First a RNA:DNA-hybrid was formed from the ops-template (T) DNA (Supplementary Table 2) and the ops-RNA (Supplementary Table 2). Stock solutions of both oligos (1 mM in 99.9% D2O) were diluted with buffer (50 mM Na2HPO4/NaH2PO4 (pH 7.5), 50 mM KCl, 0.3 mM EDTA in 99.9% D2O) by 1:1 and mixed at an equimolar ratio. The mixture was incubated for 1 min at 95 °C, then for 10 min at 70 °C, and finally cooled to room temperature within 15 min. RNAP (typically at 50–100 μM) was added at 1.3 molar excess over the hybrid, followed by 10 min incubation at room temperature. Finally, the NT-opsDNA strand (Supplementary Table 2; 1 mM stock solution in D2O) was added at a molar ratio of 1:1.3:3 (T-ops-DNA/ops-RNA-hybrid:RNAP:NT-ops-DNA) and incubated for 10 min at 37 °C. To increase the long-term stability of the complex, 2 mM DTT, 5 mM MgCl2 and 5% (v/v) d8-glycerol were added to the sample.
In vitro transcription assay
Linear templates for in vitro transcription were made by PCR and purified via a QIAquick PCR purification kit (Qiagen, Valencia, CA). For the first-round reaction, a linear template was generated by PCR of pIA349 (Supplementary Table 3) using a top biotinylated primer and a bottom primer with an EcoRI recognition site, as described in ref. 13. When indicated, the template was pre-incubated with a cleavage-deficient EcoRI Q111 mutant (at 3 μM; to ensure complete occupancy of the roadblock) in TGA2 (20 mM Tris-acetate, 20 mM Na-acetate, 2 mM Mg-acetate, 5% glycerol, 1 mM DTT, 0.1 mM EDTA, pH 7.9) for 15 min at 37 °C. The biotinylated DNA template (200 nM), RNAP holoenzyme (350 nM), ApU (100 μM) and 5 μM each CTP, GTP, and ATP were incubated with prewashed Streptavidin coated magnetic beads (Dynabeads® MyOneTM Streptavidin C1) in 40 μl volume for 15 min at 37 °C to form halted G37 ECs. WT RfaH was added at 100 nM (to ensure that all RfaH was bound to the EC), followed by a 2-min incubation at 37 °C. Unlabeled NTPs (20 µM GTP, 200 µM ATP, CTP, and UTP) and rifapentin (25 µg/ml) were added for 10 min at 37 °C. The supernatant was collected using a Magnetic Separation Stand (Promega) and purified through AutoSeq G-50 spin columns (GE Healthcare) pre-equilibrated with TGA2; 25 μl/column.
For the second-round reaction, WT ops (pIA1087) or G8C ops (pZL23) templates were prepared as described in ref. 29. The resulting linear templates contained T7A1 promoter followed by an initial 24 nt T-less transcribed region; the run-off transcript generated on these templates is 79-nt long. Linear DNA template (30 nM), holo RNAP (50 nM), ApU (100 µM), and starting NTP subsets (1 µM CTP, 5 µM ATP and GTP, 10 µCi [α32P]-GTP, 3000 Ci/mmol) were mixed in 100 µl of TGA2. Reactions were incubated for 15 min at 37 °C; thus halted ECs were stored on ice.
An equal volume of RfaH in TGA2 or supernatant from the first round (to yield 50 nM final concentrations) was added to the EC, followed by a 2-min incubation at 37 °C. Transcription was restarted by addition of nucleotides (5 µM GTP, 150 µM ATP, CTP, and UTP) and rifapentin to 25 µg/ml. Samples were removed at time points indicated in the figure and quenched by addition of an equal volume of STOP buffer (10 M urea, 60 mM EDTA, 45 mM Tris-borate; pH 8.3). Samples were heated for 2 min at 95 °C and separated by electrophoresis in denaturing 9% acrylamide (19:1) gels (7 M Urea, 0.5X TBE). The gels were dried and RNA products were visualized and quantified using FLA9000 Phosphorimaging System, ImageQuant Software, and Microsoft Excel. In vitro transcription assays were carried out in triplicates and averaged.
Model of RfaHβCTD
RfaH-NTD (PDB ID: ‘5OND’) and RfaH-CTD in the all-β state (PDB ID: ‘2LCL’) were superimposed on the structure of T. maritima NusG (PDB ID: ‘2LQ8’). No structural rearrangements were applied.
Programs
All molecular structures were visualized using The PyMOL Molecular Graphics System (Version 1.7, Schrödinger, LLC). Superpositions of protein and nucleic-acid structures were prepared with COOT75. Interaction surfaces were analyzed by the ‚protein interfaces, surfaces and assemblies’ service PISA at the European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html)76.
Supplementary Information
Acknowledgements
We thank Ramona Heißmann, and Andrea Hager for excellent technical assistance. P.K.Z. was supported by the The Elite Network Bavaria in the framework of the Elite Study Program “Macromolecular Science”. The work was supported by grant Ro 617/21-1 (P.R.) from the Deutsche Forschungsgemeinschaft, the Ludwig-Schaefer award of Columbia University (P.R.), and grant GM67153 (I.A.) from the National Institutes of Health.
Author contributions
P.K.Z., K.S., and S.H.K. carried out the NMR experiments. I.A. carried out the in vitro experiments. S.H.K., P.R., and I.A. designed and supervised research and prepared the manuscript with input from all authors.
Data availability
The source data underlying Fig. 2b, Fig. 4b, Fig. 5b, Fig. 6d, Supplementary Figure 2, and Supplementary Figure 5e are provided as a Source Data file. Other data are available form the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Journal peer review information: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Irina Artsimovitch, Email: artsimovitch.1@osu.edu.
Stefan H. Knauer, Email: stefan.knauer@uni-bayreuth.de
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-019-08567-6.
References
- 1.Werner F. A nexus for gene expression-molecular mechanisms of Spt5 and NusG in the three domains of life. J. Mol. Biol. 2012;417:13–27. doi: 10.1016/j.jmb.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo S, et al. A regulator of transcriptional elongation controls vertebrate neuronal development. Nature. 2000;408:366–369. doi: 10.1038/35042590. [DOI] [PubMed] [Google Scholar]
- 3.Hirtreiter A, et al. Spt4/5 stimulates transcription elongation through the RNA polymerase clamp coiled-coil motif. Nucleic Acids Res. 2010;38:4040–4051. doi: 10.1093/nar/gkq135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bar-Nahum G, et al. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 5.Burmann BM, et al. A NusE:NusG complex links transcription and translation. Science. 2010;328:501–504. doi: 10.1126/science.1184953. [DOI] [PubMed] [Google Scholar]
- 6.Lawson MR, et al. Mechanism for the regulated control of transcription by a universal adapter protein. Mol. Cell. 2018;71:1–12. doi: 10.1016/j.molcel.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valabhoju V, Agrawal S, Sen R. Molecular basis of NusG-mediated regulation of Rho-dependent transcription termination in bacteria. J. Biol. Chem. 2016;291:22386–22403. doi: 10.1074/jbc.M116.745364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandal SS, et al. Functional interactions of RNA-capping enzyme with factors that positively and negatively regulate promoter escape by RNA polymerase II. Proc. Natl Acad. Sci. USA. 2004;101:7572–7577. doi: 10.1073/pnas.0401493101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wier AD, Mayekar MK, Héroux A, Arndt KM, VanDemark AP. Structural basis for Spt5-mediated recruitment of the Paf1 complex to chromatin. Proc. Natl Acad. Sci. USA. 2013;110:17290–17295. doi: 10.1073/pnas.1314754110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pavri R, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernecky C, Plitzko JM, Cramer P. Structure of a transcribing RNA polymerase II-DSIF complex reveals a multidentate DNA-RNA clamp. Nat. Struct. Mol. Biol. 2017;24:809–815. doi: 10.1038/nsmb.3465. [DOI] [PubMed] [Google Scholar]
- 12.Ehara H, et al. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science. 2017;357:921–924. doi: 10.1126/science.aan8552. [DOI] [PubMed] [Google Scholar]
- 13.Kang JY, et al. Structural basis for transcript elongation control by NusG family universal regulators. Cell. 2018;173:1650–1662.e14. doi: 10.1016/j.cell.2018.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein BJ, et al. RNA polymerase and transcription elongation factor Spt4/5 complex structure. Proc. Natl Acad. Sci. USA. 2011;108:546–550. doi: 10.1073/pnas.1013828108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez-Rucobo FW, Sainsbury S, Cheung ACM, Cramer P. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J. 2011;30:1302–1310. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kyrpides NC, Woese CR, Ouzounis CA. KOW: a novel motif linking a bacterial transcription factor with ribosomal proteins. Trends Biochem. Sci. 1996;21:425–426. doi: 10.1016/S0968-0004(96)30036-4. [DOI] [PubMed] [Google Scholar]
- 17.Peters JM, et al. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 2012;26:2621–2633. doi: 10.1101/gad.196741.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saxena S, et al. Escherichia coli transcription factor NusG binds to 70S ribosomes. Mol. Microbiol. 2018;108:495–504. doi: 10.1111/mmi.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer A, et al. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 2010;17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- 20.Mooney RA, et al. Regulator trafficking on bacterial transcription units in vivo. Mol. Cell. 2009;33:97–108. doi: 10.1016/j.molcel.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gruchota J, Denby Wilkes C, Arnaiz O, Sperling L, Nowak JK. A meiosis-specific Spt5 homolog involved in non-coding transcription. Nucleic Acids Res. 2017;45:4722–4732. doi: 10.1093/nar/gkw1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bies-Etheve N, et al. RNA-directed DNA methylation requires an AGO4-interacting member of the SPT5 elongation factor family. EMBO Rep. 2009;10:649–654. doi: 10.1038/embor.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodson JR, Klupt S, Zhang C, Straight P, Winkler WC. LoaP is a broadly conserved antiterminator protein that regulates antibiotic gene clusters in Bacillus amyloliquefaciens. Nat. Microbiol. 2017;2:17003–17012. doi: 10.1038/nmicrobiol.2017.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.NandyMazumdar M, Artsimovitch I. Ubiquitous transcription factors display structural plasticity and diverse functions: NusG proteins—shifting shapes and paradigms. Bioessays. 2015;37:324–334. doi: 10.1002/bies.201400177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sevostyanova A, Belogurov GA, Mooney RA, Landick R, Artsimovitch I. The β subunit gate loop is required for RNA polymerase modification by RfaH and NusG. Mol. Cell. 2011;43:253–262. doi: 10.1016/j.molcel.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu, K. & Artsimovitch, I. A Screen for rfaH suppressors reveals a key role for a connector region of termination factor Rho. MBio8, 10.1128/mBio.00753-17 (2017). [DOI] [PMC free article] [PubMed]
- 28.Belogurov GA, Mooney RA, Svetlov V, Landick R, Artsimovitch I. Functional specialization of transcription elongation factors. EMBO J. 2009;28:112–122. doi: 10.1038/emboj.2008.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuber, P. K. et al. The universally-conserved transcription factor RfaH is recruited to a hairpin structure of the non-template DNA strand. eLlife7, e36349 (2018). [DOI] [PMC free article] [PubMed]
- 30.Belogurov GA, et al. Structural basis for converting a general transcription factor into an operon-specific virulence regulator. Mol. Cell. 2007;26:117–129. doi: 10.1016/j.molcel.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burmann BM, et al. An α helix to β barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell. 2012;150:291–303. doi: 10.1016/j.cell.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomar SK, Knauer SH, Nandymazumdar M, Rösch P, Artsimovitch I. Interdomain contacts control folding of transcription factor RfaH. Nucleic Acids Res. 2013;41:10077–10085. doi: 10.1093/nar/gkt779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi D, Svetlov D, Abagyan R, Artsimovitch I. Flipping states: a few key residues decide the winning conformation of the only universally conserved transcription factor. Nucleic Acids Res. 2017;45:8835–8843. doi: 10.1093/nar/gkx523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knauer SH, Artsimovitch I, Rösch P. Transformer proteins. Cell Cycle. 2012;11:4289–4290. doi: 10.4161/cc.22468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sprangers R, Velyvis A, Kay LE. Solution NMR of supramolecular complexes: providing new insights into function. Nat. Methods. 2007;4:697–703. doi: 10.1038/nmeth1080. [DOI] [PubMed] [Google Scholar]
- 36.Vallurupalli P, Bouvignies G, Kay LE. Studying ‘invisible’ excited protein states in slow exchange with a major state conformation. J. Am. Chem. Soc. 2012;134:8148–8161. doi: 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- 37.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 38.Artsimovitch I, Landick R. The transcriptional regulator RfaH stimulates RNA chain synthesis after recruitment to elongation complexes by the exposed nontemplate DNA strand. Cell. 2002;109:193–203. doi: 10.1016/S0092-8674(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 39.Drögemüller J, et al. Determination of RNA polymerase binding surfaces of transcription factors by NMR spectroscopy. Sci. Rep. 2015;5:16428–16441. doi: 10.1038/srep16428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo X, et al. Structural and functional analysis of the E. coli NusB-S10 transcription antitermination complex. Mol. Cell. 2008;32:791–802. doi: 10.1016/j.molcel.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramírez-Sarmiento, C. A., Noel, J. K., Valenzuela, S. L. & Artsimovitch, I. Interdomain contacts control native state switching of RfaH on a dual-funneled landscape. PLoS Comput. Biol. 11, 10.1371/journal.pcbi.1004379 (2015). [DOI] [PMC free article] [PubMed]
- 42.Nedialkov Y, Svetlov D, Belogurov GA, Artsimovitch I. Locking the non-template DNA to control transcription. Mol. Microbiol. 2018;109:445–457. doi: 10.1111/mmi.13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mooney RA, Schweimer K, Rösch P, Gottesman M, Landick R. Two structurally independent domains of E. coli NusG create regulatory plasticity via distinct interactions with RNA polymerase and regulators. J. Mol. Biol. 2009;391:341–358. doi: 10.1016/j.jmb.2009.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pufall MA, Graves BJ. Autoinhibitory domains: modular effectors of cellular regulation. Annu. Rev. Cell. Dev. Biol. 2002;18:421–462. doi: 10.1146/annurev.cellbio.18.031502.133614. [DOI] [PubMed] [Google Scholar]
- 45.Currie SL, et al. Structured and disordered regions cooperatively mediate DNA-binding autoinhibition of ETS factors ETV1, ETV4 and ETV5. Nucleic Acids Res. 2017;45:2223–2241. doi: 10.1093/nar/gkx068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Au-Yeung BB, Shah NH, Shen L, Weiss A. ZAP-70 in signaling, biology, and disease. Annu. Rev. Immunol. 2018;36:127–156. doi: 10.1146/annurev-immunol-042617-053335. [DOI] [PubMed] [Google Scholar]
- 47.Cai, Z. et al. Fatty acid DSF binds and allosterically activates histidine kinase RpfC of phytopathogenic bacterium Xanthomonas campestris pv. campestris to regulate quorum-sensing and virulence. PLoS Pathog. 13, 10.1371/journal.ppat.1006304 (2017). [DOI] [PMC free article] [PubMed]
- 48.Takemoto-Kimura S, et al. Calmodulin kinases: essential regulators in health and disease. J. Neurochem. 2017;141:808–818. doi: 10.1111/jnc.14020. [DOI] [PubMed] [Google Scholar]
- 49.Buetow L, Huang DT. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016;17:626–642. doi: 10.1038/nrm.2016.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitra P, Ghosh G, Hafeezunnisa M, Sen R. Rho protein: roles and mechanisms. Annu. Rev. Microbiol. 2017;71:687–709. doi: 10.1146/annurev-micro-030117-020432. [DOI] [PubMed] [Google Scholar]
- 51.Schmidt A, et al. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016;34:104–110. doi: 10.1038/nbt.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burmann BM, Scheckenhofer U, Schweimer K, Rösch P. Domain interactions of the transcription-translation coupling factor Escherichia coli NusG are intermolecular and transient. Biochem. J. 2011;435:783–789. doi: 10.1042/BJ20101679. [DOI] [PubMed] [Google Scholar]
- 53.Drögemüller J, et al. An autoinhibited state in the structure of Thermotoga maritima NusG. Structure. 2013;21:365–375. doi: 10.1016/j.str.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drögemüller J, et al. Thermotoga maritima NusG: domain interaction mediates autoinhibition and thermostability. Nucleic Acids Res. 2017;45:446–460. doi: 10.1093/nar/gkw1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 56.Murzin AG. Biochemistry. Metamorphic proteins. Science. 2008;320:1725–1726. doi: 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- 57.Dishman AF, Volkman BF. Unfolding the mysteries of protein metamorphosis. Acs. Chem. Biol. 2018;13:1438–1446. doi: 10.1021/acschembio.8b00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Awasthi SK, Shankaramma SC, Raghothama S, Balaram P. Solvent-induced beta-hairpin to helix conformational transition in a designed peptide. Biopolymers. 2001;58:465–476. doi: 10.1002/1097-0282(20010415)58:5<465::AID-BIP1022>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 59.Cerpa R, Cohen FE, Kuntz ID. Conformational switching in designed peptides: the helix/sheet transition. Fold. Des. 1996;1:91–101. doi: 10.1016/S1359-0278(96)00018-1. [DOI] [PubMed] [Google Scholar]
- 60.Newlove T, Konieczka JH, Cordes MHJ. Secondary structure switching in Cro protein evolution. Structure. 2004;12:569–581. doi: 10.1016/j.str.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 61.Tuinstra RL, et al. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc. Natl Acad. Sci. USA. 2008;105:5057–5062. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. The structure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature. 2009;459:726–730. doi: 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- 63.Chang YG, et al. Circadian rhythms. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science. 2015;349:324–328. doi: 10.1126/science.1260031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burgie ES, Bussell AN, Walker JM, Dubiel K, Vierstra RD. Crystal structure of the photosensing module from a red/far-red light-absorbing plant phytochrome. Proc. Natl Acad. Sci. USA. 2014;111:10179–10184. doi: 10.1073/pnas.1403096111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zamora-Carreras H, et al. Micelle-triggered β-hairpin to α-helix transition in a 14-residue peptide from a choline-binding repeat of the pneumococcal autolysin LytA. Chemistry. 2015;21:8076–8089. doi: 10.1002/chem.201500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Surewicz WK, Apostol MI. Prion protein and its conformational conversion: a structural perspective. Top. Curr. Chem. 2011;305:135–167. doi: 10.1007/128_2011_165. [DOI] [PubMed] [Google Scholar]
- 67.Burmann BM, Luo X, Rösch P, Wahl MC, Gottesman ME. Fine tuning of the E. coli NusB:NusE complex affinity to BoxA RNA is required for processive antitermination. Nucleic Acids Res. 2010;38:314–326. doi: 10.1093/nar/gkp736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Svetlov V, Artsimovitch I. Purification of bacterial RNA polymerase: tools and protocols. Methods Mol. Biol. 2015;1276:13–29. doi: 10.1007/978-1-4939-2392-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Drögemüller J, et al. Exploring RNA polymerase regulation by NMR spectroscopy. Sci. Rep. 2015;5:10825–10835. doi: 10.1038/srep10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meyer O, Schlegel HG. Biology of aerobic carbon monoxide-oxidizing bacteria. Annu. Rev. Microbiol. 1983;37:277–310. doi: 10.1146/annurev.mi.37.100183.001425. [DOI] [PubMed] [Google Scholar]
- 71.Sambrook, J. & Russel, D. W. Molecular Cloning: A Laboratory Manual Vol. 3 (Cold Spring Harbor Press, 2001).
- 72.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 73.Didenko T, Boelens R, Rüdiger SGD. 3D DOSY-TROSY to determine the translational diffusion coefficient of large protein complexes. Protein Eng. Des. Sel. 2011;24:99–103. doi: 10.1093/protein/gzq091. [DOI] [PubMed] [Google Scholar]
- 74.Tollinger M, Skrynnikov NR, Mulder FA, Forman-Kay JD, Kay LE. Slow dynamics in folded and unfolded states of an SH3 domain. J. Am. Chem. Soc. 2001;123:11341–11352. doi: 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
- 75.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source data underlying Fig. 2b, Fig. 4b, Fig. 5b, Fig. 6d, Supplementary Figure 2, and Supplementary Figure 5e are provided as a Source Data file. Other data are available form the corresponding authors upon reasonable request.